")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 15
The Pathogenic Mechanism of the ATP2C1 p.Ala109_Gln120del Mutation in Hailey–Hailey Disease
Authors Li P, Qi J, Zhou B, Ding T, Long J, Xiao H
Received 31 July 2022
Accepted for publication 22 September 2022
Published 11 October 2022 Volume 2022:15 Pages 2169—2175
DOI https://doi.org/10.2147/CCID.S384443
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Peiyao Li1,2 *, Jialin Qi3 *, Baishun Zhou,1 Ting Ding,4 Juan Long,5 Heng Xiao1
1Department of Pathology, School of Medicine, Hunan Normal University, Changsha, People’s Republic of China; 2Key Laboratory of Carcinogenesis and Cancer Invasion of Ministry of Education, China NHC Key Laboratory of Carcinogenesis, Xiangya Hospital, Central South University, Changsha, People’s Republic of China; 3Department of Pathology, Xiangya Hospital, Central South University, Changsha, People’s Republic of China; 4Department of Endocrinology, Yiyang Central Hospital, Yiyang, People’s Republic of China; 5Department of Dermatology, Hunan Children’s Hospital, Changsha, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Heng Xiao, Department of Pathology, School of Medicine, Hunan Normal University, 371 Tongzipo Road, Changsha, Hunan, 410013, People’s Republic of China, Tel +86-731-88912501, Email [email protected]
Background: Hailey–Hailey disease (HHD) is an autosomal dominant cutaneous disorder that manifests as repeated blisters and erosions on flexural or intertriginous skin areas. The calcium-transporting ATPase type 2C member 1 gene (ATP2C1) encodes the secretory pathway Ca2+/Mn2+-ATPase 1 (SPCA1), whose deficiency is responsible for HHD. An ATP2C1 splice-site mutation (c.325-2A>G, p.Ala109_Gln120del) was previously identified in a Han Chinese family with HHD.
Methods: In this study, the identified ATP2C1 splice-site mutation (c.325-2A>G, p.Ala109_Gln120del) was investigated in transfected human embryonic kidney 293 cells to analyze its pathogenic mechanism in HHD patients by using cycloheximide chase assay, CCK8 assay and in silico modeling of SPCA1 mutant.
Results: Cycloheximide chase assay showed that the degradation rate of the SPCA1 mutant was not obviously faster than that of the normal SPCA1. CCK8 assay showed that cell proliferation rates in the wild-type, A109_Q120del, and empty vector control groups all decreased in the gradient Mn2+ solutions in a dose-dependent manner. The cell proliferation rate in the wild-type was lower than that in the A109_Q120del and empty vector control (both P < 0.01), indicating overexpression of normal SPCA1 may rather induce Golgi stress, and even cell death. The cell proliferation rate in the A109_Q120del was lower than that in the empty vector control (P < 0.01), indicating that overexpression of the mutated SPCA1 may decrease its detoxification capability. Three-dimensional (3D) structure model of SPCA1 built by SWISS-MODEL and PyMOL showed that absence of the 12 amino acids from p.Ala109 to p.Gln120 in the SPCA1 mutant can cause obviously shortened transmembrane 2, which may affect correct localization of SPCA1 on the Golgi.
Conclusion: These results demonstrate that the ATP2C1 mutation (c.325-2A>G, p.Ala109_Gln120del) may cause impaired SPCA1 capability to detoxify Mn2+ and abnormal SPCA1 structure, which reveals a new side for the pathogenesis of HHD.
Keywords: ATPase secretory pathway Ca2+ transporting 1, Hailey–Hailey disease, pathogenesis, secretory pathway Ca2+/Mn2+-ATPase 1
Introduction
Hailey–Hailey disease (HHD; OMIM 169600), also known as familial benign chronic pemphigus, was first described by the Hailey brothers in 1939 and eventually named after them.1 The prevalence of HHD is approximately 1:50,000–1:40,000.1,2 It is an autosomal dominant blistering skin disease that usually develops around the age of 30 to 40 years old.3 It is characterized by recurrent blistering and erosions, usually affecting the intertriginous areas (axillae, groins, perineum, inframammary regions) and the neck.3 As the maceration progresses, the skin lesion is painful and has an unpleasant odor.4 Friction, heat, sweating, ultraviolet radiation, and microbial infections may cause the disease to worsen and persist.1,3–5 The severity of the disease varies among different families and even individuals in the same family.2 HHD may last for months or even years, and its recurrences and remissions can seriously affect patients’ quality of life.1,6 In rare cases, the skin lesions of HHD may develop into squamous cell carcinomas.4,7–10
The histopathological manifestation of HHD shows epidermal hyperplasia, which usually involves more than 50% of the epidermal thickness. Extensive loss of intercellular adhesion results in extensive spicule laxity in the upper basal layer of the epidermis, which is described as a “collapsing brick wall”. Dyskeratosis may be observed in the form of coronal protrusions and granules.11 Ultrastructural studies show that both basal and suprabasal keratin-forming cells exhibit disruption of bridging granules and internalization of the bridging granule-fiber complex. Keratin-forming filaments aggregate around the nucleus in thick electron-dense bundles, but keratinocytes remain connected by microvilli adhesion structures.5,12
The causative gene for HHD is the calcium-transporting ATPase type 2C member 1 gene (ATP2C1; OMIM 604384), which consists of 28 exons and is approximately 30 kb long.1,13 It encodes secretory pathway Ca2+/Mn2+-ATPase 1 (SPCA1),14 a calcium and manganese ion transporter on the Golgi apparatus, which is mainly responsible for intra-Golgi and cytosolic Ca2+/Mn2+ homeostasis.15,16 Failure of calcium deposition in the Golgi may be caused by the ATP2C1 gene mutations, which may be responsible for defective keratinocyte adhesion and acantholysis.17
In a previous study, an ATP2C1 splice-site mutation (c.325–2A>G, p.Ala109_Gln120del) was identified in a Han Chinese family with HHD.18 In this study, we investigated this ATP2C1 splice-site mutation further to understand its specific role in HHD pathogenesis.
Materials and Methods
Plasmid Construction
To construct the vector pcDNA3.1-ATP2C1, we artificially synthesized cDNAs encoding human ATP2C1 isoform 1a (NM_014382.3, NP_055197.2) plus flanking XhoI and KpnI sites. The ATP2C1 isoform 1a is the “canonical” one among the nine splice isoforms.19 Then, the cDNAs plus flanking sites were inserted between the XhoI and KpnI sites of pcDNA3.1(-) vector. The wild-type ATP2C1 expression clone was used as a template and corresponding pair of DNA oligomers (5’-GTATCACTGTGGAATATCGTTCAGAAAAATCTCTTG-3’ and 5’-GAACGATATTCCACAGTGATACTGACGGCATC-3’) as the primers to create the desired splice-site mutation (c.325–2A>G, p.Ala109_Gln120del). The generated cDNA clones were sequenced to ensure that no non-synonymous mutations had been introduced into the plasmids during the construction process.
Cell Culture and Transfection
Human embryonic kidney (HEK) 293 cells (Cell Bank of Chinese Academy of Sciences, Shanghai, China; identifier: CSTR:19375.09.3101HUMGNHu43) were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum. Cells were maintained at 37°C in a 95% O2/5% CO2 incubator. HEK293 cells were transfected using LipofectamineTM 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction.
Cycloheximide (CHX) Chase Assay
Cycloheximide (CHX, Sigma, USA) can inhibit protein translation, allowing protein degradation to be evaluated. HEK293 cells were treated with 100 μg/mL CHX at 48 hours post-transfection, and protein lysates were collected at time points 0-, 2-, 4-, 8-, 12-, and 16-hour exposure to CHX.20
Western Blotting
Cell samples were collected from the CHX chase assay. Proteins were extracted from each sample using a cell lysate extraction buffer containing protease inhibitor (Beyotime, China). Protein concentrations were quantified using the bicinchoninic acid method (Pierce Biotechnology, Rockford, IL, USA). Equal amounts of protein extracts were mixed with sodium dodecyl sulfate (SDS) buffer and incubated at 100°C for 5 min before loading. Total protein samples (40 μg) were subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were electrophoretically blotted onto polyvinylidene difluoride membranes (Millipore, Billerica, USA). The membranes were blocked with 5% skim milk in phosphate buffered saline containing 0.05% Tween 20 (PBS-T). The membranes were incubated overnight at 4°C with the primary antibodies, including SPCA1 (13310-1-AP, Proteintech, China, 1:500), α-tubulin rabbit polyclonal antibody (11224-1-AP, Proteintech, China, 1:1000). The membranes were washed four times with PBS-T and then incubated with the appropriate secondary antibodies at room temperature for 1 hour. Detection was performed using an enhanced chemiluminescence detection system (Advansta Corporation, Menlo Park, California, USA) as previously described.21
CCK8 Assay
To detect cell viability, 36 hours after transfection of HEK293 cells with plasmids, the cells were processed with gradient Mn2+ solutions for 24 hours. MnCl2·4H2O was dissolved in cell culture medium to obtain 4 mmol/L Mn2+ solution. The mother liquid was diluted to obtain gradient Mn2+ solutions: 0.125 mmol/L, 0.25 mmol/L, 0.5 mmol/L, 1.0 mmol/L, and 2.0 mmol/L. Then, the cell viability was detected using CCK8 assay (Dojindo Molecular Technologies, Tokyo, Japan) at 450 nm according to the manufacturer’s instruction.
In silico Modelling of SPCA1
Schematic illustration of SPCA1 model was drawn with SWISS-MODEL and PyMOL (Schrödinger, LLC).22 The ATP2C1 isoform 1a (NP_055197.2) in FASTA form was input into SWISS-MODEL to obtain predicted three-dimensional model for SPCA1. The built models were assessed with model quality measurement indexes. For instance, Global Model Quality Estimate (GMQE) is a major performance assessment index, which gives an overall model quality measurement score from 0 to 1. The higher the score is, the better expected quality can be.22
Statistical Analysis
Statistical analysis of the CHX data was evaluated using GraphPad Prism 8.0.2 software (GraphPad Software, Inc., LaJolla, CA, USA).23 Automated non-linear curve fitting was performed assuming a simple exponential one-phase decay model. The default model equation Y=(Y0)*exp(-K*X) was used to calculate half-life times (hours). Relative protein expression levels were quantified after subtracting background signals and normalized to α-tubulin loading controls and zero-time points. P values for comparisons of degradation rates and half-life times were derived from the One-Way ANOVA and Dunnett’s T3 test. P values for comparisons of relative OD values in CCK8 assay were derived from the Two-Way ANOVA and Tukey’s multiple comparison test. P < 0.05 was considered statistically significant. Line and bar graphs were drawn using GraphPad.
Results
Stability Evaluation of Mutated SPCA1
To determine whether the protein stability of mutated SPCA1 in the A109_Q120del was affected by the ATP2C1 c.325–2A>G splice-site mutation, CHX chase assay was used to evaluate the degradation rate of the mutated SPCA1. At 48 hours post-transfection, transfected HEK293 cells were treated with 100 μg/mL CHX to inhibit protein translation, allowing protein degradation to be detected by Western blotting. As shown in Figure 1, the mutated SPCA1 in the A109_Q120del was degraded at a similar rate as the SPCA1 in the wild-type and the empty vector control (P = 0.216 > 0.05). The half-life period of the mutated SPCA1 was about 21.35 hours (P = 0.544 > 0.05 vs the wild-type; P = 0.384 > 0.05 vs the empty vector control), compared to about 20.52 hours for that in the wild-type and 16.84 hours in the empty vector control, respectively.
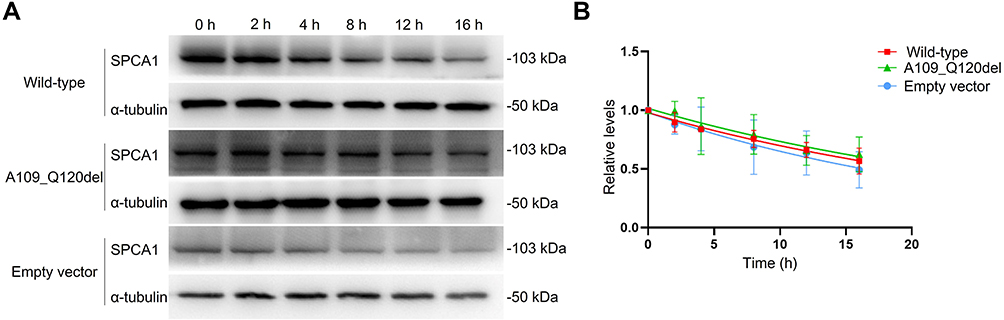 |
Figure 1 Comparison of SPCA1 degradation rates. HEK293 cells were transfected with the ATP2C1 wild-type, mutant, and empty vector control plasmids, respectively. Cycloheximide (CHX, 100 μg/mL) was added to the transfected cells at t = 0 h. Cell lysates were harvested at varying time points (t = 0, 2, 4, 8, 12 and 16 h). (A) Representative images of CHX chase assay. The mutated SPCA1 was degraded at a similar rate as the normal SPCA1 in the wild-type group or the empty vector control (P = 0.216 > 0.05). (B) A plot that shows band intensities quantified from immunoblot images of post-CHX chase assay in (A). Data were normalized to α-tubulin and zero-time control. The relative protein levels are presented as the means ± SD. The above experiments were repeated at least three times independently. |
Effect of SPCA1 on Cell Viability in MnCl2 Solutions
The transfected HEK293 cells were treated with gradient Mn2+ solutions (0, 0.125 mmol/L, 0.25 mmol/L, 0.5 mmol/L, 1.0 mmol/L, and 2.0 mmol/L) for 48 hours. CCK8 assay was conducted to determine the effect of mutated SPCA1 on cell viability. As shown in Figure 2, Mn2+ solutions inhibited HEK293 cell proliferation in the three cell groups in a dose-dependent manner. The cell proliferation rate of the wild-type was unexpectedly lower than that of the A109_Q120del and the empty vector control (both P < 0.01, multiple comparisons among groups using Tukey’s multiple comparison test), showing a more impaired detoxification capability of SPCA1 in the wild-type. The cell proliferation ability in the A109_Q120del was lower than that in the empty vector control (P < 0.01), indicating that overexpression of the SPCA1 mutant may interfere with the detoxification capability of SPCA1 to some extent.
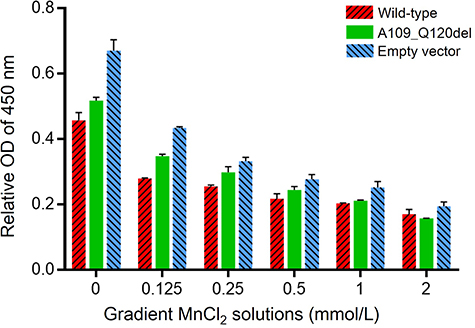 |
Figure 2 Evaluation of SPCA1 effect on cell viability in MnCl2 solutions. HEK293 cells were transfected with the ATP2C1 wild-type, mutant, and empty vector control plasmids, respectively. The transfected HEK293 cells were treated with gradient Mn2+ solutions (0, 0.125 mmol/L, 0.25 mmol/L, 0.5 mmol/L, 1.0 mmol/L, and 2.0 mmol/L). CCK8 assay was used to compare the effects of SPCA1 in the three cell groups on cell viability in gradient MnCl2 solutions. HEK293 cell proliferation rates of the three groups all decreased in the Mn2+ solutions in a dose-dependent manner. The cell proliferation rate of the wild-type was significantly lower than that of the A109_Q120del and the empty vector control (P < 0.01, Two-Way ANOVA; all P < 0.01, multiple comparisons among the three groups using Tukey’s multiple comparison test). The relative OD values of 450 nm are presented as the means ± SD. The above experiments were repeated three times independently. |
In silico Modelling of SPCA1
In the process of building in silico model for SPCA1, the SWISS-MODEL returned two predicted models. The SPCA1 model (Figure 3) with the better GMQE value was built using sarcoplasmic/endoplasmic reticulum calcium ATPase 2 isoform b (SERCA2b; OMIM 108740) as a template. The GMQE value was 0.66 (range 0–1). The ligands for Ca2+ and Mn2+ are shown in the model. Seven residues predicted to function as ligand contacts for Ca2+ are p.Val303, p.Ala304, p.Ile306, p.Glu308, p.Gln706, p.Asn738, and p.Asp742, while four residues for Mn2+ are p.Asp350, p.Thr352, p.Asp644, and p.Asp648.
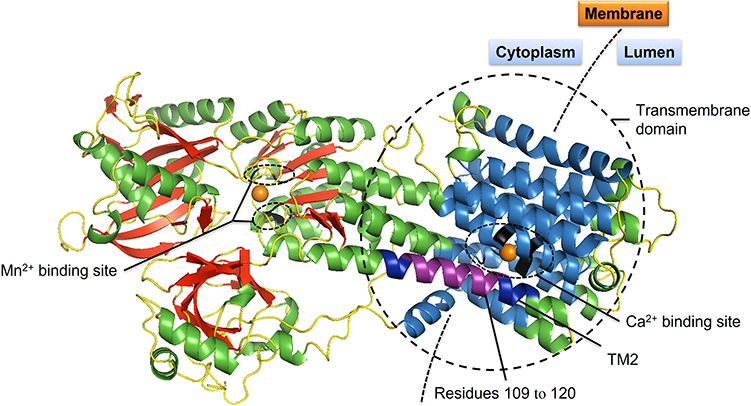 |
Figure 3 Three-dimensional (3D) structure model of SPCA1. Schematic ribbon illustration of SPCA1 is drawn with SWISS-MODEL and PyMOL (Schrödinger, LLC), using sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2b) as a template.32 Residues 109–120 are colored in purple. Transmembrane domain 2 (TM2) where the mutation p.Ala109_Gln120del located at is in dark blue. α-helices are mostly in green; β-strands in red; loops in yellow. Transmembrane domains 1 and 3–10 are in sky-blue. Ca2+ and Mn2+ are shown as orange spheres. Residues for Ca2+ binding (V303, A304, I306, E308, Q706, N738 and D742) and Mn2+ binding (D350, T352, D644 and D648) are colored in black. |
Discussion
HHD is a rare autosomal dominant genodermatosis, characterized by erosions, intraepidermal blisters and scaly erythematous plaques.24 The disease-causing factor ATP2C1 gene encodes protein SPCA1,6,25 a Golgi-localized pump, which transports Ca2+ and Mn2+ from cytosol to the Golgi lumen.24 SPCA1 consists of an actuator domain (A), a phosphorylation domain (P), a nucleotide-binding domain (N or ATP binding), ten hydrophobic transmembrane domains, five stalk helices in the cytoplasm, and five others in the intra-luminal Golgi.4,26 Mutations affecting critical domains of SPCA1, such as Ca2+/Mn2+ binding and phosphorylation domains, may cause severe SPCA1 dysfunction and damage intra-Golgi and cytosolic Ca2+/Mn2+ homeostasis.19,27 In general, the resulting SPCA1 haploinsufficiency is responsible for HHD pathogenesis.25,28
In this study, CHX chase assay showed that the ATP2C1 A109_Q120del mutant did not degrade at an accelerated rate compared with the wild-type, implying that overexpression of SPCA1 may change cytosolic or intra-Golgi Ca2+ and/or Mn2+ concentrations to some extent. The possible Ca2+ and/or Mn2+ homeostasis disequilibrium may affect SPCA1 degradation rate, because the two cations are important for appropriate glycosylation by modulating intracellular trafficking within the secretory pathway, thus, affecting protein stability.29
CCK8 assay indicated that HEK293 cell proliferation rates of the A109_Q120del, wild-type, and empty vector control all decreased in the gradient Mn2+ solutions in a dose-dependent manner. Unexpectedly, the wild-type was more vulnerable to cytotoxicity effect of the gradually increasing Mn2+ solutions, compared with the other two groups (both P < 0.01). In the transport cycle of SPCA1, the pump requires conformational changes to accomplish the delivery of ATP and Ca2+ and/or Mn2+.4 Overexpression of wild-type SPCA1 may cause obstacle for the pumps to perform conformational changes in the limited tubular noncompact zones of the Golgi membrane, where SPCA1 is normally located. Therefore, the worst cell proliferation ability in the wild-type may be caused by SPCA1 overexpression, which may induce Golgi stress, a result of high Golgi Ca2+ and/or Mn2+ level.30 Continuous Golgi stress would put the cells at risk for cell death.30 Not only lack of SPCA1 could lead to Golgi fragmentation and dysfunction of intra-Golgi transport,31 SPCA1 overexpression may also cause cytotoxicity via inducing Golgi stress. Therefore, it needs careful consideration and evaluation before we try to treat HHD by compensating for SPCA1 haploinsufficiency through improving expression of wild-type SPCA1. Overexpression of plasmids in skin lesions may produce toxic effect other than ideal therapeutic effect.
In silico modelling of SPCA1 showed that the absence of 12 residues from p.Ala109 to p.Gln120 in the ATP2C1 c.325–2A>G splice-site mutation could cause obviously shortened transmembrane helix 2 (TM2). The resulting SPCA1 structure abnormality may cause incorrect co-localization of mutated SPCA1 with the Golgi, which could lead to impaired Ca2+ and Mn2+ homeostasis.
Conclusions
In summary, in vitro studies on the ATP2C1 gene mutation (c.325–2A>G, p.Ala109_Gln120del) further indicates that the degradation rate of the mutated SPCA1 was not significantly different from that of the wild-type. The obviously shortened TM2 caused by the splice-site mutation indicates that TM2 may be essential for correct SPCA1 structure and localization. Overexpression of the SPCA1 mutant may interfere with its detoxification capability to keep intra-Golgi and cytosolic Mn2+ homeostasis. Wild-type SPCA1 overexpression in skin lesions may not be a feasible solution for the pathogenic mechanism of SPCA1 haploinsufficiency in HHD. Therefore, this study shows a new side of the pathogenic molecular basis in HHD.
Ethics Approval
This study was carried out in accordance with the Declaration of Helsinki and received approval from the Biomedical Research Ethics Committee of Hunan Normal University, Changsha, Hunan, P.R. China.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81903199, 81802871), Natural Science Foundation of Hunan Province (2021JJ40373, S2022JJQNJJ1108).
Disclosure
The authors declare that there are no competing interests associated with the manuscript.
References
1. Kellermayer R. Hailey-Hailey disease as an orthodisease of PMR1 deficiency in saccharomyces cerevisiae. FEBS Lett. 2005;579(10):2021–2025. doi:10.1016/j.febslet.2005.03.003
2. Raiko L, Siljamäki E, Mahoney MG, et al. Hailey-Hailey disease and tight junctions: claudins 1 and 4 are regulated by ATP2C1 gene encoding Ca2+/Mn2+ ATPase SPCA1 in cultured keratinocytes. Exp Dermatol. 2012;21(8):586–591. doi:10.1111/j.1600-0625.2012.01520.x
3. Missiaen L, Raeymaekers L, Dode L, et al. SPCA1 pumps and Hailey-Hailey disease. Biochem Biophys Res Commun. 2004;322(4):1204–1213. doi:10.1016/j.bbrc.2004.07.128
4. Brini M, Carafoli E. Calcium pumps in health and disease. Physiol Rev. 2009;89(4):1341–1378. doi:10.1152/physrev.00032.2008
5. Yang L, Zhang Q, Zhang S, Liu Y, Liu Y, Wang T. Generalized Hailey-Hailey disease: novel splice-site mutations of ATP2C1 gene in Chinese population and a literature review. Mol Genet Genomic Med. 2021;9(2):e1580. doi:10.1002/mgg3.1580
6. Sudbrak R, Brown J, Dobson-Stone C, et al. Hailey-Hailey disease is caused by mutations in ATP2C1 encoding a novel Ca2+ pump. Hum Mol Genet. 2000;9(7):1131–1140. doi:10.1093/hmg/9.7.1131
7. von Felbert V, Hampl M, Talhari C, Engers R, Megahed M. Squamous cell carcinoma arising from a localized vulval lesion of Hailey-Hailey disease after tacrolimus therapy. Am J Obstet Gynecol. 2010;203(3):e5–7. doi:10.1016/j.ajog.2010.06.041
8. Chun SI, Whang KC, Su WP. Squamous cell carcinoma arising in Hailey-Hailey disease. J Cutan Pathol. 1988;15(4):234–237. doi:10.1111/j.1600-0560.1988.tb00551.x
9. Holst VA, Fair KP, Wilson BB, Patterson JW. Squamous cell carcinoma arising in Hailey-Hailey disease. J Am Acad Dermatol. 2000;43(2 Pt 2):368–371. doi:10.1067/mjd.2000.100542
10. Cockayne SE, Rassl DM, Thomas SE. Squamous cell carcinoma arising in Hailey-Hailey disease of the vulva. Br J Dermatol. 2000;142(3):540–542. doi:10.1046/j.1365-2133.2000.03374.x
11. Thompson LD. Hailey-Hailey disease. Ear Nose Throat J. 2016;95(9):370. doi:10.1177/014556131609500901
12. Metze D, Hamm H, Schorat A, Luger T. Involvement of the adherens junction-actin filament system in acantholytic dyskeratosis of Hailey-Hailey disease. A histological, ultrastructural, and histochemical study of lesional and non-lesional skin. J Cutan Pathol. 1996;23(3):211–222. doi:10.1111/j.1600-0560.1996.tb01469.x
13. Richard G, Korge BP, Wright AR, et al. Hailey-Hailey disease maps to a 5 cM interval on chromosome 3q21-q24. J Invest Dermatol. 1995;105(3):357–360. doi:10.1111/1523-1747.ep12320741
14. Ikeda S, Welsh EA, Peluso AM, et al. Localization of the gene whose mutations underlie Hailey-Hailey disease to chromosome 3q. Hum Mol Genet. 1994;3(7):1147–1150. doi:10.1093/hmg/3.7.1147
15. Behne MJ, Tu CL, Aronchik I, et al. Human keratinocyte ATP2C1 localizes to the Golgi and controls Golgi Ca2+ stores. J Invest Dermatol. 2003;121(4):688–694. doi:10.1046/j.1523-1747.2003.12528.x
16. Dode L, Andersen JP, Raeymaekers L, Missiaen L, Vilsen B, Wuytack F. Functional comparison between secretory pathway Ca2+/Mn2+-ATPase (SPCA) 1 and sarcoplasmic reticulum Ca2+-ATPase (SERCA) 1 isoforms by steady-state and transient kinetic analyses. J Biol Chem. 2005;280(47):39124–39134. doi:10.1074/jbc.M506181200
17. Engin B, Kutlubay Z, Çelik U, Serdaroğlu S, Tüzün Y. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33(4):452–455. doi:10.1016/j.clindermatol.2015.04.006
18. Xiao H, Huang X, Xu H, et al. A novel splice-site mutation in the ATP2C1 gene of a Chinese family with Hailey-Hailey disease. J Cell Biochem. 2019;120(3):3630–3636. doi:10.1002/jcb.27640
19. Deng H, Xiao H. The role of the ATP2C1 gene in Hailey-Hailey disease. Cell Mol Life Sci. 2017;74(20):3687–3696. doi:10.1007/s00018-017-2544-7
20. Thorpe H, Asiri A, Akhlaq M, Ilyas M. Cten promotes epithelial-mesenchymal transition through the post-transcriptional stabilization of Snail. Mol Carcinog. 2017;56(12):2601–2609. doi:10.1002/mc.22704
21. Li P, Feng J, Liu Y, et al. Novel therapy for glioblastoma multiforme by restoring LRRC4 in tumor cells: LRRC4 inhibits tumor-infiltrating regulatory T cells by cytokine and programmed cell death 1-containing exosomes. Front Immunol. 2017;8:1748. doi:10.3389/fimmu.2017.01748
22. Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi:10.1093/nar/gky427
23. Esmail S, Kartner N, Yao Y, Kim JW, Reithmeier RAF, Manolson MF. Molecular mechanisms of cutis laxa- and distal renal tubular acidosis-causing mutations in V-ATPase a subunits, ATP6V0A2 and ATP6V0A4. J Biol Chem. 2018;293(8):2787–2800. doi:10.1074/jbc.M117.818872
24. Ton VK, Rao R. Expression of Hailey-Hailey disease mutations in yeast. J Invest Dermatol. 2004;123(6):1192–1194. doi:10.1111/j.0022-202X.2004.23437.x
25. Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet. 2000;24(1):61–65. doi:10.1038/71701
26. Roy AS, Miskinyte S, Garat A, Hovnanian A, Krzewinski-Recchi MA, Foulquier F. SPCA1 governs the stability of TMEM165 in Hailey-Hailey disease. Biochimie. 2020;174:159–170. doi:10.1016/j.biochi.2020.04.017
27. Micaroni M, Giacchetti G, Plebani R, Xiao GG, Federici L. ATP2C1 gene mutations in Hailey-Hailey disease and possible roles of SPCA1 isoforms in membrane trafficking. Cell Death Dis. 2016;7(6):e2259. doi:10.1038/cddis.2016.147
28. Chao SC, Tsai YM, Yang MH. Mutation analysis of ATP2C1 gene in Taiwanese patients with Hailey-Hailey disease. Br J Dermatol. 2002;146(4):595–600. doi:10.1046/j.1365-2133.2002.04697.x
29. Stribny J, Thines L, Deschamps A, Goffin P, Morsomme P. The human Golgi protein TMEM165 transports calcium and manganese in yeast and bacterial cells. J Biol Chem. 2020;295(12):3865–3874. doi:10.1074/jbc.RA119.012249
30. Smaardijk S, Chen J, Kerselaers S, Voets T, Eggermont J, Vangheluwe P. Store-independent coupling between the secretory pathway Ca2+ transport ATPase SPCA1 and Orai1 in Golgi stress and Hailey-Hailey disease. Biochim Biophys Acta Mol Cell Res. 2018;1865(6):855–862. doi:10.1016/j.bbamcr.2018.03.007
31. Micaroni M, Perinetti G, Berrie CP, Mironov AA. The SPCA1 Ca2+ pump and intracellular membrane trafficking. Traffic. 2010;11(10):1315–1333. doi:10.1111/j.1600-0854.2010.01096.x
32. Zhang Y, Inoue M, Tsutsumi A, et al. Cryo-EM structures of SERCA2b reveal the mechanism of regulation by the luminal extension tail. Sci Adv. 2020;6(33):eabb0147. doi:10.1126/sciadv.abb0147
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.