")
Back to Journals » International Journal of Nanomedicine » Volume 15
The Oral Administration of Lidocaine HCl Biodegradable Microspheres: Formulation and Optimization
Authors ALQuadeib BT , Eltahir EKD , Alagili MF
Received 27 October 2019
Accepted for publication 3 January 2020
Published 5 February 2020 Volume 2020:15 Pages 857—869
DOI https://doi.org/10.2147/IJN.S236273
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Bushra T ALQuadeib, Eram KD Eltahir, Modhi F Alagili
Department of Pharmaceutics, College of Pharmacy, King Saud University, Riyadh 11671, Saudi Arabia
Correspondence: Bushra T ALQuadeib
Department of Pharmaceutics, College of Pharmacy, King Saud University, P.O. Box 84428, Riyadh 11671, Saudi Arabia
Tel +966 505653169
Fax +966 118052932
Email [email protected]
Purpose: Lidocaine (LID) is a local anesthetic that is administered either by injection and/or a topical/transdermal route. However, there is a current need to develop efficacious methods for the oral delivery of LID with optimized bioavailability.
Methods: We developed oral LID biodegradable microspheres that were loaded with alginate-chitosan with different mass ratios, and characterized these microspheres in vitro. We also developed, and utilized, a simple and sensitive HPLC-tandem mass spectrometry (LC-MS-MS) method for assaying LID microspheres.
Results: The mean particle size (MPS) of the LID microspheres ranged from 340.7 to 528.3 nm. As the concentration of alginate was reduced, there was a significant reduction in MPS. However, there was no significant change in drug entrapment efficiency (DEE), or drug yield, when the alginate concentration was either increased or decreased. DSC measurements demonstrated the successful loading of LID to the new formulations. After a slow initial release, less than 10% of the LID was released in vitro within 4 h at pH 1.2. In order to evaluate nephrotoxicity, we carried out MTT assays of LID in two types of cell line (LLC-PK1 and MDCK). LID significantly suppressed the cell toxicity of both cell lines at the concentrations tested (100, 200, and 400ng/μL).
Conclusion: Experiments involving the oral delivery of LID formulations showed a significant reduction in particle size and an improvement in dissolution rate. The formulations of LID developed exhibit significantly less toxicity than LID alone.
Keywords: lidocaine, oral delivery, alginate–chitosan microspheres, in vitro characterization, dissolution rate, MTT assay
Introduction
The management perioperative/postoperative pain after any form of surgical treatment requires the administration of a local anesthetic (LA), such as lidocaine, chloroprocaine, mepivacaine, bupivacaine, or ropivacaine. Under these circumstances, the administration of LA minimizes morbidity after surgery. However, the use of LA in such circumstances is limited due to the relatively short duration of action. Lidocaine, for example, only acts for 1–2 hrs.1 LAs can be administered either by multiple injections, continuous infusion, or via a topical/transdermal (TT) route. However, these routes of administration are also associated with limitations, such as the need for an infusion device, the need for hospitalization, and the risk of infection.2,3 In addition, topical anesthetics are limited by poor permeability and slow penetration. Therefore, there is an urgent need to develop a method to deliver LAs orally in a manner that provides the best efficacy.
LID is one of the most effective and reliable hydrophilic Las, and is commonly used because of its rapid onset, intermediate action, and low systemic toxicity.4 Furthermore, LID is commonly administered intravenously, although the intravenous infusion of LID may cause a range of adverse side effects, including seizures, dizziness, nausea, vomiting, and a metallic taste. LID is rarely applied percutaneously due to poor penetration through intact skin.5
LID is commonly used as local topical anesthetic, and is widely used for dental surgery due to its rapid onset of action (between 20 s to 1 min), and intermediate duration of efficacy (5–30 min), and is often used to relieve the pain associated with mouth ulcers. LID is available as a viscous gel, or ointment; these formulations are often used to relieve pain and discomfort from a sore throat/mouth. Research shows that LID is efficiently metabolized and only trace amounts are excreted in the urine.6 Following oral ingestion, LID undergoes extensive initial metabolism, resulting in a bioavailability of approximately 30%.6
In an effort to improve and prolong the action of LA, a recent research study described the use of adjuvants for LA, including epinephrine and dexamethasone.7 Other research studies have considered the use of delivery matrices, such as liposomes, microemulsions, microspheres, microcrystals,8 cross-linked hydrogels, thermosensitive hydrogels, thermosensitive nanogels,9 nanoparticles,10 an aqueous polymer solution, a liquid polymeric matrix, a solid polymeric matrix,11 a bio-adhesive film,12 an interpenetrating polymer network (IPN) matrix,13 lipid-protein-sugar particles and fatty-acid dimer-based polymer,14 and ceramic-based granules.15 Moreover, several published articles have considered the use of oral fast disintegrating films16 and liposomal LID gels17 with regards to both topical and systemic effects. One recent study reported the oral administration of LID HCL prior to anesthesia for laparoscopy.18 Other research studies have described the oral delivery of microspheres, consisting of Alginate (ALG) and Chitosan (CS) as mucoadhesive polymers. These microspheres were successfully combined with various drugs, such as glipizide,19 insulin,20 ranitidine,21 metformin,22 diclofenac,23 and various other drugs,24 and showed improved solubility and bioavailability.
The aim of this study was to develop microspheres for the oral delivery of LID consisting of ALG and CS. We then optimized a series of new in vitro, characterized particle sizes, and used transmission electron microscopy (TEM) to determine the surface morphology of the microspheres. Finally, we investigated drug yield, drug loading, and the LID release rate in vitro.
Materials and Methods
Materials
Lidocaine HCL was provided as a gift from Aljazeera Pharmaceutical Industry (Riyadh, Saudi Arabia). Diclofenac sodium (DS) was supplied by Spimaco (Al-Qassim, Saudi Arabia) while sodium alginate (MW about 115,000), was supplied by Fluka Chemie (Buchs, Switzerland). Chitosan (poly(D-glycosamine), deacetylated chitin, high molecular weight) was supplied by Aldrich, Darmstadt, Germany. Rat embryonic cardiomyocytes (H9c2 (2–1) cells) were purchased from the American Type Culture Collection (ATCC) (Manassas, Virginia, USA, ATCC® CRL-1446™), while the LLC-PK1 cell line was purchased from Sigma Aldrich (Taufkirchen, Germany). For the assessment of in vitro nephrotoxicity, we utilized the LLC-PK1 cell line from Sigma Aldrich (Taufkirchen, Germany).
All other reagents and chemicals were of high-performance liquid chromatography (HPLC) analytical grade, and were used as received. Water was deionized and purified with a Milli-Q Reagent Grade water system (Millipore Corporation, Bedford, MX, USA).
Preparation of Lidocaine HCl Microspheres
LID microspheres were prepared using an extrusion congealing technique, described previously by Tahtat.25 In brief, solution (1) was created by mixing 2% (w/v) sodium alginate with 25 mg of lidocaine in 40 mL of distilled water at room temperature under mechanical stirring. Solution (2) was then created by dissolving 0.5% chitosan solution in 1% v/v acetic acid (10 mL); the pH was then adjusted to pH 5 using 1 M NaOH. The two solutions were then mixed until homogeneous using a high-speed stirrer (9500 rpm), and an Ultra-Turrax (Ika, Artisantg, Illinois, USA), at room temperature for 60 min. Then, calcium carbonate was added to blend the solutions at a ratio of 1:1 to alginate sodium. Next, the mixture was dropped, using a 24 G syringe needle, into 100 mL of calcium chloride solution (1% w/v) containing glacial acetic acid (10% v/v) (Figure 1).
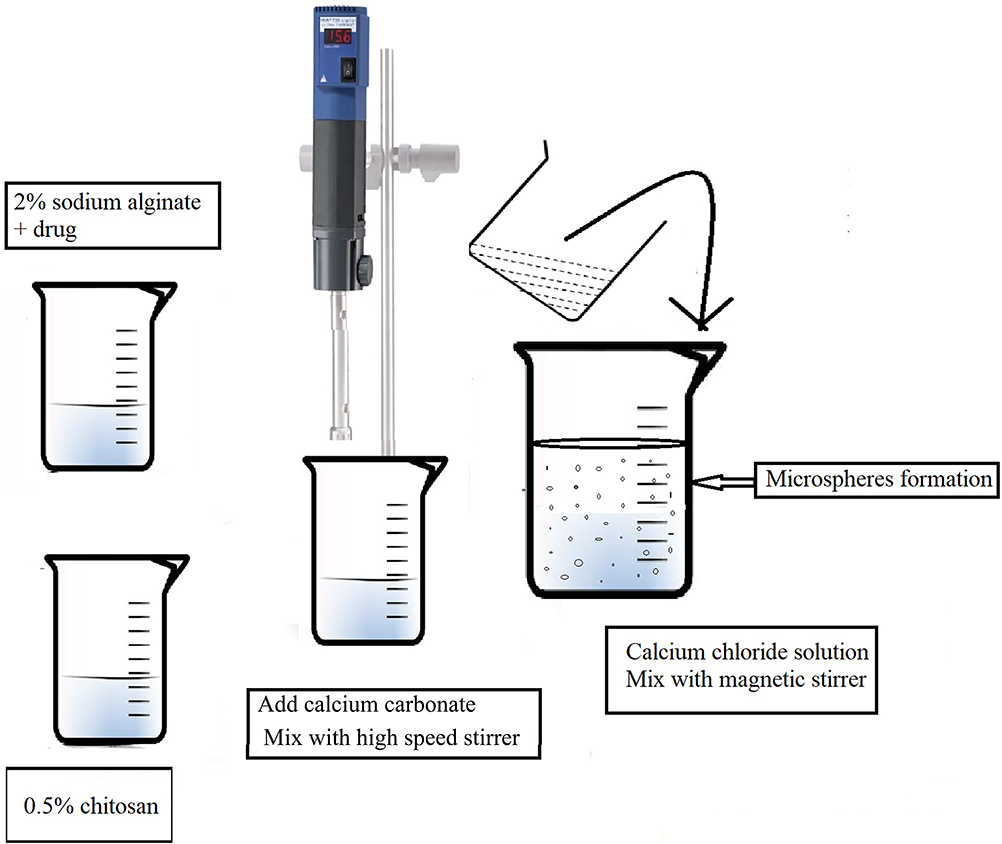 |
Figure 1 Schematic method for preparation of microspheres by extrusion congealing technique. |
In order to evaluate different mass proportions, four formulations were tested named N1, N2, N3, and N4 which contained alginate to chitosan at a weight/weight ratio of 1.0:0, 0.8:0.2, 0.7:0.3 and 0.6:0.4, respectively. Smooth and spherical microspheres beads were formed by mechanical stirring for 4 h. The microspheres were then washed with distilled water to remove any remaining calcium chloride. The wet microspheres obtained were then suspended in glutaraldehyde solution (2%) for 48 h at room temperature. Then, the microspheres were washed with hot-distilled water, air-dried, and stored at 4°C. All formulations were prepared in triplicate as a minimum.
Characterization of the Formulated Lidocaine Microspheres
Particle Size Analysis
The mean of particle size (MPS) was determined using a Zetasizer NanoZS, Malvern (Almelo, Netherland). Typically, 5 mg of the formulation samples (N1, N2, N3, and N4) were dissolved in phosphate buffer pH 7.4 (15 mL each), and sonicated prior to measurement. Measurements were taken in triplicate and the mean ± standard deviation was calculated.
Determination of Drug Entrapment Efficiency and Microsphere Yield
Yield is determined using equation (1).
(1)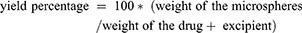
For drug entrapment efficacy (DEE), we took 5 mg of LID from each microsphere formulation, and dissolved this in 15 mL of phosphate buffer (pH 7.4) overnight. The next day, the solution was sonicated for 15 min and then analyzed using a mass tandem –liquid chromatography. All experiments were performed in triplicate. The concentration of LID was calculated using the following equation (2).
(2)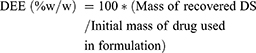
Differential Scanning Calorimetry (DSC)
Pure components, as well as drug-loaded microspheres, were measured thermally using a Perkin Elmer DSC-4000, Buckinghamshire, UK. Approximately 2 mg of each sample was weighed and placed into standard aluminum pans, which were then hermetically sealed. An empty pan was used as a reference. The heating rate was 5°C/min, with a temperature range of 30°C to 180°C, and was carried out in a closed-pan system under a stream of nitrogen gas. The apparatus was calibrated with 99.99% indium.
Transmission Electron Microscopy (TEM)
Next, we investigated the morphology of the formulated LID loaded microspheres by TEM (Jel-1010 Electron microscope, Jeol, Japan). The particulate carriers were negatively stained using uranyl acetate. The stained grid was then air dried and examined immediately under TEM.
Drug Release from Lidocaine-Loaded Biodegradable Microspheres
In vitro release tests (dissolution) were performed as reported previously,16 using US Pharmacopeia XXXII dissolution apparatus 2 (paddle), along with 900 mL medium; experiments were carried out with stirring at 75 rpm and the temperature was maintained at 37 ± 0.5°C. Dissolution media were evaluated in three different pH media (a 0.1 N hydrochloric acid solution (pH 1.2), and two citrate-phosphate buffers (pH 4.5, and 7.4)). A LID sample, equivalent to 25 mg of lidocaine, was placed on the surface of the dissolution medium. At appropriate time intervals (0, 0.5, 1, 2, 3, 4, 5, 6, 8 and 24 h), 2.0 mL samples were withdrawn from each vessel, and mixed with 1.0 mL of internal standard (IS) (10 μg/mL) and 5.0 mL of methanol:water (75:25). The solution was then filtered through a 0.22 μm Millipore membrane filter and analyzed using a validated UPLC assay, as described below. The volume was replaced each time with 2 mL of fresh medium, which was maintained at a temperature of 37 ± 0.5°C to maintain a sink condition. All samples were tested in triplicate.
Analyzing the Kinetics of Drug Release
To evaluate the drug release kinetics for LID formulations, we used several different dissolution models, including zero, first, Higuchi and Korsmeyer–Peppas order. The slope of each model represented the release rate. The most appropriate model was selected by determining the best fitting (R2) values.
Chromatographic Conditions
Drug analysis was carried out with a Waters Acquity UPLCTM system fitted with an autosampler. An Acquity UPLCTM BEH Shield RP18 (1.7 μm column, 2.1 × 50 mm, Waters Corp, Milford, MA, USA) was used for separation, and the column temperature was maintained at 25°C. The gradient elution for UPLC analysis consisted of two solvent compositions: solvent (A) (10 mM ammonium formate (pH 3 ± 0.2), containing 0.2% formic acid and 1% acetonitrile), and solvent (B) (acetonitrile containing 0.2% formic acid). The gradient elution of solvent A:B was increased from 30% to 85% of solvent A within 3.3 min, then reduced back to 30% of solvent A, with a flow rate of 0.25 mL/min. The total run time was 4.0 min. Data were collected in multi-channel analysis mode and processed using MassLynxTM V 4.1 software the TargetLynxTM V 4.1 program (Waters Corp., Milford, MA, USA) was used to analyze the data. The UPLC was connected to a triple quadrupole tandem-mass detector (Water, Corp), with a positive ion mode electrometry ionization (ESI) source for mass spectroscopy detection. The optimal mass spectrometry parameters were as follows: cone voltage = 27V; desolvation temperature = 400°C, and collision energy of 20 eV. Argon was used as the collision gas at a pressure of approximately 0.25 Pa. The optimized collision energy for lidocaine was 22 eV.
The precision and accuracy of the developed LC-MS/MS method were measured for the concentration range of 0.5–20 ng/mL; there were no significant differences between the inter- and-intra-day analysis (p>0.05) of quality control samples. A linear relationship was observed over the investigated concentration range, with a correlation coefficient of r>0.994 (n=6/day). The assay was able to detect LID concentrations without any form of modification.
In vitro Cell Viability and Cytotoxicity Assays
In order to evaluate in vitro cardiotoxicity, we used rat embryonic cardiomyocytes (H9c2 (2–1) cells). These cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, containing 4 mmol of glutamine and penicillin-streptomycin (100 U/mL) in 75 cm2 tissue culture flasks. Cells were maintained in a humidified incubator in an environment of 95% air/5% CO2, and a temperature of 37°C. For cytotoxicity and ultrastructural studies, cells were seeded in 96-well microliter plates, and 12-well plates, respectively.
In order to investigate in vitro nephrotoxicity, we used the LLC-PK1 cell line. This cell line was grown in an RPMI-1640 culture medium (Sigma St. Louis, MO) and supplemented with 10% (v/v) bovine fetal serum (Invitrogen Co Ltd, Carlsbad, CA), 100 IU of penicillin/mL, and 100 mg/mL of streptomycin (Sigma, St. Louis, MO). Cells were subsequently grown in 75 cm2 flasks (TPP, Sigma Aldrich, St. Louis, MO), and kept at 37°C in an environment containing 5% CO2.
Cells (H9c2 and LLC-PK1) were seeded into 12 well plates and suspended in 1.0 mL of medium. After incubation for 1 day, cell cultures were treated with 100, 200, and 400 ng/µL of culture medium alone (control), or with standard LID and LID formulations (N1, N2, N3, and N4), and incubated for another 2 and 24 hrs. The cells were then pelleted by centrifugation at 1000 rpm for 3 min and then supernatant discarded prior to re-suspension in 0.5 mL of fresh medium without FBS. Cells were then incubated with 500 µg/mL of MTT for a further 2 hrs at 37°C in an environment containing 5% CO2. Next, we added 800 µL of isopropanol to each well, and after 10 min, determined the absorbance at 570 nm. The number of viable cells in the treated wells was then compared to the number of viable cells in the untreated wells in order for us to determine the overall cell viability (%). Tests were performed in triplicate from three independent experiments. No ethics approval was required from the Institutional Review Board, the experiment only featured commercial cell lines.
Stability Studies
The freeze-thaw stability of the plasma samples was evaluated by exposing quality control samples to three freeze (−20°C) and thaw (room temperature) cycles for different time intervals including the 0, 1, 2, and 4 days, as well as one and 3 weeks, then additionally tested after 1 and 3 months of preparation and storage at −20°C. The samples were analyzed by a UPLC system, as described previously.
Results and Discussion
Formulation Development and Optimization
The major factor underlying the successful encapsulation and release of drugs from microspheres is the production method. In addition, a range of other factors can also have a strong impact on drug delivery rates, including the type of polymer, the molecular weight of the polymer, the nature of any excipients added to the microsphere formulation, and the size of the microspheres.
Microspheres consisting of ALG-CS polymers are formulated by three common techniques: spray-drying, extrusion, and emulsification/gelation. The extrusion and emulsification/gelation technique has already been applied to encapsulate various types of drugs in the pharmaceutical and biotechnology industry.24 More recently, alginate–chitosan loaded drugs have been developed to improve the bioavailability of a large number of drugs.19–24,26,27
Mean Particle Size, Poly Disparity Index, and Zeta Potential
The particle size (MPS) measurements for all of the developed formulations were in the nano-range (340.6 ± 21.5 to 528.3 ± 14.7 nm; Table-1). A significant reduction in MPS was observed when we reduced the amount of sodium alginate. These findings are consistent with previous literature relating to vancomycin-encapsulated chitosan–alginate microparticles.28 The size of the microspheres was not correlated with zeta potential (ZP) values, drug loading, and/or drug yield. A negative surface charge was observed for all of the measured nano-microspheres (>−29; Table 1). This finding was in agreement with previous studies using sodium alginate diclofenac microspheres, which also showed negative ZP measurements (−35 mV).23
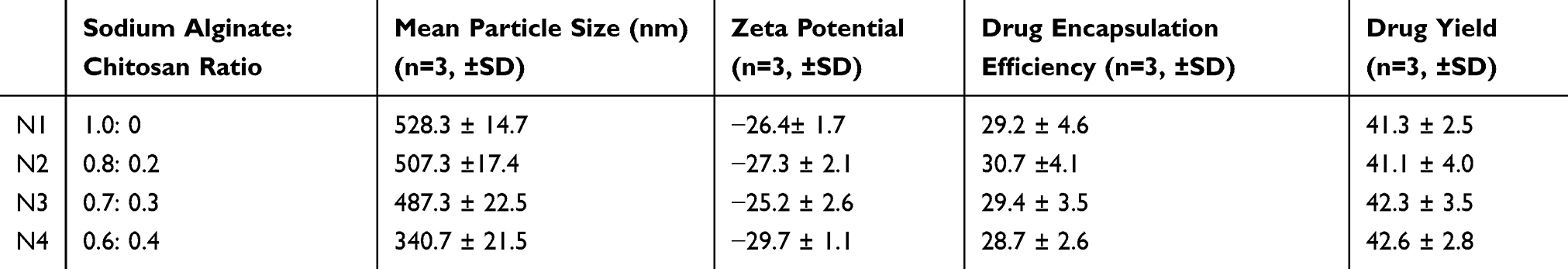 |
Table 1 Mean Particle Size, Drug Encapsulation, and Drug Yield of Developed Formulations. Each Value Is the Mean ± Standard Deviation of at Least Three Experimental Determinations |
Determination of Drug Entrapment Efficiency and Microsphere Yield
Table 1 shows the percentage yield, and drug encapsulation efficiency (DEE), of the developed formulations. There was no correlation between the concentrations of alginate, or the size of microspheres (MPS), with either the DEE or drug yield. These results are consistent with results from several previous studies.29–31
Thermal Analysis
Differential scanning calorimetry (DSC) studies of the pure LID produced an endotherm sharp peak corresponding to a melting point of 81°C, indicating that LID HCl is crystalline (Figure 2). A similar endothermic peak was reported in previous studies.32–34 Both sodium alginate and chitosan are crystalline and semi-crystalline polymers; this is because of their linear and regular structures. These compounds melt at temperatures ranging from 60°C to 100°C, due to the loss of water.32,35 The sharp drug peak was not evident in the LID formulations (N1, N2, N3, and N4); this was due to the LID being solubilized in a melted mass during the heating cycle of the DSC studies, or because the drug may have been present in its amorphous state in the polymer matrix. A similar phenomenon has been observed for tetracaine in water.36
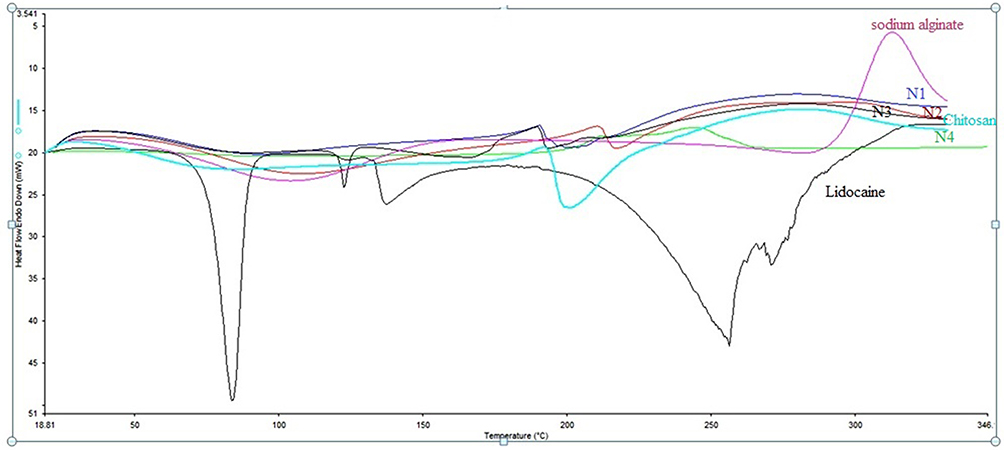 |
Figure 2 DSC Thermogram of pure Lidocaine, pure sodium alginate, pure chitosan, and the developed microspheres formulations (N1, N2, N3, and N4). |
Transmission Electron Microscopy of LID
Next, we examined the morphology of the microspheres using TEM (Figure 3). TEM images showed that the microspheres in our formulations were almost spherical, with some irregularities at the periphery of the particles. The particle size of the formulations developed, as determined by TEM, was in agreement with MPS data.
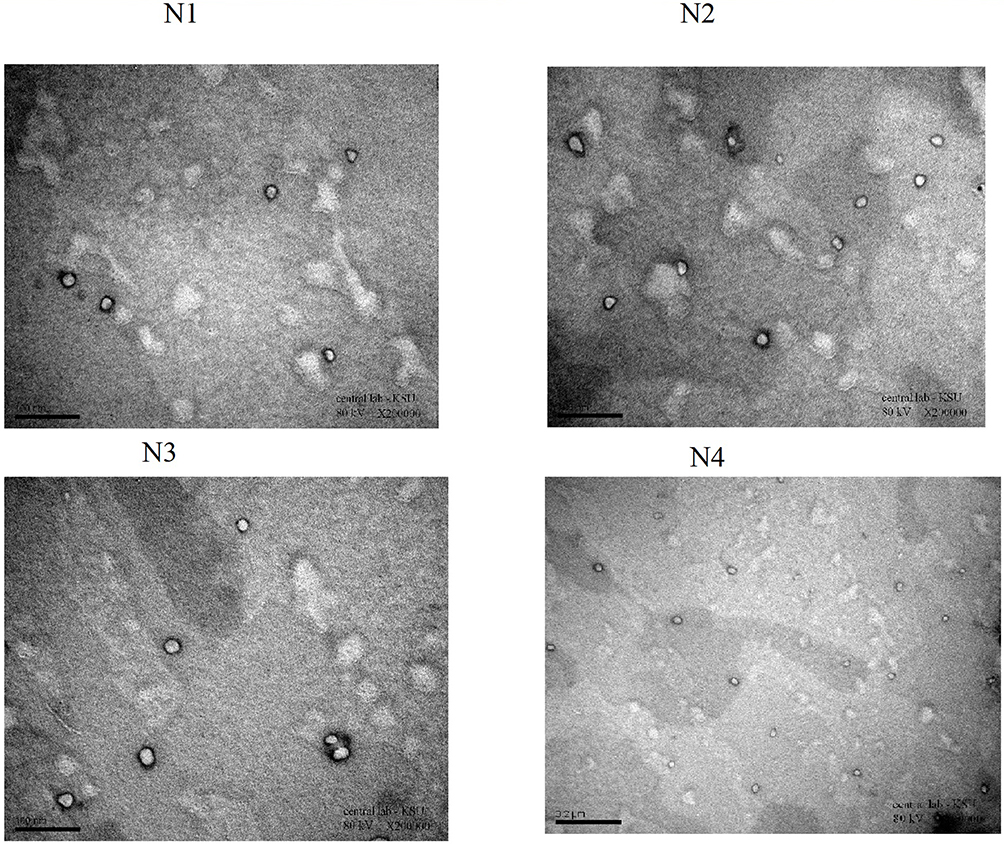 |
Figure 3 TEM images for Lidocaine microspheres formulations (N1, N2, N3, and N4). |
In vitro Release Studies
Since LID and chitosan exhibit pH-dependent solubility,37–40 the pH of the dissolution medium will affect the dissolution of the drug from the microspheres, as shown in Figure 4a, b and c, at pH 1.2 (stomach), pH 4.5 (lysosomes), and pH 7.4 (equivalent to blood pH), respectively. At all pH values, the rate of LID release from the microspheres occurred in a biphasic process. The burst effect occurred slowly (<10%) within the first 60 min; this was followed by a slower release step. The initial burst release of LID from the microspheres may be attributed to the drug being adsorbed on to the surface; the drug can then readily diffuse into the media, thus creating the observed burst release. After 60 min, we observed a sustained form of drug release, indicating that the LID had been successfully incorporated into the microspheres. Compared to low pH (pH 1.2), there was a significant enhancement in drug release between pH 4.5 and 7.4. Thus, an acidic medium appeared to yield optimal results. This is in agreement with previous literature2. The resulting alginate–chitosan microspheres exhibited a release behavior that was dependent on pH.29
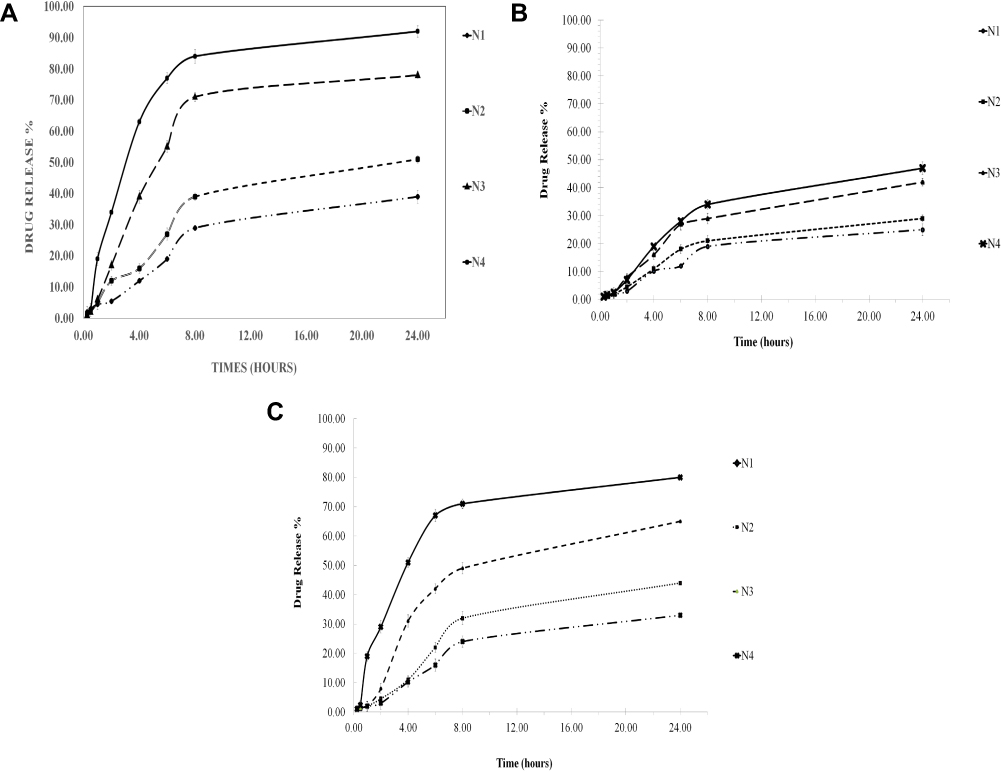 |
Figure 4 (A) Mean percent release profile LID-microspheres (± SD. N=3) in PBS (pH 1.2, 37°C) as dissolution medium from different microspheres for 24 hrs. (B) Mean percent release profile LID-microspheres (± SD. N=3) in PBS (pH 4.5, 37°C) as dissolution medium from different microspheres for 24 hrs. (C) Mean percent release profile LID-microspheres (± SD. N=3) in PBS (pH 7.4, 37°C) as dissolution medium from different microspheres for 24 hrs. |
Less than 10% of the burst release of the drug was leached from the microspheres; this was consistent across all pH values tested. Approximately 25–94% of the LID was released after 24 h, depending upon the pH conditions. The highest release rate was observed at pH 1.2, followed by pH 4.5 and pH 7.4 (Figure 3); these findings are related to the pKa values of LID. As pH increases, the ionized lidocaine diffused out of the polymeric matrix more easily (Figure 3). Similar phenomena were observed for LID chitosan, and hyaluronic acid bioadhesive transdermal,44 and for the release of sunitinib, an anticancer agent, from magnetic chitosan nanoparticles.41 In addition, the observed pH-dependent pattern was consistent with the previously described drug release behavior of indomethacin-crosslinked chitosan–Malginate microspheres, in which a higher pH caused lower rates of indomethacin release due to greater crosslinking in the microspheres.27 Protonation of the amine groups of the drug is known to improve solubility in an acidic medium. At low pH, alginate and chitosan are known to exist in a gel form, while at neutral pH, the viscous complex will swell and the gel will slowly disintegrate, thus causing the drug to be released.29
Moreover, the rate of drug release correlated with the mean particle size, and was fastest for small particles and slowest for large particles; this was the case for all pH conditions tested. The release rate of LID from microspheres was higher in the N4 treatment, followed by the N3, N2, and N1 treatments. For example, after 4 hrs at pH 1.2, 12%, 16%, 39%, and 63% of drug release was observed for the N1, N2, N3, and N4 formulations. The higher release rates correlated with lower MPS values. The same correlation was observed at both pH 4.5 and pH 7.4 (Figure 4a, b and c). The same relationship between drug loading and the rate of release was observed in a previous study.27,44
Drug Release Kinetics
The application of different mathematical models for the in vitro release of LID biodegradable beads was interpreted and evaluated by correlation coefficient (R2) analysis (Table 2). Identification of the highest correlation coefficient was used to determine the most suitable mathematical model for drug release kinetics. The Higuchi model showed a higher correlation coefficient (R2, Table 2) than the other models. Hence, the drug release profile for LID relies upon a diffusion mechanism. Hydrophilic nasal gel of LID42 showed similar results (Higuchi order).
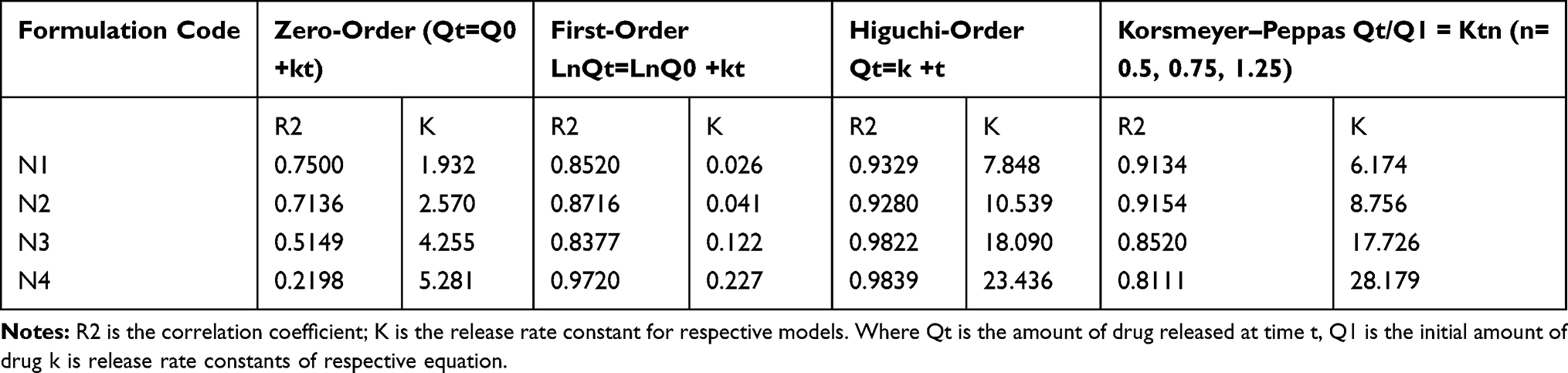 |
Table 2 Modeling of Lidocaine Release from Different Formulations |
Chromatography and Selectivity
Method Development
We developed a simple and fast UPLC assay for the preparation of LID. We found that the robustness and reproducibility of the assay improved when carried out at pH of 3 as well as the most prominent and stable fragments for LID.
Figure 5 shows MS and MS/MS traces of LID obtained from a blank, and a spiked standard solution of 10µg/mL, in the positive ion mode. All LC-MS/MS data utilized Electrospray ionization (ESI) in positive mode; in negative mode, no signal was acquired. This data was in agreement with a number of previous studies.43–48 In the MS mode, the precursor ion for LID is the protonated molecular ion [M +H]+ at m/z 235, while the daughter product ion is at m/z 86 for lidocaine (Figure 5). No interfering peak was detected at retention times that corresponded to LID. Furthermore, no interfering signals were detected from the spiked sample when drugs were frequently co-administered at the relevant drug retention times (Figure 6). This method was validated over a linear range of 10–120 µg/mL (Y= 0.0075 X+0.205, in which Y represents the area under the peak (AUP)).
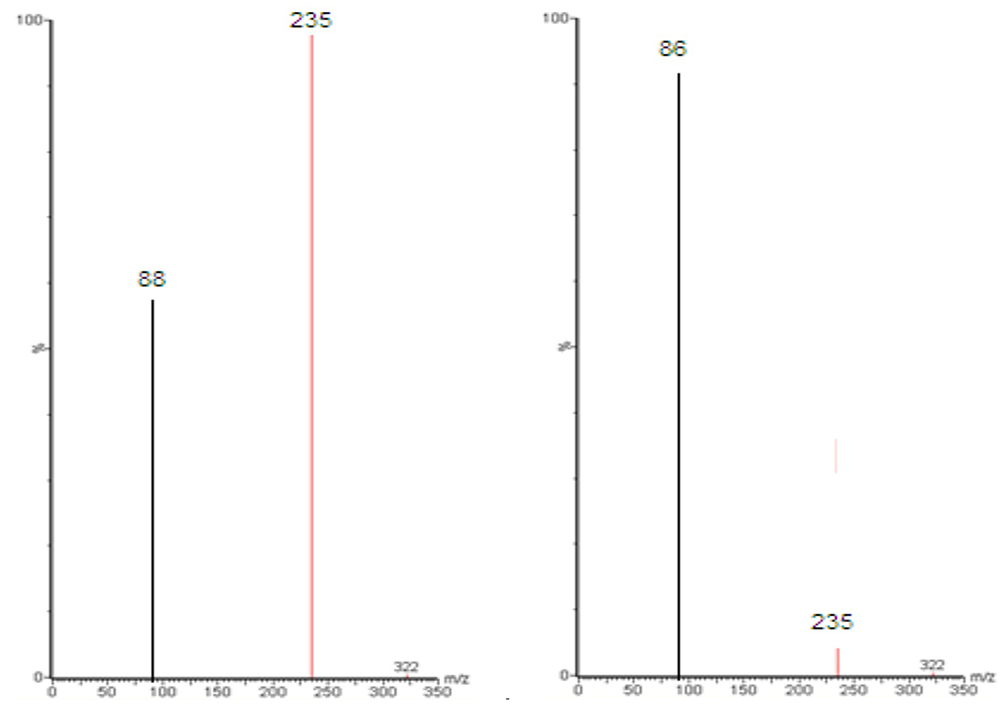 |
Figure 5 Full-scan product ion spectra of [M + H]+ of Lidocaine parent (A) drug and daughter (B). |
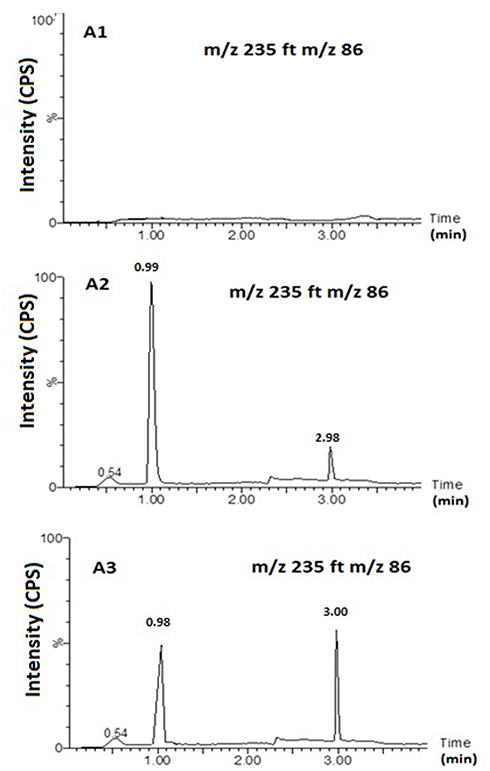 |
Figure 6 Representative MRM Chromatograms. Blank sample (A1); Spiked sample (A2;. Lidocaine (10 ng/mL) and Clopedogril (10 ng/mL)); sample form N3 formulation (A3). Column: An acquity UPLCTM BEH Shield RP18 (1.7 μm column, 2.1 × 50 mm, Waters Corp, Milford, MA, USA). |
Carry-Over
Carry-over was determined by analyzing two processed control samples after a ULOQ sample over three separate runs. We did not observe eluting peaks with areas >20% of the LLOQ when the quality control samples were directly injected after ULOQ samples. Therefore, the data met the criteria for carry-over.
In vitro Cell Viability and Cytotoxicity Assays
Both the cardio toxicity and nephrotoxicity of both LID and LID formulations (N1, N2, N3, and N4) were tested in two cell lines (H9c2 (2–1) and LLC-PK1 cells, respectively), at different concentrations (100, 200, and 400 ng/mL), for a duration of 2, 4, 6 and 24 hrs.
Previous research has shown that LID exerts cardiotoxic effects48. Figure 7a shows that pure LID is cardiotoxic, while the new LID microspheres formulations were safe. The LID microspheres formulations remained viable over different concentrations, even at 400 ng/mL (at which more than 80% of cells were viable). In addition, a dose-dependent reduction in viability was observed within the 2 hrs test. Data show that LID microsphere formulations were less toxic than the LID itself. The same observations were obtained at 4 and 6 hrs.
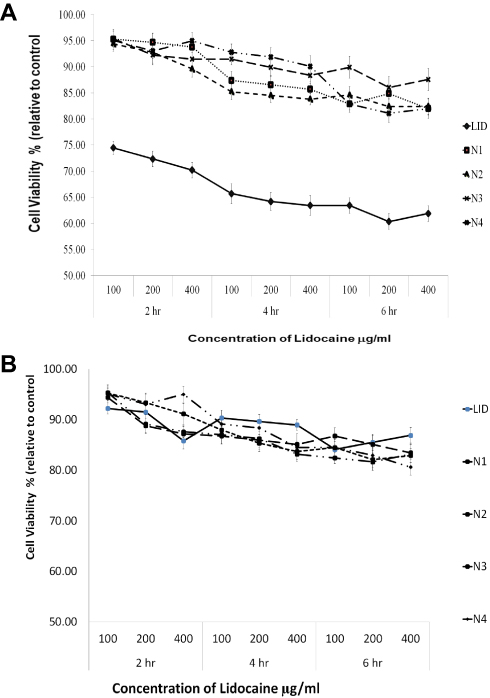 |
Figure 7 (A) The viability of H9c2 (2–1) cells after exposed to tested LID- alginate–chitosan microspheres formulations (100, 200, 400 ng/μL and control) after 2,4 and 6 hrs determined by MTT assay. Data represent mean ± SD (n=3). (B) The viability of LLC-PK1 cells after exposed to tested LID- alginate–chitosan microspheres formulations (100, 200, 400 ng/μL and control) after 2.4 and 6 hrs determined by MTT assay. Data represent mean ± SD (n=3). |
LID is known to be safe for the kidney. Figure 7b shows that the pure drug, as well as the LID microsphere formulations, were safe as cell viability exceeded 80%. Furthermore, the LID microsphere formulations were safe over a different concentration range, as cells remained viable, even at concentrations as high as 400 ng/mL.
Stability
In order to determine the best storage temperature for the microspheres, we conducted a stability study. LID and IS remained stable for up to 90 days at room temperature (samples were processed in autosampler vials at 25°C). Furthermore, these drugs remained stable for up to 3 months when stored at 4°C and −20°C. Therefore, all samples were stored at 4°C for same-day analysis by LC/MS/MS. Chromatographic analysis showed that there was no significant freeze/thaw degradation after three repetitive cycles (<10% degradation) for either LID or IS (Figure 8).
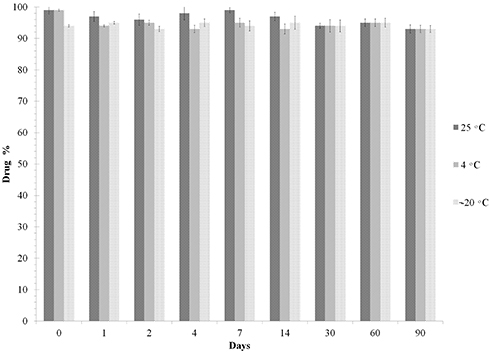 |
Figure 8 Freeze–thaw stability of LID at different temperatures. Data represent mean ± SD (n=3). |
This study has limitations that should be considered testing different concentrations of both polymers (alginate, chitosan) as well as addition of different stabilizers. Further studies are now needed to investigate the pharmacokinetic and pharmacodynamics of the formulated biodegradable LID microspheres to elucidate the specific patterns of in vivo release, and to develop the potential of the microspheres as an oral delivery system.
Conclusions
We developed a novel LID oral formulation that improves the solubility of LID and enhances the release of LID in vitro. These innovative LID microsphere formulations had a mean particle size that was in the nano range. Different ratios of alginate and chitosan were used to create different types of microsphere. We observed that MPS reduced as the concentration of alginate was reduced; there was no correlation with either drug loading or drug yield. Our data suggest that the formulations developed are viable and effective, and can be used as a novel oral delivery system for LID. Moreover, these new LID microspheres exhibited significantly less cardiac toxicity than LID alone.
Acknowledgments
This research project was supported by a grant from the Research Center of the Female Scientific and Medical Colleges, Deanship of Scientific Research, King Saud University.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Pasero C, McCaffery M. Pain Assessment and Pharmacologic Management. E-Book, Elsevier Health Sciences;2010:209–218.
2. Shafer A, Van Doze A, Shafer SL, White PF. Pharmacokinetics and pharmacodynamics of propofol infusions during general anesthesia. Anesthesiol. 1988;69:348–356. doi:10.1097/00000542-198809000-00011
3. Shukla A, Krause A, Neubert RHH. Microemulsions as colloidal vehicle systems for dermal drug delivery. part iv: investigation of microemulsion systems based on a eutectic mixture of lidocaine and prilocaine as the colloidal phase by dynamic light scattering. J Pharm Pharmacol. 2003;55:741–748. doi:10.1211/002235703765951339
4. Haas DA. An update on local anesthetics in dentistry. J Can Dent Assoc. 2002;68:546–552.
5. Campbell BJ, Rowbotham M, Stitzlein Davies P, Jacob P
6. Tucker GT, Mather LE. Clinical pharmacokinetics of local anaesthetics. Clini Pharmacokinet. 1979;4:241–278. doi:10.2165/00003088-197904040-00001
7. Shipton EA. New formulations of local anaesthetics—Part I. Anesthesiol Res Pract. 2012;2012:1–24.
8. Silva AC, Lopes CM, Lobo JMS, Amaral MH. Delivery systems for biopharmaceuticals. part II: liposomes, micelles, microemulsions and dendrimers. Curr Pharmaceut Biotechnol. 2015;16:955–965. doi:10.2174/1389201016666150817094637
9. de Araujo DR, da Silva DC, Barbosa RM, et al. Strategies for delivering local anesthetics to the skin: focus on liposomes, solid lipid nanoparticles, hydrogels and patches. Expert Opin Drug Delivery. 2013;10:1551–1563. doi:10.1517/17425247.2013.828031
10. Lockman PR, Mumper RJ, Khan MA, Allen DD. Nanoparticle technology for drug delivery across the blood-brain barrier. Drug Dev Ind. Pharm. 2002;28:1–13. doi:10.1081/DDC-120001481
11. Daryanavard SM, Jeppsson‐Dadoun A, Andersson LI, Hashemi M, Colmsjö A, Abdel‐Rehim M. Molecularly imprinted polymer in microextraction by packed sorbent for the simultaneous determination of local anesthetics: lidocaine, ropivacaine, mepivacaine and bupivacaine in plasma and urine samples. Biomed Chromatogr. 2013;27:1481–1488. doi:10.1002/bmc.2946
12. Rencher WF, inventor; Johnson and Johnson Consumer Inc, assignee. Bioadhesive pharmaceutical carrier. United States patent US5462749A. 1995 Oct 31.
13. Chung C-W, Kang JK, Yoon I-S, et al. Interpenetrating polymer network (IPN) scaffolds of sodium hyaluronate and sodium alginate for chondrocyte culture. Colloids Surf B Biointerfaces. 2011;88:711–716. doi:10.1016/j.colsurfb.2011.08.005
14. Colombo G, Padera R, Langer R, Kohane DS. Prolonged duration local anesthesia with lipid–protein–sugar particles containing bupivacaine and dexamethasone. J Biomed Mater Res B. 2005;75:458–464. doi:10.1002/jbm.a.30443
15. Jang YJ, Lee JH, Beom Seo T, Oh SH. Lidocaine/multivalent ion complex as a potential strategy for prolonged local anesthesia. Eur J Pharm Biopharm. 2017;115:113–121. doi:10.1016/j.ejpb.2017.02.007
16. Liu X, Ma X, Kun E, Guo X, Yu Z, Zhang F. Influence of lidocaine forms (salt vs. freebase) on properties of drug–eudragit® l100-55 extrudates prepared by reactive melt extrusion. Int. J. Pharmaceut. 2018:(1–2):291–302.Franz-Montan M, Baroni D, Brunetto G, Roberta V, Sobral V, Morais Gonçalves da Silva C, et al. Liposomal lidocaine gel for topical use at the oral mucosa: characterization, in vitro assays and in vivo anesthetic efficacy in humans. J Liposome Res. 2015;25:11–19. doi:10.3109/08982104.2014.911315
17. Adjepon-Yamoah KK, Scott DB, Prescott LF. Impaired absorption and metabolism of oral lignocaine in patients undergoing laparoscopy. Brit J Anaesth. 1973;45:143–147. doi:10.1093/bja/45.2.143
18. Sharma M, Choudhury PK, KUMAR SD. Formulation and in-vitro-in-vivo evaluation of alginate-chitosan microspheres of glipizide by ionic gelation method. Asian J Pharm Clin Res. 2017;10:385–390. doi:10.22159/ajpcr.2017.v10i7.18725
19. Zhang Y, Wei W, Lv P, Wang L, Ma G. Preparation and evaluation of alginate–chitosan microspheres for oral delivery of insulin. Euro J Pharm Biopharm. 2011;77:11–19. doi:10.1016/j.ejpb.2010.09.016
20. Rakesh P, Vipin K, Kanchan K. Alginate beads prepared by ionotropic gelation technique: formulation design. Res J Chem Sci. 2015;2231:606X.
21. Nayak A, Jain SK, Pandey RS. Controlling release of metformin HCL through incorporation into stomach specific floating alginate beads. Mol Pharm. 2011;8:2273–2281. doi:10.1021/mp2001395
22. Pireddu R, Sinico C, Ennas G, et al. Novel nanosized formulations of two diclofenac acid polymorphs to improve topical bioavailability. Euro J Pharm Sci. 2015;77:208–215. doi:10.1016/j.ejps.2015.06.006
23. Uyen NTT, Hamid ZAA, Tram NXT, Ahmad NB. Fabrication of alginate microspheres for drug delivery: a review. Int J Biol Macromol. In press 2020.
24. Tahtat D, Mahlous M, Benamer S, et al. Oral delivery of insulin from alginate/chitosan crosslinked by glutaraldehyde. Int J Biol Macromol. 2013;58:160–168. doi:10.1016/j.ijbiomac.2013.03.064
25. Mi F-L, Sung H-W, Shyu -S-S. Drug release from chitosan–alginate complex beads reinforced by a naturally occurring cross-linking agent. Carbohyr Polym. 2002;48:61–72. doi:10.1016/S0144-8617(01)00212-0
26. Sinha VR, Singla AK, Wadhawan S, et al. Chitosan microspheres as a potential carrier for drugs. Int J Pharm. 2004;274:1–33. doi:10.1016/j.ijpharm.2003.12.026
27. Unagolla JM, Jayasuriya AC. Drug transport mechanisms and in vitro release kinetics of vancomycin encapsulated chitosan-alginate polyelectrolyte microparticles as a controlled drug delivery system. Euro J Pharmaceut Sci. 2018;114:199–209. doi:10.1016/j.ejps.2017.12.012
28. Tønnesen HH, Karlsen J. Alginate in drug delivery systems. Drug Dev Ind Pharm. 2002;28:621–630. doi:10.1081/DDC-120003853
29. Xu Y, Zhan C, Fan L, Wang L, Zheng H. Preparation of dual crosslinked alginate–chitosan blend gel beads and in vitro controlled release in oral site-specific drug delivery system. Int J Pharm. 2007;336:329–337. doi:10.1016/j.ijpharm.2006.12.019
30. Tiwari RK, Singh L, Sharma V. Alginate micro-beads in novel drug delivery system: an overview. Int J Part Ther. 2013;5:1–13.
31. Repka MA, Gutta K, Prodduturi S, Munjal M, Stodghill SP. Characterization of cellulosic hot-melt extruded films containing lidocaine. Euro J Pharm Biopharm. 2005;59:189–196. doi:10.1016/j.ejpb.2004.06.008
32. Zhou G, Dong J, Wang Z, Li Z, Li Q, Wang B. Determination and correlation of solubility with thermodynamic analysis of lidocaine hydrochloride in pure and binary solvents. J Mol Liq. 2018;265:442–449. doi:10.1016/j.molliq.2018.06.035
33. Croisier F, Jérôme C. Chitosan-based biomaterials for tissue engineering. Euro Polym J. 2013;49:780–792. doi:10.1016/j.eurpolymj.2012.12.009
34. Al-Otoum R, Abulateefeh SR, Taha MO. Preparation of novel ionotropically crosslinked beads based on alginate-terephthalic acid composites as potential controlled release matrices. Die Pharmazie InterJ Pharm Sci. 2014;69:10–18.
35. Foldvari M. In vitro cutaneous and percutaneous delivery and in vivo efficacy of tetracaine from liposomal and conventional vehicles. Pharm Res. 1994;11:1593–1598. doi:10.1023/A:1018909821048
36. Reyes R, Duprat F, Lesage F, et al. Cloning and expression of a novel ph-sensitive two pore domain k+ channel from human kidney. J Biol Chem. 1998;273:30863–30869. doi:10.1074/jbc.273.47.30863
37. Park S-B, You J-O, Park H-Y, Haam SJ, Kim W-S. A novel ph-sensitive membrane from chitosan—teos ipn; preparation and its drug permeation characteristics. Biomaterials. 2001;22:323–330. doi:10.1016/S0142-9612(00)00187-3
38. Deng KL, Zhong HB, Tian T, Gou YB, Li Q, Dong LR. Drug release behavior of a pH/temperature sensitive calcium alginate/poly (n-acryloylglycine) bead with core-shelled structure. Express Polym Lett. 2010;4(12):773–780. doi:10.3144/expresspolymlett.2010.93
39. Hua S, Ma H, Li X, Yang H, Wang A. pH-sensitive sodium alginate/poly (vinyl alcohol) hydrogel beads prepared by combined ca2+ crosslinking and freeze-thawing cycles for controlled release of diclofenac sodium. Int J Biol Macromol. 2010;46:517–523. doi:10.1016/j.ijbiomac.2010.03.004
40. Anirudhan TS, Nair SS, Nair AS. Fabrication of a bioadhesive transdermal device from chitosan and hyaluronic acid for the controlled release of lidocaine. Carbohydr Polym. 2016;152:687–698. doi:10.1016/j.carbpol.2016.06.101
41. Karimi MH, Mahdavinia GR, Massoumi B. pH-controlled sunitinib anticancer release from magnetic chitosan nanoparticles crosslinked with κ-carrageenan. Mat Sci Eng C. 2018;91:705–714. doi:10.1016/j.msec.2018.06.019
42. Hu K-L, Mei N, Feng L, Jiang X-G. Hydrophilic nasal gel of lidocaine hydrochloride. 1st communication: preparation, formulation optimization and in vitro release study. Arzneimittel-Forschung. 2009;59:543–549. doi:10.1055/s-0031-1296442
43. Bo LD, Mazzucchelli P, Marzo A. Highly sensitive bioassay of lidocaine in human plasma by high-performance liquid chromatography– tandem mass spectrometry. J Chromatogr A. 1999;854:3–11. doi:10.1016/S0021-9673(99)00415-X
44. Abdel-Rehim M, Bielenstein M, Askemark Y, Tyrefors N, Arvidsson T. High-performance liquid chromatography–tandem electrospray mass spectrometry for the determination of lidocaine and its metabolites in human plasma and urine. J Chromatogr B Biomed Sci Appl. 2000;741:175–188. doi:10.1016/S0378-4347(00)00054-2
45. Maes A, Weiland L, Sandersen C, Gasthuys F, De Backer P, Croubels S. Determination of lidocaine and its two n-desethylated metabolites in dog and horse plasma by high-performance liquid chromatography combined with electrospray ionization tandem mass spectrometry. J Chromatogr B. 2007;852:180–187. doi:10.1016/j.jchromb.2007.01.010
46. Weijden ET, Van den Broek MPH, Ververs FFT. Easy and fast lc–ms/ms determination of lidocaine and MEGX in plasma for therapeutic drug monitoring in neonates with seizures. J Chromatogr B. 2012;881:111–114. doi:10.1016/j.jchromb.2011.11.030
47. Tonooka K, Naruki N, Honma K, et al. Sensitive liquid chromatography/tandem mass spectrometry method for the simultaneous determination of nine local anesthetic drugs. Forensic Sci Int. 2016;265:182–185. doi:10.1016/j.forsciint.2016.02.044
48. Groban L, Dolinski SY. Differences in cardiac toxicity among ropivacaine, levobupivacaine, bupivacaine, and lidocaine. Tech Reg Anesth Pain Manag. 2001;5:48–55. doi:10.1053/trap.2001.23679
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.