")
Back to Journals » Journal of Experimental Pharmacology » Volume 13
The Role of BTK Inhibition in the Treatment of Chronic Lymphocytic Leukemia: A Clinical View
Authors Tambaro FP, De Novellis D, Wierda WG
Received 26 June 2021
Accepted for publication 8 October 2021
Published 29 October 2021 Volume 2021:13 Pages 923—935
DOI https://doi.org/10.2147/JEP.S265284
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Paola Rogliani
Francesco Paolo Tambaro,1 Danilo De Novellis,1,2 William G Wierda3
1Unità Operativa di Trapianto di Midollo Osseo e Servizio Trasfusionale, Azienda Ospedaliera di Rilievo Nazionale Santobono-Pausilipon, Napoli, Italy; 2Department of Precision Medicine, University of Campania “L. Vanvitelli”, Napoli, Italy; 3Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Correspondence: William G Wierda Email [email protected]
Abstract: The B cell receptor (BCR) signaling pathway is functional and has critical cell survival implications in B cell malignancies, such as chronic lymphocytic leukemia (CLL). Orally administered small molecule tyrosine kinase inhibitors of members of the BCR signaling pathway have proven to be transformational in treatment of CLL. The first-generation inhibitor, ibrutinib, covalently binds to the C481 amino acid of Bruton’s tyrosine kinase (BTK), thereby irreversibly inhibiting its kinase activity, and interferes with the biology of the cells, ultimately resulting in CLL cell death and therapeutic response. Remissions are not deep to the point of considering discontinuation for most patients, but BTK-inhibitor-based therapy provides exceptional long-term disease control with continuous treatment. There are in-class toxicities and more selective second- and subsequent-generation agents and reversible inhibitors have been developed with the intent of reducing toxicities. Also, strategies to subvert resistance have included tighter or alternative, non-covalent, inhibitor binding. Furthermore, other strategies to deplete BTK protein, such as degraders, are in development and being tested in the clinic. Ultimately, the development and approval of these agents targeting BTK have ushered in a new era of chemotherapy-free treatments with remarkably improved survival outcomes for patients with CLL.
Keywords: chronic lymphocytic leukemia, CLL, Bruton’s tyrosine kinase, BTK, targeted therapy, ibrutinib, acalabrutinib, pirtobrutinib
Background
Chronic Lymphocytic Leukemia (CLL)
Chronic lymphocytic leukemia (CLL) is the most common leukemia in adults in Western countries, affecting primarily the aging population.1 CLL is a lymphoproliferative B-cell disorder characterized by clonal proliferation and accumulation of monoclonal B-cells co-expressing CD5, CD19, CD20 and CD23 in blood, lymph nodes, spleen, liver and bone marrow. Small lymphocytic lymphoma has the same clinical, pathologic and management features as CLL, and is distinguished by lack of lymphocytosis. Innovative knowledge of how the B-cell receptor (BCR) signaling pathway affects the biological behavior and pathophysiology of the disease led to fundamental and transformational therapeutic advances for patients with CLL/SLL.
Historic Treatments
Glucocorticoids were the first systemic treatment for CLL/SLL,2 followed by alkylating agents (chlorambucil, cyclophosphamide and bendamustine), purine analogs (fludarabine) and CD20 monoclonal antibodies (mAbs) (rituximab, ofatumumab, obinutuzumab).3 Chlorambucil was the first alkylating agent with advantages of low cost and toxicity profile, but characterized by low remission rate and by adverse long-term hematologic effects, importantly secondary myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML).4 Subsequently, purine analogs, notably fludarabine, were developed, having higher response rates compared with alkylating agents or anthracycline-based regimens.5,6 Bendamustine had improved efficacy over chlorambucil or fludarabine in 2 randomized trials.7,8 Rituximab was the first CD20 mAb evaluated against mature B-cell malignancies, including CLL/SLL.9 Other CD20 mAbs were evaluated in several trials and introduced in CLL/SLL management.10,11
Combined fludarabine, cyclophosphamide and rituximab (FCR) was studied in first-line and R/R CLL. One hundred and seventy-seven R/R patients treated with FCR achieved an overall response rate (ORR) of 73% (25% complete remission, CR).12 The single-arm study of FCR in first-line (FCR300) where 300 patients with CLL were treated, showed high ORR, CR and 6-year survival rate (95%, 72% and 77%, respectively).13 Subsequent phase III trials in first-line and R/R CLL confirmed superior progression-free survival (PFS) associated with FCR over other standard chemoimmunotherapy (CIT) regimens, but with higher associated myelosuppression.14–16
B-Cell Receptor Signaling
In normal mature B-cells, the BCR consists of a surface transmembrane immunoglobulin (Ig) receptor, associated with the Ig α (Igα, CD79A) and Ig β (Igβ, CD79B) chains (Figure 1).17 Under physiologic conditions, the BCR induces two different signals: a constitutive survival signal, which is antigen independent and involves PI3K; and a second signal induced by interaction with antigen and which promotes recruitment and activation of several intracellular signaling molecules, such as LYN, SYK, Bruton’s tyrosine kinase (BTK) and PI3K, which propagates the signal, ultimately resulting in proliferation and activation of the cells. Following interaction of the BCR with antigen, LYN and SYK kinases phosphorylate BTK, leading to sequential activation of PLCγ2, MAP kinases, AKT and NF-кB.17
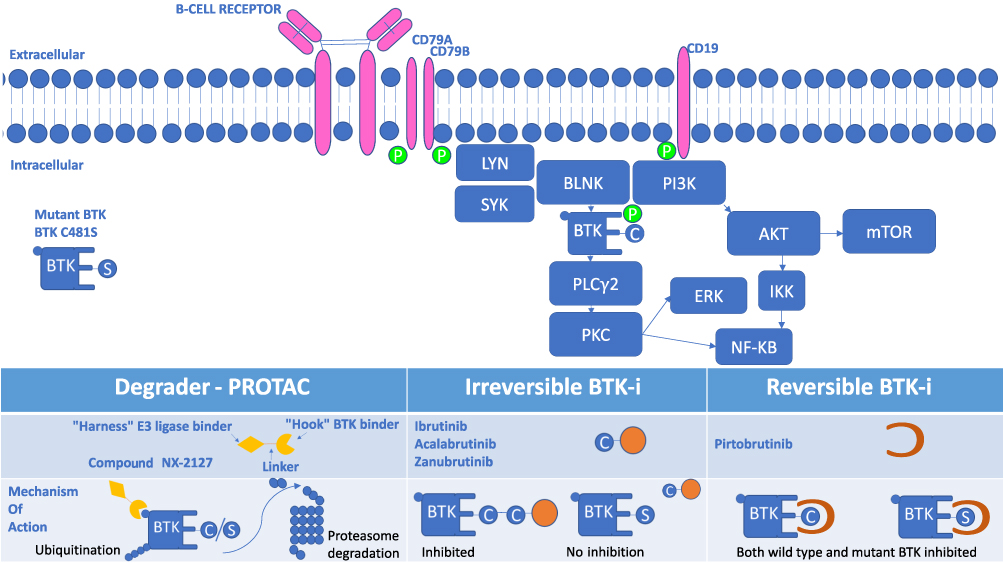 |
Figure 1 B Cell Receptor Signaling Pathway and Inhibition of BTK. Surface membrane of a B cell demonstrating surface B cell receptor and down-stream signaling molecules that participate in providing pro-survival, proliferation, migration, and apoptosis signals to the B cell, including CLL cells. Inhibitors of BTK that block this signaling are shown below, including inducers of degradation, irreversible and reversible inhibitors of BTK kinase activity. |
While CLL B-cells appear to be “locked” in a later stage of development and although they express surface immunoglobulin, generally, in vivo they are unable to further differentiate or secrete immunoglobulin following BCR binding. Downstream members of the BCR signaling pathway are expressed, able to participate in and propagate signaling, thereby having physiologic consequences. Small molecule inhibitor-targeted therapy against members of the BCR signaling pathway members with tyrosine kinase inhibitors (TKI) has been a fundamental advance in treatment for patients with CLL. Orally administered and bioavailable small molecule TKIs against BTK and PI3K have been associated with the most progress in this regard.
BTK Inhibitors
In normal B-cell physiology, BTK is an essential downstream element in signal transduction of the BCR. Stimulation during B-cell development, activates four pathways of tyrosine kinases as follows: PLCγ2, MAPK, NF-кB and serine/threonine kinase AKT, leading to B-cell proliferation and survival.18 When BTK activity is pharmacologically inhibited in CLL, signaling that allows the CLL cells to interact with the microenvironment and receive survival signals is interrupted, promoting death of the malignant lymphocytes.
BTK-i are classified as irreversible and reversible inhibitors based on how they bind BTK, and according to their different chemical structures.19 Irreversible BTK-i form a strong and irreversible covalent bond with the sulfhydryl group of Cys481 in BTK, thereby blocking the ATP-binding site and resulting in irreversible inhibition of its kinase activity (Figure 1).19 In this situation, BTK activity can only be restored by production of new BTK protein. Reversible inhibitors bind tightly to BTK, but not covalently, with binding affinity favoring the bound state in which kinase activity is also blocked by blocking ATP binding.
Both irreversible and reversible BTK-i potentially bind and inhibit other kinases which has implications in side effect and toxicity profiles, depending on which and how many other kinases are bound and inhibited (Figure 1). In theory, increasing selectivity for BTK may reduce the risk of toxicities.20
Irreversible BTK-i Features
Irreversible BTK-i currently includes several molecules with different in vitro binding selectivity profiles. The second generation BTK-i, acalabrutinib, was associated with a highly favorable selectivity profile (1.5% human wild-type kinases showed >65% inhibition). Also, tirabrutinib (2.3%) demonstrated low off-target rate. Zanubrutinib (4.3%), ibrutinib (9.4%) and spebrutinib (8.3%) had broader kinase inhibition profiles. Furthermore, acalabrutinib selectivity for BTK was high also when compared with kinases with a Cys in the same position as the Cys481 residue in BTK or to Src-family, whereas ibrutinib, spebrutinib and zanubrutinib were less selective.21
Adverse events are correlated to BTK-i specificity profile. When specificity of BTK inhibition decreases, as in the case of ibrutinib or spebrutinib, off-target activity increases against other kinases such as EGFR, ITK, JAK3, HER2 and TEC, leading to increased toxicities.22,23 Ibrutinib inhibition of EGFR kinases is correlated to mild adverse effects of rash and diarrhea, which typically do not require treatment discontinuation and can subside with dose reduction and continued therapy.24 In addition, an increased risk for bleeding is due to interaction with the TEC kinase family with inhibition of platelet aggregation.25
Abnormalities in cardiac electrical conduction, including atrial fibrillation and ventricular arrhythmia, were associated to ibrutinib and other irreversible BTK-i therapy.26,27 Risk of atrial fibrillation appears to be associated with off-target inhibition of TEC through the phosphoinositide 3-kinase (PI3K)-Akt pathway.28
One approach to reducing side effects and toxicities might be to reduce drug dose. Pharmacokinetic/pharmacodynamic studies showed that optimum target occupancy is achieved with an ibrutinib dose of 420 mg/day.29 Interestingly, reduction of ibrutinib dose may reduce PFS, as reported in subgroup analysis from the RESONATE trial.30 However, other data suggest reduced doses may be sufficient.31 Also, some toxicities do not appear to be reduced or meaningfully mitigated with drug dose reduction. Other irreversible BTK-I, such as acalabrutinib and zanubrutinib, are dosed twice daily. An important phase II trial (NCT02337829) demonstrated how acalabrutinib 100 mg twice daily (BID) was superior to 200 mg once daily (QD) in terms of ORR (95.8% vs 79.2%) and PFS (91.5% vs 87.2%); results were explained by higher BTK occupancy with twice daily dosing.32
Most current irreversible BTK-i are metabolized into active or inactive metabolites by the liver using cytochrome P450 CYP3A metabolism. Therefore, dose adjustment or avoidance when prescribing with strong or moderate CYP3A inhibitors or inducers and in patients with hepatic dysfunction may be necessary.33–37
Irreversible BTK-i Efficacy in CLL in Phase II Trials
Inhibitors of BTK are highly effective in treating B-cell malignancies, especially CLL as demonstrated in early phase II studies.38 Ibrutinib was approved by the FDA in 2014 for R/R CLL based on a single-arm trial showing excellent results in terms of ORR and duration of response in a refractory patient population.39 In addition, an excellent ORR, with durable remissions was achieved in a phase II open label multicenter RESONATE-17 trial evaluating ibrutinib in R/R CLL with del(17p).40 This supported expanding the approval label to include del(17p) and mutated TP53 CLL.41 Acalabrutinib also demonstrated promising results in previously treated CLL (ORR: 95%) in a phase II trial, prior to approval.42
Treatment Naïve CLL – Phase III BTK-i Trials
The phase III RESONATE-2 trial (NCT01722487) evaluated first-line ibrutinib in 269 patients ≥65 years with treatment-naive CLL and showed higher ORR (86% vs 35%) and significantly longer median PFS (not reached vs 18.9 months) and OS (2 years-OS: 98% vs 85%) versus chlorambucil.43 Notably, PFS was similarly favorable between patients with mutated versus unmutated IGHV and for patients with del(11q) versus non-del(11q) [excluding del(17p)]. Patients with del(17p) were excluded from this trial.
Ibrutinib was also evaluated with CD20 mAbs (rituximab or obinutuzumab) versus CIT, in three phase III multicenter first-line trials. The iLLUMINATE (obinutuzumab) (NCT02264574) and E1912 (rituximab) (NCT02048813) phase III trials compared ibrutinib-based treatment to chlorambucil-obinutuzumab and FCR, respectively in treatment-naive patients. Both trials showed improved PFS with ibrutinib-based treatment, and improved overall survival was noted in the E1912 trial.44,45 Neither of these trials evaluated BTK-i monotherapy.
In the multicenter phase-III Alliance A041202 trial, ibrutinib alone or combined with rituximab was compared with BR in 547 patients ≥65 years old with untreated CLL. A significant improvement in 2-years PFS was observed for ibrutinib (87%) and ibrutinib plus rituximab (88%) when compared with BR arm (74%).46 There was no significant difference in PFS between ibrutinib monotherapy versus combined with rituximab. Therefore, rituximab does not appear to add benefit with ibrutinib in first-line treatment.
The three-arm phase III multicenter ELEVATE-TN trial evaluated acalabrutinib versus acalabrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in 535 treatment-naive CLL patients and reported estimated 30-month PFS of 90%, 82%, and 34%, respectively.47 Several multicenter randomized phase III trials are currently ongoing to evaluate the role of irreversible BTK-i in first-line management of patients with CLL/SLL (Table 1).
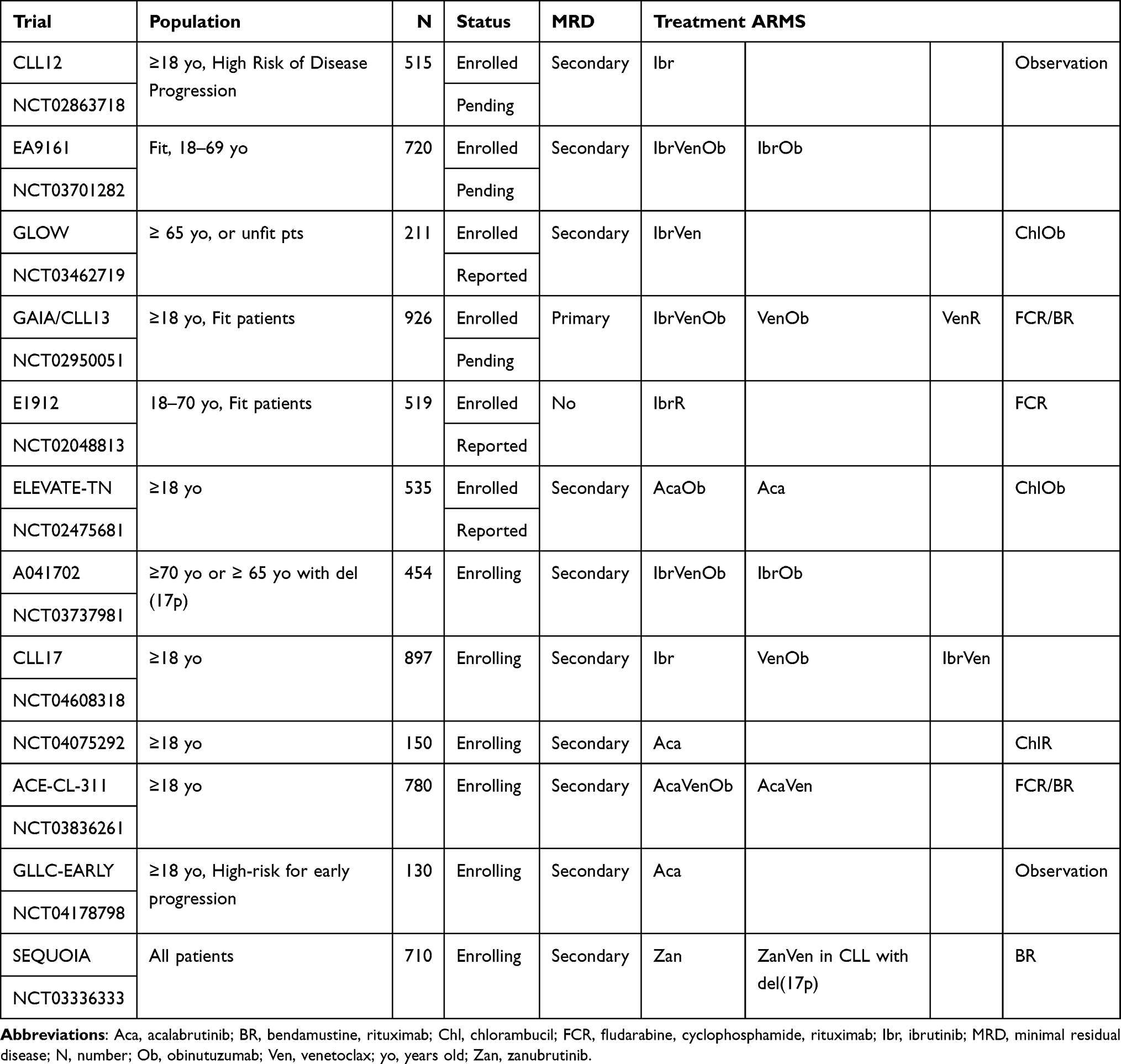 |
Table 1 Select Currently Active Phase III First-Line Trials with Irreversible BTK-i for CLL |
Relapsed/Refractory CLL– Phase III BTK-i Trials
Final analysis of the phase III RESONATE trial with 6-year follow-up confirmed longer median PFS for patients with R/R CLL treated with ibrutinib compared with ofatumumab (44.1 vs 8.1 months) with a significant difference also in high-risk subgroups of CLL patients [del(17p), mutatedTP53 or unmutated IGHV status].48 In addition, the phase III randomized HELIOS study evaluated ibrutinib plus bendamustine-rituximab (BR) versus BR in patients with R/R CLL and showed 79% vs 24% 17-month PFS (median not reached vs 13.3 months, respectively), however, clear evidence for benefit of adding BR to continuous ibrutinib is lacking.49
The randomized phase III ASCEND trial compared acalabrutinib vs investigators’ choice of either BR or idelalisib plus rituximab in 310 patients with R/R CLL/SLL and showed 88% and 68% 12-month PFS, respectively. With a median follow-up of 16 months, median PFS was significantly longer for patients treated with acalabrutinib vs investigator’s choice (not reached vs 16.5 months; p< 0.0001).50 Notably, the majority of patients on the control arm received idelalisib-based treatment. Two randomized multicenter phase III trials (NCT02477696) and ALPINE (NCT03734016) are currently ongoing to compare ibrutinib vs acalabrutinib in a setting of high-risk cytogenetic and zanubrutinib vs ibrutinib in R/R CLL/SLL, respectively.
Long-Term BTK-i Outcomes
Long-term effectiveness of ibrutinib was reported in the 5-year follow-up summary of the early phase I/II trial (NCT01105247), which showed 87% ORR (29% CR) in first-line and treatment for patients with R/R CLL, with a median duration of response of 72.8 months and 57 months, respectively; 5-year PFS rates were 92% and 44% and OS rates were 92% vs 60% in untreated and R/R CLL, respectively. In the high-risk cytogenetic subgroup, ORR was 80%, median duration of response was 31 months; median PFS and OS were 26 and 57 months, respectively. Most common adverse effects grade ≥3 were hypertension, pneumonia and bacterial infections, neutropenia, thrombocytopenia and atrial fibrillation. Major bleeding occurred in 10%. Notably, the frequency of adverse events was higher during the first-year treatment except for the incidence of atrial fibrillation that did not change during follow-up period.51 Updated results of early phase Ib/II trials (NCT02029443) where 134 patients with R/R CLL/SLL received acalabrutinib monotherapy, showed an ORR of 94%, with a median time on treatment of 41 months. Interestingly, no differences were observed between high-risk and non-high risk subgroups. Median duration of response and median PFS were not reached; estimated 45-month PFS was 62%. Diarrhea (51%) and headache (52%) were the most common mild/moderate side effects; 5% patients experienced grade ≥3 adverse events, including neutropenia (14%), pneumonia (11%), hypertension (7%), anemia (7%), and diarrhea (5%). Atrial fibrillation and major hemorrhage occurred in 7% and 5% of patients, respectively.52
Resistance to Irreversible BTK-i
Treatment with BTK-i is continuous until progression. As such, ultimately patients may come off treatment due to intolerance or loss of response and progression of disease. The most relevant BTK-I mechanism of resistance is DNA mutations in BTK catalytic site consisting of substitutions for cysteine at the residue 481 (C481); the most common substitution is serine. When this mutation occurs, the type of covalent binding changes from irreversible to reversible with consequent disruption and loss of inhibition.53
Analysis of 6 patients with R/R CLL on treatment with ibrutinib54 showed presence of mutation in the catalytic site of BTK (C481S) in 5 cases and in phosphoinositide-specific phospholipase C (PLCγ2) in 2 patients. PLCγ2 is downstream of BTK, and mutations were activating.55
Richter’s transformation (RT) is defined as CLL transformation into high-grade non‐Hodgkin (90%) or Hodgkin lymphoma (5–10%); incidence is up to 15% over the course of CLL.56 Clinical presentation is commonly with B-symptoms and rapid enlargement of lymph node sizes. Current therapeutic approaches for RT consist of CD20 mAb, anthracycline-based chemotherapy and allogeneic stem cell transplant.56 Role of BTK-i in RT treatment is still unclear. Most of the currently available information is related to ibrutinib and includes series of case reports and no randomized trials.57,58 Interestingly, acalabrutinib was evaluated in a phase I/II trial of 29 RT patients. ORR was 38% (CR: 14%), suggesting modest therapeutic activity.59 Particular attention should be paid to combined ibrutinib-nivolumab which, in a specific RT cohort of a phase I/II trial, showed an OR in 13/20 (65%) cases.60
Irreversible BTK-i Treatment Cost
Current recommendation for BTK-i treatment is until progression or unacceptable toxicity, therefore the total cost for long-term therapy can be very high. Three studies61–63 conducted in the United Kingdom and United States have tried to analyze cost-effectiveness of this therapy. Very high costs were associated with continuous and indefinite ibrutinib treatment, and drug acquisition costs seem to be a key driver in cost-effectiveness for ibrutinib across studies. In the United Kingdom, ibrutinib was considered cost-effective because of an important discount from the pharmaceutical manufacturer. Patient selection may be the most practical approach to cost containment with this treatment.64
Irreversible BTK-i off-Target Effects and Alternative Indications
Arthritis and immune-complex disease was noted in lymphoma trials with ibrutinib that was later studied in animal models to understand the underlying mechanism(s). Ibrutinib was studied in murine models of collagen-induced (CIA) and collagen antibody-induced arthritis (CAIA) showing inhibition of inflammatory cells such as synovial macrophages.65 Furthermore, Lee et al.66 confirmed an inhibitory effect of ibrutinib on autoimmune murine diseases by blocking B-lymphocyte function. Also, interleukin-2-inducible kinase (ITK), expressed by T-lymphocytes, is inhibited by ibrutinib.67 These important anti-inflammatory effects led to trials evaluating ibrutinib as a treatment for inflammatory-based conditions, such as chronic Graft versus Host Disease (cGvHD) and psoriasis.
Chronic GvHD is a potentially severe complication of allogeneic hematopoietic stem cell transplantation (HSCT) where donor lymphocytes react against recipient organs. Available therapeutic options after corticosteroid failure are very limited. There is strong evidence for the role of activated donor lymphocytes in cGvHD pathogenesis,68 therefore, ibrutinib was tested in murine cGvHD models and showed promising results in terms of reduction of B- and T-lymphocyte activation and cGvHD evolution.69 Furthermore, NOTCH signaling plays an important role in allogeneic T-cell activation during cGVHD; NOTCH inhibitors are currently being evaluated in pre-clinical models.70 Interestingly, ibrutinib was shown to downregulate NOTCH1 activity in CLL cells collected from patients during ibrutinib treatment.71 A phase I/II trial evaluated ibrutinib efficacy in patients with corticosteroid-refractory cGvHD. ORR was 67% (CR rate: 21%). Accordingly, ibrutinib was approved by FDA for treatment of cGvHD after first-line therapy.72
Pre-clinical studies showed BTK pathway is involved in neutrophilic inflammation,73 and for this reason ibrutinib was evaluated in psoriasis, a skin condition due to an infiltration of innate immune cells. In mice, ibrutinib reduced expression of psoriasis-like inflammation by downregulation of inflammatory mediators.74 Clinical studies with larger numbers of patients are needed to assess BTK-i activity in psoriasis.
Irreversible BTK-i and Coronavirus 2019 Disease
The COVID-19 pandemic is due to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).75 Clinical manifestations of COVID-19 are heterogeneous, ranging from a mild influenza-like syndrome to an acute respiratory distress syndrome (ARDS) with respiratory failure and widespread thromboembolic events.76,77
The acute lung injury in late phases of SARS-CoV-2 infection results from hyperactivation of the immune system, leading to a “cytokine storm” with elevated blood levels of IL-1β, IL-6, IL-7, IL-8 and IFN-γ.78
The rationale for use of BTK-i in viral hyperimmune conditions derives from a pre-clinical study that evaluated the effect of BTK-i in rescuing mice with lethal influenza-induced acute lung injury.79 BTK-i treatment could have strong inhibitory impact on innate inflammatory response dominated by macrophage and neutrophils.73–80
First clinical evaluations of ibrutinib in COVID-19 derive from retrospective observations. Six81 patients, with diagnosis of ibrutinib-treated Waldenstrom’s macroglobulinemia, did not experience dyspnea or hypoxia and had mild evolution of COVID-19, except for the one on treatment with reduced dose. When ibrutinib was increased to full dose in this patient, lung injury and clinical conditions progressively improved. Therefore, prospective phase II trials are currently ongoing, such as the iNSPIRE study (NCT04375397), to evaluate the role of ibrutinib in COVID-19.
Acalabrutinib was used80 in 19 hospitalized patients with severe pulmonary damage from COVID-19 and was chosen for improved tolerability profile over ibrutinib. Improvement of blood oxygenation in most patients and reduced inflammatory markers were reported. A recent update of the phase II CALAVI trial in which acalabrutinib was added to best supportive care in hospitalized patients with COVID-19 revealed that acalabrutinib did not meet the primary end point of increasing the proportion of patients alive and free of respiratory failure.82 At the moment, no data are available regarding the effectiveness of zanubrutinib. A phase II trial (NCT04382586) is currently recruiting to evaluate zanubrutinib in hospitalized patients with COVID-19.
Recent publications showed conflicting results about BTK-i efficacy in preventing severe COVID-19 in patients with CLL. Indeed, a retrospective analysis of 190 patients with CLL diagnosed with COVID-19 reported protective effects for BTK-i-based treatment83 whereas ongoing BTK-i treatment was not associated with better COVID-19 outcome, including OS in 198 CLL patients affected by symptomatic COVID-19, most of whom remained on continued ibrutinib treatment during their infection.84 Owing to the underlying immune dysfunction of CLL, with lack of immune reconstitution despite effective targeted therapy with BTK-I, there is concern for protection in patients with CLL with BNT162b2 mRNA vaccines. A prospective study that enrolled 167 patients with CLL reported an antibody response rate of 39.5% following two vaccine doses, 21 days apart. Notably, in the subgroup of BTK-i treated patients, the response rate was only 16% and in multivariable analysis, the presence of active treatment with BTK-i was not associated with antibody response.85
CLL and Patient Characteristics Associated with Resistance and Current Management of Irreversible BTK-i R/R Patients
Long-term results of early studies that evaluated ibrutinib as first-line and in R/R CLL showed patient or disease characteristics can correlate with outcomes.51
Five-year PFS was 92% and 44% with a CR rate of 29% vs 10%, in treatment-naive and R/R CLL, respectively. Clonal evolution in CLL cells and development of ibrutinib resistance is more likely in heavily pre-treated patients due to the acquisition of new genetic abnormalities, including mutations in BTK.86 Indeed, the frequency of complex karyotype and other alterations with adverse prognosis such as del(17p) and mutated TP53 is increased in patients with R/R CLL.
Interestingly, shorter PFS and OS were correlated in univariable and multivariable analyses with TP53 aberration, prior treatment, β-2 microglobulin ≥5 mg/L and lactate dehydrogenase >250 U/L in patients treated with ibrutinib in several phase II and III trials. In a prognostic model, 1 point was given for each factor and cumulative score was correlated with outcomes.87
Results with ibrutinib are highly favorable compared with CIT in patients with high-risk features with a median duration of response of 31 months in complex karyotype and in del(17p), 39 months in del(11q) R/R CLL with ibrutinib. Median PFS and OS were 26 and 57 months in del(17p) vs 31 months and 54 months in complex karyotype vs 51 months and not reached in del(11q), respectively.51 In summary, multiple prior treatments and genetic abnormalities as complex karyotype, del(17p) and mutated TP53 are associated with shorter response duration with ibrutinib and therefore considered high-risk features, but are remarkably better than was seen with CIT-based treatment in patients with these characteristics.
Patients with CLL who experience disease progression on BTK-i treatment have poor outcomes. Venetoclax is an oral inhibitor of the anti-apoptotic protein BCL2 that was approved by FDA, is a therapeutic option in such patients who fail BTK-i-based treatment. CLL cells intrinsically express high levels of BCL2, which is higher in patients with BTK-i-resistant CLL. High expression of BCL2 confers resistance to apoptosis and is involved in CLL progression.88 In a phase II study89 91 CLL patients were enrolled after ibrutinib failure. With a median follow-up of 14 months, ORR and CR rate were 65% and 9%, respectively; median PFS was 24.7 months and 12-month PFS rate was 75%.
The phase III randomized MURANO trial90 evaluated combined venetoclax and rituximab versus bendamustine and rituximab in patients with R/R CLL and demonstrated significantly improved PFS (2-year PFS: 84.9% and 36.3%) for venetoclax-based treatment. Notably, patients with high-risk genetic CLL (del(17p)/mutated-TP53) experienced PFS benefit (2-year PFS rates of 81.5% vs 27.8%, in venetoclax and rituximab and bendamustine-rituximab groups, respectively).
Optimal sequencing of targeted therapy in CLL treatment is an evolving and challenging topic and was retrospectively analyzed in a multicenter study of 683 patients.91 Results reported that, in cases of ibrutinib resistance, venetoclax or PI3K-i was superior when compared with a CIT approach and that venetoclax treatment had better outcomes when compared with idelalisib. Overall, BCL2 inhibition may be considered as an effective strategy after BTK-i resistance. In addition, since BTK mutations are common in patients who develop resistance to irreversible BTK-i, an approach to overcome BTK-i resistance was to design reversible inhibitors of BTK which do not require the C481 for binding.
Reversible BTK-i
Irreversible BTK-i covalently bind to C481 of BTK for inhibition; whereas reversible BTK-i tightly bind BTK, but non-covalently (hydrogen, ionic bonds and hydrophobic interactions) and therefore do not require C481. Non-covalent bonds are less powerful and selective than covalent interactions and potentially require longer serum half-life to enhance therapeutic activity.19 Clinical trials of reversible BTK-i are in early stages and early results are promising.
Pirtobrutinib (LOXO-305) is an oral, highly selective, reversible BTK-i tested in preclinical models92 and clinical trial. Pirtobrutinib was evaluated in a phase I/II trial of patients with B-cell malignancies; no DLTs or MTD was identified, the recommended dose of 200 mg/day appeared well-tolerated and was active, including in patients with resistance to covalent BTK-i and BCL2-i.93 In the subgroup of 121 patients with CLL and median of 4 prior treatments, the ORR was 62%. Interestingly, no differences in ORR were observed between patients with previous irreversible BTK-i resistance (53 of 79), and intolerance (22 of 42), BTK C481-mutant (17 of 24) and BTK wild-type (43 of 65) CLL.93
Another reversible inhibitor of BTK, ARQ351, also demonstrated inhibitory activity against Src and ERK kinases. In vitro, ARQ351 inhibited BTK-mediated activity including B-cell receptor (BCR) signaling, resulting in reduced cell viability, migration and NF-κB transcription. ARQ351 was more effective than ibrutinib in prolonging survival in animal models94 and also in vitro activity was observed in CLL cells harboring BTKC481S or PLCγ2 resistance mutations.94 A Phase I escalation trial recently reported preliminary clinical activity for ARQ351 in CLL.95
The selective BTK-i fenebrutinib (GDC-0853), binds the H3 region of BTK and is characterized by low rates of off-target activity.96 Fenebrutinib was very effective in vitro in blocking BTK activity with an extremely low dissociation time from BTK so that fenebrutinib duration of action might be compared to irreversible BTK-i. Pharmacokinetic analysis in rats and dogs showed excellent oral bioavailability with adequate blood exposure. Early phase I trials in which fenebrutinib was tested in the setting of autoimmune diseases97 and B-cell malignancies98 confirmed safety results without evidence of dose-limiting toxicities, with anticancer effects.
Vecabrutinib showed in vitro activity against BTKC481S.99 Vecabrutinib seems to avoid off-target activity against EGFR, resulting in lower incidence of EGFR-mediated side effects. However, clinical development of vecabrutinib was halted due to lack of clinical activity against CLL.
Accelerated BTK “Degraders”
Degradation of intracellular proteins is a fundamental process for the normal equilibrium of cell functions, such as proliferation, differentiation and apoptosis. Regulation of intracellular protein levels is a complex system involving ubiquitin-dependent proteolysis governed by 3 enzymes: ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2) and ubiquitin–protein ligases (E3). These intracellular proteins work sequentially to catalyze the bond between a ubiquitin polymer and lysine residues of substrate proteins100 which results in recognition by the multi-subunit ATP-dependent protease, proteasome, and culminates in protein degradation.101
Proteolysis-targeting chimera (PROTAC) is an emerging approach that promotes selective degradation of intracellular proteins. Small PROTAC molecules consist of three components: a target protein-binding arm (targeting arm, TA), a degradation machinery-recruiting unit (degradation arm, DA) and a linker. TA is engineered to bind target proteins whereas DA recruits an E3 ubiquitin ligase, promoting creation of ubiquitin polymers.102 This novel and innovative strategy might be used to eliminate proteins, such as BTK, including BTKC481S.
The BTK-targeting PROTAC D-04-015 consisted of pomalidomide and the BTK-ligand RN486.103 Subsequently, BTK PROTAC DD-03-171 was constructed with pomalidomide and the BTK-ligand CGI746. This compound effectively induced degradation of wild type BTK (BTKwt), and also had activity against BTKC481S, exhibiting a strong antiproliferative effect against mantle cell lymphoma cells in xenograft models.104 MT-082, composed of ibrutinib and thalidomide, degraded BTKwt and BTKC481S with antiproliferative effect on CLL cells.105 More powerful molecules, such as P13I (ibrutinib and pomalidomide) and L18I (ibrutinib and lenalidomide) were designed and led to lower nanomolar half-maximal degradation concentration (DC50). They effectively promoted degradation of BTK, reducing tumor growth of diffuse large B-cell lymphoma and HeLa cells.102–106
Preclinical work demonstrated that NX-2127 is effective BTK-targeting PROTAC which induced degradation of 50% of cellular BTK at <5 nM concentration. Interestingly, NX-2127 is more effective than ibrutinib in inhibiting proliferation of TMD8 cells harboring BTKC481Smutation.107 NX-2127 has entered phase I trial (NCT04830137).
In conclusion, significant progress has been made regarding mechanism of action, pharmacokinetic profiles and in vitro effectiveness of BTK-targeted PROTAC molecules. Proof of principle and mechanism of action for BTK-targeted PROTACs will be tested in clinical trials in B-cell malignancies, including CLL (NCT04830137).
Future of BTK-i-Based Treatment
Development and availability of BTK-i has fundamentally improved CLL management and outcomes, yet there is still room for therapeutic improvement. For example, about 30% of patients become refractory to ibrutinib; the likelihood of resistance increases with longer exposure time.29 Additionally, another 40% of patients come off treatment long-term due to intolerable side-effects. Thus, there is competing risk for discontinuation due to toxicity, which may be greater for ibrutinib than for second-generation irreversible BTK-i. Continuous and indefinite treatment with BTK-i assures optimal outcomes with monotherapy or combined with CD20 mAb.
Therapeutic progress has also come with development of the BCL2-i, venetoclax, which achieves deep and durable remission with fixed-duration treatment.90,108 Furthermore, combined ibrutinib and venetoclax is highly active, has promise for fixed duration treatment, and is very well-tolerated. These two agents are therapeutically complementary, with ibrutinib having notable activity in treating lymph node disease, and venetoclax having potent activity in clearing disease from blood and bone marrow. Particularly, a phase II trial conducted in 80 high-risk or older patients (>65 years) with treatment-naive CLL demonstrated that, after 12 cycles of fixed duration, combined therapy, 88% and 61% of patients achieved hematological CR and remission with undetectable MRD, respectively. Toxicity profile was comparable to ibrutinib and venetoclax alone.91 Negative MRD is important, especially in younger patients, because of hypothesis of treatment discontinuation. Indeed, when ibrutinib in monotherapy was discontinued without a negative MRD status, the outcome was very poor with a median OS of 3.1 months.109 Encouraging clinical trial results showed encouraging rates of complete remission (CR) and negative flow cytometry measurable residual disease (MRD) when combined therapy ibrutinib-venetoclax was evaluated in R/R CLL.110 This potent fixed-duration combination may achieve deep remission, and may thereby reduce risk for clonal evolution and development of BTKC481 resistance mutations.111 Additional benefit with CD20 mAb in this combination is under investigation. Ongoing phase III clinical trials are provided in Table 1.
An interesting alternative to continuous BTK-i treatment until progression or unacceptable toxicity might be an approach of intermittent treatment with BTK-i for a specified number of cycles and retreatment with relapse or progression. The effectiveness of this strategy may be clarified by ongoing phase II trials with combined BTK-i and CD20 mAb (NCT04505254).
Disclosure
No conflict of interest or disclosures for this work.
References
1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi:10.3322/canjclin.57.1.43
2. Freymann JG, Vander JB, Marler EA, Meyer DG. Prolonged corticosteroid therapy of chronic lymphocytic leukaemia and the closely allied malignant lymphomas. Br J Haematol. 1960;6:303–323. doi:10.1111/j.1365-2141.1960.tb06247.x
3. CLL Trialists’ Collaborative Group. Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. J Natl Cancer Inst. 1999;91:861–868.
4. Callea V, Brugiatelli M, Stelitano C, et al. Incidence of second neoplasia in patients with B-cell chronic lymphocytic leukemia treated with chlorambucil maintenance chemotherapy. Leuk Lymphoma. 2006;47:2314–2320. doi:10.1080/10428190600880977
5. Rai KR, Peterson BL, Appelbaum FR, et al. Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia. N Engl J Med. 2000;343:1750–1757. doi:10.1056/nejm200012143432402
6. Johnson S, Smith AG, Löffler H, et al. Multicentre prospective randomised trial of fludarabine versus cyclophosphamide, doxorubicin, and prednisone (CAP) for treatment of advanced-stage chronic lymphocytic leukaemia. Lancet. 1996. doi:10.1016/s0140-6736(96)91681-5
7. Niederle N, Megdenberg D, Balleisen L, et al. Bendamustine compared to fludarabine as second-line treatment in chronic lymphocytic leukemia. Ann Hematol. 2013;92:653–660. doi:10.1007/s00277-012-1660-6
8. Knauf WU, Lissichkov T, Aldaoud A, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol. 2009;27:4378–4384. doi:10.1200/JCO.2008.20.8389
9. Hagemeister F. Rituximab for the treatment of non-Hodgkin's lymphoma and chronic lymphocytic leukaemia. Drugs. 2010;70:261–272. doi:10.2165/11532180-000000000-00000
10. Bauer K, Rancea M, Roloff V, et al. Rituximab, ofatumumab and other monoclonal anti-CD20 antibodies for chronic lymphocytic leukaemia. Cochrane Database Syst Rev. 2012. doi:10.1002/14651858.CD008079.pub2
11. Cartron G, de Guibert S, Dilhuydy M-S, et al. Obinutuzumab (GA101) in relapsed/refractory chronic lymphocytic leukemia: final data from the phase 1/2 GAUGUIN study. Blood. 2014;124:2196–2202. doi:10.1182/blood-2014-07-586610
12. Wierda W, O’Brien S, Wen S, et al. Chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximab for relapsed and refractory chronic lymphocytic leukemia. J Clin Oncol. 2005;23:4070–4078. doi:10.1200/JCO.2005.12.516
13. Tam CS, O’Brien S, Wierda W, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood. 2008;112:975–980. doi:10.1182/blood-2008-02-140582
14. Robak T, Moiseev SI, Dmoszynska A, et al. Rituximab, fludarabine, and cyclophosphamide (R-FC) prolongs progression free survival in relapsed or refractory chronic lymphocytic leukemia (CLL) compared with FC alone: final results from the international randomized phase III REACH trial. Blood. 2008;112:lba–1. doi:10.1182/blood.v112.11.lba-1.lba-1
15. Fischer K, Bahlo J, Fink AM, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016;127:208–215. doi:10.1182/blood-2015-06-651125
16. Eichhorst B, Fink A-M, Bahlo J, et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2016;17:928–942. doi:10.1016/S1470-2045(16)30051-1
17. Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B cell antigen receptor signaling 101. Mol Immunol. 2004;41:599–613. doi:10.1016/j.molimm.2004.04.008
18. Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer. 2018. doi:10.1186/s12943-018-0779-z
19. Liang C, Tian D, Ren X, et al. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: a mini-review. Eur J Med Chem. 2018;151:315–326. doi:10.1016/j.ejmech.2018.03.062
20. Swinney DC. The role of binding kinetics in therapeutically useful drug action. Curr Opin Drug Discov Devel. 2009;12:31–39.
21. Kaptein A, de Bruin G, Emmelot-van Hoek M, et al. Potency and selectivity of BTK inhibitors in clinical development for B-cell malignancies. Blood. 2018;132:1871. doi:10.1182/blood-2018-99-109973
22. Kaur V, Swami A. Ibrutinib in CLL: a focus on adverse events, resistance, and novel approaches beyond ibrutinib. Ann Hematol. 2017;96:1175–1184. doi:10.1007/s00277-017-2973-2
23. Burger JA. Bruton’s tyrosine kinase (BTK) inhibitors in clinical trials. Curr Hematol Malig Rep. 2014;9:44–49. doi:10.1007/s11899-013-0188-8
24. Bond DA, Woyach JA. Targeting BTK in CLL: beyond ibrutinib. Curr Hematol Malig Rep. 2019;14:197–205. doi:10.1007/s11899-019-00512-0
25. Atkinson BT, Ellmeier W, Watson SP. Tec regulates platelet activation by GPVI in the absence of Btk. Blood. 2003;102:3592–3599. doi:10.1182/blood-2003-04-1142
26. Leong DP, Caron F, Hillis C, et al. The risk of atrial fibrillation with ibrutinib use: a systematic review and meta-analysis. Blood. 2016;128:138–140. doi:10.1182/blood-2016-05-712828
27. Cheng C, Woronow D, Nayernama A, Wroblewski T, Jones SC. Ibrutinib-associated ventricular arrhythmia in the FDA adverse event reporting system. Leuk Lymphoma. 2018;59:3016–3017. doi:10.1080/10428194.2018.1457149
28. McMullen JR, Boey EJH, Ooi JYY, et al. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood. 2014;124:3829–3830. doi:10.1182/blood-2014-10-604272
29. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi:10.1056/nejmoa1215637
30. Barr PM, Brown JR, Hillmen P, et al. Impact of ibrutinib dose adherence on therapeutic efficacy in patients with previously treated CLL/SLL. Blood. 2017;129:2612–2615. doi:10.1182/blood-2016-12-737346
31. Chen LS, Bose P, Cruz ND, et al. A pilot study of lower doses of ibrutinib in patients with chronic lymphocytic leukemia. Blood. 2018. doi:10.1182/blood-2018-06-860593
32. Sun C, Nierman P, Kendall EK, et al. Clinical and biological implications of target occupancy in CLL treated with the BTK inhibitor acalabrutinib. Blood. 2020;136:93–105. doi:10.1182/blood.2019003715
33. Bose P, Gandhi VV, Keating MJ. Pharmacokinetic and pharmacodynamic evaluation of ibrutinib for the treatment of chronic lymphocytic leukemia: rationale for lower doses. Expert Opin Drug Metab Toxicol. 2016;12:1381–1392. doi:10.1080/17425255.2016.1239717
34. Kriegsmann K, Kriegsmann M, Witzens-Harig M. Acalabrutinib, A second-generation bruton’s tyrosine kinase inhibitor. Recent Results Cancer Res. 2018. doi:10.1007/978-3-319-91439-8_14
35. Mu S, Tang Z, Novotny W, et al. Effect of rifampin and itraconazole on the pharmacokinetics of zanubrutinib (a Bruton’s tyrosine kinase inhibitor) in Asian and non-Asian healthy subjects. Cancer Chemother Pharmacol. 2020;85:391–399. doi:10.1007/s00280-019-04015-w
36. Dhillon S. Tirabrutinib: first approval. Drugs. 2020. doi:10.1007/s40265-020-01318-8
37. Li Y, Ramírez-Valle F, Xue Y, et al. Population pharmacokinetics and exposure response assessment of CC-292, a potent BTK inhibitor, in patients with chronic lymphocytic leukemia. J Clin Pharmacol. 2017;57:1279–1289. doi:10.1002/jcph.923
38. Novero A, Ravella PM, Chen Y, Dous G, Liu D. Ibrutinib for B cell malignancies. Exp Hematol Oncol. 2014;3. doi:10.1186/2162-3619-3-4.
39. De Claro RA, McGinn KM, Verdun N, et al. FDA approval: ibrutinib for patients with previously treated mantle cell lymphoma and previously treated chronic lymphocytic leukemia. Clin Cancer Res. 2015;21:3586–3590. doi:10.1158/1078-0432.CCR-14-2225
40. O’Brien S, Jones JA, Coutre SE, et al. Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): a phase 2, open-label, multicentre study. Lancet Oncol. 2016;17:1409–1418. doi:10.1016/S1470-2045(16)30212-1
41. Zenz T, Mohr J, Edelmann J, et al. Treatment resistance in chronic lymphocytic leukemia - the role of the p53 pathway. Leuk Lymphoma. 2009;50:510–513. doi:10.1080/10428190902763533
42. Byrd JC, Harrington B, O’Brien S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:323–332. doi:10.1056/nejmoa1509981
43. Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373:2425–2437. doi:10.1056/nejmoa1509388
44. Moreno C, Greil R, Demirkan F, et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20:43–56. doi:10.1016/S1470-2045(18)30788-5
45. Shanafelt TD, Wang XV, Kay NE, et al. Ibrutinib–rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381:432–443. doi:10.1056/nejmoa1817073
46. Woyach JA, Ruppert AS, Heerema NA, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379:2517–2528. doi:10.1056/nejmoa1812836
47. Sharman JP, Banerji V, Fogliatto LM, et al. ELEVATE TN: phase 3 study of acalabrutinib combined with obinutuzumab (O) or alone vs O plus chlorambucil (Clb) in patients (Pts) with treatment-naive chronic lymphocytic leukemia (CLL). Blood. 2019;134:31. doi:10.1182/blood-2019-128404
48. Munir T, Brown JR, O’Brien S, et al. Final analysis from RESONATE: up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am J Hematol. 2019;94:1353–1363. doi:10.1002/ajh.25638
49. Chanan-Khan A, Cramer P, Demirkan F, et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): a randomised, double-blind, phase 3 study. Lancet Oncol. 2016;17:200–211. doi:10.1016/S1470-2045(15)00465-9
50. Ghia P, Pluta A, Wach M, et al. Ascend: phase III, randomized trial of acalabrutinib versus idelalisib plus rituximab or bendamustine plus rituximab in relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol. 2020;38:2849–2861. doi:10.1200/JCO.19.03355
51. O’Brien S, Furman RR, Coutre S, et al. Single-agent ibrutinib in treatment-naïve and relapsed/refractory chronic lymphocytic leukemia: a 5-year experience. Blood. 2018;131:1910–1919. doi:10.1182/blood-2017-10-810044
52. Byrd JC, Wierda WG, Schuh A, et al. Acalabrutinib monotherapy in patients with relapsed/refractory chronic lymphocytic leukemia: updated phase 2 results. Blood. 2020;135:1204–1213. doi:10.1182/blood.2018884940
53. Furman RR, Cheng S, Lu P, et al. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014. doi:10.1056/nejmc1402716
54. Woyach JA, Furman RR, Liu T-M, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–2294. doi:10.1056/nejmoa1400029
55. Nishida M, Sugimoto K, Hara Y, et al. Amplification of receptor signalling by Ca2+ entry-mediated translocation and activation of PLCγ2 in B lymphocytes. EMBO J. 2003;22:4677–4688. doi:10.1093/emboj/cdg457
56. Allan JN, Furman RR. Current trends in the management of Richter’s syndrome. Int J Hematol Oncol. 2018;7:IJH09. doi:10.2217/ijh-2018-0010
57. Master S, Leary C, Takalkar A, et al. Successful treatment of Richter transformation with ibrutinib in a patient with chronic lymphocytic leukemia following allogeneic hematopoietic stem cell transplant. Case Rep Oncol. 2017;10:534–541. doi:10.1159/000477338
58. Fischer A, Bastian S, Cogliatti S, et al. Ibrutinib-induced rapid response in chemotherapy-refractory Richter’s syndrome. Hematol Oncol. 2018;36:370–371. doi:10.1002/hon.2464
59. Hillmen P, Schuh A, Eyre TA, et al. Acalabrutinib monotherapy in patients with Richter transformation from the phase 1/2 ACE-CL-001 clinical study. Blood. 2016;128:60. doi:10.1182/blood.v128.22.60.60
60. Younes A, Brody J, Carpio C, et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: a phase 1/2a study. Lancet Haematol. 2019;6:e67–e78. doi:10.1016/S2352-3026(18)30217-5
61. Mikudina B, Goodall M, Adler AI. NICE guidance on ibrutinib for previously treated chronic lymphocytic leukaemia and untreated chronic lymphocytic leukaemia in the presence of 17p deletion or TP53 mutation. Lancet Oncol. 2017;18:289–290. doi:10.1016/S1470-2045(17)30062-1
62. Sinha R, Redekop WK. Cost-effectiveness of ibrutinib compared with obinutuzumab with chlorambucil in untreated chronic lymphocytic leukemia patients with comorbidities in the United Kingdom. Clin Lymphoma Myeloma Leuk. 2018;18:e131–e142. doi:10.1016/j.clml.2017.12.005
63. Barnes JI, Divi V, Begaye A, et al. Cost-effectiveness of ibrutinib as first-line therapy for chronic lymphocytic leukemia in older adults without deletion 17p. Blood Adv. 2018;2:1946–1956. doi:10.1182/bloodadvances.2017015461
64. Harkins RA, Patel SP, Flowers CR. Cost-effectiveness of new targeted agents in the treatment of chronic lymphocytic leukemia. Cancer J. 2019. doi:10.1097/PPO.0000000000000411
65. Chang BY, Huang M, Francesco M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res Ther. 2011;13:R115. doi:10.1186/ar3400
66. Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA. 2010;107:13075–13080. doi:10.1073/pnas.1004594107
67. Dubovsky JA, Beckwith KA, Natarajan G, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122:2539–2549. doi:10.1182/blood-2013-06-507947
68. Allen JL, Tata PV, Fore MS, et al. Increased BCR responsiveness in B cells from patients with chronic GVHD. Blood. 2014;123:2108–2115. doi:10.1182/blood-2013-10-533562
69. Dubovsky JA, Flynn R, Du J, et al. Ibrutinib treatment ameliorates murine chronic graft-versus-host disease. J Clin Invest. 2014;4867–4876. doi:10.1172/JCI75328.
70. Baldoni S, Ruggeri L, Del Papa B, et al. NOTCH1 inhibition prevents GvHD and maintains GvL effect in murine models. Bone Marrow Transplant. 2021;56:2019–2023. doi:10.1038/s41409-021-01297-8
71. DelPapa B, Baldoni S, Dorillo E, et al. Decreased NOTch1 activation correlates with response to ibrutinib in chronic lymphocytic leukemia. Clin Cancer Res. 2019;25:7540–7553. doi:10.1158/1078-0432.CCR-19-1009
72. Miklos D, Cutler CS, Arora M, et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood. 2017;130:2243–2250. doi:10.1182/blood-2017-07-793786
73. Volmering S, Block H, Boras M, Lowell CA, Zarbock A. The neutrophil Btk signalosome regulates integrin activation during sterile inflammation. Immunity. 2016;44:73–87. doi:10.1016/j.immuni.2015.11.011
74. Al-Harbi NO, Nadeem A, Ahmad SF, et al. Therapeutic treatment with Ibrutinib attenuates imiquimod-induced psoriasis-like inflammation in mice through downregulation of oxidative and inflammatory mediators in neutrophils and dendritic cells. Eur J Pharmacol. 2020;877:173088. doi:10.1016/j.ejphar.2020.173088
75. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi:10.1016/S0140-6736(20)30251-8
76. Guan W, Ni Z-Y, Hu Y, et al. Clinical characteristics of Coronavirus disease 2019 in China. N Engl J Med. 2020;382:1708–1720. doi:10.1056/nejmoa2002032
77. Mondal S, Quintili AL, Karamchandani K, Bose S. Thromboembolic disease in COVID-19 patients: a brief narrative review. J Intens Care. 2020;8. doi:10.1186/s40560-020-00483-y.
78. Conti P, Ronconi G, Caraffa AL, et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents. 2020. doi:10.23812/CONTI-E
79. Florence JM, Krupa A, Booshehri LM, Davis SA, Matthay MA, Kurdowska AK. Inhibiting bruton’s tyrosine kinase rescues mice from lethal influenza-induced acute lung injury. Am J Physiol. 2018. doi:10.1152/ajplung.00047.2018
80. Roschewski M, Lionakis MS, Sharman JP, et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci Immunol. 2020;5:eabd0110. doi:10.1126/SCIIMMUNOL.ABD0110
81. Treon SP, Castillo JJ, Skarbnik AP, et al. The BTK inhibitor ibrutinib may protect against pulmonary injury in COVID-19 infected patients. Blood. 2020;135:1912–1915. doi:10.1182/BLOOD.2020006288
82. NCT. Acalabrutinib study with best supportive care versus best supportive care in subjects hospitalized with COVID-19. CALAVI (Calquence Against the Virus); 2020. Available from: https://clinicaltrials.gov/show/NCT04346199.
83. Scarfò L, Chatzikonstantinou T, Rigolin GM, et al. COVID-19 severity and mortality in patients with chronic lymphocytic leukemia: a joint study by ERIC, the European Research Initiative on CLL, and CLL Campus. Leukemia. 2020;34:2354–2363. doi:10.1038/s41375-020-0959-x
84. Mato AR, Roeker LE, Lamanna N, et al. Outcomes of COVID-19 in patients with CLL: a multicenter international experience. Blood. 2020;136:1134–1143. doi:10.1182/blood.2020006965
85. Herishanu Y, Avivi I, Aharon A, et al. Efficacy of the BNT162b2 mRNA COVID-19 vaccine in patients with chronic lymphocytic leukemia. Blood. 2021;137:3165–3173. doi:10.1182/blood.2021011568
86. Stilgenbauer S, Sander S, Bullinger L, et al. Clonal evolution in chronic lymphocytic leukemia: acquisition of high-risk genomic aberrations associated with unmutated VH, resistance to therapy, and short survival. Haematologica. 2007;92:1242–1245. doi:10.3324/haematol.10720
87. Ahn IE, Tian X, Ipe D, et al. Prediction of outcome in patients with chronic lymphocytic leukemia treated with ibrutinib: development and validation of a four-factor prognostic model. J Clin Oncol. 2021;39:576–585. doi:10.1200/JCO.20.00979
88. Marschitz I, Tinhofer I, Hittmair A, et al. Analysis of Bcl-2 protein expression in chronic lymphocytic leukemia: a comparison of three semiquantitation techniques. Am J Clin Pathol. 2000;113:219–229. doi:10.1309/491w-l1tn-ufqx-t61b
89. Jones JA, Mato AR, Wierda WG, et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018;19:65–75. doi:10.1016/S1470-2045(17)30909-9
90. Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax–rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378:1107–1120. doi:10.1056/nejmoa1713976
91. Mato AR, Hill BT, Lamanna N, et al. Optimal sequencing of ibrutinib, idelalisib, and venetoclax in chronic lymphocytic leukemia: results from a multicenter study of 683 patients. Ann Oncol. 2017;28:1050–1056. doi:10.1093/annonc/mdx031
92. Brandhuber B, Gomez E, Smith S, et al. LOXO-305, a next generation reversible BTK inhibitor, for overcoming acquired resistance to irreversible BTK inhibitors. Clin Lymphoma Myeloma Leuk. 2018. doi:10.1016/j.clml.2018.07.081
93. Mato AR, Shah NN, Jurczak W, et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): a phase 1/2 study. Lancet. 2021;397:892–901. doi:10.1016/S0140-6736(21)00224-5
94. Reiff SD, Mantel R, Smith LL, et al. The btk inhibitor arq 531 targets ibrutinib-resistant cll and Richter transformation. Cancer Discov. 2018;8:1300–1315. doi:10.1158/2159-8290.CD-17-1409
95. Woyach J, Stephens DM, Flinn IW, et al. Final results of phase 1, dose escalation study evaluating ARQ 531 in patients with relapsed or refractory B-cell lymphoid malignancies. Blood. 2019;134:4298. doi:10.1182/blood-2019-127260
96. Katewa A, Wang Y, Hackney JA, et al. Btk-specific inhibition blocks pathogenic plasma cell signatures and myeloid cell-associated damage in IFNα-driven lupus nephritis. JCI Insight. 2017;2. doi:10.1172/jci.insight.90111
97. Chan P, Yu J, Chinn L, et al. Population pharmacokinetics, efficacy exposure-response analysis, and model-based meta-analysis of fenebrutinib in subjects with rheumatoid arthritis. Pharm Res. 2020. doi:10.1007/s11095-019-2752-y
98. Byrd JC, Smith S, Wagner-Johnston N, et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget. 2018;9:13023–13035. doi:10.18632/oncotarget.24310
99. Fabian CA, Reiff SD, Guinn D, et al. Abstract 1207: SNS-062 demonstrates efficacy in chronic lymphocytic leukemia in vitro and inhibits C481S mutated Bruton tyrosine kinase. Cancer Res. 2017. doi:10.1158/1538-7445.am2017-1207
100. Ciechanover A, Orian A, Schwartz AL. Ubiquitin-mediated proteolysis: biological regulation via destruction. BioEssays. 2000;22:442–451. doi:10.1002/(SICI)1521-1878(200005)22:5<442::AID-BIES6>3.0.CO;2-Q
101. Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi:10.1146/annurev.biochem.70.1.503
102. Sun Y, Zhao X, Ding N, et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018;28:779–781. doi:10.1038/s41422-018-0055-1
103. Huang HT, Dobrovolsky D, Paulk J, et al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem Biol. 2018;25:88–99.e6. doi:10.1016/j.chembiol.2017.10.005
104. Dobrovolsky D, Wang ES, Morrow S, et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood. 2019;133:952–961. doi:10.1182/blood-2018-07-862953
105. Buhimschi AD, Armstrong HA, Toure M, et al. Targeting the C481S ibrutinib-resistance mutation in Bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry. 2018;57:3564–3575. doi:10.1021/acs.biochem.8b00391
106. Sun Y, Ding N, Song Y, et al. Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia. 2019;33:2105–2110. doi:10.1038/s41375-019-0440-x
107. Robbins DW, Kelly A, Tan M, et al. Nx-2127, a degrader of BTK and IMiD neosubstrates, for the treatment of B-cell malignancies. Blood. 2020;136:34. doi:10.1182/blood-2020-141461
108. Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17:768–778. doi:10.1016/S1470-2045(16)30019-5
109. Jain P, Keating M, Wierda W, et al. Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood. 2015. doi:10.1182/blood-2014-09-603670
110. Hillmen P, Rawstron AC, Brock K, et al. Ibrutinib plus venetoclax in relapsed/refractory chronic lymphocytic leukemia: the CLARITY study. J Clin Oncol. 2019;37:2722–2729. doi:10.1200/JCO.19.00894
111. Aw A, Brown JR. The potential combination of BCL-2 inhibitors and ibrutinib as frontline therapy in chronic lymphocytic leukemia. Leuk Lymphoma. 2017;58:2287–2297. doi:10.1080/10428194.2017.1312387
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.