")
Back to Journals » Infection and Drug Resistance » Volume 14
The Pivotal Role of Microbiota in Modulating the Neuronal–Glial–Epithelial Unit
Authors Luo S, Zhu H , Zhang J, Wan D
Received 5 October 2021
Accepted for publication 26 November 2021
Published 21 December 2021 Volume 2021:14 Pages 5613—5628
DOI https://doi.org/10.2147/IDR.S342782
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Siyu Luo,1 Huifeng Zhu,2 Junhui Zhang,3 Dong Wan1
1Department of Emergency & Critical Care Medicine, The First Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China; 2College of Pharmaceutical Sciences & Chinese Medicine, Southwest University, Chongqing, People’s Republic of China; 3Health Management Center, The First Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China
Correspondence: Dong Wan
Department of Emergency & Critical Care Medicine, The First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China
Email [email protected]
Junhui Zhang
Health Management Center, The First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China
Email [email protected]
Abstract: The enteric nervous system (ENS) consists of enteric neurons and enteric glial cells (EGCs) and controls the function of the epithelial barrier. Thus, a novel concept of neuronal–glial–epithelial unit in the gut was put forward by analogy with neuronal–glial–endothelial unit in the brain. The environment in the gastrointestinal (GI) tract is complex as it harbours millions of bacteria, which extensively attach with intestinal epithelium. The cross-talk between the neuronal–glial–endothelial unit and microbiota plays a pivotal role in modulating the epithelial barrier’s permeability, intestinal development and immune response. And evidence shows dysbiosis is the potent risk factor in the pathologic process of Parkinson’s disease (PD) and inflammatory bowel disease (IBD). In this review, we summarize the compelling results in favor of microbiota serving as the key modulator in the neuronal–glial–epithelial unit development and function, with profound effects on intestinal homeostasis.
Keywords: neuronal–glial–epithelial unit, microbiome, gut
Introduction
The enteric nervous system (ENS) is called “the second brain” in the gut, regulating the secretion, motility and immune responses. It coordinates with intestinal epithelium and constitutes both an anatomical and functional unity, which is named the neuronal–glial–endothelial unit. The unit has three major functional layers: 1) chemical barrier, 2) mechanical barrier, 3) immune barrier. Accumulating evidence demonstrates that microbiota play an essential role in regulating the neuronal–glial–endothelial unit. It fulfils two paradoxical tasks – ensuring the ENS maturation while disrupting the intestinal barrier and eliciting various diseases. This effects result from the bifactor model-environmental factors (such as birth mode, time of weaning, life style, diet) and host susceptibility gene. Thus, organized intestinal epithelium and well-developed ENS are controllable via gut microbiota.
In this review, we provide evidence that the microbiota modulates the development of the ENS and the proliferation of enteric glial cells (EGCs), manipulates the function of the neuronal–glial–epithelial unit and fine-tunes the intestinal homeostasis. We also summarize the signal molecules involved and explain how they contribute to keep the exquisite balance of the immune response. Finally, our comprehension may help attract more attention to the microbiota, and target the microbiota to understand and treat brain-gut related diseases.
The Digestive Neuronal–Glial–Epithelial Unit
Construction of the Neuronal–Glial–Epithelial Unit
The Epithelial Cells
Intestinal epithelium lines the gastrointestinal (GI) tract. It consists of a monolayer epithelial cells and reveals a morphology of projections (called villi) and invaginations (called crypts). The villi and crypts are alternately distributed in the small intestine and the villi are substituted by flat epithelial surface in the colon. The intestinal epithelial cells originate from stem cells at the base of crypts, which give rise to rapidly cycling progenitors that migrate along the crypt-villus axis, and eventually differentiate into mature type.1 The epithelial cells include enterocytes, goblet cells, Paneth cells, M (microfold) cells and tuft cells, all of which are derived from stem cells. Enterocytes play a crucial role in absorption and protection, while the goblet cells contribute to secreting mucus. Paneth cells are associated with the secretion of antimicrobial peptides (AMPs), which support the homeostasis of intestine lumen. And the maturity of immune system is assisted by M cells, a specialized cell that transmitting antigens to resident immune cells. Tuft cells are committed to transmitting signals to the immune cells and activating immune responses.2
The Enteric Nervous System
The enteric nervous system (ENS) consists of three parts: enteric neurons, enteric glial cells (EGCs), and intestinal cells of Cajal (ICCs). They form two major ganglionated structures and functional subunits – submucosal plexus (or Meissner’s plexus) and myenteric plexus (or Auerbach’s plexus). Meissner’s plexus is located in the connective tissue of submucosa, and innervates muscularis mucosae, intestinal neuroendocrine cells, glandular epithelium and submucosal blood vessels while Auerbach’s plexus is located between the circular and longitudinal muscle layers, and is associated with the contractility of the circular and longitudinal muscles.3
Though the precise roles of the ENS remain to be explored, accumulating results suggest multiple roles for the ENS in regulating the epithelial barrier, secretion, rehabilitation, motility and immune response of the gut.4 Meanwhile, emerging data shows that it communicates with the central nervous system via the brain-gut axis and plays a crucial role in disorders such as Parkinson’s disease (PD), Alzheimer’s disease (AD), traumatic brain injury (TBI), etc5 (Figure 1).
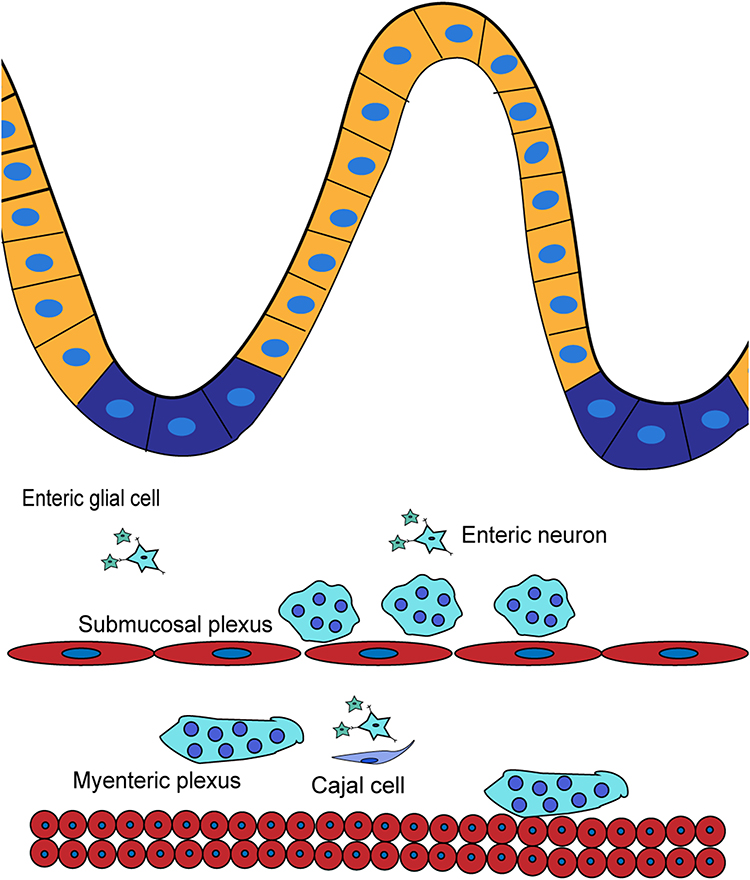 |
Figure 1 The enteric nervous system. |
Functions of the Neuronal–Glial–Epithelial Unit
Chemical Barrier
The digestive tract harbors diverse microorganisms, and the number of microorganisms among it is as many as 10.6,14 Dysbiosis and an impaired intestinal barrier contribute to intestinal inflammation, so the integrity of the intestinal barrier is of utmost importance. Mucus, rather than the intestinal epithelial cells (IECs) directly contact with the outer environment, separating the inner tissue with outer substances, and constituting the front line of the intestinal barrier. Its biosynthesis and secretion depends on goblet cells, which were considered a purely secretory cell, and have recently been shown to have a more complex function.7
Mucus is a gel-like mixture containing water, electrolytes, lipids, and proteins. 145 different proteins have been found in the mucus layer of the GI tract, serving as growth factors, structural proteins, glycoproteins and defensive proteins. Mucins are the main functional components of mucus and are responsible for the viscoelastic property. Their nature is glycoprotein, which are classified into membrane mucins, including MUC 1, 3A, 3B, 4, 11, 12, 13, 15, 16, 17, 20, 21, and secretory mucins, including MUC 2, 5AC, 5B, 6, 7, 8, 9, and 19.8 Studies show 2 types of mucus layer organization in the gastrointestinal tract: a two-layer system in the colon and glandular stomach, and a single layer system in the small intestine. Johansson et al has observed a structure of loose layer (≈100 μm thick), which could be aspirated easily, and firmly adhering to an epithelial cell layer (≈50 μm thick) in the colon of mice. Alcian blue staining showed 2 bands of MUC2, which doubled in intensity compared with the loose layer. In situ hybridization of a universal 16S ribosomal RNA detected a marked reduction of bacteria in a firm layer.9 That the inner mucus layer in deficient mice had a higher bacteria penetration score and increased infiltrated leukocytes compared to wild-type (WT) mice, suggested a susceptibility to spontaneous colitis.10 The small intestine plays a crucial role in absorbing nutrients, thus its mucus layer is unlikely to be as thick as that in the colon. Unlike special separation in the colon, small intestinal maintenance of homeostasis relies on defensive proteins such as immunoglobulin A (IgA), antimicrobial peptides (AMPs) secreted by Paneth cells, enterocytes and other immune cells.11 Vaishnava et al reported MyD88 deficient mice showed a 100-fold higher mucosal bacteria load compared with WT littermates in a culture-dependent manner as measured by quantitative polymerase chain reaction (Q-PCR) determination of total 16SrRNA gene copy number. The results were consistent with a reduction of RegIIIgγ.12 Mucus is dynamic, it is renewed every 1–2 h and moves with the peristalsis of the intestine, showing the rheological property of mucus.13 To summarize, the mucus layer is the front line of keeping bacteria away from the host tissue. A double mucus layer ensures the physical separation in the colon and the single mucus layer in the small intestine heavily depends on antibacterial proteins to protect tissues. They are biosynthesized and secreted by multiple epithelial lineages and regulated by acetylcholine (Ach) to form the chemical barrier of the intestine.14
Mechanical Barrier
The epithelial barrier is highly selective, allowing water, ions and nutrients to enter and preventing the penetration of macromolecules and microbes. It relies heavily on the apical junctional complex, a structure comprised of tight junction (TJ), adheren junction (AJ), and desmosome. It is deeply associated with epithelial permeability and apicobasal cell polarity. The structure of TJs was observed by electron microscopy firstly, showing a fused region between adjoining membranes at high magnification.15 Evidence supports a “protein” model. The development of immunological techniques allows us to identify and characterize TJ proteins, which include integral membrane proteins (claudins, occludin, junctional adhesion molecules), cytoplasmic plaque proteins, and cytoskeletal proteins.16,17 In 1993, Furuse et al described occludin, which is the first transmembrane protein of the tight junction to be observed. It directly participates in the formation of TJ strands by binding its phosphorylated cytoplasmic C terminus to cytoplasmic plaque proteins.17 But occludin is not necessarily required for the formation of TJ strands as the epithelial cells differentiated from occludin-deficient embryonic stem cells can also develop well-formed TJ strands.18 The claudin family formed the backbone of TJs by generating TJ strands. Thus, animals knocked out corresponding genes demonstrating defects in TJs function.19 There are two types of paracellular pathways, the “pore” and the “leak” pathway. In light of emerging data, claudins support the flux of solutes, determining the permeability of paracellular pathways, especially in channel formation.20 Claudin-2 constitutes a channel that permits Na+ and uncharged small molecules through, that is, the pore pathway is a size-selective and charge-selective route.21,22 The cytoplasmic plaque proteins usually refer to proteins of the ZO family (ZO1, ZO2 and ZO3). Evidence shows that ZO-1 and ZO-2 deficient cells lack expression of TJs. In addition, TJ structure and function are regulated by a distinct but overlapping subset of conserved domains.23,24 Their PDZ domains directly bind to transmembrane tight junction proteins, such as claudins, occludins, junctional adhesion molecules (JAM), and tight junction-associated marvel proteins (TAMPs), playing the roles in forming TJs and modulating the leak pathway.16,25 Adherens junctions (AJs) and desmosomes contribute to adhering adjacent cells. Overall, TJs, AJs and desmosomes strongly hold cells together, preventing bacteria and macromolecules going through. But there is no absolute occlusion as there exists a pore and leak pathway, and they all constitute the selective property of the epithelial barrier.
Immune Barrier
We have reviewed the mucus layer of the intestinal epithelial barrier and mentioned that antimicrobial peptides (AMPs) are produced by Paneth cells. Actually, both AMPs and the intestinal mucus layer constitute the innate protective mechanisms of the gut.26 The AMPs consist of lysozyme, a glycosidase hydrolyzing the 1,4-glucosidic bands of cytoderm, Secretory phospholipase A2 (sPLA2), enzymes that hydrolyze the bacterial membrane, and other molecules such as defensins, C-type lectins of the REG3 family, and cathelicidins.27 Interestingly, the payer’s patch (PP), which is covered by follicle-associated epithelium (FAE), does not secrete mucus and results in contacting with lumen bacteria, regulates the maturity of the immune system and activating mucosal T/B cells. M cells resident in gut-associated lymphoid tissues (GALT), including the payer’s patches (PPs), Isolated Lymphoid Follicles (ILF), and are associated with the initiation of the mucosal immune system.28,29 It expresses diverse receptors such as integrin β1, glycoprotein 2 (GP2), C5a receptor, and poliovirus receptor (PVR/CD155), and is specialized for antigen sampling and transcytosis.30 It’s believed that the process of transcytosis is mediated by these various receptors. Fluorescent protein (GFP) is a protein expressed in E-coli, and three-dimensional imaging analysis directly demonstrated that GP2 accumulated around GFP. In GP2-deficient mice, the count of Y. enterocditica or S. Typhimurium was reduced, suggesting GP2 was critical for type-1-piliated bacteria uptake.31 Differentiation of M cells is manipulated by the RANKL–RANK signaling. The mice with conditional deletion of the RANK gene in the intestinal epithelium had a delayed germinal center development in PPs. Meanwhile, flow cytometry suggested a reduction of IgA+ CD138+ plasma cells.32 For the purposes of immune surveillance and retaining barrier function, it’s plausible that M cells interact with underlying lymphocytes and mononuclear phagocytes via transcytosis.33 Luminal contents are captured and translated to the basolateral pocket, where the dendritic cells (DCs), T cells and B cells accumulate. In addition, diverse cytokines and chemokines are released by FAE and DCs, which recruit more T cells, B cells, and antigen presentation cells (APCs). Immunofluorescence staining showed significant expression of CXCR5 in the germinal centre, indicating that CXCR5-CXCL13 singling is essential for attracting B cells. Also, accumulation of CD45.1 positive cells (B cells) in CXCR4 deficient mice suggested that CXCR4-CXCL12 induces B cell migration into lymph nodes.34–36 However, M cells are restricted in population and spatial distribution. How bacteria interact with other epithelial cells and transmit signals to the APCs, T cells, and B cells beneath intestinal epithelium is unclear. Actually, the enterocytes, intraepithelial lymphocytes, and some DCs located within intestinal epithelium are prone to taking advantage of the toll-like receptor (TLR), which binds to pathogen-associated molecular patterns and regulates the immune barrier and immune tolerance.37 As noted, molecules, such as AMPs, IgA, and cells, in regard to enterocytes, innate immunocytes, T cells and B cells, are involved in the exquisite balance between immune tolerance and immune defense. Among them, M cells play a key role in activating cellular and humoral immunity. They transmit signals via diverse pathways and influence each other, finely tuning the immune barrier of the gut (Figure 2).
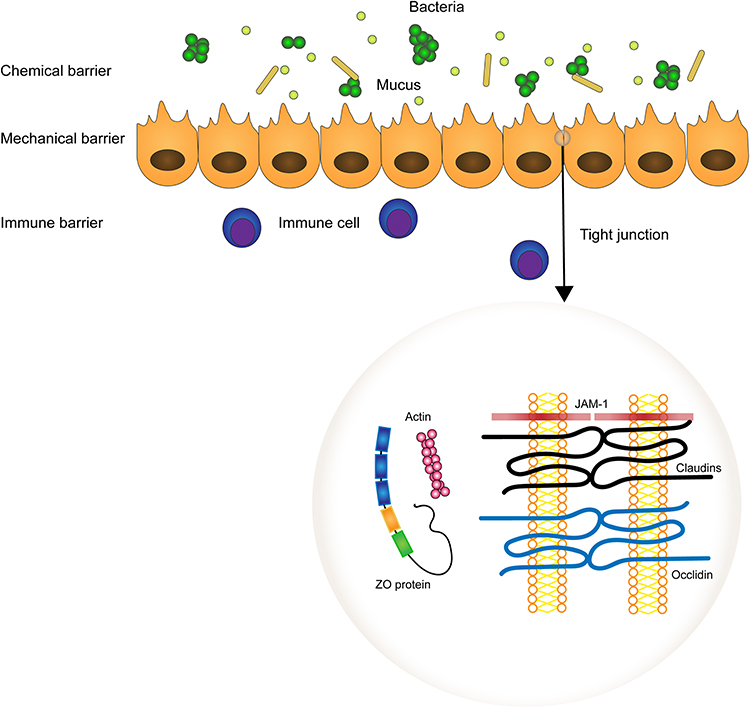 |
Figure 2 The function layers of the neuronal–glial–endothelial unit and the organization of tight junctions. Tight junctions are composed of two membrane proteins the Occludin and Claudins, the intracellular protein ZO protein, which are MAGUK family proteins ZO-1, ZO-2, and ZO-3, and the junction adhesion molecule (JAM). |
Gut Microbiota in Physiological Status
Diverse microorganisms, known as commensal bacteria, colonize in the gastrointestinal tract. Bacteroidetes, Firmicutes, Actinobacteria and Proteobacteria are dominated in healthy adult gut microbiomes.6 And these microbiota seems to be acquired, maternally and environmentally. Fetus in utero is sterile and colonization by microorganisms starts only after birth.38 Though several studies have shown detectable bacteria from amniotic fluid and umbilical cord blood, they are ascribed to contaminated samples and excessively sensitive technology.39,40 As one varietal factor influencing the development and variety composition of gut microbiota, birth mode is always the first propriety to be concerned. Take, for instance, the Bacteroides genus which could not be observed in samples from children born by cesarean section until 6–18 months after birth.41 And vaginal birth is deeply associated with Gammaproteobacteria.42 The bacterial communities develop fastest in the early 6 months of life, showing steadily increasing Enterobacteriaceae, Bifidobacteriaceae, and Clostridiaceae. But for the development, there are no one-size-fits-all disciplines as studies show a different bacterial composition in similarly aged children.41,42 That identifies that diet, sex, race, and ethnicity are equally important. Evidence suggests that maternal fish and seafood consumption increases the abundance of Streptococcus agalactiae, and Group B Streptococcus in infants, while lipid and protein intake is significantly associated with Bifidobacterium and Firmicutes.43,44 Though a healthy adult gut microbiome is considered to be stable with fewer numbers and diversity in species compared to children, these factors still influence the GI bacterial community after puberty.6 Consistent with previous studies, lifestyle makes the GI microbiome different, but Jha et al, considering the contributions of water, showed that the beginning of changes in the gut is much earlier than we thought.45 That means, the environment shapes the gut microbiome at the base of genes and this process usually continues during an individual's lifetime, keeping a dynamic balance of gut microbiome and maintaining gut homeostasis. Millions of microbes colonise our GI tract, having a profound effect on immune, metabolism, even the functions of distant organs. Thus, taking advantage of probiotics and fecal microbiota transplantation to optimize the composition of gut microbiota is promising.46–49
Dysbiosis and Disruption of the Neuronal–Glial–Epithelial Unit
Recently, dysbiosis is regarded as an initial factor accounting for a large number of diseases.50 In light of the studies, functional gastrointestinal disorders, including constipation and abdominal distension, are usually accompanied by an inflammatory state and changes of bacterial composition of the gut. In turn, a disrupted intestinal barrier, provoked immune response and bacterial translocation contribute to the progression of disease.51,52 In this regard, neurodegenerative diseases and inflammatory bowel disease (IBD) attract more attention.
Parkinson’s Disease (PD)
GI dysfunctions are common in PD and, most importantly, they represent the earliest stage of disease.51 Based on 16S rRNA amplicon sequencing, evidence shows an increase of Proteobacteria, but a decrease of Firmicutes in PD patients, indicating a pattern of pro-inflammation dysbiosis.50 In detail, Proteobacteria, Enterobacteriaceae, Christensenellaceae, Lactobacillaceae, Coriobacteriaceae, Bifidobacteriaceae, and Parabacteroides increased, and there was a decrease of Lachnospiraceae, particularly of the Roseburia genus.53 Consistent with previous studies, Qian et al demonstrated a higher abundance of Alistipes, Paraprevotella, Klebsiella, Sphingomonas, Acinetobacter, Aquabacterium, Desulfovibrio, Clostridium IV, Lachnospiracea incertae sedis, Butyricicoccus, Clostridium XVIII and Nitrososphaera in Chinese PD patients.54 Additionally, clinical data supports the higher α-diversity indexes in PD patients. Interestingly, a further study showed the total fecal bacterial counts decreased in all PD patients within 2 years, and the decreasing Bifidobacterium was associated with the progression of the disease.55 Compared to wild-type, germ-free alpha-synuclein-overexpressing (GF-ASO) mice exhibit a deficit in motor skills such as beam traversal, pole descent, and hindlimb clasping, supporting the view of dysbiosis playing a crucial role in the motor dysfunction of PD.56 The fecal marker of intestinal inflammation, calprotectin, as well as the markers of intestinal barrier, alpha-1-antitrypsin and zonulin were significantly elevated in PD patients compared to healthy controls.57 More directly, immunocytochemistry and microscopic analyses of colonic samples showed a reduced expression of ZO-1.58,59 Thus, a disrupted intestinal barrier is common in PD. Exerting studies reported the reduction of Faecalibacterium Prausnitzii was related to the state of pro-inflammation and intestinal barrier dysfunction.60 Though, quite a few studies have proved the close interplay between dysbiosis and intestinal permeability, further investigations are needed to clarify the effects of specific bacteria. Taken together, the excessively activated immune response and dysfunction of the neuronal–glial–epithelial unit is significantly related to dysbiosis, which is consistent with the view of the pro-inflammatory state and changes of inflammatory phenotype of EGCs.61 But there is no sufficiently direct data to support a clear causal relationship between the dysbiosis, dysfunction of neuronal–glial–epithelial unit and PD, and more studies are needed to investigate further62 (Figure 3).
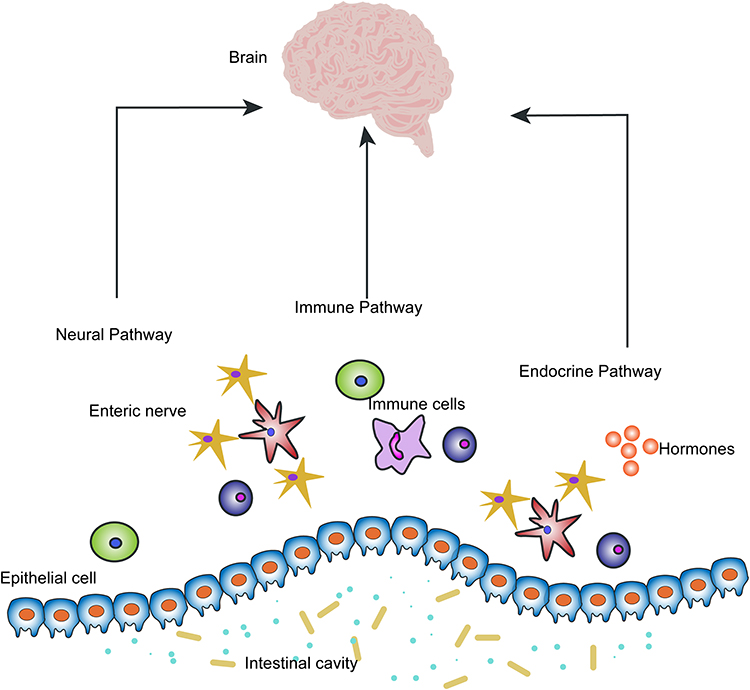 |
Figure 3 Pathways of communication between the neuronal–glial–epithelial unit and the brain. The neural pathway, immune pathway and endocrine pathway are three main pathways that exist between the gut and brain, which microbiota can modulate the gut–brain axis. |
IBD
IBD, usually referred to as Crohn’s disease (CD) and ulcerative colitis (UC), is a process involving different gastrointestinal linings, and is characterized by chronic relapsing inflammation. Though the aberrantly activated inflammation has been thought to be the key of pathology of IBD, genetic predisposition, dysbiosis, intestinal barrier dysfunction and environmental factors have recently been suggested to play a crucial role in its pathogenesis.63 It’s accepted that microbial diversity is reduced in IBD patients.64,65 In addition, the decreased relative abundance of specific bacterial taxa are also reported. In pediatric IBD patients, both abundance and diversity of bifidobacterial populations were low.66 Other studies documented the reduction of Bacteroidetes and Firmicutes (such as Faecalibacterium prausnitzii), which indicated the pro-inflammatory state.64,65,67 Conversely, Enterobacteriaceae, especially invasive E. coli (AIEC), were observed to increase in IBD individuals, which was further verified to be highly associated with CD.68,69 Despite the fact that the results are commonly accepted, we have to admit the existence of differences between individuals. Susceptibility gene deficit exacerbates colitis, accompanied by dysbiosis, which may explain the individual differences.70,71 Nevertheless, rare studies implicate the causal relationship. Furthermore, peripartum antibiotic exposure is definitely a risk factor for early onset of IBD.72,73 Another critical pathogenic link between IBD and dysbiosis is the destroyed intestinal barrier. MUC2 precursor synthesis was significantly decreased in active UC patients, suggesting that the activation of inflammation is correlated with the extent of the reduced production of MUC2.74 Besides, MUC2 deficient mice were prone to spontaneous colitis, shedding light on the protective effects of the mucosal layer.75 However, the situation seems to be different in CD because the mucosal thickness was observed to be greater in the colon and rectum, contrasting to controls.76 The paradoxical condition could be explained by the changed quality of the mucus. The proportion of sulphated mucins was shrunk in CD, which was associated with the increasing Desulfovibrio, the genus that could reduce sulphate.77,78 Although a large amount of studies focus on dysbiosis and the disrupted intestinal barrier, their relationship with inflammation remains unclear. Several studies showed results that there were no clear differences of microbiota composition between inflamed and noninflamed mucosa in IBD patients. Additionally, the dysbiosis index did not approach the controls with the treatment, though the inflammatory response and symptoms have been relieved.79,80 In short, multifactor pathogenesis leads to the onset of IBD. It’s widely accepted that dysbiosis and a disrupted intestinal barrier are highly associated with the deterioration of IBD, which may occur independent from the inflammation.
The Microbiota Controls the Neuronal–Glial–Epithelial Unit
The Microbiota Modulates the Development of ENS
On account of the capacity of controlling intestinal physiological activity independently, ENS is called “the second brain” in the gut. The intrinsic primary afferent neurons (IPANs) form synapses with enteric interneurons via their process, transmitting signals and making the ENS different from the autonomic nervous system.81 But the mice model demonstrated that ENS is a neural-crest derivative, which mainly arises from a vagal crest. The enteric neural crest cells (ENCCs) invade the foregut of mice and the wave migrates to a caudal part of the hindgut during embryonic day (E) 9.5 and E14.82,83 At E11, the gut folding inward results in the juxtaposition of the midgut and hindgut, allowing the ENCCs to enter the hindgut in a trans-mesenteric pattern.84 The glial cell line-derived nerve growth factor (GDNF) is crucial for proliferation and migration of ENCCs. It binds with RET and GFRα1, the receptors express the ENS progenitors and neurons, and modulate the prenatal ENS development and postnatal ENS proliferation.85–87 Accumulating evidence suggests that the ENS undergoes renewal and proliferation continuously after birth.88,89 Postnatal gut microbiota colonization and maturation of ENS seems to be parallel in temporal and spatial distribution, which attracts more attention on the relationship between the ENS development and GI microbiota. Germ-free (GF) rats showed an abnormal myenteric plexus with unevenly spaced ganglia neurons and thinner nerve fibers. Moreover, EGCs were eliminated in villi in the S100b immunostaining.90,91 This is of interest as Hirschsprung’s disease, a disorder associated with the GDNF-Ret signaling mutation, has similar pathological characteristics.92 It’s reasonable to presume that bacteria in the gut plays an important role in regulating the expression of GDNF and GDNF-Ret signaling. Brun et al offered compelling evidence with Tlr2/mice. They observed the decrement in immunoreactivity of GDNF in TLR2 deficient mice. In addition, the numbers of S100β+ EGCs and HuC/D+ neurons decreased in the myenteric plexus. And expression of βIII-tubulin, S100β, HuC/D, GFAP, peripherin significantly decreased in Western blot.93 An in vitro experiment further verified that stimulation of TLR2, TLR4, TLR5 and TLR9 ligands increased the expression of GDNF and corrected the neuronal phenotype.94 Similar to GF rats and TLR2 deficient mice, mice treated with antibiotics showed alerted myenteric plexus. Additionally, TLR2 immunoreactivity were observed to be enhanced, which implied the effects of the “microbiota- TLR2-axis”.95
Emerging evidence challenges the view that the ENS remains stable in adulthood. Actually, a healthy enteric nervous network is dynamic and the neural apoptosis and neurogenesis keep an exquisite balance.96 Microbiota also modulates the ENS development and renewal via 5-hydroxytryptamine (5-HT). And the neuronal function of GF mice could be reversed by microbiota colonization.97 Nestin+ cells are responsible for the supplement of myenteric neurons and the muscularis macrophages eliminate the debris by phagocytosis.89,98 5% Nestin+ cells in GF mice expressed Ki67 at 3–15 days after microbiota colonization, suggesting reserved proliferation.98 5-HT is considered to be a strongly neuroprotective and neurogenerative factor acting on ENS. 2–4 weeks after infusing 5-HT4 agonist, BrdU-labeled (BrdU is an antibody used for immunocytochemistry) enteric neurons were detected, while the number of labeled cells were small in 5-HT4 receptor knock-out (KO) mice.99 Thus, it’s reasonable that the 5-HT signal pathway may mediate ENS development and renewal. Filipe et al further elucidated the effects of microbiota in the regulating expression of 5-HT. GF mice demonstrated an absent 5-HT expression in immunohistochemistry. Moreover, mucous 5-HT is also produced by gut microbiota.98,100 In brief, microbiota plays an essential role in postnatal neuronal precursor migration and neurogenesis, and has an effect on neuroprotection. Future goals may refer to the specific targets of gut microbiota and the effects of their interaction which may participate in the pathogenesis of gut-brain associated disease.
The Microbiota Modulates the Epithelial Proliferation and Repairation
The stem cells in the base of crypts are crucial for the proliferation of epithelial cells. Nascent progenitors migrate upward towards the gut lumen and finally complete the process of differentiation and maturation.101 The gut harboring millions of microbes after birth complicates the internal luminal environment.6,38 A large amount of studies have revealed that the intestinal bacteria accounts for the disruption of the epithelial barrier and exacerbates the diseases such as IBD, PD and Alzheimer’s disease.63,102 But few of them paid attention to the effects on epithelial development. Actually, several studies have proved GF mice own an immature gut epithelia. Usually, the height of villi, depth of crypt and number of cells are applied to judge the maturity of the gut. The transit time of precursors in GF mice were approximately twice over the control group. In addition, mice in the control group showed a larger number of villus cells and deeper crypts in contrast to the GF mice.103,104 Recently, intaking probiotics offered compelling evidence that after bacteria colonization in the GF mice model, the number of crypt cells increased by an average of 5 on day 21.105 Likewise, GF mice colonized the microbiota from a poor weight gain preterm infant revealing a shorter small intestine and shrunken height of villi and depth of crypt. Furthermore, in an immunofluorescence test, the number of goblet cells, Paneth cells and Enteroendocrine cells were all detected to be less than in the control group. Insightfully, the researchers assayed the protein occludin and ZO-1, which could suggest the permeability of the epithelial barrier and the results showed a lower expression compared to the control group.106 To optimize the intestinal function, apoptotic cells are expelled and precursors are transient from the crypt stem cells to the tip, continuously. So how does the microbiota modulate the proliferation of stem cells?107 Typically, the cell cycle is regulated by multiple signal channels and proteins, and cell cycle proteins cyclin D and cyclin E are responsible for the G1 phase. The retinoblastoma (Rb) pathway and P53 pathway downregulate cyclin D and cyclin E, and promote apoptosis.108,109 Interestingly, researchers have detected that Lactobacillus casei and Bifidobacterium breve could downregulate the expression of cyclin D1 and cyclin E1. As a result, the process of proliferation was suppressed.110 Of note, Wnt/β-catenin signal pathway is classical in regulating cell proliferation. It also has been reported that after Lactobacillus intake, Wnt3 and Wnt2b were activated. Meanwhile, Ki-67 positive cells significantly increased. The process was considered to be dominated by G-protein-coupled receptor 81, a specific receptor for lactate.111 Others implicate the effects of TLRs. The interleukin-17 (IL-17) and growth factor (FGF2) were documented to alleviate the damage of intestinal epithelium in the colitis mice model.112 Moreover, variety metabolites of microbiota also play a critical role in intestinal epithelium repair and proliferation.110,113 Overall, these data strongly suggest that gut microbiota dominates the homeostasis of intestinal epithelium. They may interplay with innate immune systems and regulate the cell cycle, suppressing or activating the apoptosis and proliferation.
The Microbiota Regulates the Neuronal–Glial–Epithelial Unit Function
Microbiota is often regarded as the key in a diseases’ etiology. The pathological mechanisms may involve the increased permeability, provoked inflammation and dysfunctional enteric neuron and glia. Previous studies have reported that a disrupted epithelial barrier accompanied with dysbiosis is found via immunocytochemistry or fecal marker test in PD, which all strongly implies the close relationship between microbiota and the increased permeability.57,58 This imposes a requirement to elucidate if the dysbiosis drives the damage of the epithelial barrier. E. coli O124 K72 treatment mice had an incomplete villi, decreased expression of occludin and Muc2, which sheds light on that bacteria and leads to a disrupted epithelial barrier.114 Others documented the TLRs as mediators in increasing colonic permeability.115 Nevertheless, more bacteria showed protective effects. Bifidobacterium could reduce colon permeability by phosphorylating extracellular signal-regulated kinases (ERKs) and increasing the expression of ZO-1 and occludin.116 And others may contribute to epithelial proliferation and repair by virtue of cytokines.112,117 IL-22 and IL-17 are secreted by γδ T cells which respond to IL-23, the upstream signals triggered by lipopolysaccharide (LPS). They were reported to play an important role in AMPs secretion and protective immunity to infection. In other words, bacteria is critical to trigger the innate and adaptive immune response.118 LPS and short-chain fatty acids (SCFA) are known to activate the specific receptors and promote an inflammatory response. But evidence that TLRs and receptors of SCFA were detected on enteric neurons suggests their potential role in modulating intestinal motility and secretion.119–123 In short, bacteria is a potent stimulator in triggering intestinal inflammation, and the underlying mechanisms remain to explore.
Signal Molecules in Modulating the Neuronal–Glial–Epithelial Unit
Microbiota Metabolites
Studies on LPS, a bilayer structure from the cell wall of gram-negative bacteria, have persisted for approximately 50 years.124 It was found to provoke an immune response in septic shock firstly and was confirmed to damage the endothelial and epithelial cells in the animal model.125–127 LPS has definite effects on inflammation and often is regarded as the initial and worsening factor, so that it is often used for inflammation inducing in a mice model.128,129 It is doubtless that the increased serum LPS is markedly associated with a higher level of plasma zonulin, indicating intestinal barrier dysfunction.130 Among pattern recognition receptors (PRRs), TLR, especially TLR4 is activated by LPS. Several studies have reported LPS was connected with activated TLR4, CD14 and nuclear factor κB (NFκB).131,132 Recently, it’s acknowledged that LPS induces increased permeability and stimulates the expression of MyD88 and NFκB via activating TLR4 and its co-receptor. As a result of the inflammatory cascade, inflammatory cytokines are produced and released.133 Though TLR4 is the mainstay in mediating inflammation, it could not explain TLR4 deficient mice also having a response to LPS. NOD-like receptors (NLRs) are intracellular PRRs that bind with LPS and promote Caspase-11, triggering the secretion of IL-1β and initiating programmed cell death.127
SCFAs, mainly referring to acetate, butyrate, and propionate, are digestion products of gut microbiota, which serve a protective role in the intestinal barrier. For example, PD patients had lower fecal SCFA concentrations.60 G protein coupled receptors (GPRs) bind with SCFAs, mediating the regulation of the intestinal barrier function. Free fatty acid receptor 2 (FFAR2), also known as GPR43, and FFAR3, also termed as GPR41, which expressed on IECs, and enteric neurons, were activated by SCFA and downregulated NLRP3, suppressing the autophagy and apoptosis.123,134,135 Additionally, SCFAs repaired the intestinal epithelium and promoted proliferation by modulating the cyclin E1 and cyclin D1.110 Therefore, SCFAs optimize the stabilization of intestinal epithelium and reduce the intestinal permeability. Furthermore, it has been established that SCFAs have a prominent role in intestinal immune response. Acetate could increase intestinal IgA by acting on DCs, however, GPR43 deficient mice did not show the similar reaction.136 Though the positive effects of SCFAs have been recognized, we could not ignore that they may destabilize homeostasis of intestinal barrier sometimes. Actually, the binding of SCFAs and their receptors promoted the secretion of cytokines like IL-6 and recruited Neutrophil, eliciting an inflammatory state.137 The paradoxical effects indicate the complex role of bacteria, and further reinforced researches are undoubtedly required.
Neurotransmitter
5-HT is an important neurotransmitter in the central nervous system. Over 95% of 5-HT is biosynthesized in the gut. Enterochromaffin cells (ECs) and neurons exert tryptophan hydroxylase-1 (TPH-1) and TPH-2, the rate-limiting synthetic enzyme, respectively, to synthesize 5-HT and support the function of transmitting signals. It enters cells via serotonin reuptake transporter (SERT), stimulating both extrinsic and afferent primary neurons.138 Microbiota has been proofed to modulate the secretion and biosynthesis of 5-HT. GF mice showed a lower level of 5-HT and Yano et al demonstrated that spore-forming microbes were the key mediators.98,100 It’s widely accepted that 5-HT is markedly correlated with IBD. Mice with chronic colitis showed ECs hyperplasia and increased levels of 5-HT, which strongly indicated that 5-HT may perform a pro-inflammatory role in immune response.139,140 DCs express 5-HT receptors and are essential to initiate an innate immune response. TPH-1 deficient mice downregulate levels of IL-17, IFN-γ, and IL-12p40, which derived from DCs, suggesting the key link of DCs between 5-HT and the intestinal pro-inflammatory state.141 In light of these data, Guseva et al identified the 5-HT7 receptor that expressed on DCs was the utmost important mediator, revealing the proinflammatory effect of 5-HT depending on 5-HT7 receptor's manner.142,143 In contrast, 5-HT4 agonist ameliorated intestinal motor dysfunction, and 5-HT4 receptor deficient mice showed a longer intestinal transit time and reduced enteric neurons, which indicated the 5-HT4 receptor mediates neuroprotection and neurogenesis, promoting maturation of ENS98,99 (Figure 4).
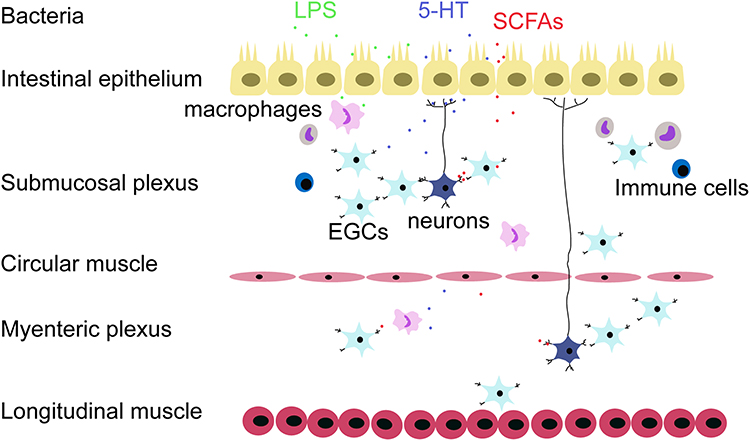 |
Figure 4 The neuronal–glial–endothelial unit is an entity which combines both anatomy and function. The epithelial cells are on the front line and ENS interspersed in the following muscular and connective tissue. Scattered immune cells, APCs, and neurons communicate with each other in favor of the complex cellular network. Various cytokines and signal molecules such as LPS, 5-HT, and SCFA target to regulate the neuronal–glial–endothelial unit activity. |
Nitric oxide (NO) is the gaseous signaling molecule regulating gut homeostasis. Traditionally, NO generates from cells with the catalysis of NO synthases (NOS). Inducible NOS, also known as NOS2, is the isozyme upregulated when inflammation occurs. However, other sources provided by dietary nitrite and nitrate-reducing bacteria, supply the NO in a NOS-independent manner.144,145 Microbiota serves as both the regulator and producer in NO synthesis. Listeria monocytogenes recognized by PRRs on macrophage, triggered the NFκB pathway and promoted the expression of NOS2.146 Consequently, abruptly elevated NO had neurotoxic effects on ENS, leading to neuronal death. Likewise, NOS2 inhibitor ameliorated this process, further verifying the prominent effects of NO signals on ENS.147
Enterogenous Hormone
Glucagon-like peptide 1 (GLP-1) is the incretin hormone secreted by enteroendocrine L-cells in response to nutrient ingestion. It activates the GLP-1 receptors that are extensively distributed in pancreatic islets, kidney, GI tract and so on, regulating cell metabolism, gastrointestinal motor and secretion patterns.148,149 Counterintuitively, after microbiota colonization, GF mice showed fat accumulation with a slower consumption rate. This GLP-1 and insulin resistance was elicited by dysbiosis, which was sensed by PRRs and mediated in the mechanism of an NO-dependent pathway.150–152 Myenteric enteric neurons expressing GLP-1 receptors were downregulated after fecal transplantation, leading to rapid intestinal motor.153
Overall, these studies are all strongly in favor of the view that microbiota modulates intestinal homeostasis by means of GLP-1.
Immune Pathway
Recently, cytokines have attracted unprecedented interest in regulating immune response and maintaining intestinal homeostasis. The IL-17 family (including IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, IL-17F) has been the most studied and much evidence is in favor of the view that microbiota modulates the intestinal barrier in the IL-17/IL-17R pathway.112,133,137,154 IL-17 serves as a protective factor in IBD, and neutralization or deficit of IL-17 was reported to lose the integrity of the intestinal barrier and aggravate inflammation. Though IL-23 is a known regulator of IL-17 and IL-22, sharing the similar characteristics of promoting an immune response with IL-17 and IL-22, IL-17 was documented to be elicited without IL-23 sometimes.155–157 IL-22 is another downstream signal of IL-23, contributing to encoding inflammatory cytokine and chemokine, synergistically with IL-17A. IL-22 was essential for AMPs secretion and proinflammatory phenotype change of lymphocytes after C. rodentium infection.158 In addition, IL-22 boosted the gene encoding of IECs proliferation and repair.117 In light of those studies, cytokines such as IL-23, IL-22, and IL-17 are essential for maintaining intestinal barrier permeability and intestinal immune response, and bacteria serves as both the target to be limited and the underlying manipulator of gut homeostasis.
Conclusions
The neuronal–glial–epithelial unit is not a simple anatomical combination of ENS and epithelial cells, but a functional entity responsible for the gut's immune response and multiple diseases such as PD and IBD. Therefore, when we target to prevent and treat some brain-gut related diseases, the intestinal cells, ENS and immune system should be considered as a whole. The microflora is a major regulator in keeping its morphological integrity and functional coordination. We reviewed current findings on how microbiota influences the micro environment of the gut, and they also convinced us the ENS and epithelial cells own the property of plasticity.
Targeting microbiota is a promising research avenue and some hypotheses have been proposed. For example, fecal bacteria transplantation is targeted at the microbiota to reshape the structure and repair the function of the gut. It was applied in animal experiments to explore Hirschsprung’s disease, PD, autism and so on. But the research objects are restricted in children and the senior and it’s far from being applied in the clinic. Based on new technical means, it is possible to explore more gastrointestinal biomarkers in the future, so that some brain-gut related diseases can be diagnosed and treated early.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81873034), the Natural Science Foundation Project of CQ CSTC (cstc2018jcyjAX0158), the Medical Research Project of Chongqing Health Commission (2015MSXM014 and 2016MSXM198) and the National Infrastructure of Chinese Genetic Resources [YCZYPT[2020]04].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Martini E, Krug SM, Siegmund B, et al. Mend your fences: the epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol. 2017;4(1):33–46. doi:10.1016/j.jcmgh.2017.03.007
2. Cardoso-Silva D, Deborah D, Itzlinger A, et al. Intestinal barrier function in gluten-related disorders. Nutrients. 2019;11(10):2325. doi:10.3390/nu11102325
3. Heiss CN, Olofsson LE. The role of the gut microbiota in development, function and disorders of the central nervous system and the enteric nervous system. J Neuroendocrinol. 2019;31(5):e12684. doi:10.1111/jne.12684
4. Million M, Larauch M. Stress, sex, and the enteric nervous system. Neurogastroenterol Motil. 2016;28(9):1283–1289. doi:10.1111/nmo.12937
5. Ma Q, Xing C, Long W, et al. Impact of microbiota on central nervous system and neurological diseases: the gut-brain axis. J Neuroinflammation. 2019;16(1):53. doi:10.1186/s12974-019-1434-3
6. Hollister EB, Riehle K, Luna RA, et al. Structure and function of the healthy pre-adolescent pediatric gut microbiome. Microbiome. 2015;3(1):36. doi:10.1186/s40168-015-0101-x
7. Allen A, Flemström G, Garne A, et al. Gastroduodenal mucosal protection. Physiol Rev. 1993;73(4):823–857. doi:10.1152/physrev.1993.73.4.823
8. Bansil R, Turner BS. The biology of mucus: composition, synthesis and organization. Adv Drug Deliv Rev. 2017;124:S0169409X17302041.
9. Johansson MEV, Mia P, Joel P, et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Gut Microbes. 2008;105(39):51–54.
10. Johansson MEV, Gustafsson JK, Holménlarsson J, et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut. 2014;63(2):281–291. doi:10.1136/gutjnl-2012-303207
11. Ayabe T, Satchell DP, Wilson CL, et al. Secretion of microbicidal α-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. 2000;1(2):113–118. doi:10.1038/77783
12. Vaishnava S, Yamamoto M, Severson KM, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334(6053):255–258. doi:10.1126/science.1209791
13. Johansson MEV. Fast renewal of the distal colonic mucus layers by the surface goblet cells as measured by in vivo labeling of mucin glycoproteins. PLoS One. 2012;7(7):e41009. doi:10.1371/journal.pone.0041009
14. Specian RD, Neutra MR. Mechanism of rapid mucus secretion in goblet cells stimulated by acetylcholine. J Cell Biol. 1980;85(3):626–640. doi:10.1083/jcb.85.3.626
15. Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. 1963;17(2):375–412. doi:10.1083/jcb.17.2.375
16. Stevenson BR, Siliciano JD, Mooseker MS, et al. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol. 1986;103(3):755–766. doi:10.1083/jcb.103.3.755
17. Furuse M, Hirase T, Itoh M, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123(6):1777–1788. doi:10.1083/jcb.123.6.1777
18. Mikio F, Kohji F, Takashi H, et al. Claudin-1 and −2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141(7):1539–1550. doi:10.1083/jcb.141.7.1539
19. Furuse M. Knockout animals and natural mutations as experimental and diagnostic tool for studying tight junction functions in vivo. Biochim Biophys Acta. 2009;1788(4):813–819. doi:10.1016/j.bbamem.2008.07.017
20. Pearce SC, Al-Jawadi A, Kishida K, et al. Marked differences in tight junction composition and macromolecular permeability among different intestinal cell types. BMC Biol. 2018;16(1):19. doi:10.1186/s12915-018-0481-z
21. Rosenthal R, Günzel D, Krug SM, et al. Claudin-2-mediated cation and water transport share a common pore. Acta Physiol. 2016;219(2):521–536. doi:10.1111/apha.12742
22. Weber CR, Liang GH, Wang Y, et al. Claudin-2-dependent paracellular channels are dynamically gated. Elife. 2015;4:e09906. doi:10.7554/eLife.09906
23. Umeda K, Ikenouchi J, Katahira-Tayama S, et al. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell. 2006;126(4):741–754. doi:10.1016/j.cell.2006.06.043
24. Rodgers LS, Tanner Beam M, Anderson JM, et al. Epithelial barrier assembly requires coordinated activity of multiple domains of the tight junction protein ZO-1. J Cell Sci. 2013;126(7):1565–1575. doi:10.1242/jcs.113399
25. Van Itallie CM, Fanning AS, Bridges A, et al. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol Biol Cell. 2009;20(17):3930. doi:10.1091/mbc.e09-04-0320
26. Antoni L, Nuding S, Weller D, et al. Human colonic mucus is a reservoir for antimicrobial peptides. J Crohn's Colitis. 2013;7(12):e652–e664. doi:10.1016/j.crohns.2013.05.006
27. Sohini M, Hooper LV. Antimicrobial defense of the intestine. Immunity. 2015;42(1):28–39. doi:10.1016/j.immuni.2014.12.028
28. Nochi T, Denton P, Wahl A, et al. Cryptopatches are essential for the development of human GALT. Cell Rep. 2013;3(6):1874–1884. doi:10.1016/j.celrep.2013.05.037
29. Haber AL, Biton M, Rogel N, et al. A single-cell survey of the small intestinal epithelium. Nature. 2017;551(7680):333–339. doi:10.1038/nature24489
30. Rouch JD, Scott A, Lei NY, et al. Development of functional microfold (M) cells from intestinal stem cells in primary human enteroids. PLoS One. 2016;11(1):e0148216. doi:10.1371/journal.pone.0148216
31. Koji H, Kazuya K, Tomonori N, et al. Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature. 2010;462(6):226–230.
32. Rios D, Wood MB, Li J, et al. Antigen sampling by intestinal M cells is the principal pathway initiating mucosal IgA production to commensal enteric bacteria. Mucosal Immunol. 2016;9(4):907–916. doi:10.1038/mi.2015.121
33. Mabbott NA, Donaldson DS, Ohno H, et al. Microfold (M) cells: important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol. 2013;6(4):666–677. doi:10.1038/mi.2013.30
34. Schmidt TH, Oliver B, Gray EE, et al. CXCR4 promotes B cell egress from Peyer’s patches. J Exp Med. 2013;210(6):1099–1107. doi:10.1084/jem.20122574
35. Kim SH, Kim YN, Jang YS. Cutting edge: LL-37-mediated formyl peptide receptor-2 signaling in follicular dendritic cells contributes to B cell activation in Peyer’s patch germinal centers. J Immunol. 2017;198(2):629–633. doi:10.4049/jimmunol.1600886
36. Carlsen HS, Baekkevold ES, F-e J, et al. B cell attracting chemokine 1 (CXCL13) and its receptor CXCR5 are expressed in normal and aberrant gut associated lymphoid tissue. Gut. 2002;51(3):364–371. doi:10.1136/gut.51.3.364
37. Wang J, Ford HR, Grishin AV. NF-|[kappa]|B-mediated expression of MAPK phosphatase-1 is an early step in desensitization to TLR ligands in enterocytes. Mucosal Immunol. 2010;3(5):523–534. doi:10.1038/mi.2010.35
38. Lim ES, Rodriguez C, Holtz LR. Amniotic fluid from healthy term pregnancies does not harbor a detectable microbial community. Microbiome. 2018;6(1):87. doi:10.1186/s40168-018-0475-7
39. Collado MC, Rautava S, Aakko J, et al. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci Rep. 2016;6(1):23129. doi:10.1038/srep23129
40. Jiménez E, Fernández L, Marín ML, et al. Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Curr Microbiol. 2005;51(4):270–274. doi:10.1007/s00284-005-0020-3
41. Moran Y, Tommi V, Heli S, et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci Transl Med. 2016;8(343):343ra381.
42. Ho TTB, Groer MW, Kane B, et al. Dichotomous development of the gut microbiome in preterm infants. Microbiome. 2018;6(1):157. doi:10.1186/s40168-018-0547-8
43. Lundgren SN, Madan JC, Emond JA, et al. Maternal diet during pregnancy is related with the infant stool microbiome in a delivery mode-dependent manner. Microbiome. 2018;6(1):109. doi:10.1186/s40168-018-0490-8
44. Grier A, Qiu X, Bandyopadhyay S, et al. Impact of prematurity and nutrition on the developing gut microbiome and preterm infant growth. Microbiome. 2017;5(1):158. doi:10.1186/s40168-017-0377-0
45. Jha AR, Davenport ER, Gautam Y, et al. Gut microbiome transition across a lifestyle gradient in himalaya. PLoS Biol. 2018;16(11):e2005396. doi:10.1371/journal.pbio.2005396
46. Liang Y, Liang S, Zhang Y, et al. Oral administration of compound probiotics ameliorates HFD-induced gut microbe dysbiosis and chronic metabolic inflammation via the G protein-coupled receptor 43 in non-alcoholic fatty liver disease rats. Probiotics Antimicrob Proteins. 2019;11(1):175–185. doi:10.1007/s12602-017-9378-3
47. Abildgaard A, Kern T, Pedersen O, et al. The antidepressant-like effect of probiotics and their faecal abundance may be modulated by the cohabiting gut microbiota in rats. Eur Neuropsychopharmacol. 2019;29(1):98–110. doi:10.1016/j.euroneuro.2018.10.011
48. Kim CJ, Walmsley SL, Raboud JM, et al. Can probiotics reduce inflammation and enhance gut immune health in people living with HIV: study Designs for the Probiotic Visbiome for Inflammation and Translocation (PROOV IT) pilot trials. HIV Clin Trials. 2016;17(4):147–157. doi:10.1080/15284336.2016.1184827
49. Paramsothy S, Kamm MA, Kaakoush NO, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet. 2017;389(10075):1218. doi:10.1016/S0140-6736(17)30182-4
50. Ali K, Green SJ, Engen PA, et al. Colonic bacterial composition in Parkinson’s disease. Mov Disorders. 2015;30(10):1351–1360. doi:10.1002/mds.26307
51. Kallol Ray C, Pablo MM, Schapira AHV, et al. International multicenter pilot study of the first comprehensive self-completed nonmotor symptoms questionnaire for Parkinson’s disease: the NMSQuest study. Mov Disorders. 2010;21(7):916–923.
52. Kelly LP, Carvey PM, Ali K, et al. Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Mov Disord. 2014;29(8):999–1009. doi:10.1002/mds.25736
53. Barichella M, Severgnini M, Cilia R, et al. Unraveling gut microbiota in Parkinson’s disease and atypical parkinsonism. Mov Disorders. 2019;34(3):396–405. doi:10.1002/mds.27581
54. Qian Y, Yang X, Xu S, et al. Alteration of the fecal microbiota in Chinese patients with Parkinson’s disease. Brain Behav Immun. 2018;70:S088915911830028X. doi:10.1016/j.bbi.2018.02.016
55. Minato T, Maeda T, Fujisawa Y, et al. Progression of Parkinson’s disease is associated with gut dysbiosis: two-year follow-up study. PLoS One. 2017;12(11):e0187307. doi:10.1371/journal.pone.0187307
56. Sampson T, Debelius J, Thron T, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016;167(6):1469. doi:10.1016/j.cell.2016.11.018
57. Schwiertz A, Spiegel J, Dillmann U, et al. Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson’s disease. Parkinsonism Relat Disord. 2018;50:104–107. doi:10.1016/j.parkreldis.2018.02.022
58. Perez-Pardo P, Dodiya HB, Engen PA, et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut. 2019;68(5):829–843. doi:10.1136/gutjnl-2018-316844
59. Clairembault T, Leclair-Visonneau L, Coron E, et al. Structural alterations of the intestinal epithelial barrier in Parkinson’s disease. Acta Neuropathol Commun. 2015;3(1):12. doi:10.1186/s40478-015-0196-0
60. Unger MM, Spiegel J, Dillmann KU, et al. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord. 2016;32:66–72. doi:10.1016/j.parkreldis.2016.08.019
61. Thomas C, Willem K, Laurène LV, et al. Enteric GFAP expression and phosphorylation in Parkinson’s disease. J Neurochem. 2014;130(6):805–815. doi:10.1111/jnc.12742
62. Sangjune K, Seung-Hwan K, Tae-In K, et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron. 2019;103(4):627–641.e7. doi:10.1016/j.neuron.2019.05.035
63. Souza HSP, De Claudio F. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016;13(1):13–27.
64. Crigottier-Gois M. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Digest World Core Med J. 2006;55(2):205–211.
65. Hai-Qin M, Ting-Ting Y, Xiao-Jing Z, et al. Fecal microbial dysbiosis in Chinese patients with inflammatory bowel disease. World J Gastroenterol. 2018;24(13):1464–1477. doi:10.3748/wjg.v24.i13.1464
66. Maukonen J, Kolho KL, Paasela M, et al. Altered fecal microbiota in paediatric inflammatory bowel disease. J Crohn's Colitis. 2015;9(12):1088. doi:10.1093/ecco-jcc/jjv147
67. Harry S, Bénédicte P, Laurie W, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105(43):16731–16736. doi:10.1073/pnas.0804812105
68. Seksik P, Rigottier-Gois L, Gramet G, et al. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut. 2003;52(2):237. doi:10.1136/gut.52.2.237
69. Darfeuille-Michaud A, Neut C, Barnich N, et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115(6):1405–1413. doi:10.1016/S0016-5085(98)70019-8
70. Ridler C. IBD: dysbiosis underlies CARD9 risk alleles in colitis. Nat Rev Gastroenterol Hepatol. 2016;13(6):316. doi:10.1038/nrgastro.2016.82
71. Margarita MM, Jérémy D, Nicolas D, et al. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut. 2014;63(1):116. doi:10.1136/gutjnl-2012-304119
72. Miyoshi J, Bobe AM, Miyoshi S, et al. Peripartum antibiotics promote gut dysbiosis, loss of immune tolerance, and inflammatory bowel disease in genetically prone offspring. Cell Rep. 2017;20(2):491. doi:10.1016/j.celrep.2017.06.060
73. Örtqvist AK, Lundholm C, Halfvarson J. Fetal and early life antibiotics exposure and very early onset inflammatory bowel disease: a population-based study. Gut. 2019;68(2):218–225. doi:10.1136/gutjnl-2017-314352
74. Tytgat KM, Vand der Wal JW, Einerhand AW, et al. Quantitative analysis of MUC2 synthesis in ulcerative colitis. Biochem Biophys Res Commun. 1996;224(2):397. doi:10.1006/bbrc.1996.1039
75. Sluis MVD, Koning BAED, Bruijn ACJMD, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131(1):117–129. doi:10.1053/j.gastro.2006.04.020
76. Pullan RD, Thomas GA, Rhodes M, et al. Thickness of adherent mucus gel on colonic mucosa in humans and its relevance to colitis. Gut. 1994;35(3):353–359. doi:10.1136/gut.35.3.353
77. Raouf AH, Tsai HH, Parker N, et al. Sulphation of colonic and rectal mucin in inflammatory bowel disease: reduced sulphation of rectal mucus in ulcerative colitis. Clin Sci. 1992;83(5):623. doi:10.1042/cs0830623
78. Lennon G, Balfe Á, Bambury N, et al. Correlations between colonic crypt mucin chemotype, inflammatory grade and Desulfovibrio species in ulcerative colitis. Colorectal Dis. 2014;16(5):O161–O169. doi:10.1111/codi.12503
79. Shaw KA, Bertha M, Hofmekler T, et al. Dysbiosis, inflammation, and response to treatment: a longitudinal study of pediatric subjects with newly diagnosed inflammatory bowel disease. Genome Med. 2016;8(1):1–13. doi:10.1186/s13073-016-0331-y
80. Forbes JD, Van DG, Bernstein CN. Microbiome survey of the inflamed and noninflamed gut at different compartments within the gastrointestinal tract of inflammatory bowel disease patients. Inflamm Bowel Dis. 2016;22(4):817.
81. Furness JB, Jones C, Nurgali K, et al. Intrinsic primary afferent neurons and nerve circuits within the intestine. Prog Neurobiol. 2004;72(2):143–164. doi:10.1016/j.pneurobio.2003.12.004
82. Kapur RP, Yost C, Palmiter RD. A transgenic model for studying development of the enteric nervous system in normal and aganglionic mice. Development. 1992;116(1):167–175. doi:10.1242/dev.116.1.167
83. Young HM, Hearn CJ, Ciampoli D, et al. A single rostrocaudal colonization of the rodent intestine by enteric neuron precursors is revealed by the expression of Phox2b, Ret, and p75 and by explants grown under the kidney capsule or in organ culture. Dev Biol. 1998;202(1):67–84. doi:10.1006/dbio.1998.8987
84. Chihiro N, Toshihiro U, Takayuki M, et al. Trans-mesenteric neural crest cells are the principal source of the colonic enteric nervous system. Nat Neurosci. 2012;15(9):1211–1218. doi:10.1038/nn.3184
85. Rodrigues DM, Li AY, Nair DG, et al. Glial cell line-derived neurotrophic factor is a key neurotrophin in the postnatal enteric nervous system. Neurogastroenterol Motil. 2011;23(2):e44–e56. doi:10.1111/j.1365-2982.2010.01626.x
86. Toshihiro U, Sanjay J, Shigenobu Y, et al. Conditional ablation of GFRalpha1 in postmigratory enteric neurons triggers unconventional neuronal death in the colon and causes a Hirschsprung’s disease phenotype. Development. 2007;134(11):2171–2181. doi:10.1242/dev.001388
87. Hongtao W, Inna H, William P, et al. The timing and location of glial cell line-derived neurotrophic factor expression determine enteric nervous system structure and function. J Neurosci. 2010;30(4):1523–1538. doi:10.1523/JNEUROSCI.3861-09.2010
88. Joseph NM, Shenghui H, Elsa Q, et al. Enteric glia are multipotent in culture but primarily form glia in the adult rodent gut. J Clin Invest. 2011;121(9):3398. doi:10.1172/JCI58186
89. Kulkarni S, Micci MA, Leser J, et al. Adult enteric nervous system in health is maintained by a dynamic balance between neuronal apoptosis and neurogenesis. Proc Natl Acad Sci USA. 2017;114(18):E3709. doi:10.1073/pnas.1619406114
90. Collins J, Borojevic R, Verdu EF, et al. Intestinal microbiota influence the early postnatal development of the enteric nervous system. Neurogastroenterol Motil. 2013;26(1):98–107. doi:10.1111/nmo.12236
91. Kabouridis P, Lasrado R, Mccallum S, et al. Microbiota controls the homeostasis of glial cells in the gut lamina propria. Neuron. 2015;85(2):289–295. doi:10.1016/j.neuron.2014.12.037
92. Angrist M, Bolk S, Halushka M, et al. Germline mutations in glial cell line-derived neurotrophic factor (GDNF) and RET in a Hirschsprung disease patient. Nat Genet. 1996;14(3):341–344. doi:10.1038/ng1196-341
93. Brun P, Giron MC, Qesari M, et al. Toll-like receptor 2 regulates intestinal inflammation by controlling integrity of the enteric nervous system. Gastroenterology. 2013;145(6):1323–1333. doi:10.1053/j.gastro.2013.08.047
94. Bruna P, Gobbo S, Caputi V, et al. Toll like receptor-2 regulates production of glial-derived neurotrophic factors in murine intestinal smooth muscle cells. Mol Cell Neurosci. 2015;68:24–35. doi:10.1016/j.mcn.2015.03.018
95. Valentina C, Ilaria M, Viviana F, et al. Antibiotic-induced dysbiosis of the microbiota impairs gut neuromuscular function in juvenile mice. Br J Pharmacol. 2017;174(20):3623–3639. doi:10.1111/bph.13965
96. Rao M, Gershon MD. Neurogastroenterology: the dynamic cycle of life in the enteric nervous system. Nat Rev Gastroenterol Hepatol. 2017;14(8):453–454. doi:10.1038/nrgastro.2017.85
97. Mcvey Neufeld KA, Perez-Burgos A, Mao YK, et al. The gut microbiome restores intrinsic and extrinsic nerve function in germ-free mice accompanied by changes in calbindin. Neurogastroenterol Motil. 2015;27(5):627–636. doi:10.1111/nmo.12534
98. Filipe DV, Estelle G, Louise MSH, et al. Gut microbiota regulates maturation of the adult enteric nervous system via enteric serotonin networks. Proc Nat Acad Sci. 2018;115(25):6458–6463. doi:10.1073/pnas.1720017115
99. Liu MT, Kuan Y-H, Wang J, et al. 5-HT4 receptor-mediated neuroprotection and neurogenesis in the enteric nervous system of adult mice. J Neurosci. 2009;29(31):9683–9699. doi:10.1523/JNEUROSCI.1145-09.2009
100. Yano J, Yu K, Donaldson G, et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. 2015;161(2):264–276. doi:10.1016/j.cell.2015.02.047
101. Neunlist M, Landeghem LV, Mahé MM, et al. The digestive neuronal-glial-epithelial unit: a new actor in gut health and disease. Nat Rev Gastroenterol Hepatol. 2013;10(2):90–100. doi:10.1038/nrgastro.2012.221
102. Pellegrini C, Antonioli L, Colucci R, et al. Interplay among gut microbiota, intestinal mucosal barrier and enteric neuro-immune system: a common path to neurodegenerative diseases? Acta Neuropathol. 2018;136(3):345–361. doi:10.1007/s00401-018-1856-5
103. Savage DC, Siegel JE, Snellen JE, et al. Transit time of epithelial cells in the small intestines of germfree mice and ex-germfree mice associated with indigenous microorganisms. Appl Environ Microbiol. 1981;42(6):996–1001. doi:10.1128/aem.42.6.996-1001.1981
104. Banasaz M, Norin E, Midtvedt T. The role of gender, age and microbial status on cell kinetics in the gastrointestinal tract of mice. Microb Ecol Health Dis. 2001;13(3):135–142. doi:10.3402/mehd.v13i3.8015
105. Banasaz M, Norin E, Holma R, et al. Increased enterocyte production in gnotobiotic rats mono-associated with Lactobacillus rhamnosus GG. Appl Environ Microbiol. 2002;68(6):3031–3034. doi:10.1128/AEM.68.6.3031-3034.2002
106. Yu Y, Lu L, Sun J, et al. Preterm infant gut microbiota affects intestinal epithelial development in a humanized microbiome gnotobiotic mouse model. AJP Gastrointest Liver Physiol. 2016;311(3):G521–G532. doi:10.1152/ajpgi.00022.2016
107. Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol. 2014;15(1):19. doi:10.1038/nrm3721
108. Duman-Scheel M, Weng L, Xin S, et al. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature. 2002;417(6886):299–304. doi:10.1038/417299a
109. Gladden AB, Diehl JA. Cell cycle progression without cyclin E/CDK2: breaking down the walls of dogma. Cancer Cell. 2003;4(3):160–162. doi:10.1016/S1535-6108(03)00217-4
110. Takahiro M, Thierry P, Béatrice R, et al. Epithelial cell proliferation arrest induced by Lactate and Acetate from Lactobacillus casei and Bifidobacterium breve. PLoS One. 2013;8(4):e63053. doi:10.1371/journal.pone.0063053
111. Lee Y-S, Kim T-Y, Kim Y, et al. Microbiota-derived lactate accelerates intestinal stem-cell-mediated epithelial development. Cell Host Microbe. 2018;24(6):833–846.e6. doi:10.1016/j.chom.2018.11.002
112. Song X, Dai D, He X, et al. Growth factor FGF2 cooperates with interleukin-17 to repair intestinal epithelial damage. Immunity. 2015;43(3):488–501. doi:10.1016/j.immuni.2015.06.024
113. Naito T, Mulet C, De Castro C, et al. Lipopolysaccharide from crypt-specific core microbiota modulates the colonic epithelial proliferation-to-differentiation balance. Mbio. 2017;8(5):e01680–01617. doi:10.1128/mBio.01680-17
114. Ren X, Zhu Y, Gamallat Y, et al. E. coli O124 K72 alters the intestinal barrier and the tight junctions proteins of Guinea pig intestine. Biomed Pharmacother. 2017;94:468–473. doi:10.1016/j.biopha.2017.07.123
115. Moyano-Porcile V, Olavarría-Ramírez L, González-Arancibia C, et al. Short-term effects of Poly(I:C) on gut permeability. Pharmacol Res. 2015;101:130–136. doi:10.1016/j.phrs.2015.06.016
116. Ewaschuk JB, Diaz H, Meddings L, et al. Secreted bioactive factors from Bifidobacterium infantis enhance epithelial cell barrier function. Am J Physiol Gastrointest Liver Physiol. 2008;295(5):G1025–1034. doi:10.1152/ajpgi.90227.2008
117. Lindemans CA, Calafiore M, Mertelsmann AM, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. 2015;528(7583):560–564. doi:10.1038/nature16460
118. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12(5):383–390. doi:10.1038/ni.2025
119. Burgueño JF, Barba A, Eyre E, et al. TLR2 and TLR9 modulate enteric nervous system inflammatory responses to lipopolysaccharide. J Neuroinflammation. 2016;13(1):187. doi:10.1186/s12974-016-0653-0
120. Prigent A, Gonzales J, Durand T, et al. Acute inflammation down-regulates alpha-synuclein expression in enteric neurons. Journal of Neurochemistry. 2019;148(6):746–760. doi:10.1111/jnc.14656
121. Murakami M, Ohta T, Ito S. Lipopolysaccharides enhance the action of bradykinin in enteric neurons via secretion of interleukin-1beta from enteric glial cells. J Neurosci Res. 2009;87(9):2095–2104. doi:10.1002/jnr.22036
122. Soret R, Chevalier J, Coppet PD, et al. Short-chain fatty acids regulate the enteric neurons and control gastrointestinal motility in rats. Gastroenterology. 2010;138(5):1772–1782. doi:10.1053/j.gastro.2010.01.053
123. Nøhr MK, Pedersen MH, Gille A, et al. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in Enteroendocrine cells vs ffar3 in enteric neurons and ffar2 in enteric leukocytes. Endocrinology. 2013;154(10):3552–3564. doi:10.1210/en.2013-1142
124. Shands JW. Evidence for a bilayer structure in gram-negative Lipopolysaccharide: relationship to toxicity. Infect Immun. 1971;4(2):167–172. doi:10.1128/iai.4.2.167-172.1971
125. Harlan JM, Harker LA, Reidy MA, et al. Lipopolysaccharide-mediated bovine endothelial cell injury in vitro. Lab Investig. 1983;48(3):269–274.
126. Ge Y, Ezzell RM, Warren HS. Localization of endotoxin in the rat intestinal epithelium. J Infect Dis. 2000;182(3):873–881. doi:10.1086/315784
127. Hagar JA, Powell DA, Aachoui Y, et al. Cytoplasmic LPS activates Caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341(6151):1250–1253. doi:10.1126/science.1240988
128. Sumbria RK, Grigoryan MM, Vasilevko V, et al. A murine model of inflammation-induced cerebral microbleeds. J Neuroinflammation. 2016;13(1):218. doi:10.1186/s12974-016-0693-5
129. de Souza Xavier Costa N, Ribeiro Júnior G, Dos Santos Alemany AA, et al. Early and late pulmonary effects of nebulized LPS in mice: an acute lung injury model. PLoS One. 2017;12(9):e0185474. doi:10.1371/journal.pone.0185474
130. Tulkens J, Vergauwen G, Van Deun J, et al. Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut. 2020;69(1):191–193. doi:10.1136/gutjnl-2018-317726
131. Plaen IGD, Tan X-D, Chang H, et al. Lipopolysaccharide activates nuclear factor kappaB in rat intestine: role of endogenous platelet-activating factor and tumour necrosis factor. Br J Pharmacol. 2000;129(2):307–314. doi:10.1038/sj.bjp.0703055
132. Guo S, Al-Sadi R, Said HM, et al. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol. 2013;182(2):375–387. doi:10.1016/j.ajpath.2012.10.014
133. Nighot M, Al-Sadi R, Guo S, et al. Lipopolysaccharide-induced increase in intestinal epithelial tight permeability is mediated by toll-like receptor 4/Myeloid differentiation primary response 88 (MyD88) activation of myosin light chain kinase expression. Am J Pathol. 2017;187(12):2698–2710. doi:10.1016/j.ajpath.2017.08.005
134. Yanhai F, Yu W, Pei W, et al. Short-chain fatty acids manifest stimulative and protective effects on intestinal barrier function through the inhibition of NLRP3 inflammasome and autophagy. Cell Physiol Biochem. 2018;49(1):190–205. doi:10.1159/000492853
135. Priyadarshini M, Kotlo KU, Dudeja PK, et al. Role of short chain fatty acid receptors in intestinal physiology and pathophysiology. Compr Physiol. 2018;8(3):1091–1115.
136. Wu W, Sun M, Chen F, et al. Microbiota metabolite short-chain fatty acid acetate promotes intestinal IgA response to microbiota which is mediated by GPR43. Mucosal Immunol. 2016;10(4):946–956. doi:10.1038/mi.2016.114
137. Kim MH, Kang SG, Park JH, et al. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology. 2013;145(2):
138. Gershon MD, Tack J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology. 2007;132(1):397–414. doi:10.1053/j.gastro.2006.11.002
139. Ghia JE, Li N, Wang H, et al. Serotonin has a key role in pathogenesis of experimental colitis. Gastroenterology. 2009;137(5):1649–1660. doi:10.1053/j.gastro.2009.08.041
140. Stavely R, Fraser S, Sharma S, et al. The onset and progression of chronic colitis parallels increased mucosal serotonin release via enterochromaffin cell hyperplasia and downregulation of the serotonin reuptake transporter. Inflamm Bowel Dis. 2018;24(5):1021–1034. doi:10.1093/ibd/izy016
141. Li N, Ghia J-E, Wang H, et al. Serotonin activates dendritic cell function in the context of gut inflammation. Am J Clin Pathol. 2011;178(2):662–671. doi:10.1016/j.ajpath.2010.10.028
142. Guseva D, Holst K, Kaune B, et al. Serotonin 5-HT7 receptor is critically involved in acute and chronic inflammation of the gastrointestinal tract. Inflamm Bowel Dis. 2014;20(9):1516–1529. doi:10.1097/MIB.0000000000000150
143. Kim JJ, Bridle BW, Ghia J-E, et al. Targeted inhibition of serotonin type 7 (5-HT7) receptor function modulates immune responses and reduces the severity of intestinal inflammation. J Immunol. 2013;190(9):4795–4804. doi:10.4049/jimmunol.1201887
144. Grishin A, Bowling J, Bell B, et al. Roles of nitric oxide and intestinal microbiota in the pathogenesis of necrotizing enterocolitis. J Pediatr Surg. 2015;51(1):13–17. doi:10.1016/j.jpedsurg.2015.10.006
145. Hyde ER, Andrade F, Vaksman Z, et al. Metagenomic analysis of nitrate-reducing bacteria in the oral cavity: implications for nitric oxide homeostasis. PLoS One. 2014;9(3):e88645. doi:10.1371/journal.pone.0088645
146. Farlik M, Reutterer B, Schindler C, et al. Nonconventional initiation complex assembly by STAT and NF-κB transcription factors regulates nitric oxide synthase expression. Immunity. 2010;33(1):25–34. doi:10.1016/j.immuni.2010.07.001
147. Venkataramana S, Lourenssen S, Miller KG, et al. Early inflammatory damage to intestinal neurons occurs via inducible nitric oxide synthase. Neurobiol Dis. 2015;75:40–52. doi:10.1016/j.nbd.2014.12.014
148. Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87(4):1409–1439. doi:10.1152/physrev.00034.2006
149. Cho YM, Fujita Y, Kieffer TJ. Glucagon-like peptide-1: glucose homeostasis and beyond. Annu Rev Physiol. 2014;76(1):535–559. doi:10.1146/annurev-physiol-021113-170315
150. Backhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Nat Acad Sci. 2004;101(44):15718–15723. doi:10.1073/pnas.0407076101
151. Grasset E, Puel A, Charpentier J, et al. A specific gut microbiota dysbiosis of Type 2 diabetic mice induces GLP-1 resistance through an enteric NO-dependent and gut-brain axis mechanism. Cell Metab. 2017;25(5):1075–1090.e5. doi:10.1016/j.cmet.2017.04.013
152. Lebrun LJ, Lenaerts K, Kiers D, et al. Enteroendocrine L cells sense LPS after gut barrier injury to enhance GLP-1 secretion. Cell Rep. 2017;21(5):1160–1168. doi:10.1016/j.celrep.2017.10.008
153. Yang M, Fukui H, Eda H, et al. Involvement of gut microbiota in the association between GLP-1/GLP-1 receptor expression and gastrointestinal motility. Am J Physiol Gastrointest Liver Physiol. 2017;312(4):G367–G373. doi:10.1152/ajpgi.00232.2016
154. Song X, Gao H, Lin Y, et al. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity. 2014;40(1):140–152. doi:10.1016/j.immuni.2013.11.018
155. Lee JS, Tato CM, Joyce-Shaikh B, et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity. 2015;43(4):727–738. doi:10.1016/j.immuni.2015.09.003
156. Ogawa A, Andoh A, Araki Y, et al. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol. 2004;110(1):55–62. doi:10.1016/j.clim.2003.09.013
157. Gaffen SL, Jain R, Garg AV, et al. The IL-23–IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14(9):585–600. doi:10.1038/nri3707
158. Zheng Y, Valdez PA, Danilenko DM, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14(3):282–289. doi:10.1038/nm1720
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.