")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 9
The potential use of biomarkers in predicting contrast-induced acute kidney injury
Authors Andreucci M, Faga T, Riccio E, Sabbatini M, Pisani A, Michael A
Received 27 January 2016
Accepted for publication 9 March 2016
Published 9 September 2016 Volume 2016:9 Pages 205—221
DOI https://doi.org/10.2147/IJNRD.S105124
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Michele Andreucci,1 Teresa Faga,1 Eleonora Riccio,2 Massimo Sabbatini,2 Antonio Pisani,2 Ashour Michael,1
1Department of Health Sciences, University “Magna Graecia” of Catanzaro, Catanzaro, 2Department of Public Health, University of Naples Federico II, Naples, Italy
Abstract: Contrast-induced acute kidney injury (CI-AKI) is a problem associated with the use of iodinated contrast media, causing kidney dysfunction in patients with preexisting renal failure. It accounts for 12% of all hospital-acquired kidney failure and increases the length of hospitalization, a situation that is worsening with increasing numbers of patients with comorbidities, including those requiring cardiovascular interventional procedures. So far, its diagnosis has relied upon the rise in creatinine levels, which is a late marker of kidney damage and is believed to be inadequate. Therefore, there is an urgent need for biomarkers that can detect CI-AKI sooner and more reliably. In recent years, many new biomarkers have been characterized for AKI, and these are discussed particularly with their use in known CI-AKI models and studies and include neutrophil gelatinase-associated lipocalin, cystatin C (Cys-C), kidney injury molecule-1, interleukin-18, N-acetyl-β-d-glucosaminidase, and L-type fatty acid-binding protein (L-FABP). The potential of miRNA and metabolomic technology is also mentioned. Early detection of CI-AKI may lead to early intervention and therefore improve patient outcome, and in future any one or a combination of several of these markers together with development in technology for their analysis may prove effective in this respect.
Keywords: radiocontrast media, acute renal failure, markers, renal injury
Introduction
Acute kidney injury (AKI) induced by contrast – known as contrast-induced AKI (CI-AKI) or contrast-induced nephropathy (CIN) – is defined as a renal impairment occurring 24–72 hours after the intravascular injection of radiographic contrast media (CM) in the absence of an alternative etiology. It is usually a nonoliguric, asymptomatic, and transient decline in renal function, generally occurring within 24 hours of contrast administration, usually peaking on the third to the fifth day and returning to the baseline within 10–14 days.
The incidence of AKI has risen in the last decade, likely as a reflection of aging populations, which has led to an increase in multiple comorbidities, such as infection-related hospitalization, and increase in use of nephrotoxic drugs, such as aminoglycosides, nonsteroidal anti-inflammatory drugs, chemotherapeutic drugs, and intravenous (IV) contrast agents.1 The use of CM has risen in recent years due to an increase in radiographic procedures, and this coupled with an aging population already suffering from diabetes and cardiovascular/renal diseases (including chronic kidney disease, hypertension, hypotension, advanced congestive heart failure, and compromised left ventricular performance) has led to an increase in the incidence of CI-AKI.2–8 CI-AKI is a common cause of hospital-acquired kidney failure, accounting for 12% of all cases.9,10
Pathophysiology of CI-AKI
The effects of CM that lead to AKI are not fully understood, although two main mechanisms, direct cellular toxicity and effects on renal hemodynamics, are believed to be involved.11–14 The direct toxic effects of CM have been studied in various cells, including renal epithelial and mesangial cells.15 The functional and structural changes observed as a result of CM action included cell death, a decrease in cell viability, and an increase in brush border and lysosomal enzyme activity;16 cellular DNA fragmentation;17 downregulation of signaling molecules involved in cell survival such as Akt and upregulation of signaling molecules in cell death such as the p38 and c-Jun N-terminal kinase members of the mitogen-activated protein kinases and the transcription factor nuclear factor κB as well as caspase activation.13,18,19 Nuclear factor κB and c-Jun N-terminal kinases are believed to be involved in the upregulation of the proinflammatory cytokine interleukin-8 (IL-8).14 It is believed that CM may induce the release of the vasoconstrictor molecules endothelin-1 and adenosine that may contribute to reduced blood flow and hence lead to hypoxic conditions in the renal parenchyma.15 The hypoxic conditions thus generated may lead to production of reactive oxygen species (ROS) that may have detrimental effects within the cells per se.20 The reaction between the ROS superoxide anion with nitric oxide will also give rise to the even more potent oxidant, peroxynitrite anion. Indeed, the administration in vivo of a recombinant manganese superoxide dismutase to rats undergoing treatment with the high-osmolar CM diatrizoate caused an improvement in the glomerular filtration rate (GFR) and a reduction in histological damage.21
Diagnosis of CI-AKI: the necessity for novel biomarkers
The diagnosis of CI-AKI is usually based on an absolute (≥0.5 mg/dL) or relative (by ≥25%) increase in serum creatinine from baseline.22,23 This is because variation in serum creatinine levels after CM application has been interpreted as indicating nephrotoxicity by contrast, even though such a variation may occur without CM administration.24,25 In some cases, contrast-induced AKI is a severe ARF with oliguria (<400 mL/24 h), requiring dialysis. In these patients, the mortality is high.25,26
It is likely that the renal injury begins immediately after CM administration and that sensitive early biomarkers could detect the kidney injury very soon,27 and to this effect much effort has been made in recent years to identify early, specific biomarkers to allow an early diagnosis of AKI and hopefully improve the patients’ outcome.28 The term biomarker (or biological marker) has been defined as any substance, structure, or process that can be measured in the body or its products and influence or predict the incidence of outcome or disease,29 while the National Institutes of Health Biomarkers Definitions Working Group defined a biomarker as a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or results of therapy.29
A desirable feature of any biomarker is that it is sensitive and quantifiable and is able to be analyzed rapidly in order to allow timely clinical decisions (interventions) to be made. In addition, it would also be beneficial if the biomarker is able to give the prognosis and check the efficacy of any therapy. In this respect, knowledge of the mechanism or pathophysiology of the disease state is important in order to identify biomarkers associated with the disease that would offer information as to the progress of the disease and efficacy of any therapy as evidenced by changes in the levels of the biomarker. The urgency for novel biomarkers in renal injury is highlighted when one considers that for a measurable change in serum creatinine to be significant it is possible that 50% of the nephrons have already been injured.30
The studied biomarkers may be an expression of kidney dysfunction, ie, a change in glomerular filtration (eg, serum creatinine, Cys-C, and microalbumin) or the result of impaired tubular function (eg, N-acetyl-β-d-glucosaminidase [NAG]) or upregulated proteins (eg, kidney injury molecule-1 [KIM-1], neutrophil gelatinase-associated lipocalin [NGAL], and IL-18) resulting from renal injury. Figure 1 shows the sites of origin of some of these potential biomarkers within the nephron of the kidney.
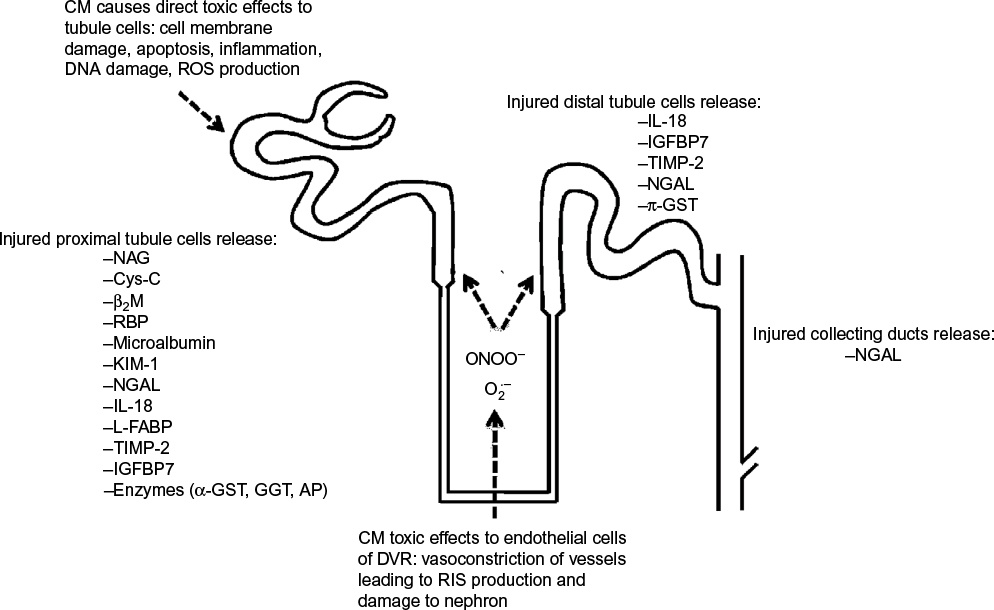 | Figure 1 Illustration of toxicity caused by CM in the nephron and sites of origin of biomarkers. Note: RIS: superoxide ( Abbreviations: CM, contrast media; RIS, reactive inflammatory species; ROS, reactive oxygen species; IL-18, interleukin-18; IGFBP7, insulin-like growth factor-binding protein 7; TIMP-2, tissue inhibitor of metalloproteinases-2; NGAL, neutrophil gelatinase-associated lipocalin; π-GST, pi-glutathione S-transferase; NAG, N-acetyl-β-d-glucosaminidase; Cys-C, cystatin C; β2M, β2-microglobulin; RBP, retinol-binding protein; KIM-1, kidney injury molecule-1; L-FABP, L-type fatty acid-binding protein; α-GST, alpha-glutathione S-transferase; GGT, gamma-glutamyl transpeptidase; AP, alkaline phosphatase; DVR, descending vasa recta. |
 ) and peroxynitrite (ONOO-).
) and peroxynitrite (ONOO-).The characteristics of an ideal biomarker have been cited by many authors,31–33 and a list of these is given in Table 1.
 | Table 1 Desirable characteristics of potential biomarkers of CI-AKI Abbreviations: CI-AKI, contrast-induced acute kidney injury; CM, contrast media; CKD, chronic kidney disease. |
Since the clinical features and the management of CI-AKI are the same as those for AKI due to other causes,34,35 the biomarkers studied to predict an early diagnosis of other types of AKI can also be used to predict CI-AKI. We have listed those biomarkers of AKI for which at least some data are available with regard to their application to CI-AKI.
Creatinine
In the worldwide literature, the renal function is usually evaluated either by measuring serum creatinine or better still by utilizing estimated GFR (calculated GFR from the creatinine clearance: Modification of Diet in Renal Disease formula,25,36 Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation,37 or Cockcroft–Gault formula38).
According to some authors,39 serum creatinine is not an adequate marker of renal function. The reasons for this concern are as follows: 1) Variation in its production from muscles and hence it may vary with age, sex, race, and weight, which affect muscle mass. Thus, daily changes in serum creatinine poorly reflect changes in kidney function in patients with AKI. 2) Creatinine undergoes secretion by renal tubular epithelium. 3) Altered metabolism of creatinine in AKI because severely ill patients are hypercatabolic. 4) Dilution of creatinine during volume overload that may be present in patients with AKI. 5) There are drugs that alter the secretion of creatinine by renal tubules. 6) Creatinine is both a late and indirect reflection of kidney damage. 7) Timed collection of urine is cumbersome and not accurate.32,40,41
Blood urea nitrogen (BUN) has also been used to diagnose AKI and, like serum creatinine, is not an ideal marker of kidney injury, its levels being dependent on nonrenal factors that are independent of kidney function.42
Cystatin C
The increase in serum creatinine after exposure to CM that leads to AKI is delayed, usually achieving a maximum level at 2–5 days after contrast exposure. Thus, the use of another marker of glomerular filtration has been suggested, namely cystatin C (Cys-C).
Cys-C (a 13 kDa protein) is present in all nucleated cells. It is filtered by glomeruli and then metabolized in proximal renal tubule cells following Megalin-mediated endocytosis.43 Unlike creatinine, Cys-C is not secreted in the urine by renal tubules. Its production occurs at a constant rate. Thus, its serum concentration is determined by glomerular filtration.
Cys-C is distributed in the extracellular fluid volume,44 while creatinine is distributed in the total body water,45 a volume which is three times larger. Thus, serum Cys-C rises more rapidly than serum creatinine when GFR decreases. The shorter t1/2 (half-life) of Cys-C explains the earlier change in its serum level compared to serum creatinine.46 Thus, serum Cys-C is better than serum creatinine as an indicator of GFR.47 Since Cys-C is not secreted by renal tubules, its appearance in the urine is attributed to AKI.
Briguori et al48 have demonstrated that a rise in serum Cys-C of <10% at 24 hours will exclude CI-AKI, while a rise ≥10% at 24 hours will be an independent predictor of 1-year major adverse events, including death and dialysis.
When AKI occurs, the reduced GFR will cause a rise in serum Cys-C, taking place much earlier than the increase in serum creatinine. In CI-AKI, serum Cys-C has been shown to peak as early as 24 hours after radiocontrast administration, thereby allowing detection of even small changes in GFR.49–52
Herget-Rosenthal et al53 have determined daily serum creatinine and serum Cys-C in 85 patients at high risk to develop AKI; 44 patients developed AKI and 41 served as controls. The authors concluded that serum Cys-C is a useful marker of AKI and may detect AKI 1–2 days earlier than serum creatinine.
It should be noted that both Cys-C and albumin are reabsorbed by Megalin-mediated endocytosis in the renal proximal tubule.43,54 This means that albuminuria inhibits the renal tubular reabsorption of Cys-C, thereby causing its urinary excretion.55 Thus, serum Cys-C cannot be used as a marker of GFR in nephrotic syndrome. Moreover, the analytic evaluation of Cys-C is modified by many associated pathologic conditions.32
The cellular injury in the renal proximal tubules will cause the appearance of Cys-C in the urine much earlier than the occurrence of histologic changes in proximal tubular epithelium. When compared with serum Cys-C, urine Cys-C appears to be an earlier biomarker of AKI.32
Microalbumin
The term microalbumin indicates the urinary albumin in a concentration that is below the threshold of detection of albumin by conventional urinary dipstick. Its value ranges between 30 mg/L and 300 mg/L. While microalbuminuria is believed to be an important marker of alteration of glomerular structure and function,56 in vivo rat studies have suggested that albuminuria may result from failure of proximal tubule cells to retrieve the protein by endocytosis during renal injury.57 Microalbuminuria has been utilized as a biomarker to investigate attenuation of CI-AKI by N-acetylcysteine.58
The occurrence of albuminuria has been shown to precede the increase of urinary NAG and serum creatinine.59
A drawback of urine albumin, as marker of AKI, is its presence also in chronic renal failure.
N-Acetyl-β-d-glucosaminidase
NAG is a lysosomal enzyme (>130 kDa) that is produced by the cells of renal proximal tubules. In healthy subjects, NAG is present in the urine in small amounts.
Because of its large molecular weight, NAG cannot be filtered by glomeruli: thus, only renal tubule disruption leads to its increase in the urine.60
Urinary NAG is increased when the renal proximal tubular cells are damaged in AKI.61 It has been demonstrated that this increase takes place in AKI 12 hours to 4 days earlier than the rise in serum creatinine.62 The very high urinary NAG in AKI is correlated with poor prognosis.63
Increased urinary excretion of NAG may also be the result of increased lysosomal activity without cell disruption.60
Ren et al64 have studied 590 patients who underwent percutaneous coronary angiography for acute coronary syndromes or stable angina. Urinary NAG, osmolality, and serum creatinine were measured before and 1 day, 2 days, and 6 days after a low-osmolality nonionic radiocontrast agent injection. CI-AKI occurred in 33 patients. In these patients, urinary NAG and serum creatinine, on days 1 and 2 after the radiocontrast injection, were significantly higher than at baseline and when compared with patients without CI-AKI. Urinary NAG levels peaked earlier and increased much more than those of serum creatinine. The authors concluded that NAG may be used as early biomarker of CI-AKI in patients undergoing coronary angiography and/or therapeutic percutaneous coronary intervention (PCI).
Urinary KIM-1
KIM-1 is a phosphatidylserine receptor that when expressed in epithelial cells enables them to recognize and phagocytose dead cells that are present in the stressed kidney.65 It has been found that the ectodomain of KIM-1 is shed into the urine of humans after proximal tubule ischemic or toxic injury. In normal subjects, KIM-1 is undetectable in the urine.66,67 Han et al68 demonstrated that urinary KIM-1 is much more elevated in ischemic acute tubular necrosis than in CI-AKI.
Following a renal tubular injury, it is upregulated and delivered into the extracellular space and into the urine.67,69
The use of urinary KIM-1 as a biomarker of AKI is based on the fact that there is no KIM-1 expression in the kidneys of healthy subjects, and on its upregulation in the apical cell membrane of tubules during AKI.67
Vijayasimha et al70 have carried out a study to evaluate whether KIM-1 allows the identification of CI-AKI earlier than the rise in serum creatinine. The study involved 100 consecutive patients with normal serum creatinine undergoing angiographic procedure. Urinary KIM-1 was assessed at 4 hours, 8 hours, and 24 hours after the angiographic procedure, while serum creatinine was measured at basal, 24 hours, and 48 hours after the procedure. CI-AKI occurred in 12% of the patients. A significant increase in urinary KIM-1 was observed after 24 hours of coronary angiography (using iodixanol or iopromide), while there were no changes in estimated GFR up to 48 hours after the angiography. Studies on urinary KIM-1 have also been performed in cases of AKI due to causes other than CM.
It has been demonstrated that urinary KIM-1 predicts AKI71 much more effectively than NGAL in subjects who underwent cardiac surgery.72
Sabbisetti et al73 have demonstrated that in surgical patients with AKI, plasma KIM-1 was higher than in patients without AKI (area under the curve [AUC] 0.96).
Liangos et al74 have shown that urinary KIM-1 and urinary NAG did predict adverse prognosis in 201 patients with AKI due to different causes.
Jost et al75 using a rat model for age and diabetes-related renal impairment found a significant increase in KIM-1 transcript levels (measured by quantitative reverse transcription polymerase chain reaction) 24 hours after administration of the iso-osmolar CM iodixanol.
KIM-1 may be detected by conventional enzyme-linked immunosorbent assay (ELISA), and a more sensitive modification of this technique was developed that requires less sample volume.74
Neutrophil gelatinase-associated lipocalin
NGAL is a 25 kDa protein associated with human neutrophil gelatinase that belongs to the superfamily of the lipocalins.76
The homodimeric form of NGAL is produced by neutrophils. But the monomeric (mainly) and the heterodimeric forms are the prevalent forms produced by tubules.77
The concentration of NGAL in normal subjects is 20 ng/mL, both in the serum and in the urine. NGAL is filtered by glomeruli and then reabsorbed by proximal tubules where it is partly degraded by megalin and partly excreted in the urine. In patients with albuminuria, there is an increase in its excretion even without tubular cell damage.
Following renal tubular cell damage, NGAL is released into the plasma and the urine; this causes a rise in its plasma and urine concentration, much earlier than the increase in serum concentration of creatinine.32 Thus, NGAL has the potential to act as a powerful and independent predictor of AKI.78–81
Many clinical studies and reviews are reported in the literature concerning the potential role of NGAL as a reliable diagnostic and prognostic biomarker of AKI, since its serum and urinary levels increase earlier and show a better sensitivity than serum creatinine. Some authors stated the plasma NGAL level to be less specific than its urinary concentration.66,80
Mishra et al78 have demonstrated that NGAL is a good biomarker of AKI in children undergoing cardiopulmonary bypass. AKI occurred in 20 out of 71 children, while serum creatinine was increased only 1–3 days after the procedure, urinary NGAL rose from 1.6 µg/L at baseline to 147 µg/L and serum NGAL increased from 3.2 µg/L at baseline to 61 µg/L only 2 hours after the cardiopulmonary bypass. Similar results have been obtained by other authors.82–86
Wagener et al87 studied 81 adult patients undergoing cardiac surgery at Columbia University Medical Center of New York; 16 (20%) developed postoperative AKI. Patients who developed AKI had significantly higher peak postoperative urinary NGAL (5,994±7,616 ng/mL, n=16) compared with patients who did not develop AKI (1,760±3,527 ng/mL, n=65; P=0.0014).
Haase-Fielitz et al,88–90 Fadel et al,91 and Krawczeski et al92 have found similar results in a prospective study of adult cardiac surgery patients.
In studies performed in critically ill patients with septic shock, Nickolas et al93 found urinary NGAL to predict AKI with an AUC of 0.95.
Martensson et al41,94 studied 65 patients admitted to the general intensive care unit (ICU) to evaluate the impact of inflammation/sepsis on the concentrations of NGAL in plasma and urine in adult ICU patients in order to obtain an early detection of AKI in patients with septic shock; 27 patients had systemic inflammatory response syndrome, severe sepsis, or septic shock without AKI and 18 patients had septic shock and concomitant AKI. Plasma NGAL was raised in patients with systemic inflammatory response syndrome, severe sepsis, and septic shock and, according to the authors, should be used with caution as a marker of AKI in ICU patients with septic shock. The urinary NGAL is more useful in predicting AKI, as its levels are normal in septic patients who do not have AKI. Similar results were reported by Constantin et al95 and, in children, by Wheeler et al.96
Studies have also been performed in transplanted patients to figure out whether NGAL may be a biomarker of renal dysfunction after transplantation.
Niemann et al97 have evaluated the possibility of using NGAL as a marker of early renal injury in patients undergoing liver transplantation. Serum NGAL increased only 2 hours after transplantation and predicted AKI in all patients.
Li et al98 investigated whether NGAL and L-FABP (liver-type fatty acid binding protein) could be good markers for an early diagnosis of AKI in liver transplanted patients. The two markers were detected in the urine of 25 patients 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, and 120 hours after the hepatic transplantation. Urinary NGAL levels were only slightly elevated at 2 hours in the transplanted subjects who were not affected by AKI while it rose and stayed high from 2 hours to 6 hours in the patients with AKI.
It has been demonstrated that NGAL is also an early, sensitive, and predictive biomarker of AKI induced by some nephrotoxic drugs.99–101
Parikh et al102 evaluated urinary NGAL and IL-18 in renal transplanted patients (who received the kidney from cadaver donor) in order to assess whether they can be early biomarkers for delayed function of the transplanted organ because of tubule cell injury. In patients who had delayed graft function, peak posttransplant serum creatinine requiring dialysis occurred at days 2–4 after transplantation, while urine NGAL and IL-18 on day 0 were already higher than in recipients of living donor kidneys or deceased donor kidneys with prompt graft function (P<0.0001).
In a prospective, multicenter, observational cohort study of deceased donor kidney transplant patients to evaluate whether urinary NGAL, IL-18, and KIM-1 could be used as biomarkers for predicting dialysis within 1 week of transplant followed by graft recovery, it was concluded that urinary NGAL and IL-18, but not KIM-1, predict the future need for dialysis.103
Thus, in addition to the usefulness of urinary NGAL in the early prediction of the occurrence of AKI, this marker, when its levels are high, has been shown to predict poor prognosis (dialysis and mortality).86,95,97,104–108
According to Hirsch et al,109 NGAL concentrations in urine and plasma are early and powerful independent predictors of CI-AKI in children undergoing elective cardiac catheterization with radiocontrast medium.
Similar results were reported by Ling et al,110 Shaker et al,111 and others41,112,113 in adult patients undergoing PCI.
Haase et al114 performed a pooled analysis of aggregate data from ten prospective observational studies of NGAL, including 2,322 critically ill patients; 19.2% of patients had increased NGAL without relevant changes in serum creatinine. Thus, they defined “subclinical AKI” when patients have increased urinary NGAL while serum creatinine is still normal. It remains unclear whether in these cases NGAL is an indicator of subclinical AKI or simply a marker of severity of illness of these patients.28
In conclusion, NGAL is a good marker of AKI, since its increase in the serum and in the urine occurs earlier than the rise in serum creatinine.66
Urinary IL-18
IL-18 is a cytokine that belongs to the IL-1 superfamily.
IL-18 is involved in acute ischemic AKI. It is doubled in the renal proximal tubules of the kidneys of AKI patients and is produced from the IL-18 precursor by the action of caspase-1.115,116
Urine levels of IL-18 are increased in acute tubular necrosis but not in prerenal azotemia. It is a sensitive and specific biomarker of AKI.117
A meta-analysis of 23 studies involving 4,512 patients has demonstrated that urine IL-18 is a good biomarker of AKI in patients undergoing cardiac surgery, in patients admitted in ICUs, and in coronary care units.118 Parikh et al102 evaluated the role of urinary NGAL and IL-18 to predict delayed graft function in kidney transplanted patients from cadaver donors: urinary IL-18 (like urinary NGAL) represents an early, predictive biomarker of delayed graft function after kidney transplantation.102 Urinary IL-18, like urinary NGAL, is an early noninvasive, accurate predictor of the need for dialysis within the first week after kidney transplantation.103
Parikh et al82 have evaluated whether IL-18 is a predictive biomarker of AKI after cardiopulmonary bypass, as they have shown for urinary NGAL.78 Their study was performed using 20 patients who developed AKI and 35 controls who did not develop AKI after cardiopulmonary bypass. Using serum creatinine as a marker, AKI was detected 2–3 days after the cardiopulmonary bypass. But, when measuring levels of urinary IL-18, it was observed that these had already increased 4–6 hours after cardiopulmonary bypass, peaking at 12 hours, and remaining markedly elevated up to 48 hours after cardiopulmonary bypass. The results indicate that IL-18, like NGAL, is an early, predictive biomarker of AKI after cardiopulmonary bypass.
The same group119 carried out a case–control study within the Acute Respiratory Distress Syndrome (ARDS) Network trial to figure out whether urinary IL-18 is an early diagnostic marker of AKI. The authors concluded that urinary IL-18 can be used for the early diagnosis of AKI and also to predict the mortality of patients who have acute respiratory distress syndrome and are in the ICU.
The same group120 also carried out a prospective, multicenter cohort study involving 311 children undergoing surgical treatment of cardiac lesions. On the basis of their results, the authors concluded that urinary IL-18 and urinary NGAL, but not plasma NGAL, are associated with the subsequent AKI and poor outcomes among children undergoing cardiac surgery.
But different results were obtained by Siew et al,121 who prospectively evaluated the capacity of urinary IL-18, measured within 24 hours of admission, to predict AKI, death, and receipt of acute dialysis in a large mixed adult population of 451 ICU patients (86 developed AKI within 48 hours of enrollment). On the basis of their results, the authors concluded that urine IL-18 is not a good marker of AKI.
In addition, studies of AKI caused by CM have yielded conflicting results with regard to IL-18 as a biomarker of injury.
He et al122 carried out a study on 180 patients who underwent coronary interventional procedures at the Department of Cardiology. Urine IL-18 levels were found to be increased in the CI-AKI patients 6–12 hours after the CM injection, compared with those in the non-CI-AKI patients (P<0.01). The authors concluded that urine IL-18 is a good marker of CI-AKI.
Ling et al110 have reached the same conclusion: urinary IL-18, like NGAL, is quite effective, in allowing an early diagnosis of CI-AKI compared with serum creatinine (P<0.05).
Different results have been obtained by Bulent Gul et al,123 who carried out a case–control study using a cohort of 157 patients admitted to hospital for elective PCI for stable angina. Fifteen patients (9.5%) had CI-AKI. No statistically significant difference was observed in urinary IL-18 in the 15 cases of CI-AKI compared with non CI-AKI patients; neither was a significant difference detected between urinary IL-18 before and after PCI. The authors concluded that urine IL-18 cannot be used as a marker of CI-AKI following intravascular injection of iodinated CM.
Hayashi and Izawa66 have also disputed the use of IL-18 as a reliable biomarker of CI-AKI.
Liver-type Fatty Acid-Binding Protein
The FABP family of proteins consists of 15 kDa proteins present in the cytoplasm of tissues with fatty acid metabolism, serving as intracellular lipid chaperones that transport lipids within the cell. In proximal tubular cells, FABPs transport free fatty acids to the mitochondria or peroxisomes where they are metabolized by β-oxidation.124 Two types of FABP are found in the kidney: liver-type (L-) FABP, located in the renal proximal convoluted and straight tubules (it can also be reabsorbed from the glomerular filtrate via megalin, a multiligand proximal tubule endocytic receptor), and heart type (H-) FABP, located in the renal distal tubules. In normal subjects, L-FABP is not detected in the urine. Kamijo et al, using a transgenic mouse model expressing human L-FABP, correlated the levels of urinary L-FABP with stress suffered by the proximal tubules and also presented clinical data correlating urinary L-FABP with severity of tubulointerstitial injury.125 Under normal conditions, L-FABP is present in the lysosomal compartment of the renal proximal convoluted and straight tubules.52
Experimental studies have demonstrated that increased urine L-FABP is a good marker of ischemic AKI.126 Thus, Negishi et al127 have carried out a study to evaluate whether urinary L-FABP could be used to monitor histological injury in AKI induced by cis-platinum injection and ischemia reperfusion. AKI of different severity was induced by cis-platinum injection and ischemia time in human L-FABP transgenic mice. Renal histological injury scores increased with both cis-platinum dose and ischemic time. In the AKI induced by cis-platinum, urinary L-FABP increased exponentially as early as 2 hours after toxin injection, while BUN increased only at 48 hours. In the AKI induced by ischemia, BUN levels increased only in the 30-minute ischemia group followed by 24 hours of reperfusion, while urinary L-FABP increased >100-fold, even in the 5 minute ischemia group followed by 1 hour of reperfusion. On the basis of their results, the authors concluded that urinary L-FABP is the best marker for early detection of histological and functional insults in ischemic and nephrotoxin-induced AKI.
L-FABP is a good marker of AKI in patients treated with cisplatin,128 in patients with septic shock,99,129 and in subjects receiving CM.130
Clinical studies have demonstrated that urinary L-FABP was elevated a few hours after cardiopulmonary bypass surgery, appearing as an independent risk indicator of AKI post-cardiac surgery, suggesting it to be a sensitive, predictive, and early biomarker of AKI after cardiac surgery.128,131 Similar results have been observed in patients admitted to the ICU with sepsis.132
Yamamoto et al126 have studied the potential role of L-FABP as a biomarker of renal ischemia following human kidney transplantation from living-related donors. Urinary L-FABP was measured in the first urine produced from 12 kidney transplant recipients immediately after reperfusion of the transplanted organ. At the same time, intravital video analysis of peritubular capillary blood flow was performed. The authors found a significant direct correlation between urinary L-FABP and both peritubular capillary blood flow and the ischemic time of the transplanted kidney (P<0.0001). These data showing an increase in urinary L-FABP after ischemic–reperfusion injury (IRI) suggest that L-FABP is a biomarker of AKI.
L-FABP may be useful when it has to be decided to perform either a combined kidney–liver transplantation or a liver transplantation alone in patients with liver disease. If urinary L-FABP is present, the kidneys are damaged, suggesting a combined kidney–liver transplantation. Under such condition, serum creatinine, in fact, does not help since it does not increase, despite there being damage to the kidney.133
Urinary levels of the L-FABP have been shown to increase in patients who develop AKI after exposure to IV CM.133–137
Bachorzewska-Gajewska et al112 studied 25 patients with normal serum creatinine who underwent PCI for unstable angina: urinary L-FABP increased significantly, like urinary NGAL, after 4 hours and remained elevated up to 48 hours.
The observations of a study of 91 hospitalized patients performed by Ferguson et al138 are also worthy of note. These authors demonstrated that urinary L-FABP is not only a good diagnostic marker of AKI (five patients had CI-AKI) but has also prognostic significance, since its urinary levels were significantly higher in patients with a poor outcome, defined as the requirement for renal replacement therapy.
However, some authors deny any predictive value of L-FABP for CI-AKI.66 In addition, Li et al,98 who studied the behavior of NGAL and L-FABP in liver transplant recipients, concluded that L-FABP is not a marker of AKI in liver transplant recipients.
Netrins
Netrins belong to the laminin-related family. There are three secreted netrins (netrin-1, -3, and -4).139 Netrins are located in the peritubular capillaries of the kidneys. During IRI, netrin-1 is highly concentrated within the renal tubular epithelial cells.140 In various mice models of AKI (IRI and toxin-induced injury), levels of netrin-1 were found to be induced in renal tubules, and its urinary levels dramatically increased. Urinary netrin-1 increased early (within a few hours) and dramatically and returned to baseline within 24 hours, and in some cases preceded BUN and creatinine by 24 hours.141 It can be measured in urine by sodium dodecyl sulfate polyacrylamide gel electrophoresis, Western blots, and commercially available ELISA kits.
Midkine
Midkine (MK) is a 13 kDa growth factor with various biological roles, including inflammation;37 it regulates cell growth and survival, migration, and antiapoptotic activity in nephrogenesis and development.142
In the mouse kidneys, MK expression is found in the proximal tubular cells where it is tentatively believed to play a role in promoting the survival of these cells143 and is also present in endothelial cells.144 In a mouse model of IRI, it was found that MK levels were increased threefold in proximal tubular cells, leading to neutrophil recruitment and resulting in an inflammatory reaction and tubulointerstitial damage.143 It was believed that superoxide anions produced during the IRI procedure led to the upregulation of MK, and it is feasible that MK expression could be altered during CI-AKI. Malyszko et al145 investigated whether MK could represent an early biomarker of CI-AKI in 89 patients with normal serum creatinine levels, who underwent PCI for angina, using the iso-osmolar CM iodixanol. Serum MK was evaluated before and 2 hours, 4 hours, 8 hours, 24 hours, and 48 hours after the administration of the radiocontrast medium; serum creatinine was assessed before and 24 hours and 48 hours after CM injection. CI-AKI – defined as an increase in serum creatinine by >25% of the baseline 48 hours after PCI – occurred in 10% of the patients. In these patients with CI-AKI, the authors found a significant rise in serum MK after only 2 hours (P<0.001 compared to the baseline values); MK remained significantly higher 4 hours after the administration of CM to return to the baseline values after 24 hours. In this same study, NGAL levels were significantly higher at 2 hours (serum NGAL) or 4 hours (urinary NGAL) after PCI. Cys-C was higher at 8 hours and 24 hours after PCI in patients with CI-AKI.
Cell cycle arrest markers
A recent study by Kashani et al146 identified the urine insulin-like growth factor-binding protein 7 (IGFBP7) and the tissue inhibitor of metalloproteinases-2 (TIMP-2), as both involved in cell cycle arrest.147,148 Kashani et al146 carried out a prospective, multicenter investigation in which the two new biomarkers of AKI – IGFBP7 and TIMP-2 – were found in the urine of adult patients at high risk of AKI. The study had two phases: discovery phase and validation phase. In the discovery phase, blood and urine samples were obtained from the patients of three cohorts (all three from ICUs, after which the data were then pooled) with the purpose of identifying new biomarkers of AKI among 340 proteins. In the validation phase, a fourth cohort was called Sapphire study146 and was made up of 35 ICUs (total of 744 subjects) in North America (460 subjects) and Europe (284 subjects). AKI occurred in 14% of Sapphire subjects. The two biomarkers IGFBP7 and TIMP-2 from discovery were validated. Urine IGFBP7 and TIMP-2 together demonstrated an AUC of 0.80, much greater than that of all known markers of AKI, none of which achieved an AUC >0.72. The authors conclude that both are excellent biomarkers of AKI and provide additional information over other clinical variables. Cell cycle arrest may result as a consequence of cellular damage, in particular DNA damage, thus preventing the cell from entering cell division. Cellular DNA damage by CM has been reported;17 and therefore, it is feasible that the CM may upregulate these cell cycle arrest markers in renal tubule cells and may be detected in urine after CM-induced renal injury.
It should be noted that while some other authors have confirmed that TIMP-2 and IGFBP7 are good markers of AKI,149–153 other authors have denied their usefulness as markers of AKI.154
Alpha-glutathione S-transferase and pi-glutathione S-transferase
These two enzymes are present in many organs, kidneys included. They cannot be found in the urine of normal subjects. Following renal injury, alpha-glutathione S-transferase (α-GST) is detected in the tubular proximal cells, pi-glutathione S-transferase (π-GST) in the distal tubular cells.60
In the prospective study of Westhuyzen et al,62 urine samples from 26 patients who were admitted to the ICU were collected twice daily for up to 7 days. Four patients (15.4%) developed AKI (ie, at least 50% increase in serum creatinine that occurred between 12 hours and 4 days after the admission). The two tubular enzymes, α-GST and π-GST, in the urine indexed to urine creatinine gave an AUC of 0.893 and 0.929, respectively. The authors concluded that urinary tubular enzymes are useful in predicting AKI. However, in an in vivo study using various rat models of hypertension, diabetes, and nephropathy,155 no significant increase in α-GST was observed after IV administration with the low-osmolar CM (LOCM) iohexol.
Gamma-glutamyl transpeptidase and alkaline phosphatase
Gamma-glutamyl transpeptidase (GGT) and alkaline phosphatase are two enzymes on the brush border of the renal proximal tubules, which appear in the urine when damage of the brush border occurs.60
Westhuyzen et al62 have studied four cases of AKI out of 26 consecutive patients who were admitted to the ICU. The markers GGT, alkaline phosphatase, NAG, α-GST, and π-GST were increased in the subjects with AKI when compared with patients who did not develop AKI (P<0.05), on admission and they remained elevated at 24 hours. The levels of GGT in the urine were very high in 22 patients injected with the high-osmolar CM diatrizoate and in 12 patients with the LOCM iohexol, 24 hours after CM administration, while there was no increase in serum creatinine.156 A more recent study set out to investigate the predictive value of GGT for risk of CI-AKI in patients who underwent primary PCI. Patients were divided into three groups depending on the levels of GGT measured in the patients on admission (group 1: GGT <19 U/L, group 2: GGT 19–33 U/L, and group 3: GGT >33 U/L). The study found that CIN incidence was significantly higher in group 3 and concluded that GGT was a significant predictor of CI-AKI.157
β2-Microglobulin
β2-Microglobulin (β2M) is an 11.8 kDa protein that is filtered by glomeruli and reabsorbed by the renal proximal tubules.158 While low levels of β2M are found in urine and serum of normal subjects, they rise after renal injury due to decreased reabsorbance by the damaged tubules.
In a study of 68 cases of multiple myeloma undergoing computerized tomography scan, 12 patients who had received an LOCM suffered from CI-AKI within 7 days.159 It was observed that β2M levels correlated with the occurrence of CI-AKI, but other parameters used in the study, namely albumin level and BUN–creatinine ratio, did not.
Retinol-binding protein
Retinol-binding protein is a 21 kDa protein that is filtered by glomeruli and is reabsorbed by proximal tubules. It has been shown to be a good marker of AKI.160 It has been used as a marker in assessing prophylactic treatments for CI-AKI.161
MicroRNA molecules as potential biomarkers of CI-AKI
MicroRNA (miRNA) molecules are involved in proliferation, differentiation, and death of cells as well as in inflammation. It is reasonable to suggest therefore that they may be implicated in the pathogenesis of CI-AKI. The identification of such molecules will enable a further understanding of the pathogenesis of kidney injury as well as serving as biomarkers of the injury. One advantage of using miRNAs as biomarkers is their stability in serum, urine, and saliva,162 with some reports suggesting that they may be stable in urine samples after several freeze–thaw cycles and even up to 24 hours at room temperature,163 while others have reported a significant level of viability of miRNA molecules after 5 days of storage at 4°C.164 A disadvantage is that miRNA levels in body fluids are low and require sensitive and specialized tools for analysis.
The miR-21 has been extensively studied and found to play a role in cell proliferation and downregulation of apoptosis after renal IRI and inflammation.165–168 Serum and urine levels of miR-21 also predicted the progression of AKI in cardiac surgery patients.169 In an in vivo mouse model study, renal ischemia reperfusion caused the increase of several miRNA molecules in plasma and kidneys when compared with sham-treated mice, at 3 hours, 6 hours and 24 hours following the ischemic injury.170 These molecules were correlated with plasma creatinine and histological observations of tubular degeneration and necrosis. In particular, the mRNA targets of one of these molecules, miR-1897-3p, included that of nuclear casein kinase and cyclin-dependent kinase substrate 1. Targets of nuclear casein kinase and cyclin-dependent kinase substrate 1 have been implicated in renal injury, inflammation, and apoptosis.170 Another study utilized serum samples from normal subjects and patients with AKI; miRNA molecules could be considered as markers of AKI.171 Notably, several of the miRNA molecules had already been associated with kidney injury: in proximal tubule adhesion and trafficking during IRI (miR-127), in an in vivo mouse model of IRI and in patients with immunoglobulin A nephropathy (miR-146a), and in progression of chronic kidney disease (miR-29a). In addition, several of the miRNA molecules could also be correlated with AKI severity according to the Acute Kidney Injury Network classification.171 Two groups have found several miRNAs whose serum levels were increased in patients with CI-AKI.172,173 Future aims could be directed toward identifying miRNAs that are expressed specifically in the kidney174 and to correlate their changes with kidney injury due to use of CM.
Metabolomic studies to identify biomarkers
Metabolomics may be simply defined as the study and measurement of metabolites present in the cell, tissue, or organism. The last few years has seen a number of studies investigating changes in metabolites accompanying AKI, especially with respect to the use of nephrotoxins such as cisplatin.175,176 A recent study investigated changes in the metabolic profiles in a mouse model of IRI, in which a period of renal ischemia was followed by 2 hours to 7 days of reperfusion,177 using gas chromatography/mass spectrometry and liquid chromatography/mass spectrometry analysis. There were changes in metabolites related to energy and purine metabolism as well as osmotic regulation and inflammation. Changes in a wide range of metabolites may be considered together, giving rise to a signature associated with the injury. In this respect, the authors reported such signatures of inflammation: changes in prostaglandins, increased catabolism of tryptophan via the kynurenate pathway, and changes in arginine metabolism, which has also been associated with renal failure.178 Of note, an increase in the metabolite 3-indoxyl sulfate was observed early on during reperfusion, and it could serve as a potential biomarker, given that it has also been reported as increasing in other models of toxin-induced renal failure.176 In another study, changes were noted in urinary metabolites of patients undergoing computerized tomography scan scan and administered with the LOCM iohexol,179 but the study did not set out to associate any of these changes with CI-AKI.
Biomarker measurement and quantification
The discovery of novel potential biomarkers brings with it fresh challenges for their accurate, rapid, and reproducible measurements. Nephelometry and turbidometry have been used to measure some of these markers (Table 2) and improved particle enhanced turbidometric and nephelometric immunoassays have improved analysis in terms of reduction in sample volume required and time. The protein biomarkers have also been analyzed by the use of ELISA and by Western blotting. The traditional ELISA has several disadvantages including the fact that only one antigen can be detected per assay plate, relatively large volumes of sample are required, the need to optimize the volumes of sample in order for the protein to be measured within an accepted range, and it may take several hours to obtain a result. An advantage however is that it is easy to perform with relatively little technical expertise required. Similar disadvantages are also inherent with Western blotting, which may take as long as the traditional ELISA to yield results, although some companies have developed equipment that can reduce turnaround time. Recent advances in multiplex technologies can reduce the requirement for sample amount and time for analysis while being able to analyze many molecules together.180 These technologies include the use of micrometer plastic beads impregnated with fluorescent dyes and coated with a specific antibody to an antigen of interest (biomarker). Many sets of these beads with each set having a specific antibody to a specific antigen (biomarker) will be combined with the biological sample to be tested (urine and serum), followed by combining with a mixture of detection antibodies (for each biomarker of interest) which have been labeled with a reporter dye different from the dyes within the beads. Then, the bead biomarker–reporter mixture is analyzed through an analytical flow chamber. Another multiplexed assay is based upon the standard 96-well microtiter plate, which offers the possibility of using preexisting equipment for analysis. It should still be noted that reproducibility of data is still dependent on the source of antibodies used and may be subject to batch-to-batch variation.
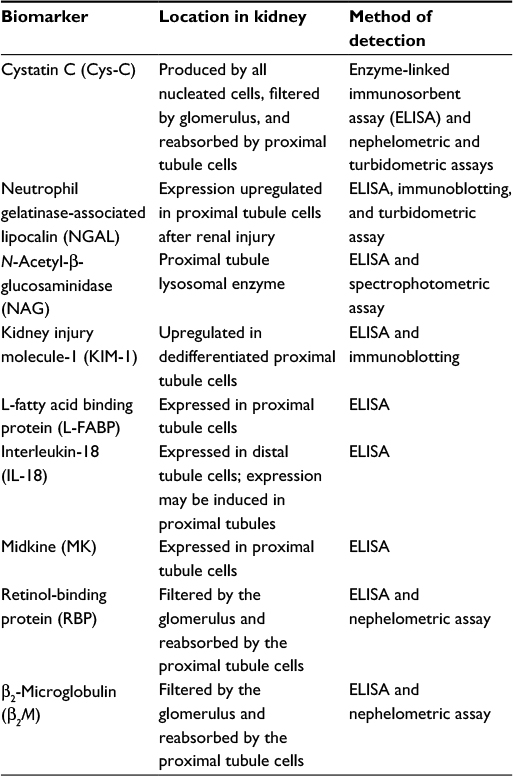 | Table 2 Potential biomarkers of contrast-induced acute kidney injury Notes: Particle-enhanced turbidometric and nephelometric immunoassays are relatively quick, avoiding the need for sample pretreatment and allowing use of less sample. Similarly, the development of bead-based multiplex immunoassays will allow the detection of several molecules at the same time compared with conventional ELISA. |
MiRNA molecule expression may be studied by microarray analysis, real-time polymerase chain reaction, in situ hybridization, solution hybridization, and Northern blotting. Of these, quantitative reverse transcription polymerase chain reaction is the most accurate and sensitive. However, the analysis of miRNAs can be a challenge and time consuming, requiring their isolation from samples and the use of specific reagents and analytical instruments. These techniques will also require a degree of technical ability. Similarly, the study of metabolomics depends on sophisticated equipment such as nuclear magnetic resonance spectroscopy and the already mentioned gas chromatography/mass spectrometry and liquid chromatography/mass spectrometry, which also require skilled operators. In addition, samples may need to be prepared prior to analysis, which will lengthen the procedure, but analysis of metabolites may be aided by the existence of public databases such as the Human Metabolome Database.
Some ideas of potential treatment/prevention of CI-AKI
Once CI-AKI has been established, there are limited treatment options available. The management of CI-AKI is the same as that for AKI due to other causes.5,10,35
What is important is prevention.
The first general rule of prevention is that in any patient undergoing any radiographic procedure, the occurrence of renal injury should be detected as early as possible, and this may be achieved by using several sensitive early biomarkers that may include serum creatinine, Cys-C, NGAL, KIM-1, IL-18, and L-FABP, and to repeat any preventive measures in the few days following the use of CM. This may need to be done even though no robust evidence exists to support the reliability of any specific biomarker in clinical studies. The diagnosis of AKI is even more difficult in the elderly because of their clinical conditions favoring the occurrence of CI-AKI: dehydration, due to impaired sensation of thirst4 and tendency to salt depletion following abnormal renal salt losses associated with insufficient salt intake and reduction in “effective” circulating blood volume, that aggravate renal vasoconstriction, thereby predisposing to AKI.26 The effective circulating blood volume may be defined as the relative fullness of the arterial tree as determined by cardiac output, peripheral vascular resistance, and total blood volume.5 A reduction in the effective circulating blood volume in the elderly may be due to congestive heart failure, compromised left ventricle systolic performance, and prolonged hypotension; under such circumstances, renal vasoconstriction is accentuated, thereby making renal ischemia more severe.3,181
Measures to prevent CI-AKI that may be useful for physicians include: 1) discontinuation of potentially nephrotoxic drugs (aminoglycosides, vancomycin, amphotericin B, metformin, and nonsteroidal anti-inflammatory drugs2; 2) choice of the least nephrotoxic radiocontrast agent (iohexol is less nephrotoxic than diatrizoate; iodixanol and iopamidol seem the least nephrotoxic agents10; 3) use of the lowest dosage possible of CM (some formulas have been suggested to calculate the dosage that is least dangerous for the kidney182–185; 4) adequate hydration of the patient186,187 by IV infusion of 0.9% saline at a rate of ~1 mL/kg body weight/hr or 154 mEq/L infusion of sodium bicarbonate as a bolus of 3 mL/kg bw/hr for 1 hour;188 5) short-duration pretreatment with antioxidants, such as N-acetylcysteine (an oral dose of 600 mg twice daily the day before and the day of procedure10 or an IV dose of 150 mg/kg over half an hour before the procedure or 50 mg/kg administered over 4 hours;189 6) Nebivolol (5 mg/d for 1 week or 5 mg every 24 hours for 4 days).190,191
Summary and future perspectives
CI-AKI is a leading cause of hospital-acquired kidney failure and no doubt will continue to increase with the increase in use of radiological procedures. Serum creatinine is still utilized as marker of kidney function, even though it is a late marker of renal injury and its measurement may be cumbersome and its levels may not reflect kidney damage. Hence, there is an urgent need for new markers, and many promising serum and urinary biomarkers have been identified that may be able to detect CI-AKI prior to the expected rise in serum creatinine, including Cys-C, NGAL, KIM-1, IL-18, and L-FABP, and may give a diagnosis in hours rather than days. However, there are still reports questioning the accuracy and reliability of these new biomarkers in the context of AKI, and more studies are required to clarify their effectivity. There is certainly a need for well-designed, multicenter clinical trials enrolling many patients in order to clarify the potential of a particular biomarker in different sets of patients (age group: elderly adult, adult, child, infant, neonate; and those presenting other disease states: diabetes, cardiovascular disease, etc). In addition, it may be the case that more biomarkers should be quantified in tandem in order to properly evaluate the extent of renal injury. In this respect, newer technologies may be useful in allowing rapid and accurate measurements of several molecules simultaneously, such as the development of bead-based multiplex immunoassays, and further developments in technology will in turn be spurred on by clinical developments in diagnosis of the disease. We can also look forward to other approaches in discovering potential biomarkers such as the use of miRNA molecules and metabolomic analyses, which may however require sophisticated and expensive equipment together with greater technical expertise, but this will be outweighed by the potential benefits. The hope for the future is that this will allow for more timely and successful intervention and lead to improved patient care.
Disclosure
Dr Ashour Michael is currently a recipient of a research grant from University “Magna Graecia” of Catanzaro, Italy. The other authors report no conflicts of interest in this work.
References
Yong K, Dogra G, Boudville N, Pinder M, Lim W. Acute kidney injury: controversies revisited. Int J Nephrol. 2011;2011:762634. | ||
Andreucci M, Solomon R, Tasanarong A. Side effects of radiographic contrast media: pathogenesis, risk factors, and prevention. Biomed Res Int. 2014;2014:741018. | ||
Andreucci M, Faga T, Pisani A, Sabbatini M, Michael A. Acute kidney injury by radiographic contrast media: pathogenesis and prevention. Biomed Res Int. 2014;2014:362725. | ||
Andreucci VE, Russo D, Cianciaruso B, Andreucci M. Some sodium, potassium and water changes in the elderly and their treatment. Nephrol Dial Transplant. 1996;11(suppl 9):9–17. | ||
Andreucci VE, Fuiano G, Russo D, Andreucci M. Vasomotor nephropathy in the elderly. Nephrol Dial Transplant. 1998;13(suppl 7):17–24. | ||
Fuiano G, Mancuso D, Indolfi C, et al. Early detection of progressive renal dysfunction in patients with coronary artery disease. Kidney Int. 2005;68(6):2773–2780. | ||
Andreucci M. Contrast media and nephrotoxicity: a molecular conundrum. G Ital Nefrol. 2011;28(4):355. | ||
Andreucci M. Side effects of radiographic contrast media. Biomed Res Int. 2014;2014:872574. | ||
Nash K, Hafeez A, Hou S. Hospital-acquired renal insufficiency. Am J Kidney Dis. 2002;39(5):930–936. | ||
Gleeson TG, Bulugahapitiya S. Contrast-induced nephropathy. AJR Am J Roentgenol. 2004;183(6):1673–1689. | ||
Heyman SN, Brezis M, Epstein FH, Spokes K, Silva P, Rosen S. Early renal medullary hypoxic injury from radiocontrast and indomethacin. Kidney Int. 1991;40(4):632–642. | ||
Humes HD, Hunt DA, White MD. Direct toxic effect of the radiocontrast agent diatrizoate on renal proximal tubule cells. Am J Physiol. 1987;252(2 pt 2):F246–F255. | ||
Andreucci M, Faga T, Russo D, et al. Differential activation of signaling pathways by low-osmolar and iso-osmolar radiocontrast agents in human renal tubular cells. J Cell Biochem. 2014;115(2):281–289. | ||
Andreucci M, Faga T, Lucisano G, et al. Mycophenolic acid inhibits the phosphorylation of NF-kappaB and JNKs and causes a decrease in IL-8 release in H2O2-treated human renal proximal tubular cells. Chem Biol Interact. 2010;185(3):253–262. | ||
Michael A, Faga T, Pisani A, et al. Molecular mechanisms of renal cellular nephrotoxicity due to radiocontrast media. Biomed Res Int. 2014;2014:249810. | ||
Andersen KJ, Christensen EI, Vik H. Effects of iodinated x-ray contrast media on renal epithelial cells in culture. Invest Radiol. 1994;29(11):955–962. | ||
Hizoh I, Strater J, Schick CS, Kubler W, Haller C. Radiocontrast-induced DNA fragmentation of renal tubular cells in vitro: role of hypertonicity. Nephrol Dial Transplant. 1998;13(4):911–918. | ||
Andreucci M, Fuiano G, Presta P, et al. Radiocontrast media cause dephosphorylation of Akt and downstream signaling targets in human renal proximal tubular cells. Biochem Pharmacol. 2006;72(10):1334–1342. | ||
Andreucci M, Lucisano G, Faga T, et al. Differential activation of signaling pathways involved in cell death, survival and inflammation by radiocontrast media in human renal proximal tubular cells. Toxicol Sci. 2011;119(2):408–416. | ||
Pisani A, Riccio E, Andreucci M, et al. Role of reactive oxygen species in pathogenesis of radiocontrast-induced nephropathy. Biomed Res Int. 2013;2013:868321. | ||
Pisani A, Sabbatini M, Riccio E, et al. Effect of a recombinant manganese superoxide dismutase on prevention of contrast-induced acute kidney injury. Clin Exp Nephrol. 2014;18(3):424–431. | ||
Thomsen HS, Morcos SK. Contrast media and the kidney: European Society of Urogenital Radiology (ESUR) guidelines. Br J Radiol. 2003;76(908):513–518. | ||
Mehran R, Nikolsky E. Contrast-induced nephropathy: definition, epidemiology, and patients at risk. Kidney Int Suppl. 2006;100:S11–S15. | ||
Newhouse JH, Kho D, Rao QA, Starren J. Frequency of serum creatinine changes in the absence of iodinated contrast material: implications for studies of contrast nephrotoxicity. AJR Am J Roentgenol. 2008;191(2):376–382. | ||
Andreucci M, Faga T, Pisani A, Sabbatini M, Michael A. The choice of the iodinated radiographic contrast media to prevent contrast-induced nephropathy. Adv Nephrol. 2014;2014:11. Article ID 691623. | ||
Andreucci M, Federico S, Andreucci VE. Edema and acute renal failure. Semin Nephrol. 2001;21(3):251–256. | ||
Sirota JC, Klawitter J, Edelstein CL. Biomarkers of acute kidney injury. J Toxicol. 2011;2011:328120. | ||
Ostermann M, Philips BJ, Forni LG. Clinical review: biomarkers of acute kidney injury: where are we now? Crit Care. 2012;16(5):233. | ||
Strimbu K, Tavel JA. What are biomarkers? Curr Opin HIV AIDS. 2010;5(6):463–466. | ||
Mussap M, Noto A, Fanos V, Van Den Anker JN. Emerging biomarkers and metabolomics for assessing toxic nephropathy and acute kidney injury (AKI) in neonatology. Biomed Res Int. 2014;2014:602526. | ||
Endre ZH, Walker RJ, Pickering JW, et al. Early intervention with erythropoietin does not affect the outcome of acute kidney injury (the EARLYARF trial). Kidney Int. 2010;77(11):1020–1030. | ||
Charlton JR, Portilla D, Okusa MD. A basic science view of acute kidney injury biomarkers. Nephrol Dial Transplant. 2014;29(7):1301–1311. | ||
Ostermann M, Forni LG. Measuring biomarkers of acute kidney injury during renal replacement therapy: wisdom or folly? Crit Care. 2014;18(3):155. | ||
Briguori C, Colombo A, Violante A, et al. Standard vs double dose of N-acetylcysteine to prevent contrast agent associated nephrotoxicity. Eur Heart J. 2004;25(3):206–211. | ||
Andreucci VE, Fuiano G, Stanziale P, Andreucci M. Role of renal biopsy in the diagnosis and prognosis of acute renal failure. Kidney Int Suppl. 1998;66:S91–S95. | ||
Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130(6):461–470. | ||
Levey AS, Stevens LA, Schmid CH, et al; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration). A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604–612. | ||
Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41. | ||
Bragadottir G, Redfors B, Ricksten SE. Assessing glomerular filtration rate (GFR) in critically ill patients with acute kidney injury – true GFR versus urinary creatinine clearance and estimating equations. Crit Care. 2013;17(3):R108. | ||
Andreucci M, Faga T, Michael A. The renal problems in X-ray based imaging techniques using iodinated radiographic contrast agents. Open Access J Sci Technol. 2015;3:16. Article ID 101145. | ||
Simsek A, Tugcu V, Tasci AI. New biomarkers for the quick detection of acute kidney injury. ISRN Nephrol. 2013;2013:394582. | ||
Luke RG. Uremia and the BUN. N Engl J Med. 1981;305(20):1213–1215. | ||
Kaseda R, Iino N, Hosojima M, et al. Megalin-mediated endocytosis of cystatin C in proximal tubule cells. Biochem Biophys Res Commun. 2007;357(4):1130–1134. | ||
Tenstad O, Roald AB, Grubb A, Aukland K. Renal handling of radiolabelled human cystatin C in the rat. Scand J Clin Lab Invest. 1996;56(5):409–414. | ||
Schloerb PR. Total body water distribution of creatinine and urea in nephrectomized dogs. Am J Physiol. 1960;199:661–665. | ||
Sjostrom P, Tidman M, Jones I. The shorter T1/2 of cystatin C explains the earlier change of its serum level compared to serum creatinine. Clin Nephrol. 2004;62(3):241–242. | ||
Briguori C, Quintavalle C, Donnarumma E, Condorelli G. Novel biomarkers for contrast-induced acute kidney injury. Biomed Res Int. 2014;2014:568738. | ||
Briguori C, Visconti G, Rivera NV, et al. Cystatin C and contrast-induced acute kidney injury. Circulation. 2010;121(19):2117–2122. | ||
Kuwabara T, Mori K, Mukoyama M, et al. Urinary neutrophil gelatinase-associated lipocalin levels reflect damage to glomeruli, proximal tubules, and distal nephrons. Kidney Int. 2009;75(3):285–294. | ||
Hvidberg V, Jacobsen C, Strong RK, Cowland JB, Moestrup SK, Borregaard N. The endocytic receptor megalin binds the iron transporting neutrophil-gelatinase-associated lipocalin with high affinity and mediates its cellular uptake. FEBS Lett. 2005;579(3):773–777. | ||
Mishra J, Ma Q, Prada A, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003;14(10):2534–2543. | ||
Mishra J, Mori K, Ma Q, et al. Amelioration of ischemic acute renal injury by neutrophil gelatinase-associated lipocalin. J Am Soc Nephrol. 2004;15(12):3073–3082. | ||
Herget-Rosenthal S, Marggraf G, Husing J, et al. Early detection of acute renal failure by serum cystatin C. Kidney Int. 2004;66(3):1115–1122. | ||
Amsellem S, Gburek J, Hamard G, et al. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J Am Soc Nephrol. 2010;21(11):1859–1867. | ||
Nejat M, Hill JV, Pickering JW, Edelstein CL, Devarajan P, Endre ZH. Albuminuria increases cystatin C excretion: implications for urinary biomarkers. Nephrol Dial Transplant. 2012;27(suppl 3):iii96–iii103. | ||
Ferguson MA, Vaidya VS, Bonventre JV. Biomarkers of nephrotoxic acute kidney injury. Toxicology. 2008;245(3):182–193. | ||
Russo LM, Sandoval RM, McKee M, et al. The normal kidney filters nephrotic levels of albumin retrieved by proximal tubule cells: retrieval is disrupted in nephrotic states. Kidney Int. 2007;71(6):504–513. | ||
Levin A, Pate GE, Shalansky S, et al. N-acetylcysteine reduces urinary albumin excretion following contrast administration: evidence of biological effect. Nephrol Dial Transplant. 2007;22(9):2520–2524. | ||
Rached E, Hoffmann D, Blumbach K, Weber K, Dekant W, Mally A. Evaluation of putative biomarkers of nephrotoxicity after exposure to ochratoxin a in vivo and in vitro. Toxicol Sci. 2008;103(2):371–381. | ||
de Geus HR, Betjes MG, Bakker J. Biomarkers for the prediction of acute kidney injury: a narrative review on current status and future challenges. Clin Kidney J. 2012;5(2):102–108. | ||
Waring WS, Moonie A. Earlier recognition of nephrotoxicity using novel biomarkers of acute kidney injury. Clin Toxicol. 2011;49(8):720–728. | ||
Westhuyzen J, Endre ZH, Reece G, Reith DM, Saltissi D, Morgan TJ. Measurement of tubular enzymuria facilitates early detection of acute renal impairment in the intensive care unit. Nephrol Dial Transplant. 2003;18(3):543–551. | ||
Chew SL, Lins RL, Daelemans R, Nuyts GD, De Broe ME. Urinary enzymes in acute renal failure. Nephrol Dial Transplant. 1993;8(6):507–511. | ||
Ren L, Ji J, Fang Y, et al. Assessment of urinary N-acetyl-beta-glucosaminidase as an early marker of contrast-induced nephropathy. J Int Med Res. 2011;39(2):647–653. | ||
Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest. 2008;118(5):1657–1668. | ||
Hayashi M, Izawa H. Recent prophylactic strategies and novel biomarkers for contrast-induced acute kidney injury. OA Nephrol. 2014;2:1. | ||
Bonventre JV. Kidney injury molecule-1 (KIM-1): a urinary biomarker and much more. Nephrol Dial Transplant. 2009;24(11):3265–3268. | ||
Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int. 2002;62(1):237–244. | ||
Ichimura T, Bonventre JV, Bailly V, et al. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem. 1998;273(7):4135–4142. | ||
Vijayasimha M, Vijaya Padma V, Mujumdar SKD, Satyanarayana PVV. Kidney injury molecule-1: a urinary biomarker for contrast induced acute kidney injury. Adv Life Sci Technol. 2013;15:33–40. | ||
Koyner JL, Vaidya VS, Bennett MR, et al. Urinary biomarkers in the clinical prognosis and early detection of acute kidney injury. Clin J Am Soc Nephrol. 2010;5(12):2154–2165. | ||
Han WK, Wagener G, Zhu Y, Wang S, Lee HT. Urinary biomarkers in the early detection of acute kidney injury after cardiac surgery. Clin J Am Soc Nephrol. 2009;4(5):873–882. | ||
Sabbisetti VS, Waikar SS, Antoine DJ, et al. Blood kidney injury molecule-1 is a biomarker of acute and chronic kidney injury and predicts progression to ESRD in type I diabetes. J Am Soc Nephrol. 2014;25(10):2177–2186. | ||
Liangos O, Perianayagam MC, Vaidya VS, et al. Urinary N-acetyl-beta-(D)-glucosaminidase activity and kidney injury molecule-1 level are associated with adverse outcomes in acute renal failure. J Am Soc Nephrol. 2007;18(3):904–912. | ||
Jost G, Pietsch H, Sommer J, et al. Retention of iodine and expression of biomarkers for renal damage in the kidney after application of iodinated contrast media in rats. Invest Radiol. 2009;44(2):114–123. | ||
Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268(14):10425–10432. | ||
Cai L, Rubin J, Han W, Venge P, Xu S. The origin of multiple molecular forms in urine of HNL/NGAL. Clin J Am Soc Nephrol. 2010;5(12):2229–2235. | ||
Mishra J, Dent C, Tarabishi R, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet. 2005;365(9466):1231–1238. | ||
Paragas N, Qiu A, Hollmen M, Nickolas TL, Devarajan P, Barasch J. NGAL-Siderocalin in kidney disease. Biochim Biophys Acta. 2012;1823(9):1451–1458. | ||
Haase M, Bellomo R, Devarajan P, Schlattmann P, Haase-Fielitz A; NGAL Meta-analysis Investigator Group. Accuracy of neutrophil gelatinase-associated lipocalin (NGAL) in diagnosis and prognosis in acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis. 2009;54:1012–1024. | ||
Ronco C. Biomarkers for acute kidney injury: is NGAL ready for clinical use? Crit Care. 2014;18(6):680. | ||
Parikh CR, Mishra J, Thiessen-Philbrook H, et al. Urinary IL-18 is an early predictive biomarker of acute kidney injury after cardiac surgery. Kidney Int. 2006;70(1):199–203. | ||
Portilla D, Dent C, Sugaya T, et al. Liver fatty acid-binding protein as a biomarker of acute kidney injury after cardiac surgery. Kidney Int. 2008;73(4):465–472. | ||
Dent CL, Ma Q, Dastrala S, et al. Plasma neutrophil gelatinase-associated lipocalin predicts acute kidney injury, morbidity and mortality after pediatric cardiac surgery: a prospective uncontrolled cohort study. Crit Care. 2007;11(6):R127. | ||
Bennett M, Dent CL, Ma Q, et al. Urine NGAL predicts severity of acute kidney injury after cardiac surgery: a prospective study. Clin J Am Soc Nephrol. 2008;3(3):665–673. | ||
Devarajan P. Biomarkers for the early detection of acute kidney injury. Curr Opin Pediatr. 2011;23(2):194–200. | ||
Wagener G, Jan M, Kim M, et al. Association between increases in urinary neutrophil gelatinase-associated lipocalin and acute renal dysfunction after adult cardiac surgery. Anesthesiology. 2006;105(3):485–491. | ||
Haase-Fielitz A, Bellomo R, Devarajan P, et al. Novel and conventional serum biomarkers predicting acute kidney injury in adult cardiac surgery – a prospective cohort study. Crit Care Med. 2009;37(2):553–560. | ||
Haase M, Bellomo R, Matalanis G, Calzavacca P, Dragun D, Haase-Fielitz A. A comparison of the RIFLE and Acute Kidney Injury Network classifications for cardiac surgery-associated acute kidney injury: a prospective cohort study. J Thorac Cardiovasc Surg. 2009;138(6):1370–1376. | ||
Haase-Fielitz A, Bellomo R, Devarajan P, et al. The predictive performance of plasma neutrophil gelatinase-associated lipocalin (NGAL) increases with grade of acute kidney injury. Nephrol Dial Transplant. 2009;24(11):3349–3354. | ||
Fadel FI, Abdel Rahman AM, Mohamed MF, et al. Plasma neutrophil gelatinase-associated lipocalin as an early biomarker for prediction of acute kidney injury after cardio-pulmonary bypass in pediatric cardiac surgery. Arch Med Sci. 2012;8(2):250–255. | ||
Krawczeski CD, Woo JG, Wang Y, Bennett MR, Ma Q, Devarajan P. Neutrophil gelatinase-associated lipocalin concentrations predict development of acute kidney injury in neonates and children after cardiopulmonary bypass. J Pediatr. 2011;158(6):1009.e1–1015.e1. | ||
Nickolas TL, O’Rourke MJ, Yang J, et al. Sensitivity and specificity of a single emergency department measurement of urinary neutrophil gelatinase-associated lipocalin for diagnosing acute kidney injury. Ann Intern Med. 2008;148(11):810–819. | ||
Martensson J, Bell M, Oldner A, Xu S, Venge P, Martling CR. Neutrophil gelatinase-associated lipocalin in adult septic patients with and without acute kidney injury. Intensive Care Med. 2010;36(8):1333–1340. | ||
Constantin JM, Futier E, Perbet S, et al. Plasma neutrophil gelatinase-associated lipocalin is an early marker of acute kidney injury in adult critically ill patients: a prospective study. J Crit Care. 2010;25(1):176.e1–176.e6. | ||
Wheeler DS, Devarajan P, Ma Q, et al. Serum neutrophil gelatinase-associated lipocalin (NGAL) as a marker of acute kidney injury in critically ill children with septic shock. Crit Care Med. 2008;36(4):1297–1303. | ||
Niemann CU, Walia A, Waldman J, et al. Acute kidney injury during liver transplantation as determined by neutrophil gelatinase-associated lipocalin. Liver Transpl. 2009;15(12):1852–1860. | ||
Li Y, Zhu M, Xia Q, et al. Urinary neutrophil gelatinase-associated lipocalin and L-type fatty acid binding protein as diagnostic markers of early acute kidney injury after liver transplantation. Biomarkers. 2012;17(4):336–342. | ||
Tsuchimoto A, Shinke H, Uesugi M, et al. Urinary neutrophil gelatinase-associated lipocalin: a useful biomarker for tacrolimus-induced acute kidney injury in liver transplant patients. PLoS One. 2014;9(10):e110527. | ||
Gaspari F, Cravedi P, Mandala M, et al. Predicting cisplatin-induced acute kidney injury by urinary neutrophil gelatinase-associated lipocalin excretion: a pilot prospective case-control study. Nephron Clin Pract. 2010;115(2):c154–c160. | ||
Wasilewska A, Zoch-Zwierz W, Taranta-Janusz K, Michaluk-Skutnik J. Neutrophil gelatinase-associated lipocalin (NGAL): a new marker of cyclosporine nephrotoxicity? Pediatr Nephrol. 2010;25(5):889–897. | ||
Parikh CR, Jani A, Mishra J, et al. Urine NGAL and IL-18 are predictive biomarkers for delayed graft function following kidney transplantation. Am J Transplant. 2006;6(7):1639–1645. | ||
Hall IE, Yarlagadda SG, Coca SG, et al. IL-18 and urinary NGAL predict dialysis and graft recovery after kidney transplantation. J Am Soc Nephrol. 2010;21(1):189–197. | ||
Yang HN, Boo CS, Kim MG, Jo SK, Cho WY, Kim HK. Urine neutrophil gelatinase-associated lipocalin: an independent predictor of adverse outcomes in acute kidney injury. Am J Nephrol. 2010;31(6):501–509. | ||
de Geus HR, Bakker J, Lesaffre EM, le Noble JL. Neutrophil gelatinase-associated lipocalin at ICU admission predicts for acute kidney injury in adult patients. Am J Respir Crit Care Med. 2011;183(7):907–914. | ||
Siew ED, Ware LB, Gebretsadik T, et al. Urine neutrophil gelatinase-associated lipocalin moderately predicts acute kidney injury in critically ill adults. J Am Soc Nephrol. 2009;20(8):1823–1832. | ||
Cruz DN, de Cal M, Garzotto F, et al. Plasma neutrophil gelatinase-associated lipocalin is an early biomarker for acute kidney injury in an adult ICU population. Intensive Care Med. 2010;36(3):444–451. | ||
Bagshaw SM, Bennett M, Haase M, et al. Plasma and urine neutrophil gelatinase-associated lipocalin in septic versus non-septic acute kidney injury in critical illness. Intensive Care Med. 2010;36(3):452–461. | ||
Hirsch R, Dent C, Pfriem H, et al. NGAL is an early predictive biomarker of contrast-induced nephropathy in children. Pediatr Nephrol. 2007;22(12):2089–2095. | ||
Ling W, Zhaohui N, Ben H, et al. Urinary IL-18 and NGAL as early predictive biomarkers in contrast-induced nephropathy after coronary angiography. Nephron Clin Pract. 2008;108(3):c176–c181. | ||
Shaker OG, El-Shehaby A, El-Khatib M. Early diagnostic markers for contrast nephropathy in patients undergoing coronary angiography. Angiology. 2010;61(8):731–736. | ||
Bachorzewska-Gajewska H, Poniatowski B, Dobrzycki S. NGAL (neutrophil gelatinase-associated lipocalin) and L-FABP after percutaneous coronary interventions due to unstable angina in patients with normal serum creatinine. Adv Med Sci. 2009;54(2):221–224. | ||
Bachorzewska-Gajewska H, Malyszko J, Sitniewska E, Malyszko JS, Dobrzycki S. Neutrophil-gelatinase-associated lipocalin and renal function after percutaneous coronary interventions. Am J Nephrol. 2006;26(3):287–292. | ||
Haase M, Devarajan P, Haase-Fielitz A, et al. The outcome of neutrophil gelatinase-associated lipocalin-positive subclinical acute kidney injury: a multicenter pooled analysis of prospective studies. J Am Coll Cardiol. 2011;57(17):1752–1761. | ||
Melnikov VY, Ecder T, Fantuzzi G, et al. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J Clin Invest. 2001;107(9):1145–1152. | ||
Edelstein CL, Hoke TS, Somerset H, et al. Proximal tubules from caspase-1-deficient mice are protected against hypoxia-induced membrane injury. Nephrol Dial Transplant. 2007;22(4):1052–1061. | ||
Parikh CR, Jani A, Melnikov VY, Faubel S, Edelstein CL. Urinary interleukin-18 is a marker of human acute tubular necrosis. Am J Kidney Dis. 2004;43(3):405–414. | ||
Liu Y, Guo W, Zhang J, et al. Urinary interleukin 18 for detection of acute kidney injury: a meta-analysis. Am J Kidney Dis. 2013;62(6):1058–1067. | ||
Parikh CR, Abraham E, Ancukiewicz M, Edelstein CL. Urine IL-18 is an early diagnostic marker for acute kidney injury and predicts mortality in the intensive care unit. J Am Soc Nephrol. 2005;16(10):3046–3052. | ||
Parikh CR, Coca SG, Thiessen-Philbrook H, et al. Postoperative biomarkers predict acute kidney injury and poor outcomes after adult cardiac surgery. J Am Soc Nephrol. 2011;22(9):1748–1757. | ||
Siew ED, Ikizler TA, Gebretsadik T, et al. Elevated urinary IL-18 levels at the time of ICU admission predict adverse clinical outcomes. Clin J Am Soc Nephrol. 2010;5(8):1497–1505. | ||
He H, Li W, Qian W, et al. Urinary interleukin-18 as an early indicator to predict contrast-induced nephropathy in patients undergoing percutaneous coronary intervention. Exp Ther Med. 2014;8(4):1263–1266. | ||
Bulent Gul CB, Gullulu M, Oral B, et al. Urinary IL-18: a marker of contrast-induced nephropathy following percutaneous coronary intervention? Clin Biochem. 2008;41(7–8):544–547. | ||
Sweetser DA, Heuckeroth RO, Gordon JI. The metabolic significance of mammalian fatty-acid-binding proteins: abundant proteins in search of a function. Annu Rev Nutr. 1987;7:337–359. | ||
Kamijo A, Sugaya T, Hikawa A, et al. Urinary excretion of fatty acid-binding protein reflects stress overload on the proximal tubules. Am J Pathol. 2004;165(4):1243–1255. | ||
Yamamoto T, Noiri E, Ono Y, et al. Renal L-type fatty acid – binding protein in acute ischemic injury. J Am Soc Nephrol. 2007;18(11):2894–2902. | ||
Negishi K, Noiri E, Doi K, et al. Monitoring of urinary L-type fatty acid-binding protein predicts histological severity of acute kidney injury. Am J Pathol. 2009;174(4):1154–1159. | ||
Negishi K, Noiri E, Sugaya T, et al. A role of liver fatty acid-binding protein in cisplatin-induced acute renal failure. Kidney Int. 2007;72(3):348–358. | ||
Nakamura T, Sugaya T, Koide H. Urinary liver-type fatty acid-binding protein in septic shock: effect of polymyxin B-immobilized fiber hemoperfusion. Shock. 2009;31(5):454–459. | ||
Manabe K, Kamihata H, Motohiro M, Senoo T, Yoshida S, Iwasaka T. Urinary liver-type fatty acid-binding protein level as a predictive biomarker of contrast-induced acute kidney injury. Eur J Clin Invest. 2012;42(5):557–563. | ||
Katagiri D, Doi K, Honda K, et al. Combination of two urinary biomarkers predicts acute kidney injury after adult cardiac surgery. Ann Thorac Surg. 2012;93(2):577–583. | ||
Doi K, Noiri E, Maeda-Mamiya R, et al. Urinary L-type fatty acid-binding protein as a new biomarker of sepsis complicated with acute kidney injury. Crit Care Med. 2010;38(10):2037–2042. | ||
Thurman JM, Parikh CR. Peeking into the black box: new biomarkers for acute kidney injury. Kidney Int. 2008;73(4):379–381. | ||
Nakamura T, Sugaya T, Node K, Ueda Y, Koide H. Urinary excretion of liver-type fatty acid-binding protein in contrast medium-induced nephropathy. Am J Kidney Dis. 2006;47(3):439–444. | ||
Kato K, Sato N, Yamamoto T, Iwasaki YK, Tanaka K, Mizuno K. Valuable markers for contrast-induced nephropathy in patients undergoing cardiac catheterization. Circ J. 2008;72(9):1499–1505. | ||
Vijayasimha M, Vijaya Padma V, Majumdar SKD, Satyanarayana PVV, Yadav A. Kidney injury molecule-1: a urinary biomarker for contrast induced acute kidney injury. Transw Med J. 2013;1(1):22–26. | ||
Nozue T, Michishita I, Mizuguchi I. Predictive value of serum cystatin C, β2-microglobulin, and urinary liver-type fatty acid-binding protein on the development of contrast-induced nephropathy. Cardiovasc Interv Ther. 2010;25(2):85–90. | ||
Ferguson MA, Vaidya VS, Waikar SS, et al. Urinary liver-type fatty acid-binding protein predicts adverse outcomes in acute kidney injury. Kidney Int. 2010;77(8):708–714. | ||
Barallobre MJ, Pascual M, Del Rio JA, Soriano E. The Netrin family of guidance factors: emphasis on Netrin-1 signalling. Brain Res Brain Res Rev. 2005;49(1):22–47. | ||
Wang W, Reeves WB, Ramesh G. Netrin-1 and kidney injury. I. Netrin-1 protects against ischemia-reperfusion injury of the kidney. Am J Physiol Renal Physiol. 2008;294(4):F739–F747. | ||
Reeves WB, Kwon O, Ramesh G. Netrin-1 and kidney injury. II. Netrin-1 is an early biomarker of acute kidney injury. Am J Physiol Renal Physiol. 2008;294(4):F731–F738. | ||
Muramatsu T. Midkine and pleiotrophin: two related proteins involved in development, survival, inflammation and tumorigenesis. J Biochem. 2002;132(3):359–371. | ||
Sato W, Kadomatsu K, Yuzawa Y, et al. Midkine is involved in neutrophil infiltration into the tubulointerstitium in ischemic renal injury. J Immunol. 2001;167(6):3463–3469. | ||
Kosugi T, Yuzawa Y, Sato W, et al. Midkine is involved in tubulointerstitial inflammation associated with diabetic nephropathy. Lab Invest. 2007;87(9):903–913. | ||
Malyszko J, Bachorzewska-Gajewska H, Koc-Zorawska E, Malyszko JS, Kobus G, Dobrzycki S. Midkine: a novel and early biomarker of contrast-induced acute kidney injury in patients undergoing percutaneous coronary interventions. Biomed Res Int. 2015;2015:879509. | ||
Kashani K, Al-Khafaji A, Ardiles T, et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit Care. 2013;17(1):R25. | ||
Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803(1):55–71. | ||
Verhagen HJ, de Leeuw DC, Roemer MG, et al. IGFBP7 induces apoptosis of acute myeloid leukemia cells and synergizes with chemotherapy in suppression of leukemia cell survival. Cell Death Dis. 2014;5:e1300. | ||
Wetz AJ, Richardt EM, Wand S, et al. Quantification of urinary TIMP-2 and IGFBP-7: an adequate diagnostic test to predict acute kidney injury after cardiac surgery? Crit Care. 2015;19:3. | ||
Yamashita T, Doi K, Hamasaki Y, et al. Evaluation of urinary tissue inhibitor of metalloproteinase-2 in acute kidney injury: a prospective observational study. Crit Care. 2014;18(6):716. | ||
Meersch M, Schmidt C, Van Aken H, et al. Urinary TIMP-2 and IGFBP7 as early biomarkers of acute kidney injury and renal recovery following cardiac surgery. PLoS One. 2014;9(10):e93460. | ||
Bihorac A, Chawla LS, Shaw AD, et al. Validation of cell-cycle arrest biomarkers for acute kidney injury using clinical adjudication. Am J Respir Crit Care Med. 2014;189(8):932–939. | ||
Gocze I, Koch M, Renner P, et al. Urinary biomarkers TIMP-2 and IGFBP7 early predict acute kidney injury after major surgery. PLoS One. 2015;10(3):e0120863. | ||
Bell M, Larsson A, Venge P, Bellomo R, Martensson J. Assessment of cell-cycle arrest biomarkers to predict early and delayed acute kidney injury. Dis Markers. 2015;2015:158658. | ||
Rouse RL, Stewart SR, Thompson KL, Zhang J. Kidney injury biomarkers in hypertensive, diabetic, and nephropathy rat models treated with contrast media. Toxicol Pathol. 2013;41(4):662–680. | ||
Parvez Z, Ramamurthy S, Patel NB, Moncada R. Enzyme markers of contrast media-induced renal failure. Invest Radiol. 1990;25(suppl 1):S133–S134. | ||
Oksuz F, Yarlioglues M, Cay S, et al. Predictive value of gamma-glutamyl transferase levels for contrast-induced nephropathy in patients with ST-segment elevation myocardial infarction who underwent primary percutaneous coronary intervention. Am J Cardiol. 2015;116(5):711–716. | ||
Bernier GM. beta 2-Microglobulin: structure, function and significance. Vox Sang. 1980;38(6):323–327. | ||
Pahade JK, LeBedis CA, Raptopoulos VD, et al. Incidence of contrast-induced nephropathy in patients with multiple myeloma undergoing contrast-enhanced CT. AJR Am J Roentgenol. 2011;196(5):1094–1101. | ||
Bernard AM, Vyskocil AA, Mahieu P, Lauwerys RR. Assessment of urinary retinol-binding protein as an index of proximal tubular injury. Clin Chem. 1987;33(6):775–779. | ||
Sadat U, Walsh SR, Norden AG, Gillard JH, Boyle JR. Does oral N-acetylcysteine reduce contrast-induced renal injury in patients with peripheral arterial disease undergoing peripheral angiography? A randomized-controlled study. Angiology. 2011;62(3):225–230. | ||
Weber JA, Baxter DH, Zhang S, et al. The microRNA spectrum in 12 body fluids. Clin Chem. 2010;56(11):1733–1741. | ||
Lorenzen JM, Volkmann I, Fiedler J, et al. Urinary miR-210 as a mediator of acute T-cell mediated rejection in renal allograft recipients. Am J Transplant. 2011;11(10):2221–2227. | ||
Mall C, Rocke DM, Durbin-Johnson B, Weiss RH. Stability of miRNA in human urine supports its biomarker potential. Biomark Med. 2013;7(4):623–631. | ||
Li YF, Jing Y, Hao J, et al. MicroRNA-21 in the pathogenesis of acute kidney injury. Protein Cell. 2013;4(11):813–819. | ||
Sabbatini M, Santillo M, Pisani A, et al. Inhibition of Ras/ERK1/2 signaling protects against postischemic renal injury. Am J Physiol Renal Physiol. 2006;290(6):F1408–F1415. | ||
Andreucci M, Michael A, Kramers C, et al. Renal ischemia/reperfusion and ATP depletion/repletion in LLC-PK(1) cells result in phosphorylation of FKHR and FKHRL1. Kidney Int. 2003;64(4):1189–1198. | ||
Andreucci M, Fuiano G, Presta P, et al. Downregulation of cell survival signalling pathways and increased cell damage in hydrogen peroxide-treated human renal proximal tubular cells by alpha-erythropoietin. Cell Prolif. 2009;42(4):554–561. | ||
Du J, Cao X, Zou L, et al. MicroRNA-21 and risk of severe acute kidney injury and poor outcomes after adult cardiac surgery. PLoS One. 2013;8(5):e63390. | ||
Bellinger MA, Bean JS, Rader MA, et al. Concordant changes of plasma and kidney microRNA in the early stages of acute kidney injury: time course in a mouse model of bilateral renal ischemia-reperfusion. PLoS One. 2014;9(4):e93297. | ||
Aguado-Fraile E, Ramos E, Conde E, et al. A pilot study identifying a set of microRNAs as precise diagnostic biomarkers of acute kidney injury. PLoS One. 2015;10(6):e0127175. | ||
Gutierrez-Escolano A, Santacruz-Vazquez E, Gomez-Perez F. Dysregulated microRNAs involved in contrast-induced acute kidney injury in rat and human. Ren Fail. 2015;37(9):1498–1506. | ||
Sun S, Zhang T, Cai Z, et al. Circulating miRNA-30a,-30e,-188 as Early Biomarkers for CI-AKI diagnosis. Circulation. 2015;132(Suppl 3):A12855. Available from: http://circ.ahajournals.org/content/132/Suppl_3/A12855. Accessed July 15, 2016. | ||
Kito N, Endo K, Ikesue M, Weng H, Iwai N. miRNA profiles of tubular cells: diagnosis of kidney injury. Biomed Res Int. 2015;2015:465479. | ||
Portilla D, Schnackenberg L, Beger RD. Metabolomics as an extension of proteomic analysis: study of acute kidney injury. Semin Nephrol. 2007;27(6):609–620. | ||
Uehara T, Horinouchi A, Morikawa Y, et al. Identification of metabolomic biomarkers for drug-induced acute kidney injury in rats. J Appl Toxicol. 2014;34(10):1087–1095. | ||
Wei Q, Xiao X, Fogle P, Dong Z. Changes in metabolic profiles during acute kidney injury and recovery following ischemia/reperfusion. PLoS One. 2014;9(9):e106647. | ||
Sabbatini M, Pisani A, Uccello F, et al. Arginase inhibition slows the progression of renal failure in rats with renal ablation. Am J Physiol Renal Physiol. 2003;284(4):F680–F687. | ||
Diercks DB, Owen KP, Tolstikov V, Sutter ME, Kline JA. Urinary metabolomic analysis to detect changes after intravenous, non-ionic, low osmolar iodinated radiocontrast for computerized tomographic imaging. West J Emerg Med. 2014;15(2):152–157. | ||
Tighe P, Negm O, Todd I, Fairclough L. Utility, reliability and reproducibility of immunoassay multiplex kits. Methods. 2013;61(1):23–29. | ||
Andreucci M, Faga T, Pisani A, Sabbatini M, Michael A. Pathogenesis of acute renal failure induced by iodinated radiographic contrast media. Austin J Nephrol Hypertens. 2014;1(1):id1005. | ||
Cigarroa RG, Lange RA, Williams RH, Hillis LD. Dosing of contrast material to prevent contrast nephropathy in patients with renal disease. Am J Med. 1989;86(6 pt 1):649–652. | ||
Laskey WK, Jenkins C, Selzer F, et al. Volume-to-creatinine clearance ratio: a pharmacokinetically based risk factor for prediction of early creatinine increase after percutaneous coronary intervention. J Am Coll Cardiol. 2007;50(7):584–590. | ||
Keaney JJ, Hannon CM, Murray PT. Contrast-induced acute kidney injury: how much contrast is safe? Nephrol Dial Transplant. 2013;28(6):1376–1383. | ||
Gurm HS, Dixon SR, Smith DE, et al. Renal function-based contrast dosing to define safe limits of radiographic contrast media in patients undergoing percutaneous coronary interventions. J Am Coll Cardiol. 2011;58(9):907–914. | ||
Mueller C. Prevention of contrast-induced nephropathy with volume supplementation. Kidney Int Suppl. 2006;100:S16–S19. | ||
Balemans CE, Reichert LJ, van Schelven BI, van den Brand JA, Wetzels JF. Epidemiology of contrast material-induced nephropathy in the era of hydration. Radiology. 2012;263(3):706–713. | ||
Masuda M, Yamada T, Mine T, et al. Comparison of usefulness of sodium bicarbonate versus sodium chloride to prevent contrast-induced nephropathy in patients undergoing an emergent coronary procedure. Am J Cardiol. 2007;100(5):781–786. | ||
Baker CS, Wragg A, Kumar S, De Palma R, Baker LR, Knight CJ. A rapid protocol for the prevention of contrast-induced renal dysfunction: the RAPPID study. J Am Coll Cardiol. 2003;41(12):2114–2118. | ||
Avci E, Yesil M, Bayata S, Postaci N, Arikan E, Cirit M. The role of nebivolol in the prevention of contrast-induced nephropathy in patients with renal dysfunction. Anadolu Kardiyol Derg. 2011;11(7):613–617. | ||
Gunebakmaz O, Kaya MG, Koc F, et al. Does nebivolol prevent contrast-induced nephropathy in humans? Clin Cardiol. 2012;35(4):250–254. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.