")
Back to Journals » Infection and Drug Resistance » Volume 15
The Relevance of Host Gut Microbiome Signature Alterations on de novo Fatty Acids Synthesis in Patients with Multi-Drug Resistant Tuberculosis
Authors Shi J, Gao G, Yu Z, Wu K, Huang Y, Wu LP, Wu Z, Ye X, Qiu C, Jiang X
Received 6 May 2022
Accepted for publication 19 August 2022
Published 21 September 2022 Volume 2022:15 Pages 5589—5600
DOI https://doi.org/10.2147/IDR.S372122
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Jichan Shi,1,* Gexin Gao,2,* Zhijie Yu,3,* Kaihuai Wu,4 Youquan Huang,5 Lian-Peng Wu,6 Zhengxing Wu,1 Xinchun Ye,1 Chaochao Qiu,1 Xiangao Jiang1
1Department of Infectious Disease, Wenzhou Central Hospital, The Dingli Clinical Institute of Wenzhou Medical University, Wenzhou, Zhejiang, 325000, People’s Republic of China; 2Department of Nursing School, Wenzhou Medical University, Wenzhou, Zhejiang, 325000, People’s Republic of China; 3Department of Hematology, Wenzhou Key Laboratory of Hematology, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325000, People’s Republic of China; 4Departments of Infectious Diseases, Taishun People’s Hospital, Wenzhou, 325000, People’s Republic of China; 5Departments of Infectious Diseases, Yongjia People’s Hospital, Wenzhou, 325000, People’s Republic of China; 6Department of Laboratory, Wenzhou Central Hospital, The Dingli Clinical Institute of Wenzhou Medical University, Wenzhou, Zhejiang, 325000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiangao Jiang, Department of Infectious Disease of Wenzhou Central Hospital, The Dingli Clinical Institute of Wenzhou Medical University, 252 Baili East Road, Lucheng District, Wenzhou, Zhejiang, 325000, People’s Republic of China, Tel +86 13587691822, Fax +86 577 88070007, Email [email protected]
Background: Tuberculosis (TB) is still the single pathogen infectious disease with the largest number of deaths worldwide. The relationship that intestinal microbiota disorder and de novo fatty acid synthesis metabolism have with disease progression in multi-drug resistant TB (MDR-TB) has not yet been fully studied.
Objective: To investigate the effects of long periods of MDR-TB, pre-extensively drug-resistant TB (pre-XDR-TB), or rifampicin-resistant TB (RR-TB) on gut microbiome dysbiosis and advanced disease.
Methods: The sample was chosen between March 2019 and September 2019 in Wenzhou Central Hospital and comprised 11 patients with pre-XDR-TB, 23 patients with RR-TB, and 28 patients with MDR-TB. Healthy individuals were chosen as the control group (CK group). An overnight fast blood sample was drawn via venipuncture into tubes without anticoagulant. For analysis, 300 mg of faeces from patients from the same group was mixed and analysed using DNA extraction, NGS sequencing, and bioinformatics. A QIAamp Fecal DNA Mini Kit was used to isolate the DNA. The extracted DNA was stored at − 20°C.
Results: Advanced TB was concurrent with an elevated level of the proportions of acetyl-CoA carboxylase (ACC1) to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and fatty acid synthase (FASN) to GAPDH in de novo fatty acids synthesis, and Eubacterium, Faecalibacterium, Roseburia, and Ruminococcus were increased significantly in RR-TB patients compared to healthy individuals, whereas their abundance in the pre-XDR-TB and MDR-TB groups showed little change in comparison with the control group. Proteobacteria levels were greatly increased in the RR-TB and MDR-TB patient groups but not in the patients with pre-XDR-TB or the healthy subjects. The pre-XDR-TB group exhibited alterations of the intestinal microbiome: coliform flora showed the highest abundance of Verrucomicrobiales, Enterobacteriales, Bifidobacteriales and Lactobacillales. De novo fatty acids synthesis was enhanced in patients and was associated with the gut microbiome dysbiosis induced by the antimicrobials, with Bacteroidetes, Bacteroidales, and Bacteroidaceae displaying the most important correlations on a phylum, order, and family level, respectively.
Conclusion: The progression to advanced TB was observed to be a result of the interaction between multiple interrelated pathways, with gut–lung crosstalk potentially playing a role in patients with drug-resistant TB.
Keywords: multi-drug resistant TB disease, gut commensal, microbial imbalance, de novo fatty acids synthesis, microbiome biosignature alterations
Background
Tuberculosis (TB) is the main cause of death from a single infectious disease, ranking before HIV/AIDS. It usually affects the lungs (TB), but it can also affect other parts of the body (extrapulmonary TB).1 Drug-resistant TB is an ongoing public health threat. In 2018, there were approximately 500,000 new cases of rifampicin-resistant TB (RR-TB), 78% of which were multi-drug resistant TB (MDR-TB).2
Studies on the effect of pulmonary and intestinal flora imbalance on Mycobacterium TB have provided relatively limited evidence.3 Antibiotics are the main cause of all microbiome disorders; however, first-line TB antibacterial agents, such as pyrazinamide and isoniazid, are prodrugs that are only activated in Mycobacterium TB.4,5 The direct dysregulated effect of Mycobacterium TB on the lung-gut microbial axis remains to be determined. Important indicator phyla and species, such as Bacteroides, Firmicutes, and Verrucomicrobia, in healthy patients and patients with TB should be tracked to determine their role and the potential benefits they could provide for fighting, slowing down, or preventing the pathogenesis of TB.6
Antibiotics are still the main and most powerful factor causing ecological disorders because they indiscriminately destroy all bacteria, both beneficial and harmful.7 Rifampicin produces more extensive changes in intestinal flora imbalance, indicating that the type of TB antimicrobial agent used can affect the degree of flora imbalance.8 Another important issue regarding the interaction between antibiotics and microbiota is the transformation of metabolic pathways that affect fatty acids biosynthesis. In patients receiving treatment, fatty acids oxidation and biosynthesis decreased, indicating that the function/metabolism of anti-TB drugs had an impact on intestinal microbes.9 These findings illustrate the relationship between the metabolic kinetics of intestinal function and the pathogenesis of TB as the consumption of important nutrients may cause the host to lose the necessary components to resist TB.10
A growing body of evidence highlights the significance of free fatty acids in human health, including their interaction with the metabolic states and the immune system.11 An altered serum fatty acid profile has been found to be related to several metabolic conditions, although the underlying mechanism remains unclear. Recent studies also reveal a link between the microbiota in the gut or lung and the host’s metabolism.12 However, although most of these studies13,14 focus on different clinical conditions, there is a lack of evidence concerning the role these mediators play in patients with drug-resistant, extensively drug-resistant (XDR-TB), or MDR-TB. In addition, the underlying cause of the change in the composition of the fatty acids pool is still not clear.
Based on these, it is hypothesised that changes in the composition of the intestinal flora and metabolic activities may be related to changes in the level of nascent fatty acid synthesis, which in turn may be related to inflammation or certain mediators of the immune system. To understand the combined effects of these factors in MDR-TB and pre-XDR-TB, the relationship between selected microbiota and serum nascent fatty acid synthesis levels was examined. The main purpose of this study is to evaluate whether specific gut microbial characteristics are related to changes in the level of nascent fatty acid synthesis in patients with drug-resistant TB, XDR-TB, and MDR-TB.
Methods
Ethical Approval
This research has been approved by the Ethics Committee of Wenzhou Central Hospital and was republished on 7 April 2020 (L2020-02-003X) in accordance with the Chinese Declaration. All participants were fully informed and signed an informed consent form before taking part in this study.
Subjects
Sixty-two subjects were recruited from the patient population of Wenzhou Central Hospital in China and divided into two groups, one of patients with TB and the other comprising healthy individuals. The patient group was further divided into three subgroups. The demographic characteristics of the included population are summarised below. Of the 62 patients with active TB, 11 had XDR-TB, 23 had drug-resistant TB, and 28 had MDR-TB. The subjects’ basic information is presented in Table 1.
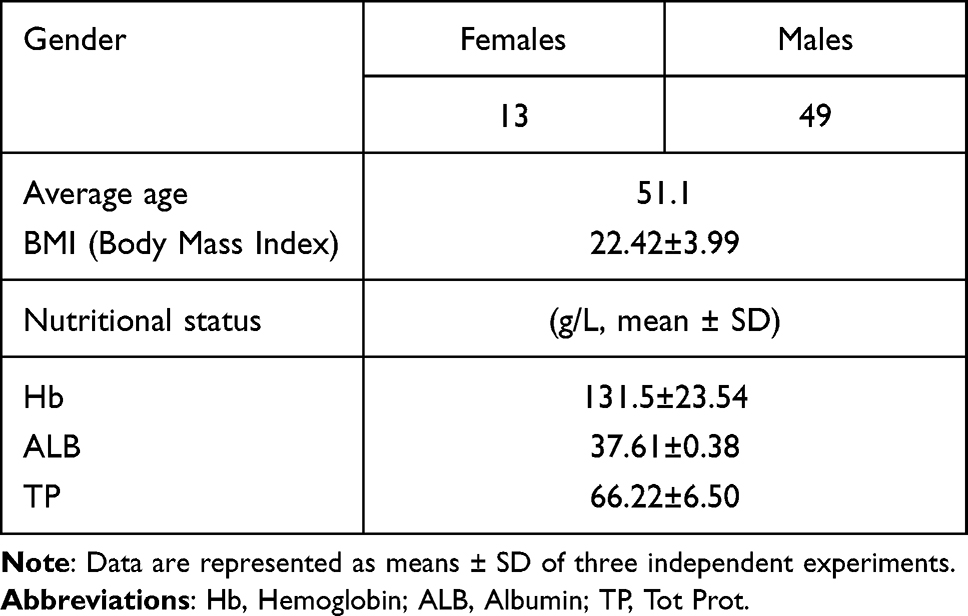 |
Table 1 Basic information of 62 patients with active TB |
Probability sampling was used to select five healthy individuals. Their families were asked about their previous exposure to TB or TB symptoms. All participants were examined by a well-trained doctor. At the start of the experiment and in the second, fourth, and sixth month, they were screened for any potential diseases and infections. The age and gender of the participants were equivalent. On 10 May 2019, the healthy participants, comprising three men and two women, had a physical examination at Wenzhou Central Hospital. Their basic information is exhibited in Table 2.
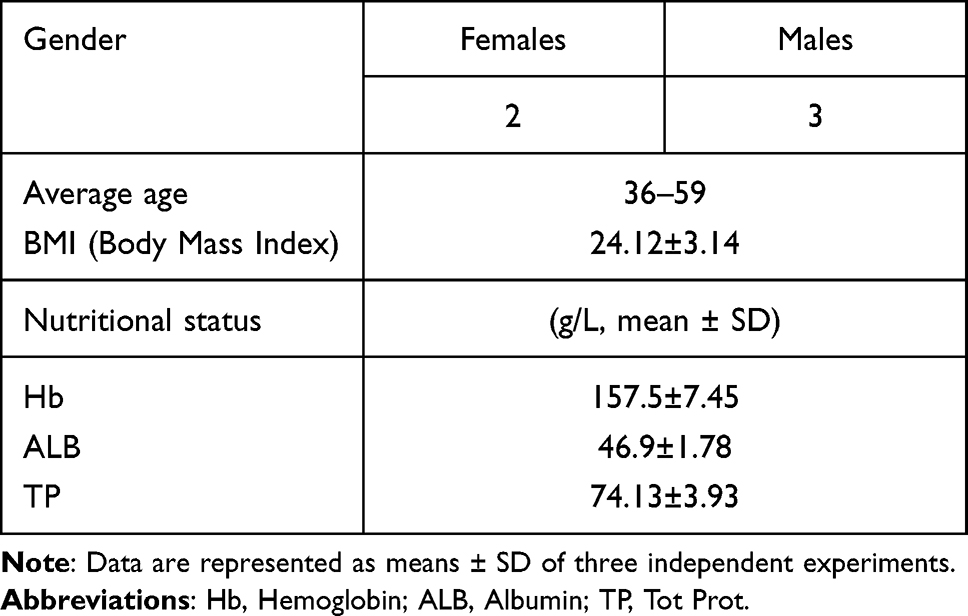 |
Table 2 Basic information of healthy individuals |
TB samples were collected from the confirmed TB patients, who were being treated in TB centres and hospitals. A sample was taken from each participant at the start of anti-TB treatment and three months after the start of treatment. All these patients received anti-TB treatment in accordance with the World Health Organization guidelines followed by the Tuberculosis Center of Wenzhou Central Hospital.
Study Design
The sample selected included 11 former XDR-TB patients, 23 drug-resistant TB patients, and 28 MDR-TB patients who were being treated at Wenzhou Central Hospital between March 2019 and September 2019. The exclusion criteria were at least one of the following: chronic kidney disease, mechanical ventilation, intestinal obstruction, perioperative period, pregnant, positive for AIDS, cancer diagnosis, or changed metabolic conditions and current (in the past six months) use of metabolic agents, probiotics, or antibiotics. The sample was divided into two groups: healthy individuals (CK group) and patients with drug-resistant TB, pre-XDR-TB, and MDR-TB. Venipuncture without anticoagulant was used to obtain fast overnight coagulation. After clotting, the serum was collected and stored at −80°C. A basic serum biochemical analysis was performed using standardized procedures. Stool samples were collected for analysis before and after the experiment. During processing, these were stored at −80°C for observation without interruption.
Stool Sample and Faecal DNA Extraction
To collect the samples, sterile stool sample tubes, frozen biohazard bags, sample collection instructions, foam coolers, and ice packs were given to the subjects’ caregivers. These caregivers were instructed to collect stool samples within one day of each time point, namely at two and eight weeks, and immediately store them in frozen biohazard bags. The samples were then frozen and stored in a refrigerator at −80°C until analysis. For analysis, 300 mg of faeces collected from the same group of individuals was mixed and analyzed via DNA extraction, high-throughput sequencing, and bioinformatics. Following the manufacturer’s instructions, the QIAamp Fecal DNA Mini Kit (Qiagen, USA) was used to isolate metagenomic DNA from the mixed faeces. The extracted DNA was divided into four test tubes to avoid multiple gels and was then stored at −20°C.
Characteristics of Intestinal Microbiota
High-throughput sequencing and intestinal bacterial bioinformatics analysis methods were used to analyse the characteristics of the intestinal microbiota. The high-throughput intestinal genomic DNA that was extracted was divided into appropriate sizes. DNA fragments were connected into complete DNA molecules. After the primer was hydrolyzed, the primer on the lead chain became a completely new chain to realize bridge amplification.15 RDP classifier software (v2.10.1), which is based on the Bergey classification method, was used to classify the processed sequences for species classification, and the naive Bayesian allocation algorithm was used to calculate the p-value for each sequence, with the ranking being performed at different levels. The classification results were mostly reliable (P>0.8). Using Bergey’s Systematic Classification, the dominant bacteria were classified according to their phylum, order, and family.
The empirical research methods used by this study are mainly in accordance with the bioinformatics analysis of microbial identification, evolution, and drug resistance of high-throughput sequencing.16,17
Assay of mRNA Expression of CPT1, GAPDH, FASN, and ACC1
A spin column blood total RNA purification kit, which was purchased from Sangon Biotech, Shanghai, China, was used to isolate the total RNA from the blood samples, and a BeyoRT™ III First Strand cDNA Synthesis Kit, which was purchased from Beytime, Shanghai, China, was used to reverse transcribe the extracted RNA into cDNA. The qRT-PCR method was used to detect the relative mRNA abundance, and the primer sequences can be seen in Table 3. The reaction conditions were 95°C for 5 minutes, 95°C for 15 seconds, 60°C for 30 seconds, 72°C for 30 seconds, and 40 cycles. All samples were repeated in triplicate. To evaluate the success of the PCR amplification, a melting curve analysis was performed at the end of the amplification cycle. The expression level was calculated via the ΔCt method normalized to the internal reference gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression.
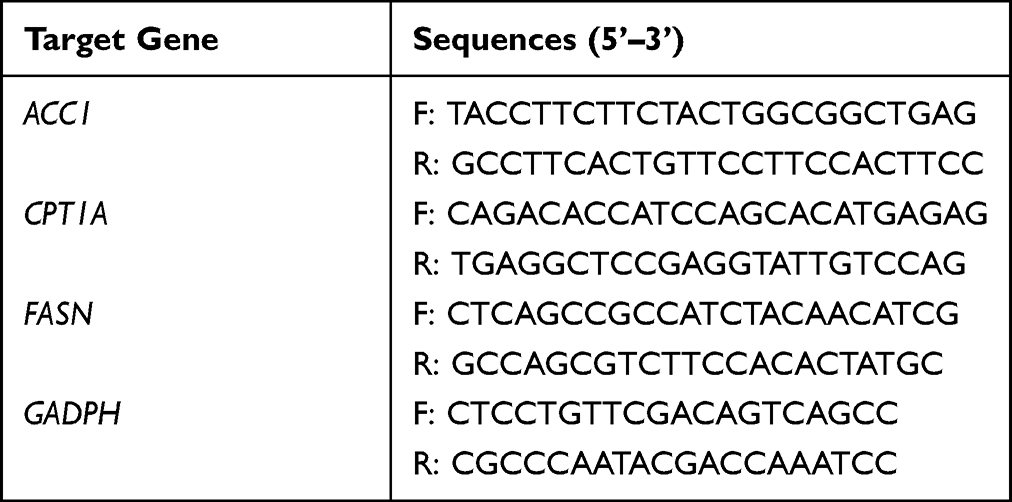 |
Table 3 Primer Sequences for Real-Time PCR |
Statistical Analysis
A one-way variance A test was used to determine the statistically significant relationship between the CK group and the patient group. P ≤ 0.05 demonstrated a significant difference. Statistical analysis software, SPSS (version 13.0) was used to perform the data analysis.
Results
Faecal Bacterial Communities in Patients with TB Compared to Healthy Individuals
A total of 62 patients with TB, aged between 36 and 59 years, was included in the scope of this study. Of these patients, 49 (79%) were males and 13 (21%) were females. The mean BMI of these patients was 24.12 kg/m2. All patients were monitored for three consecutive months. As shown in Figure 1, the diversity of the samples from the patients with RR-TB, pre-XDR-TB, and MDR-TB and the healthy participants was sufficient, which indicates that the sampling was reasonable.
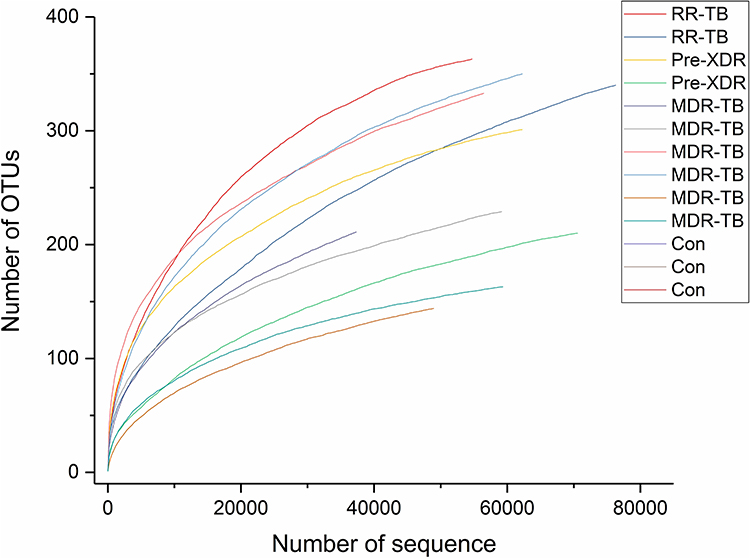 |
Figure 1 Diversity of gut microbiota in each patients with RR-TB, Pre-XDR, MDR-TB and healthy individuals. Abbreviations: MDR-TB, multi-drug resistant TB; pre-XDR-TB, pre-extensively drug-resistant TB; RR-TB, rifampicin-resistant TB, Con, control. |
The α-diversity of each subject, which is a measure of species richness, including chao1, ACE, and the Shannon and Simpson index, was different for the control group and the subject sample with TB. Chao1 and the ACE index represent the gut microbiome varieties in the groups of patients with RR-TB, pre-XDR-TB, and MDR-TB and the group of healthy participants. The results concerning the total variety of bacteria range from those with RR-TB having the most variety to those with pre-XDR-TB to those with MDR-TB and finally to the healthy participants. The bacteria varieties found in the results of the different groups of patients with TB were quite different, while the healthy participants’ results were the least variable in terms of gut microbiome varieties. In contrast, the trend shown by the Shannon and Simpson index was the reverse, with it increasing from those with RR-TB to those with pre-XDR-TB to those with MDR-TB and finally to the healthy participants. Based on the principal analysis, the Shannon and Simpson diversity index suggests that the bacterial diversity of the intestinal microbiome is impacted by treatment (Figure 2).
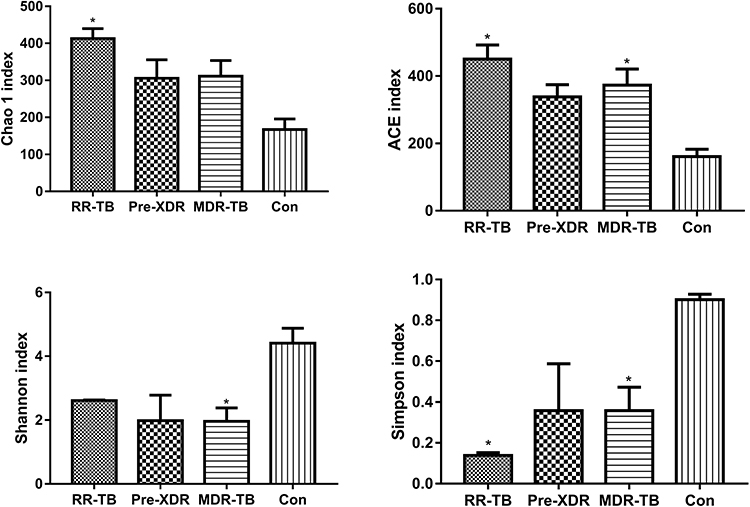 |
Figure 2 Variety differences of gut microbiome (different treatments vs healthy individuals). Abbreviations: MDR-TB, multi-drug resistant TB; pre-XDR-TB, pre-extensively drug-resistant TB; RR-TB, rifampicin-resistant TB, Con, control. Notes: Data are represented as means ± SD of three independent experiments. *p < 0.05. |
The relative abundance of the intestinal microbiome distributions among the RR-TB, pre-XDR-TB, and MDR-TB patients was also studied and compared with those of the healthy participants at different levels. At the phylum level, the proportion of Firmicutes in the gut increased significantly in the patients with RR-TB compared to the healthy participants, whereas their prevalence in the pre-XDR-TB and MDR-TB groups showed little difference to the CK group (Figure 3).
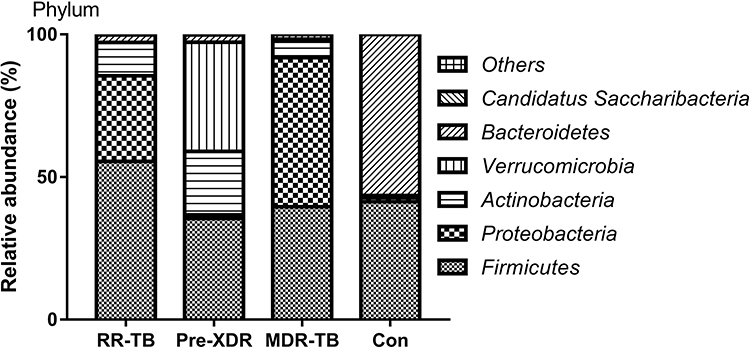 |
Figure 3 Distribution of gut microbiome in different subjects at the phylum level. Abbreviations: MDR-TB, multi-drug resistant TB; pre-XDR-TB, pre-extensively drug-resistant TB; RR-TB, rifampicin-resistant TB, Con, control. |
The Compositions of the Patients’ Gut Microbiome Profiles Were Shifted by the Exposure to Multiple Drugs
Great differences were also found between the participants at the order level linked to the different drug resistance status of the groups, some of which appeared in the RR-TB, pre-XDR-TB, or MDR-TB patients. These signature changes may correlate with the patients’ metabolism. The intestinal bacteria showing the biggest shifts in the patient group at the order level was Bacteroidales (Figure 4). The prevalence of Enterobacterials was strongly positively correlated with enhanced relative multi-drug resistance in the RR-TB and MDR-TB patients. As shown in Figure 4, the patients with increased resistance TB tended to have more abundant Enterobacterials. A similar but weaker correlation was observed between Clostidiales in the gut microbiome and an increase in multi-drug resistance (Figure 4). The beneficial gut microbiota detected were Bacteroidales (65%), Clostridiales (32%), and Selenomonadales (1%), which accounted for more than 97% of the total bacteria in healthy individuals at the order level. This was quite different from the RR-TB (51%), pre-XDR-TB (26%), and MDR-TB (37%) patient groups at treatment initiation, but their levels increased over time during anti-TB treatment. The main bacteria seen in the patients with MDR-TB at the order level were Bacteroidales, Bifidobacteriales, Erysipelotrichales, Coriobacteriales, Clostridiales, and Selenomonadales. The gut microbiome distributions in the patients with RR-TB were similar to those with MDR-TB and only differentiated by their abundance. Both had the same order of Bifidobacteriales, Clostridiales, and Enterobacterials. In addition, interestingly, the patients with pre-XDR-TB had alterations of the intestinal microbiome and coliform flora and showed the highest abundance of Verrucomicrobiales (41%), which is responsible for the interaction of the treatments and corollary resulting from the multiple drugs. The same seemed to be the case for Enterobacteriales, Bifidobacteriales, and Lactobacillales. In particular, the disappearance of Bacteroidales was seen in the patients with RR-TB, pre-XDR-TB, and MDR-TB, which differed slightly from the subjects being treated (Figure 4).
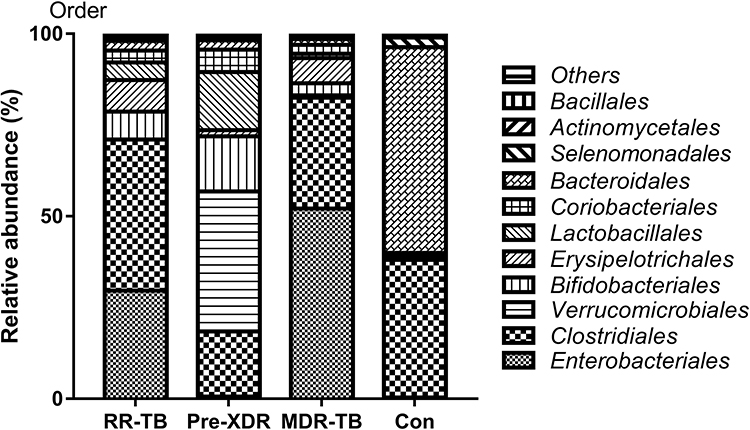 |
Figure 4 Distribution of gut microbiome in different subjects at the order level. Abbreviations: MDR-TB, multi-drug resistant TB; pre-XDR-TB, pre-extensively drug-resistant TB; RR-TB, rifampicin-resistant TB, Con, control. |
Early studies in this field have shown that faecal samples contain a relatively high abundance of pathogens (called dominant bacteria), indicating an increased risk of blood infection due to these dominant bacteria.18 The relative gut microbiome variety of the patient groups at the family level was also compared. The abundance of Lachnospiraceae, Ruminococcaceae, Bifidobacteriaceae, and Prevotellaceae in the intestinal flora of the CK group was greater than that of the patient groups (Figure 5). As noted previously, the presence of the Bacteroidaceae bacteria in the intestine has been shown to have great advantages for body health. This was characterised by the increased number of common pathogens causing nosocomial infection, for example Enterobacteriaceae.19 When the samples of the patient group and the control group were collected and analysed, it was found that the study samples were attenuated with Ruminococcaceae and Bifidobacteriaceae and enriched with Erysipelotrichaceae, Enterobacteriaceae, Lachnospiraceae, and Enterobacteriaceae. Interestingly, all these taxa were dominant in the patients, but the populations of Verrucomicrobiaceae were dominant in the pre-XDR-TB group. These results show that the gut microbiome of those in the patient groups underwent great changes. Clear multi-drug-induced microbiomic changes were observed in most participants, excluding individual differences.
 |
Figure 5 Distribution of gut microbiome in different subjects at the family level. Abbreviations: MDR-TB, multi-drug resistant TB; pre-XDR-TB, pre-extensively drug-resistant TB; RR-TB, rifampicin-resistant TB, Con, control. |
De novo Synthesis of Fatty Acids in the Patient Groups and the Healthy Participants
To address the relationship between gut microbial populations and the de novo synthesis of fatty acids in the patient groups and the healthy participants in this study, the quantitative and qualitative differences in the mRNA expression of CPT1 (carnitine palmitoyl transferase 1), GAPDH, FASN (fatty acid synthase), and ACC1 (acetyl-CoA carboxylase 1) among the study subjects were investigated (Figure 6). These are the key enzymes in the pathway of the de novo synthesis of fatty acids. As shown in Figure 6, differences were observed in the mRNA expression of CPT1, GAPDH, FASN, and ACC1 in the de novo synthesis of fatty acids. It was found that the proportions of ACC1 to GAPDH and FASN to GAPDH showed similar tendencies in the study’s subjects, becoming gradually less in the following order: MDR-TB > pre-XDR-TB > RR-TB > healthy participants.
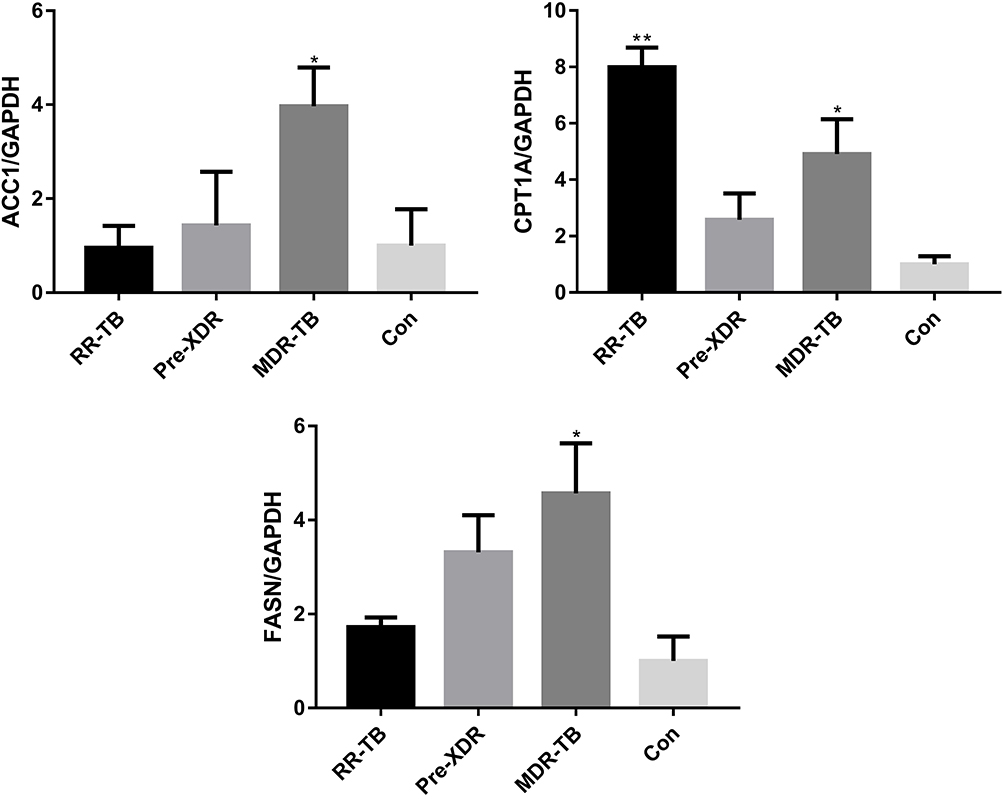 |
Figure 6 Differences of mRNA expression of CPT1, GAPDH, FASN and ACC1 in de novo synthesis of fatty acids. Abbreviations: MDR-TB, multi-drug resistant TB; pre-XDR-TB, pre-extensively drug-resistant TB; RR-TB, rifampicin-resistant TB, Con, control. Notes: Data are represented as means ± SD of three independent experiments.*p < 0.05 and **p < 0.01. |
It can be speculated that the symbiotic balance between the gut microbiome and the host might to some extent promote the proportions of ACC1 to GAPDH and FASN to GAPDH in de novo fatty acids synthesis in patients with multi-drug resistance (Figure 6). However, it is highly probable that some other factor affects the microbiota, for example the immune system of the host.
From this sample analysis, it can be said that the enhancement of de novo fatty acids synthesis was probably caused by the large variety of gut microbiome, with Bacteroidetes, Bacteroidales, and Bacteroidaceae being the most abundant in the healthy participants but also the most variable at a phylum, order, and family level, respectively. CPT1A is the rate-limiting enzyme for fatty acids oxidation, catalysing the esterification of long-chain acyls with carnitine to form acylcarnitine and allowing fatty acid moiety to be transported into the mitochondrial matrix to finally generate adenosine triphosphate.20 A CPT1A to GAPDH axis was required for common augmentation of the de novo fatty acids synthesis responses in the study’s subjects, especially in the patients with RR-TB (Figure 6), which further demonstrates that CPT1A is necessary for reduced immunity. Therefore, it can be concluded that this study found that de novo fatty acids synthesis was enhanced in the patients involved in its investigations and that this had something to do with the gut microbiome dysbiosis induced by antimicrobials, with Bacteroidetes, Bacteroidales, and Bacteroidaceae being the most important correlations at a phylum, order, and family level.
Discussion
The structure of the gut microbiome plays an important role in maintaining the intestinal environment. Under normal conditions, the bacterial structure of the human body is relatively stable and does not provide any indication about the host. Beneficial bacteria account for 70% in the intestines of healthy people and 25% in the intestines of ordinary people.21,22 The intestinal flora can regulate lipids metabolism, and an intestinal flora imbalance can trigger and promote low-grade chronic inflammation, affect the structure and function of the mucosal barrier, and regulate the mucosal barrier system.23
The Firmicutes include many genera belonging to Clostridium clusters IV and XIV,24,25 with some prominent members being Eubacterium, Faecalibacterium, Roseburia, and Ruminococcu. High levels of Proteobacteria appeared in the patients with RR-TB and MDR-TB, but not in the patients with pre-XDR-TB or the healthy participants. Interestingly, Verrucomicrobia was produced abundantly in the patients with pre-XDR-TB, but this was not explored in the other groups. In addition, Bacteroidetes, including bacteria belonging to the genera Bacteroides and Prevotella, were only found in the healthy participants.26 The major genus belonging to the phylum Actinobacteria in the human gut is Bifidobacterium, which plays an extraordinary regulatory role in the prevention and treatment of metabolic disease shifts.27
So far, few studies have covered the biological significance of antimicrobials and the mechanism of fatty acids metabolism. The profiles and compositions of FASN in serum are an important impact factor in the regulation of fatty acids absorption and metabolism, but little attention has been paid to investigating the de novo synthesis of fatty acids in patients with RR-TB, pre-XDR-TB, and MDR-TB.
This study found that the abundance of Bacteroidetes, Bacteroidales, and Bacteroidaceae, as the most important correlations at a phylum, order, and family level at 1–3%, 1–2%, and 0–2%, respectively, was significantly lower in the faeces of the patient groups than in the CK group. The proportions of ACC1 to GAPDH and FASN to GAPDH showed similar tendencies in the study’s subjects, reducing gradually in the following order: MDR-TB > pre-XDR-TB > RR-TB > healthy participants. The proportions in the faeces of the patient groups varied and were higher than in the CK group, demonstrating that antimicrobials can modulate the gut microbiome composition and enhance de novo fatty acids synthesis. The pathway for interactions of de novo fatty acids synthesis and the gut microbiome is summarised in Figure 7.
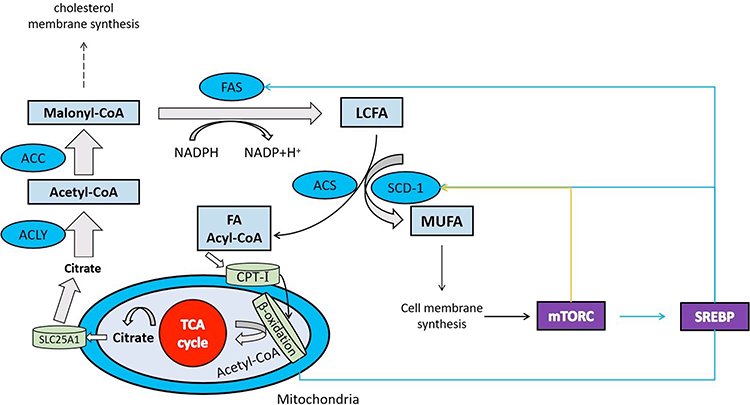 |
Figure 7 Recommended pathway of de novo synthesis of fatty acids. Abbreviations: FAS, Fatty acid synthesis; TCA, Tricarboxylic acid cycle; mTORC, Mammalian target of rapamycin; SREBP, Sterol-regulatory element binding proteins; LCFA, Long- Chain Fatty Acid. |
ACC1 encodes the acetyl-CoA carboxylase and regulates the de novo biosynthesis of fatty acids,28 while FASN is a key enzyme in lipids synthesis and is the only enzyme in the human body capable of converting metabolised sugar into the fatty acid palmitate.29 This saturated fatty acid building block is used to make longer chains, creating polyunsaturated fatty acids used by the cell for energy production and is itself an important component of cellular signal transduction pathways. GAPDH has multiple functions independent of its role in energy metabolism.24 Although increased GAPDH gene expression and enzymatic function have been associated with cell proliferation, conditions such as oxidative stress can impair GAPDH catalytic activity and lead to cellular aging and apoptosis.
Symbiotic gut bacteria can positively influence human immunity, energy expenditure, drug metabolism, and cognitive function, resulting in health benefits. This study has found that the role of intestinal microbiota in health and in disease is different. At present, the research and development of microbial functional genomics and innovative anti-infection drugs are being carried out. For example, the anti-TB drug might have an impact on the composition of the intestinal microbiome. A recent study reports that the α-diversity of intestinal microbiota will decrease after a Mycobacterium TB infection. Further, the decreased α-diversity of intestinal microbiota has been generally observed in the relative abundance of species in Bacteroides.30
Conclusions
This study examined (1) the relationship between intestinal flora and various levels of MDR-TB, (2) the potential for the use of microbiome biosignature alterations to discriminate MDR-TB disease from active TB patients, and (3) the short-term effects of short-term TB antibiotic therapy on the intestinal flora and health of patients based on the alterations of de novo fatty acids synthesis metabolism in patients with MDR-TB, as well as a possible link between shifts of de novo fatty acids synthesis metabolism and TB recrudescence following successful cure. A recommended pathway of de novo synthesis of fatty acids was also discussed, whereby the gut microbiome could serve as a target for clinical manipulation to have an impact on MDR-TB.
In general, this study compares patients with healthy participants, providing a theoretical basis for larger well-designed trials. It is possible that the provision of anti-TB drugs to different patients with drug-resistant forms of the disease could negatively impact the clinical outcomes for chronically ill patients due to the discriminative alterations of the gut microbiome and de novo fatty acids synthesis shifts. In addition, further studies need to be conducted to confirm the factors associated with increased susceptibility to MDR-TB, including changes in de novo fatty acids synthesis and intestinal microbiota caused by long-term anti-TB antibiotic therapy. This may speed up the treatment of TB and improve the cure rate, and it could be a breakthrough in the treatment of drug-resistant TB.
Abbreviations
TB, tuberculosis; MDR-TB, multi-drug resistant TB; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; FASN, fatty acid synthase; ACC1, acetyl-CoA carboxylase 1; pre-XDR-TB, pre-extensively drug-resistant TB; RR-TB, rifampicin resistant TB.
Data Sharing Statement
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Wenzhou Central Hospital. All participants were informed and signed an informed consent form before being included in the study.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
Basic Public Welfare Projects of Zhejiang Province, China (LGF18H010003). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
None of the authors reported a conflict of interest related to the study.
References
1. World Health Organization. Global Tuberculosis Report. Geneva: World Health Organization; 2019:13–24.
2. Asante J, Osei Sekyere J. Understanding antimicrobial discovery and resistance from a metagenomic and metatranscriptomic perspective: advances and applications. Environ Microbiol Rep. 2019;1–25. doi:10.1111/1758-2229.12735
3. Dumas A, Corral D, Colom A, et al. The host microbiota contributes to early protection against lung colonization by Mycobacterium tuberculosis. Front Immunol. 2018;9:2656. doi:10.3389/fimmu.2018.02656
4. Osei Sekyere J, Reta MA, Maningi NE, Fourie PB. Antibiotic resistance of Mycobacterium tuberculosis complex in Africa: a systematic review of current reports of molecular epidemiology, mechanisms and diagnostics. J Infect. 2019;79:550–571. doi:10.1016/j.jinf.2019.10.006
5. Osei Sekyere J, Asante J. Emerging mechanisms of antimicrobial resistance in bacteria and fungi: advances in the era of genomics. Future Microbiol. 2018;13:241–262. doi:10.2217/fmb-2017-0172
6. Hu Y, Yang Q, Liu B, et al. Gut microbiota associated with pulmonary tuberculosis and dysbiosis caused by anti-tuberculosis drugs. J Infect. 2019;78(4):317–322. doi:10.1016/j.jinf.2018.08.006
7. Yu H. Enhancement of nitrogen and phosphorus removal from eutrophic water by annual ryegrass bombarded with low energy ions. Environ Sci Pollution Res. 2014;21(16):9617–9625.
8. Wipperman MF, Fitzgerald DW, Juste MAJ, et al. Antibiotic treatment for tuberculosis induces a profound dysbiosis of the microbiome that persists long after therapy is completed. Sci Rep. 2017;7(1):10767. doi:10.1038/s41598-017-10346-6
9. Maji A, Misra R, Dhakan DB, et al. Gut microbiome contributes to impairment of immunity in pulmonary tuberculosis patients by alteration of butyrate and propionate producers. Environ Microbiol. 2018;20:402–419. doi:10.1111/1462-2920.14015
10. Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. GPCR-mediated signaling of metabolites. Cell Metab. 2017;25(4):777–796. doi:10.1016/j.cmet.2017.03.008
11. Kim YS, Lee HM, Kim JK, et al. PPAR-alpha activation mediates innate host defense through induction of lipid catabolism. J Immunol. 2017;198:3283–3295. doi:10.4049/jimmunol.1601920
12. Hauptmann M, Schaible UE. Linking microbiota and respiratory disease. FEBS Lett. 2016;590:3721–3738. doi:10.1002/1873-3468.12421
13. Santo CE, Caseiro C, Martins MJ, et al. Gut microbiota, in the halfway between nutrition and lung function. Nutrients. 2021;13(5):1716. doi:10.3390/nu13051716
14. Lai HC, Lin TL, Chen TW, et al. Gut microbiota modulates COPD pathogenesis: role of anti-inflammatory Parabacteroides goldsteinii lipopolysaccharide. Gut. 2021;2021:
15. Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi:10.1038/nature09944
16. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi:10.1093/bioinformatics/btq461
17. Eren AM, Zozaya M, Taylor CM, Dowd SE, Martin DH, Ferris MJ. Exploring the diversity of Gardnerella vaginalis in the genitourinary tract microbiota of monogamous couples through subtle nucleotide variation. PLoS One. 2011;6:e26732. doi:10.1371/journal.pone.0026732
18. Marchesi JR, Adams DH, Fava F, et al. The gut microbiota and host health: a new clinical frontier. Gut. 2016;65:330–339. doi:10.1136/gutjnl-2015-309990
19. Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi:10.1038/nature11053
20. Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306. doi:10.1038/nature17038
21. Henao-Mejia J, Elinav E, Jin C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi:10.1038/nature10809
22. Fredborg M, Theil PK, Jensen BB, Purup S. G protein-coupled receptor120 (GPR120) transcription in intestinal epithelial cells is significantly affected by bacteria belonging to the Bacteroides, Proteobacteria, and Firmicutes phyla. J Anim Sci. 2012;90(Suppl. 4):10–12. doi:10.2527/jas.53792
23. Baay-Guzman GJ, Duran-Padilla MA, Rangel-Santiago J. Dual role of hypoxia-inducible factor 1 alpha in experimental pulmonary tuberculosis: its implication as a new therapeutic target. Future Microbiol. 2018;13:785–798. doi:10.2217/fmb-2017-0168
24. Namasivayam S, Kauffman KD, McCulloch JA, et al. Correlation between disease severity and the intestinal microbiome in Mycobacterium tuberculosis-infected rhesus macaques. mBio. 2019;10:18–19. doi:10.1128/mBio.01018-19
25. Bravo M, Combes T, Martinez FO, et al. Lactobacilli isolated from wild boar (Sus scrofa) antagonize Mycobacterium bovis Bacille Calmette-Guerin (BCG) in a species-dependent manner. Front Microbiol. 2019;10:1663. doi:10.3389/fmicb.2019.01663
26. Rodriguez JM, Murphy K, Stanton C, et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb Ecol Health Dis. 2015;26:26050. doi:10.3402/mehd.v26.26050
27. Nicholson JK, Holmes E, Kinross J, et al. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–1267. doi:10.1126/science.1223813
28. Baud S, Guyon V, Kronenberger J, et al. Multifunctional acetyl-CoA carboxylase 1 is essential for very long chain fatty acid elongation and embryo development in Arabidopsis. Plant J. 2003;33:75–86. doi:10.1046/j.1365-313X.2003.016010.x
29. Vaca Jacome AS, Rabilloud T, Schaeffer-Reiss C, et al. N-terminome analysis of the human mitochondrial proteome. Proteomics. 2015;15:2519–2524. doi:10.1002/pmic.201400617
30. Bae B-I, Hara MR, Cascio MB, et al. Mutant Huntingtin: nuclear translocation and cytotoxicity mediated by GAPDH. Proc Nat Acad Sci. 2006;103:3405–3409. doi:10.1073/pnas.0511316103
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.