")
Back to Journals » Hepatic Medicine: Evidence and Research » Volume 8
The pathophysiology of thrombocytopenia in chronic liver disease
Authors Mitchell O, Feldman D, Diakow M, Sigal S
Received 1 August 2015
Accepted for publication 4 December 2015
Published 15 April 2016 Volume 2016:8 Pages 39—50
DOI https://doi.org/10.2147/HMER.S74612
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Gerry Lake-Bakaar
Oscar Mitchell,1 David M Feldman,1,2 Marla Diakow,1 Samuel H Sigal3
1Department of Medicine, 2Division of Gastroenterology and Liver Diseases, New York University School of Medicine, Langone Medical Center, New York, 3Division of Gastroenterology and Liver Diseases, Department of Medicine, Montefiore Medical Center, Albert Einstein College of Medicine, Bronx, NY, USA
Abstract: Thrombocytopenia is the most common hematological abnormality encountered in patients with chronic liver disease (CLD). In addition to being an indicator of advanced disease and poor prognosis, it frequently prevents crucial interventions. Historically, thrombocytopenia has been attributed to hypersplenism, which is the increased pooling of platelets in a spleen enlarged by congestive splenomegaly secondary to portal hypertension. Over the past decade, however, there have been significant advances in the understanding of thrombopoiesis, which, in turn, has led to an improved understanding of thrombocytopenia in cirrhosis. Multiple factors contribute to the development of thrombocytopenia and these can broadly be divided into those that cause decreased production, splenic sequestration, and increased destruction. Depressed thrombopoietin levels in CLD, together with direct bone marrow suppression, result in a reduced rate of platelet production. Thrombopoietin regulates both platelet production and maturation and is impaired in CLD. Bone marrow suppression can be caused by viruses, alcohol, iron overload, and medications. Splenic sequestration results from hypersplenism. The increased rate of platelet destruction in cirrhosis also occurs through a number of pathways: increased shear stress, increased fibrinolysis, bacterial translocation, and infection result in an increased rate of platelet aggregation, while autoimmune disease and raised titers of antiplatelet immunoglobulin result in the immunologic destruction of platelets. An in-depth understanding of the complex pathophysiology of the thrombocytopenia of CLD is crucial when considering treatment strategies. This review outlines the recent advances in our understanding of thrombocytopenia in cirrhosis and CLD.
Keywords: cirrhosis, thrombocytopenia, thrombopoietin
Introduction
Thrombocytopenia is the most common hematological abnormality encountered in patients with chronic liver disease (CLD),1 occurring in 64%–84% of patients with cirrhosis or fibrosis.2 Among patients undergoing bone marrow biopsies for thrombocytopenia of unknown etiology, the prevalence of cirrhosis is as high as 35%.3 In addition to being an indicator of advanced disease,4 thrombocytopenia is associated with a poorer prognosis,1 and it frequently prevents patients from receiving crucial interventions such as medications, as well as invasive diagnostic or therapeutic procedures.5
Historically, thrombocytopenia has been attributed to hypersplenism, namely, the increased pooling of platelets in a spleen enlarged by congestive splenomegaly secondary to portal hypertension.6 Over the past decade, however, there have been significant advances in the understanding of thrombopoiesis, which, in turn, has led to an improved understanding of thrombocytopenia in cirrhosis. Multiple factors contribute to the development of thrombocytopenia in the cirrhotic patient and these can broadly be divided into those leading to decreased production, splenic sequestration, and increased destruction (Figure 1). This review outlines the recent advances in our understanding of the pathophysiology of thrombocytopenia in cirrhosis.
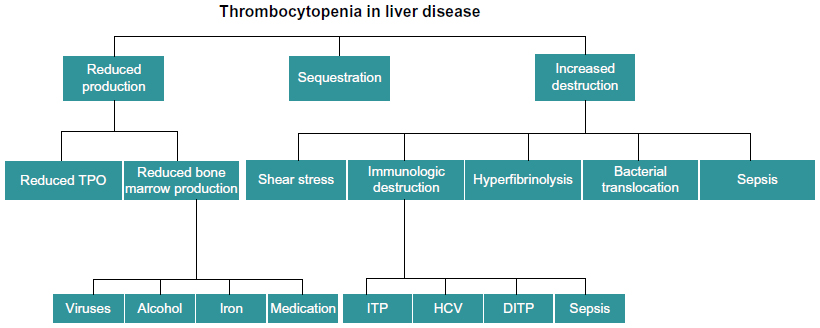 | Figure 1 Factors that contribute to the development of thrombocytopenia in patients with cirrhosis. |
Decreased platelet production
Platelet production can be decreased due to depressed thrombopoietin (TPO) levels and direct bone marrow suppression.
Thrombopoietin
Hepatic production of TPO plays a pivotal role in thrombopoiesis (Figure 2). In 1990, the oncogene v-mpl was identified from the murine myeloproliferative leukemia virus, which was capable of immortalizing bone marrow hematopoietic cells from different lineages.7 In 1992, the human homologue, c-mpl, was cloned, and sequence data revealed that it encoded a protein that was homologous to members of the hematopoietic receptor superfamily.8 Antisense oligodeoxynucleotides of c-mpl were shown to selectively inhibit megakaryocyte colony formation, demonstrating that c-mpl regulated thrombopoiesis.9
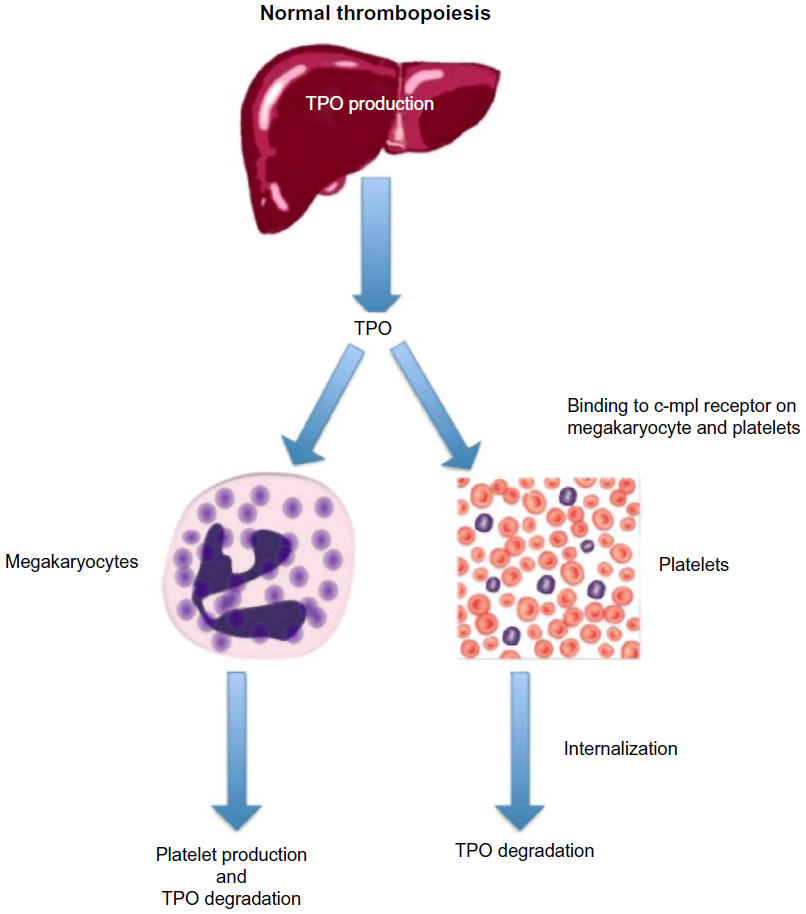 | Figure 2 Normal thrombopoiesis. |
The ligand for c-mpl, TPO, was cloned in 1994. It has a 353-amino acid transmembrane domain with two extracellular cytokine receptor domains and two intracellular cytokine receptor box motifs.10 TPO is the major regulator of megakaryocytopoiesis, and it regulates both platelet production and maturation. It is a glycoprotein (GP) that shares significant amino acid sequence homology with erythropoietin (EPO).11 TPO is primarily made in the liver by both parenchymal cells and sinusoidal endothelial cells and is secreted into the circulation at a constant rate.12
After binding to the surface of platelets and megakaryocytes through the c-mpl receptor,13 TPO is internalized and destroyed, thereby reducing further platelet and megakaryocyte exposure to the hormone.14 Stimulation of the TPO receptor results in activation of a number of signaling pathways via Janus kinase type 2 (JAK2) and tyrosine kinase 2 (TYK2).15 Mitogen-activated protein kinase activation subsequently leads to changes in gene expression, promoting progression of stem cells along the megakaryocytic pathway, megakaryocyte maturation, and subsequent release of normally functioning platelets into the peripheral circulation.16 Because the circulating level of TPO is inversely correlated to the platelet mass, low platelet counts lead to higher TPO levels due to decreased degradation (Figure 3). The increased exposure of undifferentiated bone marrow cells to TPO leads to their differentiation into megakaryocytes and maturation. This increased platelet cell mass, in turn, binds increasing amounts of TPO, reducing its circulation level, and leading to decreased platelet production. This negative feedback mechanism is highlighted by the observation that mice genetically altered to be defective in c-mpl have low platelet and megakaryocyte numbers and elevated TPO levels.17
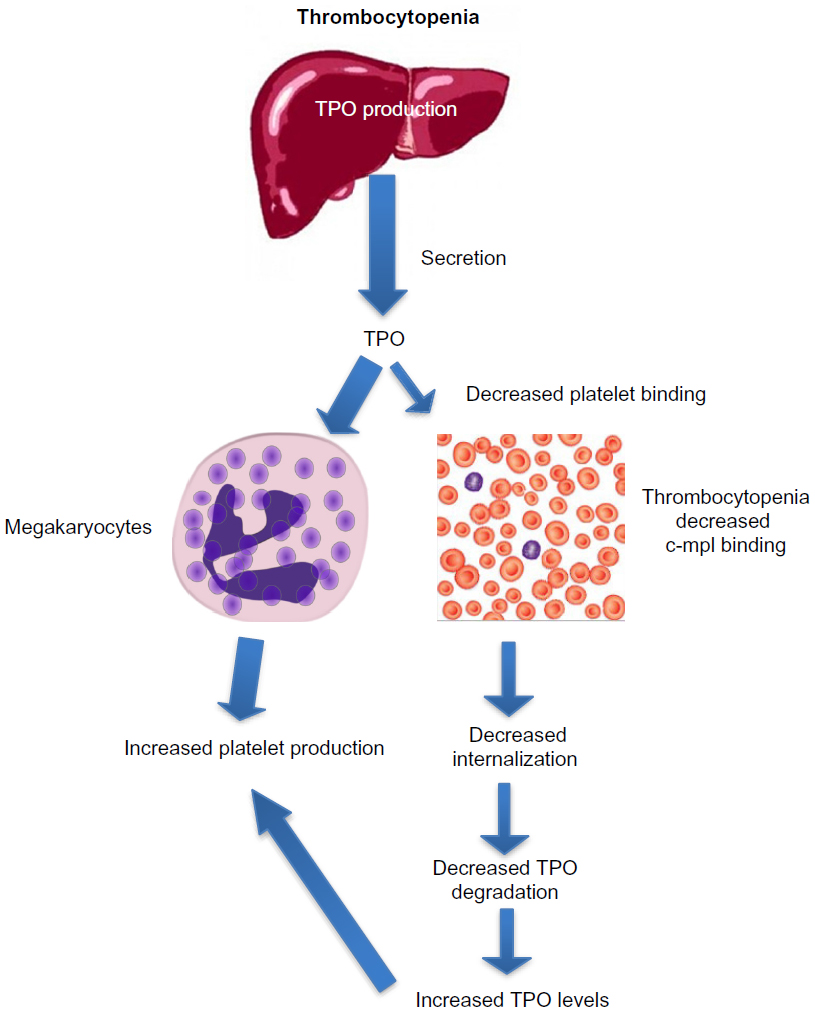 | Figure 3 Increased thrombopoietin levels in thrombocytopenia lead to increased platelet production. |
Decreased hepatic production of TPO is a critical factor in the development of thrombocytopenia in cirrhosis (Figure 4). The prevalence and severity of thrombocytopenia correlate with and parallel the severity of underlying liver disease, particularly, the extent of fibrosis.18 The prevalence of thrombocytopenia is higher in patients with Stages 3 and 4 fibrosis when compared to patients with Stages 0–2 fibrosis (64% vs 6%).2 There is an inverse relationship between TPO levels and liver function, as assessed by tests that measure liver function, such as the indocyanine green retention and aminopyrine breath tests.19 Cirrhotic patients with thrombocytopenia have lower levels of circulating TPO than those with normal platelet counts.20 The key role played by TPO in thrombocytopenia of CLD is highlighted by the interaction between TPO and platelets during the perioperative period of liver transplantation: TPO levels are often undetectable in patients with cirrhosis before transplantation and rise immediately posttransplantation, which is followed by a rise in peripheral platelet count and normalization of both TPO levels and platelet count in most patients within 14 days.21
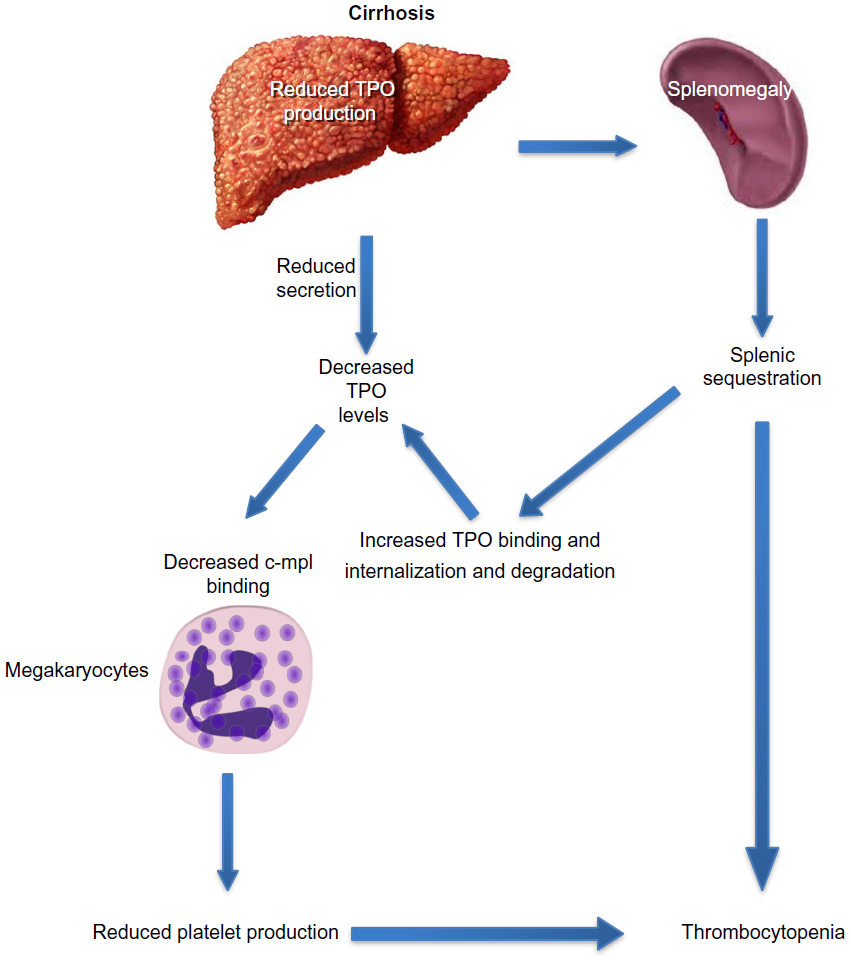 | Figure 4 Decreased thrombopoietin levels in cirrhosis lead to decreased platelet production. |
Bone marrow suppression
Inadequate production of platelets due to bone marrow suppression in selected cases may also play a crucial role in the development of thrombocytopenia in cirrhosis.22 Possible etiologies include suppression by viruses, alcohol, iron overload, and medications.
Viruses
Hepatitis A virus, hepatitis B virus, and hepatitis C virus (HCV) directly inhibit the growth and differentiation of human bone marrow progenitor cells in vitro.23 Thrombocytopenia is especially common in patients infected with HCV through a variety of mechanisms, one of which is direct bone marrow suppression.24 Patients with HCV without splenomegaly show depressed platelet production,25 and production increases after successful treatment of the infection.26
Alcohol
Thrombocytopenia occurs frequently in alcoholics through a direct effect on the bone marrow.27 Alcohol reduces platelet life span and leads to ineffective megakaryopoiesis.27 Following alcohol withdrawal, platelet counts rise within 5–7 days and normalize in a few weeks.28
Iron
The status of iron stores is an important factor in thrombopoiesis, especially in determining the response to EPO. Thrombocytosis is a common presentation of iron deficiency anemia,29 and it has been suggested that it serves as a protective mechanism by increasing the coagulation capacity in conditions with chronic bleeding.30 Repletion of iron deficiency in renal failure and inflammatory bowel disease, in contrast, leads to a decrease in platelet levels, occasionally with precipitous reductions and the development of thrombocytopenia.31 Experimental studies indicate that thrombocytosis in iron deficiency is due to an increased commitment of hematopoietic progenitors to the megakaryocytic lineage with accelerated differentiation that is for the most part independent of EPO and TPO.30 The elevated EPO levels that are a normal physiological response to anemia,32 however, do affect platelet production but in a biphasic response. An early but transient increase in platelet count followed by development of thrombocytopenia may be observed with EPO treatment due to a functional iron deficiency.33
The liver serves a central role in iron storage and functions as the main site of synthesis of iron transport proteins. Hepcidin, the principal iron regulatory hormone, is produced in hepatocytes34 and is secreted into circulation, wherein it binds ferroportin in macrophages and enterocytes, inducing internalization and degradation of ferroportin and inhibiting iron export.35 Excess iron or inflammation triggers increased hepcidin expression,36 resulting in decreased enterocyte iron absorption and reduced iron release from macrophages. Increased hepcidin expression has been implicated in anemia of inflammation,37 whereas decreased hepcidin expression plays an important role in hemochromatosis.38
Iron overload associated with spur cell hemolytic anemia is common in advanced cirrhosis, occurring through several mechanisms.39 Prohepcidin expression is reduced in proportion to the severity of liver disease,40 leading to decreased hepcidin levels and increased iron absorption. Spur cell hemolytic anemia is caused by a combination of altered red blood cell (RBC) membrane composition, oxidative damage, decreased RBC membrane fluidity that leads to decreased RBC survival, and hemolytic anemia41 and further contributes to increased iron absorption and hepatic iron loading.42 Experimentally, the iron status is a major determinant of the platelet response to EPO. Compared to animals with depletion of iron stores, animals with iron overloading show a more pronounced degree of thrombocytopenia due to competition between erythroid and megakaryocytic development pathways of stem cells in the absence of the protection afforded by iron deficiency.33
Medications
Cirrhotic patients are exposed to a plethora of drugs that have the potential to cause drug-induced thrombocytopenia (DITP) through multiple mechanisms that include both direct bone marrow suppression and immunological platelet destruction. Examples of medications commonly prescribed to the cirrhotic patient and that are associated with impaired thrombopoiesis include azathioprine, antibiotics, and interferon (IFN).
Azathioprine is a purine antimetabolite used as an immunosuppressive agent to treat a range of autoimmune disorders, including chronic autoimmune hepatitis. Its mechanism of action in blocking purine synthesis hinders proliferation of several cell lines, with its most pronounced effect being on lymphocytes. The most common, but often serious, side effect of this agent is bone marrow suppression, an effect that is dose dependent. Although only 5% of patients taking azathioprine show bone marrow toxicity, which can include thrombocytopenia in up to 2%,43 effective therapy is frequently not possible in cirrhotic patients with baseline thrombocytopenia. Beta-lactam antibiotics and fluoroquinolones have also been proposed as potential causes of thrombocytopenia, acting through bone marrow suppression.44
IFN-based therapies were until recently the standard of care for patients with chronic hepatitis C. Because dose-dependent thrombocytopenia is a frequent side effect of IFN, baseline thrombocytopenia in cirrhotic patients frequently prevented them from receiving effective therapy. IFN-induced bone marrow toxicity and resultant cytopenias were a common reason for treatment discontinuation or cessation.45 The mechanism for the development of IFN-induced thrombocytopenia is multifactorial and includes direct impairment of late-stage megakaryocytopoiesis46 and altered TPO levels. IFN inhibits the expression of transcription factors regulating late-stage megakaryocytopoiesis and impairs thrombopoiesis by preventing cytoplasmic maturation of megakaryocytes and preventing platelet production.46 It is also associated with both a blunted TPO response to thrombocytopenia and a direct reduction in TPO levels.47 Patients with advanced liver disease especially lack an appropriate compensatory increase in TPO in response to thrombocytopenia.47
Splenic sequestration
Historically, thrombocytopenia in cirrhosis was attributed to increased pooling of platelets in an enlarged spleen.6 The term hypersplenism was first used in 1909 to describe the presence of splenomegaly in patients with hemolytic anemia. The concept subsequently evolved to describe a distinct clinical syndrome of splenic hyperactivity associated with splenomegaly, a reduction in one or more peripheral cell types, an appropriately proliferative bone marrow response, and potential for reversal with splenectomy.48 Congestive splenomegaly develops as a result of portal hypertension and is characterized by a redistribution of blood flow and platelets from the circulating pool to the splenic pool.49 As a result, splenomegaly leads to thrombocytopenia by sequestration, and there is an inverse relationship between spleen size and platelet count.18 Because the sequestrated platelets are still capable of removing TPO from the circulation, they further contribute to the development of thrombocytopenia by lowering TPO levels.50
Increased platelet destruction
Increased platelet destruction occurs in cirrhosis through increased shear stress leading to an increased rate of platelet aggregation, immunologic destruction, increased fibrinolysis, bacterial translocation, and infection.
Shear stress
Shear stress, or the level of fluid stress applied to platelets and plasma components within the vasculature, provokes platelet aggregation. Under conditions of excessive high fluid shear stress, ultralarge von Willebrand factor (ULVWF) undergoes a conformational transition from a globular state to an extended chain conformation that is more adhesive to platelets.51,52 This leads to aggregated complexes within the vasculature and thrombus formation. ADAMTS13 (a disintegrin-like and metalloprotease with thrombospondin type 1 motif 13) is a shear-dependent metalloproteinase produced by hepatic stellate cells, which cleaves unusually large vWF.53,54 High levels of shear stress both enhance vWF–platelet aggregation and promote cleavage of vWF by ADAMTS13.55
The levels and activity of ADAMTS13 are reduced in patients with advanced cirrhosis56 due to enhanced consumption of ADAMTS13, presence of inflammatory cytokines, and the presence of an ADAMTS13 plasma inhibitor.57 Decreased levels of ADAMTS13 result in the accumulation of UL-VWFM, which, in turn, enhances high shear-stress-induced platelet aggregation. Low platelet counts in cirrhosis parallel-depressed levels of ADAMTS13 activity.56 Finally, TPO can also prime platelet aggregation in conditions of high shear stress such as portal hypertension and congestive splenomegaly.58
Immunologic destruction
Immune-mediated destruction involving antiplatelet antibodies is frequently present in cirrhosis. Among patients with CLD of diverse etiologies, up to 64% have platelet-associated anti-GP antibodies that are primarily directed against the GP IIb–IX complex.59 Lower platelet counts in patients with cirrhosis are correlated with both larger spleen volumes and higher levels of platelet-associated immunoglobulin G (PAIgG).60 In a study utilizing kinetic radio-labeled platelet techniques, platelet levels were directly correlated with platelet survival times and inversely correlated with PAIgG levels and splenic volumes.61 Specific situations in which immune-mediated thrombocytopenia are encountered include idiopathic thrombocytopenia purpura (ITP), chronic hepatitis C, infection, and medications.
Idiopathic thrombocytopenia purpura
ITP, also known as immune or autoimmune thrombocytopenia purpura, is an autoimmune disease characterized by thrombocytopenia caused by the interaction of PAIg with platelet antigens.62 In “classic” ITP, autoantibodies are predominantly, but not exclusively, produced against platelet GP IIb/IIIa and Ib/IX.63 Antibody-coated platelets are recognized by macrophages in the spleen and liver and removed from the circulation.64 Splenomegaly is typically not present, and patients usually respond well to immunosuppression.65
Autoimmune liver diseases (autoimmune hepatitis and primary biliary cirrhosis [PBC]) are frequently associated with other autoimmune conditions. Approximately 50% of patients with PBC are affected by at least one additional autoimmune disease, which may include ITP.66 Up to 40% of patients with PBC have raised levels of PAIgG,66 and there are case reports of patients with autoimmune-related liver disease with ITP.67
Viral infections have been associated with ITP, especially HCV. Up to 30% of patients with ITP without evidence of advanced liver disease are seropositive for HCV.68 The rate of ITP among patients infected with HCV is 30.2 per 100,000 person-years compared to 18.5 per 100,000 person-years for non-HCV-infected individuals.69 HCV-related ITP thrombocytopenia may be severe, usually affecting women, and usually has a good response to corticosteroids.70 Finally, ITP has also been described in association with several other viral infections, including cytomegalovirus and Epstein–Barr Virus.71
Chronic hepatitis C
Chronic infection with HCV can lead to thrombocytopenia through multiple mechanisms, as summarized by Weksler.72 Chronic HCV infection is associated with a plethora of autoimmune disorders. Approximately 38% of patients with HCV infection exhibit at least one immune-mediated, extrahepatic manifestation during the course of their disease.73 Patients with CLD due to HCV develop a thrombocytopenia that parallels the severity of their disease and is mirrored by increasing titers of PAIg.25,74 HCV can interact directly with platelets to bind platelet membranes through multiple cell surface receptors.75 Anti-HCV antibodies then coat the surface-associated HCV, ultimately leading to phagocytosis of antibody-coated platelets and accelerated platelet destruction by the reticuloendothelial system.75 The binding of HCV to platelets may also induce neoantigens on the platelet surface or drive alterations in the platelet membrane GPs, contributing to autoantibody formation against platelet membrane GPs, such as GPIIb/IIIa, and subsequent development of ITP.76 Finally, HCV is intimately related to cryoglobulinemia, and cryoglobulins might play a role in immune complex formation and accelerated platelet clearance.70
Immune-mediated drug-induced thrombocytopenia
Due to the multiple medications that cirrhotic patients receive, DITP is commonly encountered. In DITP, drug-dependent antibodies bind to specific platelet GPs in the presence of the offending drug. Platelet counts decrease within 5–7 days after exposure to a causative agent and rise within 10 days of cessation.77 Causes of DITP include antibiotics (eg, cephalosporins, linezolid, and octreotide).20–24 The hallmark of immune-related DITP is severe thrombocytopenia (platelet count: <30×109/L), often accompanied by petechiae and mucocutaneous bleeding.78
IFN therapy can rarely induce autoimmune ITP. IFN-induced autoimmune ITP has been reported to develop after 4 weeks to 12 months of therapy,79 and even after the completion of therapy.80 In contrast to the dose-dependent thrombocytopenia caused by IFN-induced bone marrow suppression, IFN-induced autoimmune ITP can cause precipitous decreases in platelet levels; it usually responds to immunosuppression.79
Heparin-induced thrombocytopenia (HIT) is one of the most commonly encountered causes of DITP. Following exposure to heparin, platelet factor 4 (PF4) forms complexes with the negatively charged heparin molecules.81 These complexes are highly immunogenic and result in the formation of HIT antibodies and subsequent aggregation of platelets with PF4/heparin complexes.82 HIT is more common in patients treated with unfractionated heparin and occurs with a frequency of 3%–6% after 7 days.83 In contrast to the aforementioned “classic” drug-induced immune thrombocytopenia, HIT is characterized usually by a moderate degree of thrombocytopenia (median platelet count nadir, ~50×109/L), and a high risk of venous and/or arterial thrombosis, rather than bleeding.84
Increased fibrinolysis
The liver plays a pivotal role in the fibrinolytic system and is responsible for sustaining a balance between bleeding and thrombosis to maintain homeostasis. The liver is important in both the production of multiple factors involved in the process and clearance of breakdown products. Under normal circumstances, deposition of fibrin within the vascular system triggers the conversion of plasminogen into the active enzyme plasmin, which then degrades fibrin and liberates fibrin and fibrinogen degradation products into the circulation.85 This plasminogen-to-plasmin conversion is driven by tissue plasminogen activator (t-PA) and opposed by plasminogen activator inhibitor (PAI).86 Alpha-2-antiplasmin is among the major inhibitors of plasmin and fibrinolysis.87 Thrombin-activatable fibrinolysis inhibitor (TAFI) inhibits recruitment of plasminogen to thrombi, slowing fibrinolysis.88
Fibrinolysis is increased in cirrhosis. There is a reduced production of clotting and inhibitory factors, as well as decreased clearance of activated factors, leading to accelerated intravascular coagulation. There is also decreased clearance of t-PA and PAI-1 from the circulation89 and decreased hepatic synthesis of alpha-2-antiplasmin and TAFI.90 As a result, there is a rebalanced state between pro- and antifibrinolytic factors, which leads to hyperfibrinolysis in up to 30%–46% of patients with end-stage liver disease.91 This hypercoagulable state with excessive platelet consumption plays a role in the development of thrombocytopenia in cirrhosis and is supported by studies of platelet kinetics analysis.92 A retrospective autopsy study of patients with liver disease found that platelet counts in patients with thrombotic complications were lower in those without thrombosis, further suggesting that increased thrombosis consumes platelets.93 Antifibrinolytic agents, such as tranexamic acid, aprotinin, and epsilon-aminocaproic acid, have been shown to reduce intraoperative bleeding in liver transplantation, as well as in cirrhotic patients with bleeding associated with hyperfibrinolysis.94–97
Bacterial translocation
Bacterial translocation associated with endotoxemia is common in cirrhosis and can accelerate platelet consumption and the development of thrombocytopenia. High levels of circulating endotoxins are observed in cirrhosis, even in those not clinically infected.98 Kalambokis and Tsianos99 first postulated the role of endotoxin in the pathophysiology of thrombocytopenia in cirrhosis: intestinal bacterial overgrowth and altered gut permeability allow bacterial translocation of microorganisms from the intestinal lumen into the portal circulation.100 Impairment of the reticuloendothelial system,101 along with portosystemic shunting, accounts for its presence in the systemic circulation.102 Endotoxin accelerates the release of proinflammatory cytokines (tumor necrosis factor-alpha [TNF-α] and interleukins [IL-3, IL-6, and IL-11]).103 The various cytokines are important regulators of inflammation, cell growth, and maturation; they have key roles in thrombopoiesis and are elevated in cirrhotic patients in proportion to the degree of liver disease.104 In patients with alcoholic cirrhosis, endotoxin levels are significantly higher among those with thrombocytopenia than in those without thrombocytopenia, and platelet counts are inversely correlated with endotoxin levels.105
Endotoxin stimulates B-cell activity and production of IgG, including PAIgG, which increases the removal of platelets from the circulation.99 It contributes to thrombocytopenia by triggering disseminated intravascular coagulation (DIC), platelet activation, aggregation, and platelet toll-like receptors.106 Platelet consumption from activation of platelet–monoctye aggregates is induced by endotoxin,107 and endotoxemia impairs ADAMTS13 activity and promotes thrombotic complications and thrombocytopenia.57
TNF-α and other inflammatory cytokines suppress hepatic production of TPO,105 inhibit the growth and differentiation of megakaryocytes,108 and induce platelet apoptosis.109 Finally, TNF-α induces vascular nitric oxide production,110 which is the main mediator for the development of portal hypertension, and suppresses TPO production.111
Cirrhosis may also predispose patients to an excessive response to lipopolysaccharide (LPS), a component of cell walls of Gram-negative bacteria, which directly increases platelet aggregation in animal models. In experimental animals and in human cells from cirrhotic patients ex vivo, LPS induces higher levels of TNF-α and IL-6 than noncirrhotic controls.112
Bacterial infection
Thrombocytopenia commonly develops in patients with infection, especially sepsis. In a retrospective review of all patients admitted to a Medical Intensive Care Unit with severe sepsis or septic shock, thrombocytopenia developed in 47.6% of patients.113 Infection is more common in patients with cirrhosis than in the general population. The overall incidence of infection in patients with liver disease has been estimated to be up to 47%.114 Multiple sources of infection are common in advanced cirrhosis, including spontaneous bacterial peritonitis, urinary tract infection, and pneumonia. Patients with cirrhosis have an increased risk of developing sepsis, sepsis-induced organ failure, and sepsis-related death.115
Mechanisms by which sepsis lead to thrombocytopenia include intensification of the adverse effects of endotoxemia. TNF-α is increased in patients with sepsis, and TNF-α levels are higher in patients with sepsis with thrombocytopenia.116 TNF-α released during infection can trigger a DIC-like picture with hyperfibrinolysis with increased platelet activation and adhesion to endothelium.117 TNF-α triggers platelet activation and amplifies platelet response to collagen in vitro.118 Activation of the coagulation system in sepsis results in fibrin clot formation and the consumption of platelets.119
Immune mechanisms have also been implicated.120 PF4 forms immune complexes with heparin and other polyanions, in addition to binding bacteria, exposing neoantigen(s), and inciting antibody formation. Specifically, PF4 has been demonstrated to bind the negatively charged LPS on Gram-negative bacteria.121 This PF4/LPS complex is immunogenic and can elicit cross-reacting antibodies against the PF4/heparin complex, resulting in a spontaneous HIT-like picture. Accordingly, anti-PF4/heparin antibody titers are higher in patients with bacteremia.122 Finally, endogenous heparinoids can also be detected in cirrhotic patients in the setting of infection and disappear following its resolution. Although they are associated with impaired coagulation, an effect on platelet count has not been detected.123
Conclusion
Thrombocytopenia is common in CLD of all etiologies and is a complicated and multifactorial phenomenon. Recent advances in elucidating the pathways of platelet production and consumption have led to significant improvements in our understanding of thrombocytopenia in CLD. An in-depth understanding of the pathophysiology of the thrombocytopenia of CLD is crucial when considering treatment strategies.
Acknowledgment
The authors are grateful to Nadia Nieves for assistance with development of the figures.
Disclosure
Samuel Sigal received research support from GSK. Oscar Mitchell, David Feldman, and Marla Diakow report no conflicts of interest in this work.
References
Qamar AA, Grace ND, Groszmann RJ, et al. Incidence, prevalence, and clinical significance of abnormal hematologic indices in compensated cirrhosis. Clin Gastroenterol Hepatol. 2009;7(6):689–695. | |
Bashour FN, Teran JC, Mullen KD. Prevalence of peripheral blood cytopenias (hypersplenism) in patients with nonalcoholic chronic liver disease. Am J Gastroenterol. 2000;95(10):2936–2939. | |
Sheikh MY, Raoufi R, Atla PR, et al. Prevalence of cirrhosis in patients with thrombocytopenia who receive bone marrow biopsy. Saudi J Gastroenterol. 2012;18(4):257–262. | |
Poynard T, Bedossa P. Age and platelet count: a simple index for predicting the presence of histological lesions in patients with antibodies to hepatitis C virus. METAVIR and CLINIVIR Cooperative Study Groups. J Viral Hepat. 1997;4(3):199–208. | |
Hayashi H, Beppu T, Shirabe K, et al. Management of thrombocytopenia due to liver cirrhosis: a review. World J Gastroenterol. 2014;20(10):2595–2605. | |
Aster RH. Pooling of platelets in the spleen: role in the pathogenesis of “hypersplenic” thrombocytopenia. J Clin Invest. 1966;45(5):645–657. | |
Wendling F, Tambourin P. The oncogene V-MPL, a putative truncated cytokine receptor which immortalizes hematopoietic progenitors. Nouv Rev Fr Hematol. 1991;33(2):145–146. | |
Vigon I, Mornon JP, Cocault L, et al. Molecular cloning and characterization of MPL, the human homolog of the v-mpl oncogene: identification of a member of the hematopoietic growth factor receptor superfamily. Proc Natl Acad Sci U S A. 1992;89(12):5640–5644. | |
Methia N, Louache F, Vainchenker W, et al. Oligodeoxynucleotides antisense to the proto-oncogene c-mpl specifically inhibit in vitro megakaryocytopoiesis. Blood. 1993;82(5):1395–1401. | |
Lok S, Kaushansky K, Holly RD, et al. Cloning and expression of murine thrombopoietin cDNA and stimulation of platelet production in vivo. Nature. 1994;369(6481):565–568. | |
de Sauvage FJ, Hass PE, Spencer SD, et al. Stimulation of megakaryocytopoiesis and thrombopoiesis by the c-Mpl ligand. Nature. 1994;369(6481):533–538. | |
Stoffel R, Wiestner A, Skoda RC. Thrombopoietin in thrombocytopenic mice: evidence against regulation at the mRNA level and for a direct regulatory role of platelets. Blood. 1996;87(2):567–573. | |
Fielder PJ, Gurney AL, Stefanich E, et al. Regulation of thrombopoietin levels by c-mpl-mediated binding to platelets. Blood. 1996;87(6):2154–2161. | |
Saur SJ, Sangkhae V, Geddis AE, et al. Ubiquitination and degradation of the thrombopoietin receptor c-Mpl. Blood. 2010;115(6):1254–1263. | |
Sattler M, Durstin MA, Frank DA, et al. The thrombopoietin receptor c-MPL activates JAK2 and TYK2 tyrosine kinases. Exp Hematol. 1995;23(9):1040–1048. | |
Rouyez MC, Boucheron C, Gisselbrecht S, et al. Control of thrombopoietin-induced megakaryocytic differentiation by the mitogen-activated protein kinase pathway. Mol Cell Biol. 1997;17(9):4991–5000. | |
Gurney AL, Carver-Moore K, de Sauvage FJ, et al. Thrombocytopenia in c-mpl-deficient mice. Science. 1994;265(5177):1445–1447. | |
Kawasaki T, Takeshita A, Souda K, et al. Serum thrombopoietin levels in patients with chronic hepatitis and liver cirrhosis. Am J Gastroenterol. 1999;94(7):1918–1922. | |
Giannini E, Botta F, Borro P, et al. Relationship between thrombopoietin serum levels and liver function in patients with chronic liver disease related to hepatitis C virus infection. Am J Gastroenterol. 2003;98(11):2516–2520. | |
Peck-Radosavljevic M, Zacherl J, Meng YG, et al. Is inadequate thrombopoietin production a major cause of thrombocytopenia in cirrhosis of the liver? J Hepatol. 1997;27(1):127–131. | |
Martin TG 3rd, Somberg KA, Meng YG, et al. Thrombopoietin levels in patients with cirrhosis before and after orthotopic liver transplantation. Ann Intern Med. 1997;127(4):285–288. | |
Koike Y, Yoneyama A, Shirai J, et al. Evaluation of thrombopoiesis in thrombocytopenic disorders by simultaneous measurement of reticulated platelets of whole blood and serum thrombopoietin concentrations. Thromb Haemost. 1998;79(6):1106–1110. | |
Zeldis JB, Mugishima H, Steinberg HN, et al. In vitro hepatitis B virus infection of human bone marrow cells. J Clin Invest. 1986;78(2):411–417. | |
Wang CS, Yao WJ, Wang ST, et al. Strong association of hepatitis C virus (HCV) infection and thrombocytopenia: implications from a survey of a community with hyperendemic HCV infection. Clin Infect Dis. 2004;39(6):790–796. | |
Bordin G, Ballare M, Zigrossi P, et al. A laboratory and thrombokinetic study of HCV-associated thrombocytopenia: a direct role of HCV in bone marrow exhaustion? Clin Exp Rheumatol. 1995;13(Suppl 13): S39–S43. | |
Garcia-Suarez J, Burgaleta C, Hernanz N, et al. HCV-associated thrombocytopenia: clinical characteristics and platelet response after recombinant alpha2b-interferon therapy. Br J Haematol. 2000;110(1):98–103. | |
Lindenbaum J, Lieber CS. Hematologic effects of alcohol in man in the absence of nutritional deficiency. N Engl J Med. 1969;281(7):333–338. | |
Cowan DH, Hines JD. Thrombocytopenia of severe alcoholism. Ann Intern Med. 1971;74(1):37–43. | |
Gross S, Keefer V, Newman AJ. The platelets in iron-deficiency anemia. I. The response to oral and parenteral iron. Pediatrics. 1964;34:315–323. | |
Evstatiev R, Bukaty A, Jimenez K, et al. Iron deficiency alters megakaryopoiesis and platelet phenotype independent of thrombopoietin. Am J Hematol. 2014;89(5):524–529. | |
Choi SI, Simone JV. Platelet production in experimental iron deficiency anemia. Blood. 1973;42(2):219–228. | |
de Klerk G, Rosengarten PC, Vet RJ, et al. Serum erythropoietin (EST) titers in anemia. Blood. 1981;58(6):1164–1170. | |
Loo M, Beguin Y. The effect of recombinant human erythropoietin on platelet counts is strongly modulated by the adequacy of iron supply. Blood. 1999;93(10):3286–3293. | |
Park CH, Valore EV, Waring AJ, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276(11):7806–7810. | |
Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093. | |
Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276(11):7811–7819. | |
Geddis AE, Kaushansky K. Cross-reactivity between erythropoietin and thrombopoietin at the level of Mpl does not account for the thrombocytosis seen in iron deficiency. J Pediatr Hematol Oncol. 2003;25(11):919–920. [author reply 920]. | |
Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33(1):21–22. | |
Deugnier Y, Turlin B, le Quilleuc D, et al. A reappraisal of hepatic siderosis in patients with end-stage cirrhosis: practical implications for the diagnosis of hemochromatosis. Am J Surg Pathol. 1997;21(6):669–675. | |
Jaroszewicz J, Rogalska M, Flisiak R. Serum prohepcidin reflects the degree of liver function impairment in liver cirrhosis. Biomarkers. 2008;13(5):478–485. | |
Allen DW, Manning N. Abnormal phospholipid metabolism in spur cell anemia: decreased fatty acid incorporation into phosphatidylethanolamine and increased incorporation into acylcarnitine in spur cell anemia erythrocytes. Blood. 1994;84(4):1283–1287. | |
Pascoe A, Kerlin P, Steadman C, et al. Spur cell anaemia and hepatic iron stores in patients with alcoholic liver disease undergoing orthotopic liver transplantation. Gut. 1999;45(2):301–305. | |
Connell WR, Kamm MA, Ritchie JK, et al. Bone marrow toxicity caused by azathioprine in inflammatory bowel disease: 27 years of experience. Gut. 1993;34(8):1081–1085. | |
Dutta TK, Badhe BA. Ciprofloxacin-induced bone marrow depression. Postgrad Med J. 1999;75(887):571–573. | |
Yamane A, Nakamura T, Suzuki H, et al. Interferon-alpha 2b-induced thrombocytopenia is caused by inhibition of platelet production but not proliferation and endomitosis in human megakaryocytes. Blood. 2008;112(3):542–550. | |
Sata M, Yano Y, Yoshiyama Y, et al. Mechanisms of thrombocytopenia induced by interferon therapy for chronic hepatitis B. J Gastroenterol. 1997;32(2):206–210. | |
Peck-Radosavljevic M, Wichlas M, Pidlich J, et al. Blunted thrombopoietin response to interferon alfa-induced thrombocytopenia during treatment for hepatitis C. Hepatology. 1998;28(5):1424–1429. | |
Jandl JH, Aster RH. Increased splenic pooling and the pathogenesis of hypersplenism. Am J Med Sci. 1967;253(4):383–398. | |
Morris PW, Patton TB, Balint JA, et al. Portal hypertension, congestive splenomegaly, and portacaval shunt. Gastroenterology. 1962;42:555–559. | |
Rios R, Sangro B, Herrero I, et al. The role of thrombopoietin in the thrombocytopenia of patients with liver cirrhosis. Am J Gastroenterol. 2005;100(6):1311–1316. | |
Siedlecki CA, Lestini BJ, Kottke-Marchant KK, et al. Shear-dependent changes in the three-dimensional structure of human von Willebrand factor. Blood. 1996;88(8):2939–2950. | |
Arya M, Anvari B, Romo GM, et al. Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex: studies using optical tweezers. Blood. 2002;99(11):3971–3977. | |
Lian EC. Pathogenesis of thrombotic thrombocytopenic purpura: ADAMTS13 deficiency and beyond. Semin Thromb Hemost. 2005; 31(6):625–632. | |
Tsai HM. Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood. 1996;87(10):4235–4244. | |
Ruggeri ZM. Mechanisms of shear-induced platelet adhesion and aggregation. Thromb Haemost. 1993;70(1):119–123. | |
Uemura M, Fujimura Y, Matsumoto M, et al. Comprehensive analysis of ADAMTS13 in patients with liver cirrhosis. Thromb Haemost. 2008;99(6):1019–1029. | |
Uemura M, Fujimura Y, Ko S, et al. Pivotal role of ADAMTS13 function in liver diseases. Int J Hematol. 2010;91(1):20–29. | |
Oda A, Miyakawa Y, Druker BJ, et al. Thrombopoietin primes human platelet aggregation induced by shear stress and by multiple agonists. Blood. 1996;87(11):4664–4670. | |
Pereira J, Accatino L, Alfaro J, et al. Platelet autoantibodies in patients with chronic liver disease. Am J Hematol. 1995;50(3):173–178. | |
Sanjo A, Satoi J, Ohnishi A, et al. Role of elevated platelet-associated immunoglobulin G and hypersplenism in thrombocytopenia of chronic liver diseases. J Gastroenterol Hepatol. 2003;18(6):638–644. | |
Aoki Y, Hirai K, Tanikawa K. Mechanism of thrombocytopenia in liver cirrhosis: kinetics of indium-111 tropolone labelled platelets. Eur J Nucl Med. 1993;20(2):123–129. | |
Macchi L, Clofent-Sanchez G, Marit G, et al. PAICA: a method for characterizing platelet-associated antibodies – its application to the study of idiopathic thrombocytopenic purpura and to the detection of platelet-bound c7E3. Thromb Haemost. 1996;76(6):1020–1029. | |
van Leeuwen EF, van der Ven JT, Engelfriet CP, et al. Specificity of autoantibodies in autoimmune thrombocytopenia. Blood. 1982;59(1):23–26. | |
Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med. 2002;346(13):995–1008. | |
George JN, Woolf SH, Raskob GE, et al. Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood. 1996;88(1):3–40. | |
Bassendine MF, Collins JD, Stephenson J, et al. Platelet associated immunoglobulins in primary biliary cirrhosis: a cause of thrombocytopenia? Gut. 1985;26(10):1074–1079. | |
Arakawa Y, Amaki S, Miyakawa H, et al. PBC-AIH overlap syndrome with concomitant ITP and Hashimoto’s disease with positivity for anti-centromere antibody. J Gastroenterol. 2004;39(5):490–495. | |
Pawlotsky JM, Bouvier M, Fromont P, et al. Hepatitis C virus infection and autoimmune thrombocytopenic purpura. J Hepatol. 1995;23(6):635–639. | |
Chiao EY, Engels EA, Kramer JR, et al. Risk of immune thrombocytopenic purpura and autoimmune hemolytic anemia among 120 908 US veterans with hepatitis C virus infection. Arch Intern Med. 2009;169(4):357–363. | |
Misiani R, Bellavita P, Fenili D, et al. Hepatitis C virus infection in patients with essential mixed cryoglobulinemia. Ann Intern Med. 1992;117(7):573–577. | |
Steeper TA, Horwitz CA, Moore SB, et al. Severe thrombocytopenia in Epstein-Barr virus-induced mononucleosis. West J Med. 1989;150(2):170–173. | |
Weksler BB. Review article: the pathophysiology of thrombocytopenia in hepatitis C virus infection and chronic liver disease. Aliment Pharmacol Ther. 2007;26(Suppl 1):13–19. | |
Mayo MJ. Extrahepatic manifestations of hepatitis C infection. Am J Med Sci. 2003;325(3):135–148. | |
Nagamine T, Ohtuka T, Takehara K, et al. Thrombocytopenia associated with hepatitis C viral infection. J Hepatol. 1996;24(2):135–140. | |
Hamaia S, Li C, Allain JP. The dynamics of hepatitis C virus binding to platelets and 2 mononuclear cell lines. Blood. 2001;98(8):2293–2300. | |
Panzer S, Seel E. Is there an increased frequency of autoimmune thrombocytopenia in hepatitis C infection? A review. Wien Med Wochenschr. 2003;153(19–20):417–420. | |
Visentin GP, Liu CY. Drug-induced thrombocytopenia. Hematol Oncol Clin North Am. 2007;21(4):685–696, vi. | |
George JN, Raskob GE, Shah SR, et al. Drug-induced thrombocytopenia: a systematic review of published case reports. Ann Intern Med. 1998;129(11):886–890. | |
Dourakis SP, Deutsch M, Hadziyannis SJ. Immune thrombocytopenia and alpha-interferon therapy. J Hepatol. 1996;25(6):972–975. | |
Arena R, Cecinato P, Lisotti A, et al. Severe immune thrombocytopenia after peg-interferon-alpha2a, ribavirin and telaprevir treatment completion: a case report and systematic review of literature. World J Hepatol. 2015;7(12):1718–1722. | |
Alberio L, Kimmerle S, Baumann A, et al. Rapid determination of anti-heparin/platelet factor 4 antibody titers in the diagnosis of heparin-induced thrombocytopenia. Am J Med. 2003;114(7):528–536. | |
Newman PM, Chong BH. Heparin-induced thrombocytopenia: new evidence for the dynamic binding of purified anti-PF4-heparin antibodies to platelets and the resultant platelet activation. Blood. 2000;96(1):182–187. | |
Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med. 2007;357(6):580–587. | |
Warkentin TE. Drug-induced immune-mediated thrombocytopenia – from purpura to thrombosis. N Engl J Med. 2007;356(9):891–893. | |
Wiman B, Collen D. Molecular mechanism of physiological fibrinolysis. Nature. 1978;272(5653):549–550. | |
Hersch SL, Kunelis T, Francis RB Jr. The pathogenesis of accelerated fibrinolysis in liver cirrhosis: a critical role for tissue plasminogen activator inhibitor. Blood. 1987;69(5):1315–1319. | |
Collen D. Identification and some properties of a new fast-reacting plasmin inhibitor in human plasma. Eur J Biochem. 1976;69(1):209–216. | |
Bajzar L. Thrombin activatable fibrinolysis inhibitor and an antifibrinolytic pathway. Arterioscler Thromb Vasc Biol. 2000;20(12):2511–2518. | |
Leiper K, Croll A, Booth NA, et al. Tissue plasminogen activator, plasminogen activator inhibitors, and activator-inhibitor complex in liver disease. J Clin Pathol. 1994;47(3):214–217. | |
Van Thiel DH, George M, Fareed J. Low levels of thrombin activatable fibrinolysis inhibitor (TAFI) in patients with chronic liver disease. Thromb Haemost. 2001;85(4):667–670. | |
Kujovich JL. Hemostatic defects in end stage liver disease. Crit Care Clin. 2005;21(3):563–587. | |
Stein SF, Harker LA. Kinetic and functional studies of platelets, fibrinogen, and plasminogen in patients with hepatic cirrhosis. J Lab Clin Med. 1982;99(2):217–230. | |
Ikura Y, Ohsawa M, Okada M, et al. The significance of platelet consumption in the development of thrombocytopenia in patients with cirrhosis. Am J Med Sci. 2013;346(3):199–203. | |
Boylan JF, Klinck JR, Sandler AN, et al. Tranexamic acid reduces blood loss, transfusion requirements, and coagulation factor use in primary orthotopic liver transplantation. Anesthesiology. 1996;85(5):1043–1048. [discussion 1030A–1031A]. | |
Porte RJ, Molenaar IQ, Begliomini B, et al. Aprotinin and transfusion requirements in orthotopic liver transplantation: a multicentre randomised double-blind study. EMSALT Study Group. Lancet. 2000; 355(9212):1303–1309. | |
Dalmau A, Sabate A, Koo M, et al. The prophylactic use of tranexamic acid and aprotinin in orthotopic liver transplantation: a comparative study. Liver Transpl. 2004;10(2):279–284. | |
Gunawan B, Runyon B. The efficacy and safety of epsilon-aminocaproic acid treatment in patients with cirrhosis and hyperfibrinolysis. Aliment Pharmacol Ther. 2006;23(1):115–120. | |
Lumsden AB, Henderson JM, Kutner MH. Endotoxin levels measured by a chromogenic assay in portal, hepatic and peripheral venous blood in patients with cirrhosis. Hepatology. 1988;8(2):232–236. | |
Kalambokis G, Tsianos EV. Endotoxaemia in the pathogenesis of cytopenias in liver cirrhosis. Could oral antibiotics raise blood counts? Med Hypotheses. 2011;76(1):105–109. | |
Wiest R, Garcia-Tsao G. Bacterial translocation (BT) in cirrhosis. Hepatology. 2005;41(3):422–433. | |
Kuratsune H, Koda T, Kurahori T. The relationship between endotoxin and the phagocytic activity of the reticuloendothelial system. Hepatogastroenterology. 1983;30(3):79–82. | |
Garcia-Tsao G, Wiest R. Gut microflora in the pathogenesis of the complications of cirrhosis. Best Pract Res Clin Gastroenterol. 2004; 18(2):353–372. | |
Deviere J, Content J, Denys C, et al. Excessive in vitro bacterial lipopolysaccharide-induced production of monokines in cirrhosis. Hepatology. 1990;11(4):628–634. | |
Lin RS, Lee FY, Lee SD, et al. Endotoxemia in patients with chronic liver diseases: relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J Hepatol. 1995; 22(2):165–172. | |
Kalambokis GN, Mouzaki A, Rodi M, et al. Rifaximin improves thrombocytopenia in patients with alcoholic cirrhosis in association with reduction of endotoxaemia. Liver Int. 2012;32(3):467–475. | |
Itoh H, Cicala C, Douglas GJ, et al. Platelet accumulation induced by bacterial endotoxin in rats. Thromb Res. 1996;83(6):405–419. | |
Panasiuk A, Zak J, Kasprzycka E, et al. Blood platelet and monocyte activations and relation to stages of liver cirrhosis. World J Gastroenterol. 2005;11(18):2754–2758. | |
Jelkmann W, Wolff M, Fandrey J. Modulation of the production of erythropoietin by cytokines: in vitro studies and their clinical implications. Contrib Nephrol. 1990;87:68–77. | |
Li J, Xia Y, Bertino AM, et al. The mechanism of apoptosis in human platelets during storage. Transfusion. 2000;40(11):1320–1329. | |
Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992;6(12):3051–3064. | |
Schobersberger W, Hoffmann G, Fandrey J. Nitric oxide donors suppress erythropoietin production in vitro. Pflugers Arch. 1996;432(6):980–985. | |
Gustot T, Durand F, Lebrec D, et al. Severe sepsis in cirrhosis. Hepatology. 2009;50(6):2022–2033. | |
Venkata C, Kashyap R, Farmer JC, et al. Thrombocytopenia in adult patients with sepsis: incidence, risk factors, and its association with clinical outcome. J Intensive Care. 2013;1(1):9. | |
Caly WR, Strauss E. A prospective study of bacterial infections in patients with cirrhosis. J Hepatol. 1993;18(3):353–358. | |
Foreman MG, Mannino DM, Moss M. Cirrhosis as a risk factor for sepsis and death: analysis of the National Hospital Discharge Survey. Chest. 2003;124(3):1016–1020. | |
Wang YQ, Wang B, Liang Y, et al. Role of platelet TLR4 expression in pathogensis of septic thrombocytopenia. World J Emerg Med. 2011;2(1):13–17. | |
Gawaz M, Dickfeld T, Bogner C, et al. Platelet function in septic multiple organ dysfunction syndrome. Intensive Care Med. 1997;23(4):379–385. | |
Pignatelli P, De Biase L, Lenti L, et al. Tumor necrosis factor-alpha as trigger of platelet activation in patients with heart failure. Blood. 2005;106(6):1992–1994. | |
Warkentin TE, Aird WC, Rand JH. Platelet-endothelial interactions: sepsis, HIT, and antiphospholipid syndrome. Hematology Am Soc Hematol Educ Program. 2003:497–519. | |
Kalambokis G, Tsianos EV. Thrombocytopenia associated with chronic liver disease: effects of rifaximin on platelet count. Am J Gastroenterol. 2010;105(12):2705–2707. | |
Krauel K, Hackbarth C, Furll B, et al. Heparin-induced thrombocytopenia: in vitro studies on the interaction of dabigatran, rivaroxaban, and low-sulfated heparin, with platelet factor 4 and anti-PF4/heparin antibodies. Blood. 2012;119(5):1248–1255. | |
Pongas G, Dasgupta SK, Thiagarajan P. Antiplatelet factor 4/heparin antibodies in patients with gram negative bacteremia. Thromb Res. 2013;132(2):217–220. | |
Montalto P, Vlachogiannakos J, Cox DJ, et al. Bacterial infection in cirrhosis impairs coagulation by a heparin effect: a prospective study. J Hepatol. 2002;37(4):463–470. | |
Spleen [webpage on the Internet]. Redondo Beach, CA: Pacific Health and Wellness; 2014. Available from: http://phaws.com/spleen/. Accessed February 18, 2016. | |
Liver and Cirrhosis [webpage on the Internet]. Seattle, WA; Cognition Studio; Available from: http://cognitionstudio.com/. Accessed March 23, 2016. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.