")
Back to Journals » Drug Design, Development and Therapy » Volume 9
The pitfall of the transient, inconsistent anticancer capacity of antiestrogens and the mechanism of apparent antiestrogen resistance
Authors Suba Z
Received 29 May 2015
Accepted for publication 3 July 2015
Published 6 August 2015 Volume 2015:9 Pages 4341—4353
DOI https://doi.org/10.2147/DDDT.S89536
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Zsuzsanna Suba
National Institute of Oncology, Surgical and Molecular Tumor Pathology Centre, Budapest, Hungary
Abstract: Although antiestrogens have been available for breast cancer therapy since the early 1970s, neither their inconsistent anticancer capacity nor the developing antiestrogen resistance of tumors can be fully understood. Although clinical and experimental investigations revealed many tiny details concerning the link between estrogen signaling and tumor development, they yielded fairly controversial findings. Estrogen receptor (ER) overexpression in tumor cells induced by estrogen treatment was erroneously regarded as a promoter of DNA damage, genomic instability, and tumor growth. Similarly, compensatory ER overexpression caused by antiestrogen treatment or estrogen withdrawal was mistakenly evaluated as a key for rapid tumor growth attributed to acquired antiestrogen resistance. Nevertheless, ER upregulation induced by estrogen treatment is a physiologic process even in tumor cells, whereas in the case of antiestrogen administration, it is a contraregulatory action to defend the endangered estrogen signaling. Upregulation of estrogen signaling displays a unique dichotomy, ensuring the survival and safe proliferative activity of healthy cells, while inducing apoptotic death of malignant tumor cells. Analysis of the fairly controversial results justifies that whatever type of available endocrine therapies may be used, including estrogen, antiestrogen treatment, or oophorectomy, an extreme upregulation of ER signaling seems to be the crucial mechanism of successful prevention and treatment for breast cancer. The inconsistent therapeutic effects of antiestrogen administration may be explained by the different genetic capacities of patients for the compensatory upregulation of ER and aromatase enzyme expressions. The weaker the defensive counteraction against the inhibition of estrogen signaling, the poorer is the prognosis of the disease. De novo or acquired antiestrogen resistance of tumors may be associated with the missing capacity of patients for the extreme upregulation of estrogen signaling or with the exhaustion of defensive counteractions in cases that previously showed good reactivity. High-dose estrogen treatment is capable of restoring ER signaling and anticancer capacity even after heavy exposure to antiestrogen therapy.
Keywords: estrogen, estrogen signaling, estrogen receptor, breast cancer, BRCA gene mutation, estrogen hypersensitivity
Introduction
While improvements in detection and treatment measures are continuously introduced in developed countries, we are perhaps missing something big in how we try to prevent and treat breast cancer in the global context.1
For over 120 years, the highly ambiguous correlations among ovarian hormones and both the promotion and prevention of breast cancer have been debated.
In 1896, a favorable tumor response in a premenopausal patient with metastatic breast cancer was reported after oophorectomy.2 This result suggested that ovarian estrogen synthesis may be the fuel of breast cancer development, since oophorectomy seemed to be an appropriate therapeutic measure, presumably by means of withdrawal of the ovarian hormones. Some years later, all known ovariectomized breast cancer cases had been assembled in the UK, and a 30% tumor response rate was established.3 Despite the fairly low effectiveness of this first clinical trial for breast cancer treatment, a 30% response rate is to be regarded as a standard result of endocrine therapy ever since. Tumor responses achieved via oophorectomy proved, however, to be transient, and no responses in the majority of breast cancer cases were established. The limited anticancer capacity of estrogen withdrawal led to a search for a different approach to mammary carcinogenesis.
It was reported in 1944 that high doses of a synthetic estrogen, diethylstilbestrol (DES) were able to produce a 30% tumor response rate among women with metastatic breast cancer, presenting a surprising turn in breast cancer care.4 Thus, the effectiveness of high-dose DES therapy, as new paradox medication against breast cancer for >5 years after menopause, was inconsistent as in the case of the removal of circulating estrogens by oophorectomy. Nevertheless, in this new method, patients were not exposed to the risk of surgery for obtaining a doubtful, transient tumor response.
High-dose DES proved to be the standard of care for the treatment of metastatic breast cancer in postmenopausal patients from the early 1960s and remained the preferred treatment until the introduction of tamoxifen, a nonsteroidal antiestrogen compound.5
During the fight against breast cancer, there was a second turn of events leading to the development of tamoxifen in the early 1970s. It was the first compound nominated as an antiestrogen or more exactly, a selective estrogen receptor modulator (SERM) capable of binding to estrogen receptors (ERs).6 The aim of tamoxifen medication was to inhibit the presumed cancer promoter signaling pathways of ERs. Aromatase inhibitors were also introduced for the treatment of breast cancer, since the maintenance of low estrogen concentrations supposedly inhibits the proliferative activity of tumor cells.7
Both SERMs and aromatase inhibitors remained the choice of treating and preventing hormone receptor-positive breast cancers in spite of their controversial therapeutic results.8–10 Nevertheless, the mechanism of transient anticancer capacity of antiestrogens is still not fully understood, and systemic investigations are called for regarding the consistently developing de novo and acquired antiestrogen resistance of tumors associated with the promotion of tumor growth.
The usage of quite opposite hormonal manipulations; namely synthetic estrogens and antiestrogens could achieve equally low rates of inconsistent tumor regression even among the targeted ER-positive breast cancer cases. The uncertain and weak results of both opposing efforts suggest that the biologic mechanisms affecting the correlation between estrogen signaling and tumor development are fairly misinterpreted.11
Despite enormous scientific efforts, today’s women have a one in eight lifetime risk of developing breast cancer, with an increasing probability of the cancer development occurring after menopause. Recent reports show that there is a slight decrease in breast cancer mortality (~2% per year), but the incidence rates are stable.12–14 The worldwide incidence rate is, however, increasing by ~3% per annum and deaths by 1.8% per annum.15 Since current approaches to the struggle against breast cancer do not work as might be expected, alternative concepts should be considered.1
Since estrogen signaling has a crucial role in DNA stabilization and surveillance of cell proliferation, the therapeutic impact of antiestrogens against breast cancers seems to be an unexpected contradiction.11 Moreover, the acquired antiestrogen resistance of tumors characterized in turn by tumor growth stimulation instead of tumor inhibition is regarded as an unexpected switch, and the cellular mechanisms behind these changes are unknown in spite of thorough investigations.
The aim of the present work is to examine the sources of the ambiguous results of antiestrogen administration in breast cancer prevention and therapy by means of a comprehensive analysis of the most relevant data of clinical–epidemiologic and experimental studies. Moreover, the mechanism of the so-called antiestrogen resistance of tumors was investigated by a review of intermingled findings caused by the concomitant inhibitory effects of antiestrogens and the counteractive upregulation of estrogen signaling.
Fundamental significance of estrogen signaling
Estrogen signaling possesses the most relevant biologic significance since it was favored by evolution as a means of high integration between somatic and reproductive functions.16
There are three types of estrogen hormones circulating in the blood stream: estradiol (E2), estrone (E1), and estriol (E3), of which the most effective and abundant is estradiol. Estrogen-activated receptor isoforms such as ER alpha and ER beta confer the estrogen signal by means of classic genomic and nongenomic pathways. In the nucleus, ERs may act as ligand-activated transcription factor proteins in the promoter region of target genes. They can also regulate gene expression without binding to DNA via protein–protein interaction with nuclear transcription factors. Moreover, estrogen action also has rapid, nongenomic signaling cascades via cell membrane-associated ERs. The complexity of estrogen signaling includes not only liganded but also nonliganded activation of ERs.16
Above all, estrogen-activated ER beta is responsible for cell growth, while the role of activated ER alpha is crucial in the course of cell proliferation.17 Comprehensive analysis of experimental and clinical–epidemiologic results suggests that ER-alpha signaling is the chief safeguard of genome stability in strong interplay with DNA controlling and repairing systems, such as BRCA genes and their protein products.11 The significance of the expression and transcriptional activity of ER beta in the mechanisms of DNA repairing processes needs further investigations.18
ER isoforms exhibit strong cross talk and interplay, by which means all privileged healthy cellular functions are under exquisite surveillance, and cellular health may be ensured in both resting and proliferative biologic systems. At the same time, estrogen signaling recognizes and destroys malignant tumor cells by means of apoptotic mechanisms.11
The role of interplay between ER expression and estrogen concentration in the maintenance of appropriate estrogen signaling
The synthesis of estrogens and the function of the ER signal transduction pathways have unique, exclusive significance as compared to other hormonal mechanisms. The concentration of other hormones and biologic players is within a narrow physiologic range, since either lower or higher hormone levels induce endocrine diseases. These well-known regulatory rules were mistakenly adapted to estrogens, supposing that both estrogen deficiency and hyperestrogenism are associated with the development of serious diseases, including malignancies.
Interplay between estrogen levels and ER expressions has crucial role in the maintenance of appropriate estrogen signaling, which is the prerequisite of cellular health in mammalians. When estrogen signaling is jeopardized by genetic alterations and other endogenous or exogenous factors, defensive counteractions are recruited, such as increased ER expression and estrogen synthesis, so as to maintain the pivotal cellular estrogen surveillance.11,19
ER expression and transcriptional activity are upregulated by both decreased and increased estrogen levels
Both low and high estrogen levels promote enhanced expression and transcriptional activity of ERs aiming to maintain the crucial cellular estrogen surveillance.11
In animal experiments, ovariectomy-associated estrogen withdrawal significantly increased the levels of ER-alpha expression in the uterus, kidney, and cerebral cortex of female rats, while a 12-week treatment of ovariectomized rats with 17-beta estradiol was capable of restoring the previous ER-alpha mRNA level.20
In women, low estrogen levels are high risks of breast cancer.21,22 Estrogen depletion may be counteracted by increased expressions of ERs, aiming to strengthen cellular estrogen sensitivity for the better utilization of available hormone supply. In benign proliferative breast lesions, the higher expression of ER alpha showed close correlation with the later development of breast cancer as compared to tumors with lower ER density.23 Reactive increase in the ER expression of hyperplasic mammary cells may be a defensive counteraction against the dangers of low estrogen supply, but the insufficiency of this counteraction may result in cancer development.11
Interaction between decreased estrogen levels and increased ER expressions was observed even in tumor cells. In a breast cancer cell line, long-term estradiol deprivation induced estrogen hypersensitivity by overexpression of ERs. Hypersensitivity may be characterized by the ability of tumor cells to respond to levels of estrogens at concentrations 2–3 logs lower than required to stimulate wild-type cells.24 In tumor cells, estrogen hypersensitivity upregulates estrogen signaling and its apoptotic activity even in an estrogen-deficient milieu.11
SERM treatment of ER-alpha-positive tumors is a chemical block of available ERs inhibiting the transduction of estrogen signaling. Raloxifene or tamoxifen treatment strongly stimulated the tumor growth of antiestrogen-resistant MCF-7/Ral. When a 9-week raloxifene or tamoxifen treatment of tumors was followed by a 5-week estradiol treatment, estradiol statistically significantly reduced the size of tumors earlier stimulated by raloxifene or tamoxifen pretreatment.25
These observations justify that the completion of the antiestrogen blockade of estrogen signaling leads to antiestrogen-induced tumor growth, which may be counteracted by estradiol treatment via induction of ER overexpression (estrogen hypersensitivity) in tumor cells. This process seems to be an unexpected kamikaze action of tumor cells, since the restored upregulation of estrogen signaling results in their apoptotic death.
Extreme physiologic increase in estrogen concentrations in pregnancy also promotes overexpression of ERs, resulting in a self-generating upregulation of both estrogen signaling and DNA-stabilizer systems. These interactions ensure the safe estrogen-mediated transcriptional activity on target genes in rapidly proliferating maternal and fetal structures.11
In pregnancy, abundant estradiol supply was shown to promote uterine blood flow, rapid myometrial growth, and breast growth at term, mediated by the increased expression of myometrial and mammary ERs.26 Estrogen-mediated upregulation of ER expression in pregnancy may explain why defensive estrogen effects are prolonged and powerful in multiparous women.11
Estrogen treatment increases the ER expression and transcriptional activity even in tumor cells. Two ER-positive breast cancer cell lines (ZR 75-1 and HCC 1500) were treated by four types of estrogens: estrone, estradiol, estriol, and estetrol, and all four elicited significantly increased ER expressions as compared to untreated controls.27 The authors erroneously concluded that the estrogen-induced ER overexpression increases the proliferative activity of tumor cells instead of having an apoptotic impact.
Estradiol treatment is able to increase both nongenomic and genomic ER expression and estrogen signaling in ER-alpha-positive breast cancer cell line. Estradiol binding can rapidly increase the expression and transcriptional activity of membrane-associated ER alpha via the phosphatidylinositol 3-kinase/Akt system through nongenomic pathway. Moreover, estradiol activation of Akt may interact with nuclear ER alpha as well, promoting its expression and activity.28 These observations justify the estradiol-induced upregulation of ER signaling, however, in tumor cells; the increased transcriptional activity of ERs induces apoptotic death instead of proliferation.11
Estrogen synthesis is upregulated by both decreased and increased ER expressions
Low ER expression and/or defective function of ERs is usually counteracted by increased estrogen synthesis in both men and women. On the other hand, an abundant ER expression may be associated with a dangerous defect of estrogen signaling requiring compensatory increase in estrogen synthesis.19,29
Compensatory hyperestrogenism occurs as a feedback mechanism against defective ER signaling caused by mutations of ER regulator genes (ESRs). Severe mutation of ESRs was reported in a 28-year-old man exhibiting extremely high estrogen levels, grave signs of insulin resistance, obesity, and premature cardiovascular disease.30 Inherited estrogen resistance was also reported in the case of an 18-year-old girl presenting sky-high estrogen levels in her blood, and at the same time, the classic symptoms of estrogen deficiency, such as delayed puberty, were also observed.31 Laboratory examinations revealed that 240 times the normal estrogen level was required to get a response out of her ERs.
In BRCA-mutation carrier women, the clinical signs of defective estrogen signaling may be coupled with high but occasionally insufficient estrogen levels. In such cases, instead of being an endogenous pathogenic factor, hyperestrogenism may be regarded as a compensatory defense mechanism against defective ER expression and transcriptional activity.11,19 BRCA1/2-gene mutations seem to be imbalanced defects, crudely inhibiting the upregulation of ER expression and liganded transcriptional activity. At the same time, ER-repressor functions become predominant and induce susceptibility to specific cancer types, such as breast cancer.11
During healthy pregnancy, abundant ER expression in proliferating maternal and fetal structures upregulates the synthesis of both the BRCA protein and aromatase enzyme. Extreme increase in estrogen and BRCA protein production ensures the safeguarding of cell proliferation and all cellular mechanisms.11
Parity is regarded as a protective factor against breast cancer development, while nulliparity is a high risk of breast malignancies.32 In the resting breast of parous women, significantly lower ER-alpha expression was detectable as compared to nulliparous subjects.33 In parous women, appropriate estrogen levels and safety estrogen signaling are linked with relatively lower ER-alpha expression in mammary cells. By contrast, in hormonally challenged nulliparous women, the increased ER-alpha synthesis of mammary cells calls for a higher estrogen concentration because of the dangerous deficiency of the estrogen supply.
Controversial results of antiestrogen use in breast cancer prevention and treatment
The development of ER-blocker tamoxifen in the early 1970s was based on the erroneous concept that excessive estrogen signaling leads to unrestrained tumor proliferation in strongly hormone-responsive sites, such as the breasts.
Tamoxifen and the further developed ER-blocker compounds had become widely used as adjuvant therapy for ER-positive breast cancers.34,35 Clinical trials were conducted for the comparison of the therapeutic results of DES and tamoxifen treatment in women with advanced or recurrent breast cancer. It was established that the rate of tumor responses to tamoxifen was similarly low compared to DES, but the advantage of tamoxifen over synthetic estrogen seemed to be equivocal due to the apparently lower incidence rate of toxic side effects.8,36 Natural estrogens were not easily available for therapeutic use, and the toxic side effects obtained with synthetic DES therapy led to the abandonment of treatment with all estrogenic compounds.37
Use of selective blockers of ER signaling in the therapy of breast cancer yielded fairly ambiguous results. Primarily successful tumor regression was exhibited in only 40%–50% of targeted women with ER-positive breast cancers, while the majority of cases could not react positively, which was designated as de novo antiestrogen resistance.38 Considering the whole population of breast cancer patients, these results correspond exactly to the “magic” biologic response rate of 30%, achieved by either synthetic estrogen therapy or oophorectomy. Moreover, a large proportion of earlier responsive breast cancers may acquire secondary resistance during tamoxifen therapy, leading to rapid progression of the disease.39 Tamoxifen may elicit common side effects, which can be occasionally life threatening, such as stroke, pulmonary emboli, and malignancies at certain sites, particularly in the endometrium, attributed to the anomalous estrogen agonist activities of this compound.40
The ambiguous behavior of tamoxifen in the clinical practice was explained by its duality, as it is not a pure antiestrogen but has both agonistic and antagonistic activities affecting ER alpha. Tamoxifen was characterized as being like Dr Jekyll, when tumor regression was achieved, attributed to its antiestrogenic activity, while possibly becoming Mr Hyde when the presumed estrogenic activity resulted in toxic side effects and endometrial cancer coupled with progression of breast tumor.38 By contrast, being aware of the anticancer capacity of estrogen signaling, the presumed estrogenic behavior of tamoxifen emerged as an inducer of tumor regression, while the predominance of its antiestrogenic impact results in rapid tumor progression.41
Another group of antiestrogens is known as aromatase inhibitors. They block the activity of P450 aromatase enzyme, which converts steroid precursors and androgens to estrogen, causing estrogen deprivation in both healthy tissues and tumors.35 Use of aromatase inhibitors in breast cancer cases seemed to be safer than tamoxifen, because it induces lower rates of endometrial toxicity and thromboembolic complications. In postmenopausal women, the side effects of aromatase inhibitors are numerous, including hot flashes, vaginal dryness, arthralgia, decreased bone mineral density, and an increased bone fracture rate.42
While using an aromatase inhibitor, pure estrogen withdrawal was the presumed therapeutic mechanism against breast cancer, excluding the possibility of aberrant estrogenic actions.43 Nevertheless, de novo or acquired resistance to aromatase inhibitor treatment was also observed in postmenopausal patients with advanced breast cancer.44 Compensatory upregulation of estrogen signaling in estrogen-deficient milieu was erroneously regarded as a strong promoter of tumor cell survival and proliferation justifying antiestrogen resistance.45,46
Lately, antiestrogens are being strongly recommended for breast cancer prevention as well, in spite of the controversial results of their use in tumor therapy.10 In BRCA1 mutation carriers, however, with high risk of familiar breast cancer, antiestrogen administration proved frequently to be ineffective or quite deteriorative.47,48 Based on these data, it was presumed that BRCA1 mutation-linked mammary cancers are distinct diseases arising in a hormonally independent manner.
The clinical experiences have once again led back to the recommendation of preventive and therapeutic oophorectomy for premenopausal BRCA-mutation carriers aiming to achieve an appropriate estrogen deprivation. With this concept, the circle of a 120 years’ fight against breast cancer has closed, since we have arrived back to the year 1896, the dawn of breast cancer research.2
Searching for the mechanisms of antiestrogen resistance
During the past decades, resolution of the mechanism of antiestrogen resistance and its overturn became the most important area of breast cancer research. The anticancer capacity of antiestrogens is mistakenly accepted as evidence; consequently, the presumed changes leading to the apparent antiestrogen resistance of tumors seem to be highly controversial.
Many advances have been made in our understanding of the biology of the nuclear ERs and membrane-associated ERs, which may help to study how resistance to antiestrogens develops. Such mechanisms may include increased or defective expression and transcriptional activity of ERs, occurrence of mutant ER isoforms, changes in the ligand-independent transcriptional activity of ERs, alterations in coregulator proteins, altered postreceptor events, and the different influences of ERs on cellular growth factor signal transduction pathways.49
Numerous controversial data are reported on the momentary expression and activity of ERs and further biologic factors participating in the extremely complex estrogen signaling during antiestrogen treatment. These studies disregard the dynamic struggle between ER inhibitor and activator mechanisms and the stage of developing antiestrogen resistance.
In antiestrogen-treated breast cancer cell lines and in breast cancer models, a counteractive overexpression of ER alpha was erroneously evaluated as troubling estrogen hypersensitivity and a key for estrogen-independent growth justifying antiestrogen resistance.46,50 In vitro studies on acquired resistance of tumor cells against estrogen withdrawal indicated that the acquisition of hypersensitivity to estrogenic stimulation may be a key mechanism for restoration of estrogen signaling and presumed tumor cell survival.24
Surprisingly, in antiestrogen-resistant MCF-7 cell lines, raloxifene or tamoxifen treatment strongly stimulated tumor growth in spite of the abundant expression of targeted ER alpha in tumor cells.25 This finding may be explained by the complete antiestrogen occupancy of abundant ERs and the loss of capacity for new ER expressions.
In conclusion, acquired estrogen hypersensitivity attributed to ER overexpression is a response to either estrogen withdrawal or ER blockade and serves as maintenance of crucial estrogen signaling, resulting in apoptotic death of tumor cells.11 Measurements exhibiting abundant ER expression in antiestrogen-resistant tumor cell lines do not reflect the distribution between estrogen- and tamoxifen-bound receptors. The higher the number of tamoxifen-bound ERs, the stronger is the blockade of estrogen signaling, which is erroneously regarded as antiestrogen resistance.
Reduced or altered ER-alpha phosphorylation was also presumed to be associated with antiestrogen resistance in breast cancer models, in spite of the high expression of ER alpha.50,51 In total, 19 phosphorylation sites have been identified in ER alpha, defining different transcriptional activities. Phosphorylation at S167, S118, and S282 sites is beneficial for tamoxifen-induced tumor response according to reported experimental and clinical data, while tamoxifen resistance is likely to occur when S104/S106 or S305 is phosphorylated.51
Interestingly, the grade of phosphorylation activity at Ser167 site of ERs in breast tumors of tamoxifen-treated patients proved to be an excellent predictor of a good prognosis of the disease.44 Since Ser167 phosphorylation is the most important element of the physiologic transcriptional activity of ERs, this work unconsciously justified that compensatory maintenance of good estrogen signaling is indicative of longer disease-free survival as well as overall survival even among antiestrogen-treated breast cancer patients.
Activation of ESR1-gene mutations affecting the ligand-binding domain of ER alpha was also proposed as a key mechanism in the acquired antiestrogen resistance of metastatic breast cancers.52 Point mutations of ESR1 gene are relatively frequent and result in slightly variant forms of ERs. Gene polymorphism studies revealed that postmenopausal estrogen loss may amplify the earlier hidden slight defects of ERs being associated with increased risk for various diseases, such as breast cancer.29,53 There may be a strong parallelism between postmenopausal estrogen loss and the antiestrogenic inhibition of estrogen signaling, resulting in manifested defects of cellular estrogen surveillance in ESR1-gene mutation carriers.
Cross talk between ER and growth factor receptor (GFR) pathways is also regarded as a cause of antiestrogen therapy resistance in breast cancer cases.54,55 In one form of resistance to estrogen deprivation therapy, the tumor becomes supersensitive to low residual estrogen concentrations by ER overexpression and presumably because of the associated upregulation of mitogen-activated protein kinase. By contrast, in tumors, overexpressing human epidermal growth factor receptor 2 (HER2), acquired resistance to estrogen deprivation therapy involves the loss of ER activity and ER-regulated genes.54 These apparently controversial observations may be explained by the different stages of antiestrogen-induced changes in estrogen signaling; from the compensatory upregulation to the complete loss of ER expression and ER-regulated pathways.
In ER-positive breast cancer xenografts, both epidermal growth factor receptor (EGFR) and HER2 expressions were increased with tamoxifen treatment and markedly increased when tumors became antiestrogen resistant.56 It was established that upregulation of both EGFR and HER2 expressions mediated tamoxifen resistance in ER-positive breast cancers, while genomic ER functions were continuously suppressed by tamoxifen treatment.
In clinical practice, patients with ER-positive, HER-2 overexpressing tumors exhibited higher rates of recurrence and fatal spread after tamoxifen therapy as compared to those who did not receive the agent.57,58 The elevated insulin-like growth factor-I receptor signaling also rendered MCF-7 cells highly resistant to antiestrogens, although tumor cell lines were under continuous suppression of ER transcriptional activity by antiestrogens.59 Considering that both ER-alpha expression and transcriptional activity is downregulated by excessive GF administration,60 antiestrogen blockade of ERs is plausibly strengthened by increased GFR signaling.
A dynamic inverse relationship was revealed between the expression of ERs and GFRs, which supplied a potential of restoring ER expression in apparently ER-negative tumor cells by inhibition of growth factor signaling.61 This recognition would directly lead to the next step: a possibility for the estrogen treatment of ER-positive breast tumors.62
In conclusion, the vast majority of research articles directly or indirectly justify the fact that antiestrogen treatment may be effective against breast cancer only if the crucial estrogen signaling is upregulated by the chemical attack. The development of apparent antiestrogen resistance may be attributed to the exhaustive quiescence of earlier abundant ER expression and aromatase synthesis, leading to complete artificial estrogen resistance and uncontrolled tumor growth.
Successful estrogen treatment of postmenopausal breast cancer patients after heavy exposure to antiestrogen therapy
At the end of the past century, transfection experiments were performed by the insertion of exogenous ERs into ER-negative tumor cells.63 The reexpression of ERs activated a number of estrogen-regulated genes, and treatment with estrogen surprisingly led to growth inhibition instead of the increased proliferative activity of tumor cells. This experimental finding was the dawn of a new epoch in estrogen-related cancer research.
Working groups in the US and Europe started to examine the antitumor effect of high-dose estrogen in postmenopausal patients with advanced breast cancer, who were becoming resistant to estrogen deprivation.37,64 From the early 2000s, further researchers tried to clarify the mechanisms of antiestrogen resistance of tumors and the secret of the anticancer capacity of estrogens in patients with antiestrogen resistant, advanced breast cancer.65–69 In clinical practice, physiologic estrogen-induced apoptosis is successfully applied for breast cancer prevention and treatment following estrogen deprivation.70
ER overexpression in tumor cells treated with either estrogens27,28 or antiestrogens46,50 may be explained by the fundamental regulatory capacity of estrogens. These apparently contradictory treatments can similarly upregulate abundant ER expressions and transcriptional activities. Estrogen-induced upregulation of estrogen signaling is a physiologic process, while in case of antiestrogen administration, it may be regarded as a counteraction for the defense of endangered cellular estrogen surveillance. Increased estrogen signaling displays a unique dichotomy effect: it safeguards the survival and proliferative activity of healthy cells, while induces apoptotic death of malignant tumor cells.11
In patients with breast cancer, estrogen administration is capable of exerting self-generating, increased ER expression and estrogen synthesis as well so as to achieve the strong upregulation of estrogen signaling and apoptotic tumor cell damage. Paradoxically, antiestrogen treatment may also provoke compensatory ER overexpression in tumor cells and extreme estrogen synthesis of the patients so as to restore the apoptotic capacity of estrogen signaling (Figure 1). When these counteractions are sufficient, tamoxifen administration seems to be deceivingly effective, resulting in transient tumor regression.
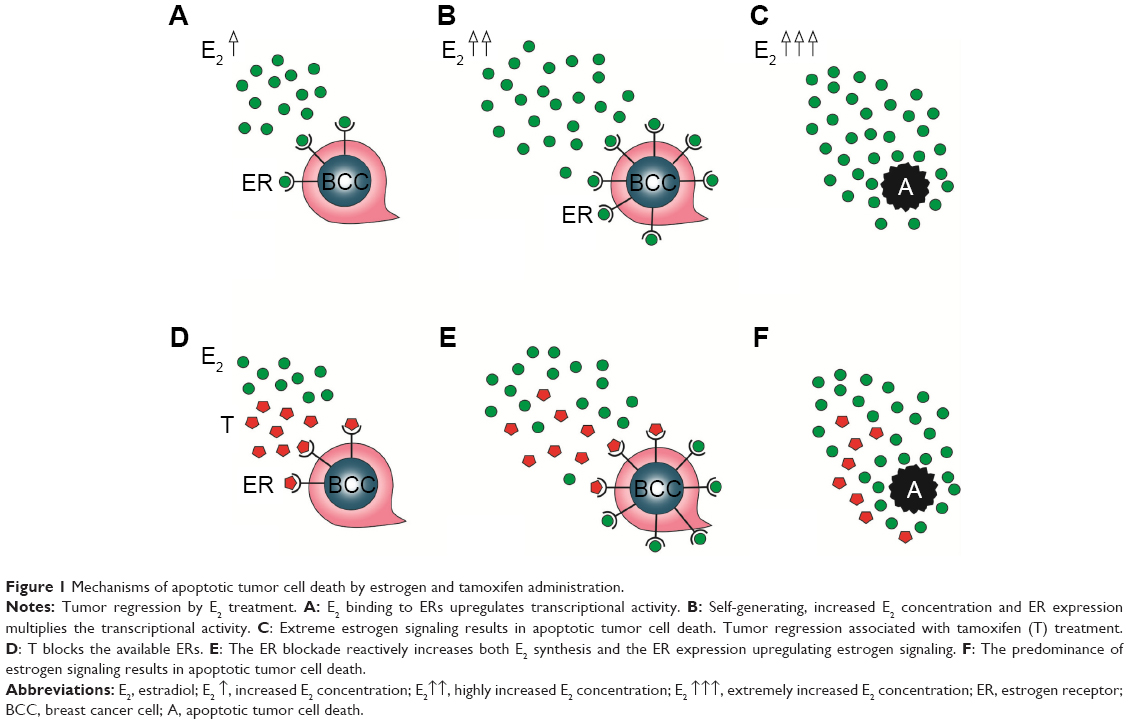 | Figure 1 Mechanisms of apoptotic tumor cell death by estrogen and tamoxifen administration. |
By contrast, primary insufficiency or exhaustion of the defensive counteractions in patients treated with antiestrogens may lead to the complete blocking of estrogen signaling, since the patient has limited capacities for extreme ER expression and estrogen synthesis. In such cases, the result is unrestrained proliferative activity of the tumor cells, and the rapid tumor spread is mistakenly evaluated as de novo or acquired antiestrogen resistance. Nevertheless, high-dose estrogen treatment is capable of restoring the suppressed estrogen signaling even after heavy exposure to antiestrogen treatment. Exogenous and newly synthesized estrogens are in competition with tamoxifen for binding sites on newly expressed, abundant ERs, and the higher the concentration of estrogens, the higher is the probability of successful defeat of tamoxifen. The triumph of estrogens leads to apoptotic tumor cell death and clinical regression of the disease (Figure 2).
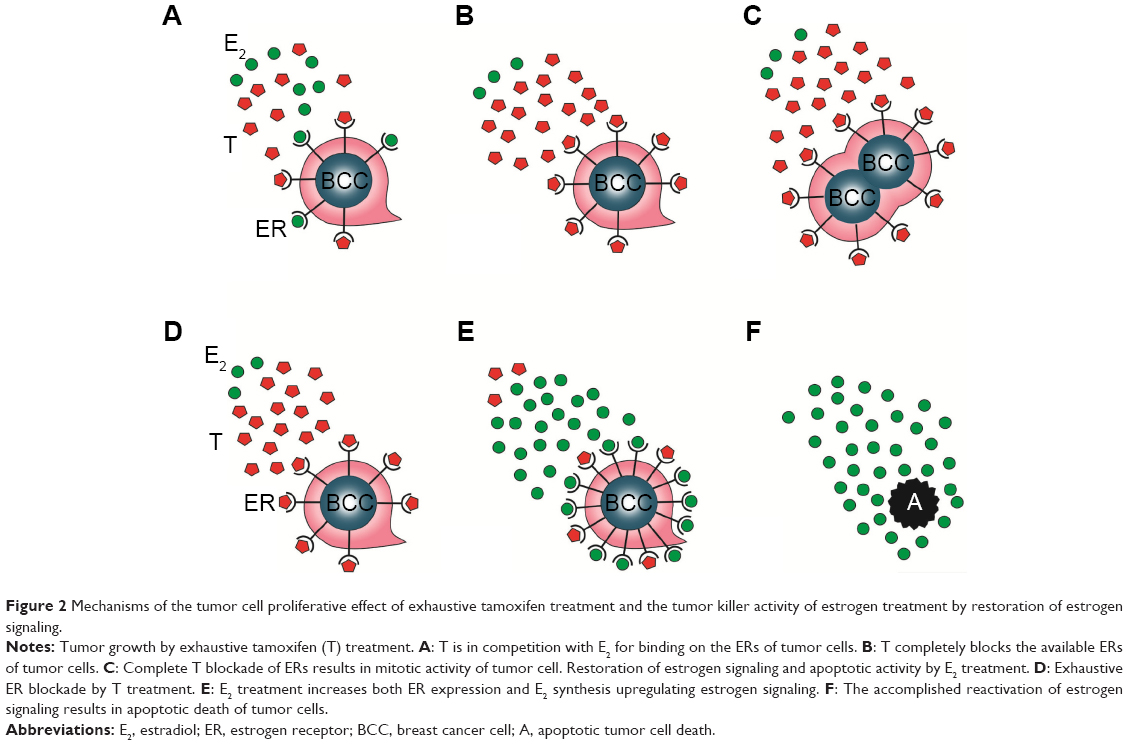 | Figure 2 Mechanisms of the tumor cell proliferative effect of exhaustive tamoxifen treatment and the tumor killer activity of estrogen treatment by restoration of estrogen signaling. |
In conclusion, during long-term antiestrogen treatment, the upregulation of both estrogen and ER synthesis exhibits gradual exhaustion and the development of complete antiestrogen blockade of estrogen signaling results in rapid tumor spread. Fortunately, high-dose estrogen treatment is capable of restoring the estrogen signaling even after exhaustive antiestrogen therapy with the achievement of rapid tumor regression.
The phases of antiestrogen treatment induced changes in estrogen signaling among genetically proficient women
Preventive tamoxifen treatment has reduced sporadic, ER-positive breast cancer development in a near 50% of the cases.71 In such cases, there are no mutations in the genes participating in estrogen signaling and the associated DNA stabilizer systems, and antiestrogen use may provoke extreme compensatory mechanisms for upregulation and restoration of attacked estrogen signaling. The clinical result is transient tumor prevention or regression, which is mistakenly attributed to a beneficial antiestrogen effect, while in the background, the upregulation of estrogen signaling exerts anticancer capacity.
There are three phases of antiestrogen administration in genetically proficient breast cancer cases, treated by either ER blocker or aromatase inhibitor, which can be characterized by good tumor regression, stagnation of tumor growth, and aggressive tumor spread (Table 1).
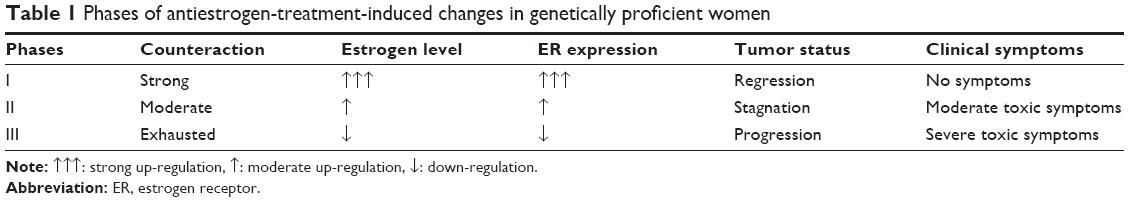 | Table 1 Phases of antiestrogen-treatment-induced changes in genetically proficient women |
In the first phase of antiestrogen treatment, mutation-free patients seem to be clinically healthy at phenomenal level, and tumor regression is the predominant clinical finding. At the same time, thorough laboratory and genetic examinations may reveal inhibitory effects of estrogen signaling in depth, together with transitorily effective defensive counteractions. These patients construe the “magic” 30% of overall breast cancer cases exhibiting transient good response to antiestrogen therapy.
There are reports on embarrassing data that suggest direct correlations between preserved estrogen signaling and the good anticancer capacity of antiestrogens. During this first period, estrogen signaling may exhibit compensatory upregulation in tamoxifen-treated premenopausal patients experiencing regular cycles and ovulatory activity with the potential to become pregnant.72 Moreover, in breast cancer biopsy specimens, active estrogen signaling characterized by intense phosphorylation at Ser(167) of ERs predicted longer disease-free survival and overall survival for the patients.44 In sequential biopsies of large primary breast tumors, measurement of aromatase content before and during effective treatment with aromatase inhibitor showed a surprisingly marked, counteractive increase in enzyme activity.73 These results support that the provoked compensatory upregulation of estrogen signaling may be in correlation with successful tumor regression instead of an antiestrogenic effect.
In the second phase of antiestrogen treatment, the compensatory actions are still operating but are not enough to restore the proper estrogen signaling and the DNA safeguarding capacity. In this phase, the stagnation of tumor growth is the main clinical experience. Patients may exhibit diverse toxic symptoms of estrogen deficiency, and at the same time, laboratory and genetic investigations may explore a wealthy cavalcade of failures in the transcriptional mechanism of ERs, and defensive but insufficient counteractions, such as hyperestrogenism.11
Tamoxifen treatment induces artificial estrogen resistance in women. These patients mimic the chaotic findings, which are characteristic of cases suffering from the genetic failures of ER expression and/or transcriptional activity coupled with counteractive defense mechanisms, such as extreme estrogen synthesis.31 In tamoxifen-treated cases, very high compensatory estrogen levels and even an increased ER expression may be associated with the symptoms of ER blockade-induced estrogen resistance, such as multiple ovarian cysts74 or endometrial hyperplasia.75 The authors mistakenly attributed these complications to the reactive hyperestrogenism, and luteinizing hormone-releasing treatment was erroneously recommended for the suppression of residual ovarian function so as to achieve proper hypoestrogenic status.75
On the other hand, exhaustive aromatase inhibitor treatment in breast cancer cases mimics the symptoms of aromatase deficiency syndrome deriving from the genetic defect of aromatase synthesis.76 In the case of this artificial aromatase deficiency, patients may exhibit even an increased counteractive expression in aromatase enzyme synthesis besides the overexpression of ERs. In the second phase of aromatase inhibitor treatment, apparent resistance to aromatase inhibitors is mistakenly regarded as an increased cross talk between intensified ER and GFR signaling pathways.45
In the third phase of antiestrogen treatment, both extreme ER expression and aromatase synthesis are exhausted, and the completion of estrogen signal blockage results in rapid growth and metastatic spread of breast cancer leading to the death of the patient. At the same time, diverse toxic effects of estrogen deficiency and accidentally developing cancers at different sites, particularly in the endometrium, may be diagnosed. This phase of complete antiestrogen blockade of estrogen signaling is mistakenly referred to as acquired antiestrogen resistance.
In the remaining half of patients, the key for ineffective antiestrogen medication against ER-positive breast cancers may be the manifestation of earlier hidden point mutations affecting either estrogen signaling or the associated gene stabilizer systems.11 These cases do not have sufficient extra capacities for defensive ER overexpression and increased estrogen synthesis against the artificial inhibition of estrogen signaling. In such patients, the failure of tumor prevention or regression by antiestrogen treatment is erroneously regarded as de novo resistance against antiestrogen treatment.
Dangers of antiestrogen treatment in women with BRCA gene mutations
In women with BRCA gene mutations, antiestrogens exhibited a very low degree of protection against familiar breast cancer risk by treatment with either ER blockade or aromatase inhibition.47,48 These unfavorable effects were erroneously explained by the presumed hormonally independent development of BRCA-mutation linked cancers.47
In BRCA gene mutation carriers, the genetically defined serious defects of ER expression and transcriptional activity as well as the baseline increased estrogen synthesis may explain the antiestrogen refractory status. The increased compensatory estrogen concentration in BRCA-mutation carriers may frequently be enough for the restoration of genetically defective ER signaling; however, the additional provocation of antiestrogen treatment may exhaust the defensive increase in aromatase synthesis.11
In conclusion, in BRCA-mutation carrier women, the momentarily compensated genetic defects of estrogen signaling are in transient equilibrium and may strongly be distressed by antiestrogen treatment.
Oophorectomy as erroneous breast cancer prevention strategy in BRCA gene mutation carriers
In BRCA-mutation carrier women, the failures of prophylactic approach by antiestrogens led to the practice of oophorectomy and breast tissue removal as preferable cancer preventive measures.77 Oophorectomy seems to be an effective means of reducing the risk of breast cancer in carriers of BRCA mutations.78
Possibility of cancer prophylaxis in BRCA-mutation carrier women by oophorectomy seems to be paradoxical, particularly in premenopausal cases. In such patients, there is a permanent fight against the defective estrogen signaling by means of increased estrogen synthesis, and the abrupt estrogen depletion of oophorectomy may be a stronger shock in these cases than in BRCA-proficient women.
Nevertheless, in BRCA-mutation carriers, the brutalism of oophorectomy may be more promising, but not a good method for cancer prevention as compared to highly toxic antiestrogen treatment. Removal of the ovaries does not provoke any further decrease in the low baseline level of ER expression as compared to the tamoxifen blockade of ERs. Moreover, the cessation of estrogen synthesis is restricted to the missing ovaries after their removal, while aromatase inhibitors block the activity of aromatase enzyme at all extraovarian sites. The shock of preterm menopause may transiently induce extreme extraovarian estrogen synthesis in BRCA-mutation carrier cases, but the possibility of delayed adaptation to the drastic changes is highly risky in terms of cancer initiation. Moreover, the initially provoked excessive extraovarian estrogen synthesis may calm down later and the patient remains defenseless.
Considering the health disadvantage of estrogen withdrawal even in BRCA-proficient cases, neither surgical nor chemical ovarian suppression may be the appropriate choice for cancer prevention and therapy in BRCA-mutation carriers. By contrast, exogenous estrogen treatment has protective effect against breast cancer even in women with BRCA gene mutation, while the anticancer effect requires higher hormone doses depending on the resistance grade of ERs.11,19,79
Conclusion
In conclusion, it can be established that whatever type of available endocrine therapies may be used, including antiestrogen, estrogen treatment, or oophorectomy, a provoked upregulation of ER signaling seems to be the key mechanism of successful prevention and treatment for ER-positive breast cancer. Satisfactory upregulation of ER expression and aromatase enzyme activity may highly increase estrogen signaling and results in apoptotic death of tumor cells.
In breast cancer cases, there are great differences in the apparent cancer-treating activities of antiestrogen administration, which may be explained by the different reserve capacities for upregulation of inhibited estrogen signaling. In patients having no genetic defects affecting either estrogen signaling or DNA stabilizer systems, antiestrogen treatment may provoke extremely upregulated estrogen signaling, which results in transiently good tumor regression. Later, the heavy exposure to antiestrogen treatment may cause exhaustion of the defensive mechanisms associated with the stagnation and even progression of tumor growth. This phase is mistakenly regarded as acquired antiestrogen resistance.
In breast cancer cases, having earlier compensated genetic point mutations affecting the estrogen signaling or other gene stabilizer systems, antiestrogens cannot provoke sufficient counteractions. The upregulation of ER expression and/or estrogen synthesis is not enough to compensate the inhibition of estrogen signaling. In such cases, the absence of tumor regression is erroneously regarded as de novo antiestrogen resistance.
In genetically heavily challenged breast cancer cases, such as BRCA gene mutation carriers, the defective capacity for ER expression and the counteractive stimulation of estrogen synthesis allow limited possibility for further defense against antiestrogen treatment. By contrast, high-dose estrogen treatment has strong upregulative impact on estrogen signaling in both genetically proficient and challenged patients and exhibits anticancer capacity even after exhaustion by heavy exposure to antiestrogen administration.
A lesson should be drawn from the 4 decades of antiestrogen use, recognizing that human willpower should not interfere with the still poorly understood gene regulatory mechanisms. The question should also be raised, whether we can ever achieve a complete understanding of the marvelous network of estrogen signaling and other DNA stabilizer systems?
Disclosure
The author reports no conflicts of interest in this work.
References
Seymour CB, Mothersill C. Breast cancer causes and treatment: where are we going wrong? Breast Cancer (Dove Med Press). 2013;5:111–119. | ||
Beatson GT. On the treatment of inoperable cases of carcinoma of the mamma: suggestions for a new method of treatment, with illustrative cases. Lancet. 1896;2:104–107. | ||
Boyd S. On oophorectomy in cancer of the breast. BMJ. 1900;2:1161–1187. | ||
Haddow A, Watkinson JM, Paterson E, Koller PC. Influence of synthetic oestrogens on advanced malignant disease. BMJ. 1944;2(393–398):1944. | ||
Kennedy BJ. Hormone therapy for advanced breast cancer. Cancer. 1965;18:1551–1557. | ||
Jordan VC, Dowse LJ. Tamoxifen as an antitumour agent: effect on oestrogen binding. J Endocrinol. 1976;68:297–303. | ||
Buzdar AU, Jonat W, Howell A, et al. Anastrozole versus megestrol acetate in the treatment of postmenopausal women with advanced breast carcinoma: results of a survival update based on a combined analysis of data from two mature phase III trials. Arimidex Study Group. Cancer. 1998;83(6):1142–1152. | ||
Cole MP, Jones CT, Todd ID. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br J Cancer. 1971;25(2):270–275. | ||
Baum M, Budzar AU, Cuzick J, et al; ATAC Trialists’ Group. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomized trial. Lancet. 2002;359(9324):2131–2139. | ||
Colditz GA, Bohlke K. Priorities for the primary prevention of breast cancer. CA Cancer J Clin. 2014;64(3):186–194. | ||
Suba Z. DNA stabilization by the upregulation of estrogen signaling in BRCA gene mutation carriers. Drug Design Devel Ther. 2015;9:2663–2675. | ||
Mukhtar TK, Yeates DR, Goldacre MJ. Breast cancer mortality trends in England and the assessment of the effectiveness of mammography screening: population-based study. J R Soc Med. 2013;106:234–242. | ||
Forouzanfar MH, Foreman KJ, Delossantos AM, et al. Breast and cervical cancer in 187 countries between 1980 and 2010: a systematic analysis. Lancet. 2011;378:1461–1484. | ||
Autier P, Boniol M, Gavin A, Vatten LJ. Breast cancer mortality in neighbouring European countries with different levels of screening but similar access to treatment: trend analysis of WHO mortality database. BMJ. 2011;343:d4411. | ||
Ginsberg O, Love RR. Breast cancer: a neglected disease for the majority of affected women worldwide. Breast J. 2011;17:289–295. | ||
Maggi A. Liganded and unliganded activation of estrogen receptor and hormone replacement therapies. Biochim Biophys Acta. 2011;1812(8):1054–1060. | ||
Helguero LA, Faulds MH, Gustafsson JA, Haldosén LA. Estrogen receptors alfa (ERalpha) and beta (ERbeta) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene. 2005;24(44):6605–6616. | ||
Litwiniuk MM, Roznowski K, Filas V, et al. Expression of estrogen receptor beta in the breast carcinoma of BRCA1 mutation carriers. BMC Cancer. 2008;8:100. | ||
Suba Z. Diverse pathomechanisms leading to the breakdown of cellular estrogen surveillance and breast cancer development: new therapeutic strategies. Drug Design Devel Ther. 2014;8:1381–1390. | ||
Mohamed MK, Abdel-Rahman AA. Effect of long-term ovariectomy and estrogen replacement on the expression of estrogen receptor gene in female rats. Eur J Endocrinol. 2000;142(3):307–314. | ||
Suba Z. Interplay between insulin resistance and estrogen deficiency as co-activators in carcinogenesis. Pathol Oncol Res. 2012;18(2):123–133. | ||
Suba Z. Circulatory estrogen level protects against breast cancer in obese women. Recent Pat Anticancer Drug Discov. 2013;8(2):154–167. | ||
Shaaban AM, Sloane JP, West CR, Foster CS. Breast cancer risk in usual ductal hyperplasia is defined by estrogen receptor-alpha and Ki-67 expression. Am J Pathol. 2002;160(2):597–604. | ||
Santen RJ, Song RX, Zhang Z, et al. Long-term estradiol deprivation in breast cancer cells up-regulates growth factor signaling and enhances estrogen sensitivity. Endocr Relat Cancer. 2005;12:S61–S73. | ||
Liu H, Lee ES, Gajdos C, et al. Apoptotic action of 17beta-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst. 2003;95(21):1586–1597. | ||
Schumacher A, Costa SD, Zenclussen AC. Endocrine factors modulating immune responses in pregnancy. Front Immunol. 2014;5:196. | ||
Liu S, Ruan X, Schultz S, et al. Oestetrol stimulates proliferation and oestrogen receptor expression in breast cancer cell lines: comparison of four oestrogens. Eur J Contracept Reprod Health Care. 2015;20(1):29–35. | ||
Stoica GE, Franke TF, Moroni M, et al. Effect of estradiol on estrogen receptor-alpha gene expression and activity can be modulated by the ErbB2/PI 3-K/Akt pathway. Oncogene. 2003;22(39):7998–8011. | ||
Suba Z. Low estrogen exposure and/or defective estrogen signaling induces disturbances in glucose uptake and energy expenditure. J Diabet Metab. 2013;4:272–281. | ||
Smith EP, Boyd J, Frank GR, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331:1056–1061. | ||
Quaynor SD, Stradtman EW, Kim HG, et al. Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant. N Engl J Med. 2013;369(2):164–171. | ||
Britt K, Ashworth A, Smalley M. Pregnancy and the risk of breast cancer. Endocr Relat Cancer. 2007;14(4):907–933. | ||
Asztalos S, Gann PH, Hayes MK, et al. Gene expression patterns in the human breast after pregnancy. Cancer Prev Res (Phila). 2010;3(3):301–311. | ||
Jordan VC, Patel R, Lewis-Wambi JS, Swaby RF. By looking back we can see the way forward: enhancing the gains achieved with antihormone therapy. Breast Cancer Res. 2008;10(suppl 4):S16. | ||
Lin NU, Winer EP. Advances in adjuvant endocrine therapy for postmenopausal women. J Clin Oncol. 2008;26:798–805. | ||
Ingle JN, Ahmann DL, Green SJ, et al. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. N Engl J Med. 1981;304(1):16–21. | ||
Ingle JN. Estrogen as therapy for breast cancer. Breast Cancer Res. 2002;4(4):133–136. | ||
Hayes DF. Tamoxifen: Dr Jekyll and Mr Hyde? J Natl Cancer Inst. 2004;96:895–897. | ||
Osborne CK. Tamoxifen in the treatment of breast cancer. N Engl J Med. 1998;339:1609–1618. | ||
Braithwaite RS, Chlebowski RT, Lau J, George S, Hess R, Col FN. Meta-analysis of vascular and neoplastic events associated with tamoxifen. J Gen Intern Med. 2003;18:937–947. | ||
Suba Z. Failures and controversies of the antiestrogen treatment of breast cancer. In: Suba Z, editor. Estrogen Prevention for Breast Cancer. (Chap 6). New York: Nova Science Publishers Inc; 2013:105–125. | ||
Howell A, Cuzick J, Baum M, et al; ATAC Trialists’ Group. Results of the ATAC (arimidex, tamoxifen, alone or in combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet. 2005;365(9453):60–62. | ||
Simpson ER, Dowsett M. Aromatase and its inhibitors: significance for breast cancer therapy. Recent Prog Horm Res. 2002;57:317–338. | ||
Jiang J, Sarwar N, Peston D, et al. Phosphorylation of estrogen receptor-alpha at Ser167 is indicative of longer disease-free and overall survival in breast cancer patients. Clin Cancer Res. 2007;13(19):5769–5776. | ||
Dowsett M, Martin LA, Smith I, Johnston S. Mechanisms of resistance to aromatase inhibitors. J Steroid Biochem Mol Biol. 2005;95(1–5):167–172. | ||
Tolhurst RS, Thomas RS, Kyle FJ, et al. Transient over-expression of estrogen receptor-α in breast cancer cells promotes cell survival and estrogen-independent growth. Breast Cancer Res Treat. 2011;128(2):357–368. | ||
Gorski JJ, Kennedy RD, Hosey AM, Harkin DP. The complex relationship between BRCA1 and ERalpha in hereditary breast cancer. Clin Cancer Res. 2009;15(5):1514–1518. | ||
Miller WR. Aromatase inhibitors: mechanism of action and role in the treatment of breast cancer. Semin Oncol. 2003;30:3–11. | ||
Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer. 2004;11:643–658. | ||
Kuske B, Naughton C, Moore K, et al. Endocrine therapy resistance can be associated with high estrogen receptor alpha (ERalpha) expression and reduced ERalpha phosphorylation in breast cancer models. Endocr Relat Cancer. 2006;13(4):1121–1133. | ||
de Leeuw R, Neefjes J, Michalides R. A role for estrogen receptor phosphorylation in the resistance to tamoxifen. Int J Breast Cancer. 2011;2011:232435. | ||
Robinson DR, Wu YM, Vats P, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45(12):1446–1451. | ||
Shin A, Kang D, Nishio H, et al. Estrogen receptor alpha gene polymorphisms and breast cancer risk. Breast Cancer Res Treat. 2003;80:127–131. | ||
Osborne CK, Shou J, Massarweh S, Schiff R. Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res. 2005;11(2 pt 2):865s–870s. | ||
Clarke R, Liu MC, Bouker KB, et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene. 2003;22(47):7316–7339. | ||
Massarweh S, Osborne CK, Creighton CJ, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68(3):826–833. | ||
Yamauchi H, Stearns V, Hayes DF. When is a tumor marker ready for prime time? A case study of c-erbB-2 as a predictive factor in breast cancer. J Clin Oncol. 2001;19(8):2334–2356. | ||
De Placido S, De Laurentiis M, Carlomagno C, et al. Twenty-year results of the Naples GUN randomized trial: predictive factors of adjuvant tamoxifen efficacy in early breast cancer. Clin Cancer Res. 2003;9(3):1039–1046. | ||
Zhang Y, Moerkens M, Ramaiahgari S, et al. Elevated insulin-like growth factor 1 receptor signaling induces antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling routes. Breast Cancer Res. 2011;13(3):R52. | ||
Stoica A, Saceda M, Doraiswamy VL, Coleman C, Martin MB. Regulation of estrogen receptor-alpha gene expression by epidermal growth factor. J Endocrinol. 2000;165(2):371–378. | ||
Massarweh S, Schiff R. Resistance to endocrine therapy in breast cancer: exploiting estrogen receptor/growth factor signaling crosstalk. Endocr Relat Cancer. 2006;13(1):515–524. | ||
Suba Z. Triple-negative breast cancer risk in women is defined by the defect of estrogen signaling: preventive and therapeutic implications. Onco Targets Ther. 2014;7:147–164. | ||
Levenson AS, Jordan VC. Transfection of human estrogen receptor (ER) cDNA into ER-negative mammalian cell lines. J Steroid Biochem Mol Biol. 1994;51(5–6):229–239. | ||
Lønning PE. Stepwise estrogen suppression manipulating the estrostat. J Steroid Biochem Mol Biol. 2001;79(1–5):127–132. | ||
Jordan VC, Lewis-Wambi JS, Patel RR, Kim H, Ariazi EA. New hypotheses and opportunities in endocrine therapy: amplification of oestrogen-induced apoptosis. Breast. 2009;18(suppl 3):S10–S17. | ||
Lønning PE. Additive endocrine therapy for advanced breast cancer back to the future. Acta Oncol. 2009;48:1092–1101. | ||
Ellis MJ, Gao F, Dehdashti F, et al. Lower-dose versus high dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: a phase-2 randomized study. JAMA. 2009;302(774–780):2009. | ||
Lobanova YS, Scherbakov AM, Shatskaya VA, Evteev VA, Krasil’nikov MA. NF-kappaB suppression provokes the sensitization of hormone-resistant breast cancer cells to estrogen apoptosis. Mol Cell Biochem. 2009;324(1–2):65–71. | ||
Mahtani RL, Stein A, Vogel CL. High-dose estrogen as salvage hormonal therapy for highly refractory metastatic breast cancer: a retrospective chart review. Clin Ther. 2009;31(pt 2):2371–2378. | ||
Jordan VC. The new biology of estrogen-induced apoptosis applied to treat and prevent breast cancer. Endocr Relat Cancer. 2015;22(1):R1–R31. | ||
Folkerd EJ, Martin LA, Kendall A, Dowsett M. The relationship between factors affecting endogenous oestradiol levels in postmenopausal women and breast cancer. J Steroid Biochem Mol Biol. 2006;102:250–255. | ||
Jordan VC, Fritz NF, Langan-Fahey S, Thompson M, Tormey DC. Alteration of endocrine parameters in premenopausal women with breast cancer during long-term adjuvant therapy with tamoxifen as the single agent. J Natl Cancer Inst. 1991;83(20):1488–1491. | ||
Miller WR, O’Neill J. The importance of local estrogen synthesis in the breast. Steroids. 1987;50:537–548. | ||
Mourits MJ, de Vries EG, Willemse PH, et al. Ovarian cysts in women receiving tamoxifen for breast cancer. Br J Cancer. 1999;79(11–12):1761–1764. | ||
Madeddu C, Gramignano G, Kotsonis P, Paribello F, Macciò A. Ovarian hyperstimulation in premenopausal women during adjuvant tamoxifen treatment for endocrine-dependent breast cancer: a report of two cases. Oncol Lett. 2014;8(3):1279–1282. | ||
Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab. 1995;80(12):3689–3698. | ||
Meijers-Heijboer H, Brekelmans CT, Menke-Pluymers M, et al. Use of genetic testing and prophylactic mastectomy and oophorectomy in women with breast or ovarian cancer from families with a BRCA1 or BRCA2 mutation. J Clin Oncol. 2003;21:1675–1681. | ||
Eisen A, Lubinski J, Klijn J, et al. Breast cancer risk following bilateral oophorectomy in BRCA1 and BRCA2 mutation carriers: an international case-control study. J Clin Oncol. 2005;23:7491–7496. | ||
Suba Z. Defective estrogen signaling is the highest risk for breast cancer. In: Watanabe HS, editor. Horizons in Cancer Research. Volume 59 (Chap 3). New York: Nova Science Publishers Inc; 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.