")
Back to Journals » Journal of Inflammation Research » Volume 15
The NLRP3 Inflammasome as a Novel Therapeutic Target for Cardiac Fibrosis
Authors Fan J, Ren M, Adhikari BK , Wang H, He Y
Received 12 April 2022
Accepted for publication 1 July 2022
Published 7 July 2022 Volume 2022:15 Pages 3847—3858
DOI https://doi.org/10.2147/JIR.S370483
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Jiwen Fan,1 Meng Ren,2 Binay Kumar Adhikari,3 Haodong Wang,1 Yuquan He1
1Department of Cardiology, China-Japan Union Hospital of Jilin University, Changchun, People’s Republic of China; 2Department of Medical Oncology, Jilin Provincial Cancer Hospital, Changchun, People’s Republic of China; 3Department of Cardiology, Nepal Armed Police Force (APF) Hospital, Kathmandu, Nepal
Correspondence: Yuquan He, Department of Cardiology, China-Japan Union Hospital of Jilin University, No. 126 Xiantai Road, Changchun, People’s Republic of China, Tel +86 15604411532, Email [email protected]
Abstract: Cardiac fibrosis often has adverse cardiovascular effects, including heart failure, sudden death, and malignant arrhythmias. However, there is no targeted therapy for cardiac fibrosis. Inflammation is known to play a crucial role in the disorder, and the NLR pyrin domain-containing-3 (NLRP3) inflammasome is closely associated with innate immunity. Therefore, further understanding the pathophysiological role of the inflammasome in cardiac fibrosis may provide novel strategies for the prevention and treatment of the disorder. The aim of this review was to summarize the present knowledge of NLRP3 inflammasome-related mechanisms underlying cardiac fibrosis and to suggest potential targeted therapy that could be used to treat the condition.
Keywords: NLRP3 inflammasome, cardiac fibrosis, AIM2, ASC, caspase-1
Introduction
Cardiac fibrosis is a common pathological feature of several cardiovascular diseases, leading to increased stiffness of the ventricular wall and impaired cardiac function.1–3 Inflammation is a crucial factor in cardiac fibrosis.4 Ischemia, hypoxia, and metabolic changes resulting from multiple causes lead to the infiltration of inflammatory cells and elevated levels of pro-inflammatory cytokines and chemokines.4,5 Interactions with damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns(PAMPs) can lead to activation and oligomerization of NLRP3 followed by recruitment of the apoptotic speck protein containing a caspase recruitment domain (ASC) and pro-caspase-1.6,7 The activated caspase-1 subsequently cleaves pro-IL-1β and pro-IL-18 to yield mature IL-1β and IL-18. In addition, caspase-1 triggers pyroptosis by cleaving Gasdermin D (GSDMD), resulting in the binding of the GSDMD N-terminal domain to phospholipids in the cell membrane, forming a GSDMD pore.8,9 NLRP3 is an important mediator of both autoimmunity and myocardial fibrosis.10–12 Therefore, elucidating the mechanism by which the NLRP3 inflammasome influences cardiac fibrosis and summarizing potential treatments for targeting the inflammasome may suggest promising therapeutic strategies for the prevention and treatment of cardiac fibrosis.
The NLRP3 Inflammasome
The inflammatory response is a normal physiological activity of the body in response to the invasion of external pathogenic microorganisms.13 The inflammasome plays a key r role in the inflammatory response.14,15 Five primary inflammasomes are currently known; these include the NLRP1, NLRP3, NLRC4, IPAF, and AIM2 inflammasomes. Among them, NLRP3 is the best-studied.16–18
The NLRP3 inflammasome is a multiprotein complex formed by the receptor protein NLRP3, the bridging protein ASC, and the pro-caspase-1 effector.19 The NLRP3 component is made up of three domains: a central nucleotide-binding NACHT domain (NOD domain), a C-terminal leucine-rich repeat (LRR), and an N-terminal pyrin domain (PYD). The NACHT domain contains an ATP-binding site that mediates NLRP3 activation and IL-1βprocessing. NLRP3 senses signals from pathogens, such as bacteria, and binds to and cleaves pro-caspase-1 through the bridging protein ASC. On the one hand, the activated caspase-1 then cleaves GSDMD to form N-GSDMD which, in turn, binds to cell membrane phospholipids to form GSDMD pores, triggering pyroptosis (Figure 1A); on the other hand, activated caspase-1 also cleaves pro-IL-1β and pro-IL-18 to the generate the mature IL-1β and IL-18, which subsequently induce an inflammatory response (Figure 1B).20
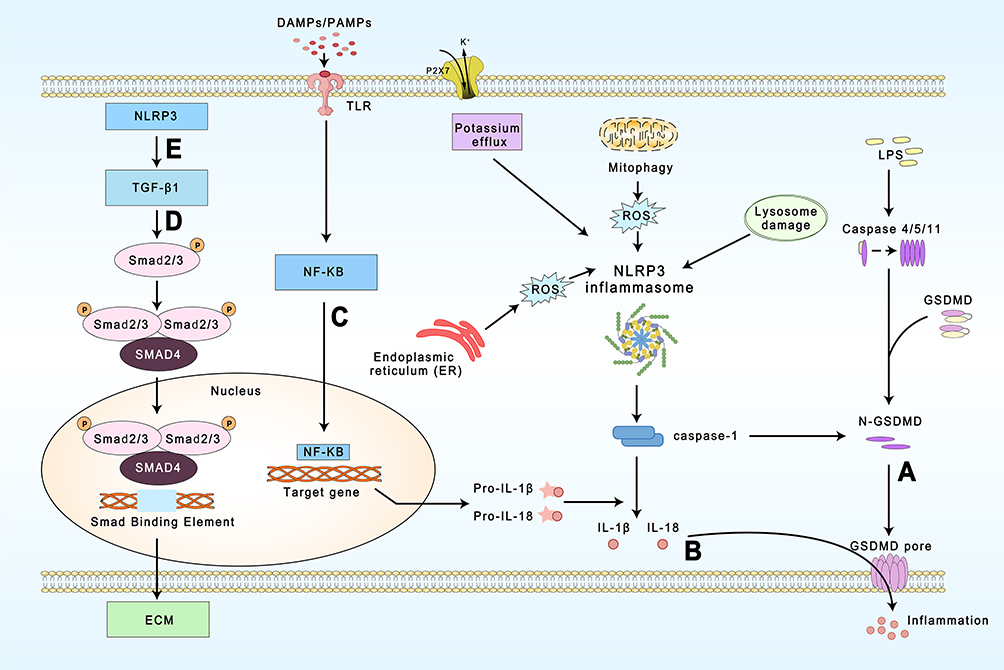 |
Figure 1 Regulation and function of NLRP3 inflammasome during cardiac fibrosis. (A) Caspase-1 activated by NLRP3 as well as caspase-4,5 and caspase-11 in the non-classical pathway are able to trigger pyroptosis. (B) Caspase-1 cleaves and activates pro-IL-1β and pro-IL-18, and is released extracellularly through the GSDMD pore. (C) The priming stage of NLRP3 inflammasome. (D) and (E) TGF-β/Smad is an important pathway leading to cardiac fibrosis and NLRP3 is capable of regulating it. |
Inflammasome activation is dependent on a stringent regulatory mechanism that includes priming and activation.20 The priming stage is the first step.12 Recognition of microbial ligands by Toll-like receptors (TLRs) and the stimulation of cytokines such as TNFα trigger nuclear factor kappa (NF-κB) to upregulate NLRP3, pro-caspase-1, and pro-IL-1 transcription (Figure 1C).21,22 A study by Toldo et al found that the priming stage is required for NLRP3 inflammasome activation and that even overexpression of NLRP3 (constitutively active) cannot activate caspase-1 when pro-caspase-1 is insufficient.23 The second step is the activation of the inflammasome complex by the oligomerization of NLRP3 and the recruitment of ASC and pro-caspase-1.24 Different endogenous and exogenous signals can induce the assembly of different inflammasomes. For example, AIM2 is a class of DNA receptors with a C-terminal HIN200 structural domain that recognizes and binds autologous or heterologous DNA.16 NLRP3 inflammasome activation pathways are more complex and are classified according to the source of the activation signal: 1) the induction of K+ efflux through P2X7 receptors by extracellular ATP;25,26 2) the generation of reactive oxygen species (ROS) through PAMP and DAMP activation;27 3) the formation of intracellular crystalline or granular structures leading to lysosomal rupture and the release of enzymes such as tissue protease B;28 4) endoplasmic reticulum (ER) stress; 5) autophagy dysfunction;29 6) Ca2+ overload. The most common activation mechanism is K+ efflux resulting from bacterial toxins and particulate matter.30 Several recent studies have found that NEK7 plays a key role in this process31,32 and that NEK7 knockout in mice inhibits both NLRP3 inflammasome activation and IL-1β production.33
The Golgi apparatus plays an important role in the transportation of newly synthesized proteins from the ER and is also closely involved in innate immunity.34 Interruption of ER-Golgi transport has been found to block both NLRP3 inflammasome assembly and caspase-1 activation.35,36 After activation, NLRP3 interacts with the ER sterol regulatory element-binding protein 2 (SREBP2) and the SREBP cleavage-activating protein (SCAP) to form the NLRP3-SCAP-SREBP2 triplex. After entry of NLRP3-SCAP-SREBP2 into the cis-Golgi network (CGN), NLRP3 dynamically traverses the Golgi to the trans-Golgi network (TGN), where it is phosphorylated by protein kinase D (PKD).37 In addition, a variety of NLRP3 activators can cause the breakdown of the TGN, and phosphatidylinositol-4-phosphate (Ptdlns4P) on the dispersed TGN (dTGN) becomes the “site” of NLRP3 inflammasome aggregation and assembly. In addition to phosphorylating NLRP3, PKD can activate NLRP3 through phosphatidylinositol-4 kinase 3β.38
Pathogenesis of Cardiac Fibrosis
Cardiac fibrosis is an alteration of myocardial cells and the myocardial interstitium caused by a variety of pathological factors. It leads to abnormalities in cardiac function and metabolism and is commonly seen at the end-stage of several cardiovascular diseases.2,3,39 Cardiac fibrosis results from both the differentiation of cardiac fibroblasts and excessive accumulation of extracellular matrix (ECM), leading to myocardial stiffness and reduced compliance of the ventricular wall.40,41 Cardiac fibroblasts are major matrix-producing cells, and represent one of the largest cell populations in normal mammalian hearts and are closely involved in the maintenance of normal cardiac function as well as cardiac remodeling in pathological states.42,43 The presence of fibroblasts in the healthy heart suggests that they may play a role in homeostasis in vivo. The fibroblasts are activated after cardiac injury, leading to their differentiation into myofibroblasts, which are the cells principally responsible for pathological cardiac remodeling.44 Fibrogenic signaling cascades are triggered by fibrogenic growth factors (including TGF-β and PDGFs), cytokines (including TNFα, IL-1, IL-6, and IL-10), and neurohumoral pathway components binding to surface receptors and the subsequent activation of downstream signaling cascades.45 Current research has revealed a close connection between the well-studied TGF-β/Smad signaling pathway and cardiac fibrosis, affecting both the secretion and degradation of ECM components and myofibroblast differentiation (Figure 1D).46 In addition, the involvement of the innate immune system in the regulation of heart functioning and remodeling is well-documented.47
The NLRP3 Inflammasome in the Development of Cardiac Fibrosis
Cardiac fibrosis is a pathological condition common to a variety of cardiovascular diseases and is characterized by excessive ECM deposition resulting in tissue damage and organ dysfunction. The NLRP3 inflammasome has been associated with the development of cardiac fibrosis.36 Increasing experimental evidence indicates that NLRP3 inflammasome expression is elevated in fibrotic cardiac tissue and, conversely, that cardiac fibrosis is mitigated by inflammasome inhibition.48,49 Louwe et al reported that transplantation of Nlrp3−/− bone marrow in mice after myocardial infarction attenuated cardiac remodeling compared with wild-type bone marrow, suggesting that the absence of the NLRP3 inflammasome in hematopoietic cells reduces adverse remodeling.50 Previous studies have demonstrated that TGF-β stimulation regulates NLRP3 inflammasome activation.28,51 However, recent research has found that TGF-β signaling pathways are regulated by NLRP3, rather than the reverse situation of TGF-β regulating NLRP3 (Figure 1E). Cáceres et al reported that serelaxin, which targets the expression of both TLR-4 and the NLRP3 inflammasome, reduced the levels of TGF-β1 and IL-1β in cardiac myofibroblasts.52 Research in this area is still in a preliminary stage, and further information is needed to clarify these interactions in cardiac fibrosis. The selective NLRP3 inhibitor MCC950 has been shown to be effective in treating a variety of inflammatory diseases in which NLRP3 is involved.53 A study by Gao et al found that in mice with induced myocardial infarction, MCC950 was able to reduce cardiac fibrosis and improve cardiac function by inhibiting NLRP3 inflammasome expression in cardiac fibroblasts.54 Animal studies have shown that angiotensin II activates the NLRP3 inflammasome, leading to cardiac fibrosis and the inflammatory response, and that these pathological changes can be reversed by MCC950.55
Hypertension is also one of the major diseases that endanger human health. Persistent hypertension can lead to left ventricular hypertrophy and myocardial fibrosis, which is a determinant of heart failure and a risk to human health. NLRP3 plays an important role in the pathogenesis of hypertensive cardiac fibrosis (Figure 2). A study by Lv et al found that the expression of NLRP3 and IL-1β was significantly elevated in cardiac tissues in mouse model of angiotensin 2(Ang II) infusion-induced hypertension, with a facilitative effect on the conversion of fibroblasts to myofibroblasts.56 Triptolide, an immunomodulator, exerts dose-dependent anti-cardiac fibrosis effects by inhibiting both the activation of the inflammasome and the release of IL-1β in AngII-stimulated cardiac fibrosis.57
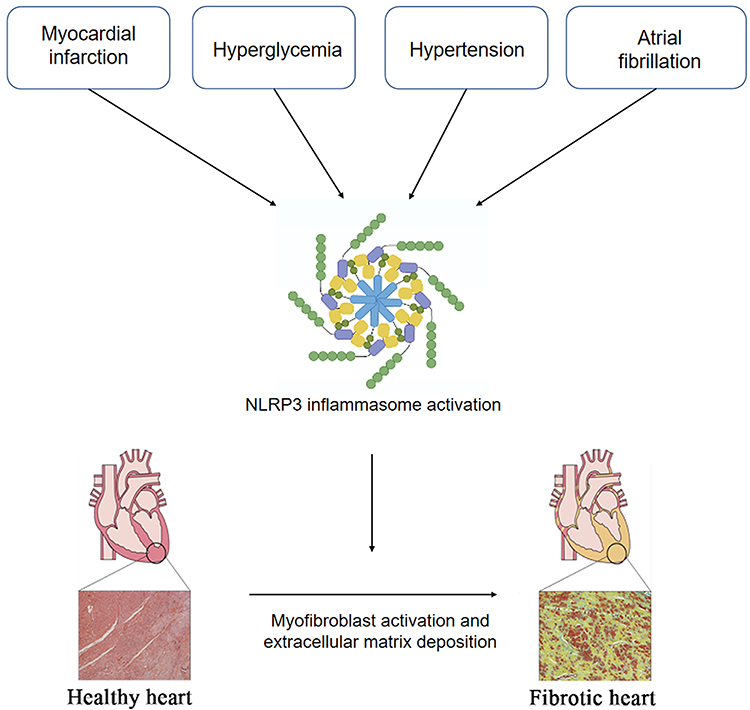 |
Figure 2 NLRP3 inflammasome activation in cardiovascular diseases to promote cardiac fibrosis. Under pathological conditions, such as myocardial infarction, hypertension, and hyperglycemia, atrial fibrillation, NLRP3 inflammasome in cardiomyocytes and fibroblasts are activated, collaboratively promoting the inflammatory cascade and pyroptosis. The resultant cardiac inflammation triggers myofibroblast activation and cardiac fibrosis. |
The rupture of unstable coronary atherosclerotic plaques is the main cause of acute myocardial infarction, and the ischemic necrotic myocardium can release large amounts of ATP and oxidative stress products (reactive oxygen species) to activate the NLRP3 inflammasome. Kawaguchi et al examined myocardial tissue from patients with myocardial infarction and found that the main inflammatory cell infiltrates at infarction site were macrophages and neutrophils and that the ASC expression levels were significantly elevated in these cells.58 As ASC is a component of the NLRP3 inflammasome, this increased expression suggests that NLRP3 inflammasomes in immune cell infiltrates are involved in pathological processes in the myocardial tissue after myocardial infarction.59 Kawaguchi et al treated isolated cultured cardiomyocytes and cardiac fibroblasts from neonatal mice with LPS and found that LPS significantly induced IL-1β production in cardiac fibroblasts from wild-type mice while reducing its production in the cardiac fibroblasts of ASC-/- mice. In contrast, LPS did not affect IL-1β production in cardiomyocytes.58 Sandanger et al observed the expression of NLRP3, IL-1β, and IL-18 mRNA in the ventricular myocardia of mice after the induction of myocardial infarction by ligation of the coronary arteries, and found that the expression levels of NLRP3, IL-1β, and IL-18 mRNA were significantly higher in the myocardial infarction group compared with the sham group and that this elevation was mainly associated with cardiac fibroblasts. In addition, they also created an ischemia-reperfusion model using NLRP3-knockout mice, observing significantly improved cardiac function and reduced ischemic damage in the knockout mice compared with their wild-type counterparts.60 These in vivo experimental studies suggest that NLRP3 inflammasomes play an important role in myocardial infarction and ischemia-reperfusion injury, and that intervention in the processes of NLRP3 inflammasome activation may provide useful clinical applications for improving myocardial fibrosis and cardiac remodeling after myocardial ischemia-reperfusion injury (Figure 2).
The inflammasome has also been found to be activated by hyperglycemia and to contribute significantly to the cardiac fibrosis typical of diabetic cardiomyopathy (DCM)(Figure 2).61–63 Hyperglycemia leads to a dramatic elevation in mitochondrial ROS generation and reduces the body’s antioxidant capacity.64,65 Under normal conditions, the oxidoreductase thioredoxin (TRX) binds to the thioredoxin-binding protein (TXNIP), inhibiting its activity. Overproduction of ROS leads to the dissociation of the TXNIP-TRX complex, and the subsequent binding of TXNIP to NLRP3 to induce inflammasome assembly.66,67 A study by Che et al suggested that inhibition of the NLRP3 inflammasome significantly ameliorated cardiac function and collagen production in diabetic mice.68 Knockdown of dual oxidase 1 (DUOX1) has been observed to inhibit the ROS-dependent pyroptosis pathway and reverse activin A-induced cardiac fibrosis.69 Zhang et al showed that hyperglycemia-induced overproduction of ROS and activation of P2X7R increased NLRP3 inflammasome expression and that this activation of the NLRP3 inflammasome activation was essential for collagen synthesis.70 In contrast, H3 relaxin effectively inhibited both ROS production and P2X7R activation and attenuated myocardial fibrosis. In addition, a study by Yao et al found that combined treatment with syringin and tilianin in diabetic rats prevented mitochondrial membrane depolarization and ROS production, while decreasing NLRP3 expression and improving cardiac function.71
Atrial fibrillation(AF) is the most common cardiac arrhythmia, and its prevalence in the population increase annually.72 Although the mechanisms responsible for AF have not been fully elucidated. Recent studies have found an association between AF, NLRP3 inflammasome and cardiac fibrosis and AF (Figure 2).73–75 Expression of the NLRP3 inflammasome has been found to be elevated in the atrial cardiomyocytes of individuals with paroxysmal and chronic AF. Yao et al established a cardiomyocyte-specific knock-in mouse model expressing constitutively active NLRP3 (CM-KI) and found that CM-KI mice developed spontaneous premature atrial beats and induced atrial fibrillation that were able to be reversed by the NLRP3 inhibitor MCC950.76 In addition, obesity is a known risk factor for atrial fibrillation and is associated with an enhanced inflammatory response.77–79 Recent studies have shown that obesity-induced atrial arrhythmias are driven by atrial NLRP3 inflammasomes, providing a molecular link between obesity-induced atrial fibrillation and NLRP3 inflammasome activation.80 Qiu et al reported that salvianolate may attenuates atrial fibrillation and interstitial fibrosis by suppressing the TXNIP/NLRP3 inflammasome signaling pathway in post-MI rats.75
Autophagy is associated with NLRP3 inflammasome-mediated cardiac fibrosis. Autophagy is a cellular self-digestion mechanism for the cell’s own metabolic needs and the recycling of organelles and other cellular material.81 Autophagy negatively regulates inflammasome activation by suppressing intracellular ROS production.82 Autophagy was found to be impaired in a mouse model of cardiac injury with isoproterenol injection and ligation of the left anterior descending artery, leading to reduced cardiac fibrosis; antifibrotic treatment with aspirin significantly ameliorated cardiac fibrosis, while rapamycin, an autophagy promoter, was able to counteract cardiac fibroblast promotion, suggesting that autophagy plays an important role in the development of cardiac fibrosis.83
Pyroptosis is a form of programmed cell death, characterized by persistent cell distension resulting in rupture of the cell membrane and the discharge of cellular contents, triggering an inflammatory reaction.84,85 The classical pyroptotic pathway is activated by the presence of pathogens, bacteria, and other signals, and leads to the interaction of NLRP3 with the ASC N-terminal PYD domain and the recruitment and cleavage of pro-caspase-1.84 The cleaved caspase-1 product then promotes the secretion of IL-1β and IL-18 into the extracellular space, leading to inflammatory necrosis of the cell. The nonclassical pyroptotic pathway is triggered by other members of the caspase family, namely, caspases-4 and −5 in humans and caspase-11 in mice.86,87 Pyroptosis is also capable of leading to cardiac fibrosis.88,89 Clinical studies have shown that both structural changes in the heart and the presence of fibrosis after myocardial infarction severely interfere with the treatment of heart failure.90,91 Nie et al found that hydrogen gas inhalation by rats with myocardial infarction blocked NLRP3-mediated pyroptosis and ameliorated myocardial infarction-induced cardiac remodeling and fibrosis.64 Luo et al reported that silencing the NLRP3 gene in a rat model of type 2 diabetes reduced cardiomyocyte pyroptosis and cardiac fibrosis, together with slowing the progression of DCM.92 Furthermore, NLRP3 derived from the mitochondria in cardiac fibroblasts was able to promote fibrotic signaling, regulate mitochondrial ROS production, and modulate fibroblast differentiation by a novel mechanism independent from the inflammasome.28 To summarize, pyroptosis has the potential to be a therapeutic target for the treatment of cardiac fibrosis in the future.
The NLRP3 Inflammasome as a Target for Pharmacological Inhibition
In the previous sections of this review, we have discussed the mediation of inflammatory responses by the NLRP3 inflammasome and its role in the development of cardiac fibrosis. This evidence suggests that targeting the NLRP3 inflammasome and its associated factors may be effective for treating cardiac fibrosis (Table 1).
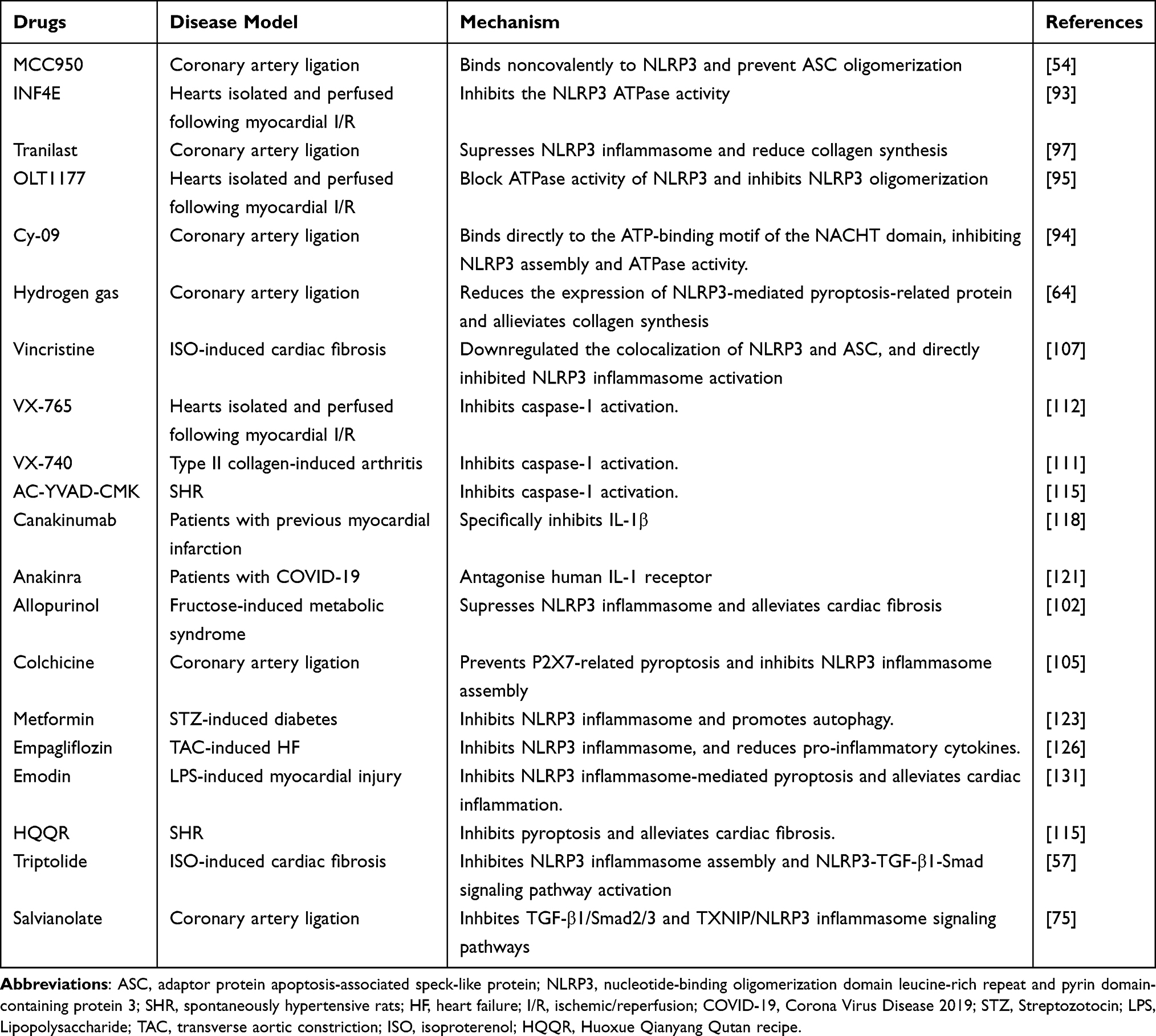 |
Table 1 Cardiac Fibrosis Therapies Targeting the NlRP3 Inflammasome Pathway |
NLRP3 Inhibitors
Research over the past few decades has identified various agents that block NLRP3 activation. These include MCC950, INF4E, Tranilast, OLT1177, Cy-09, JC-124, allopurinol, colchicine, vincristine, and hydrogen gas.93 MCC950 is a selective inhibitor of the NLRP3 inflammasome and does not affect NLRP1 and AIM2 inflammasomes.53 It inhibits ATPase activity and prevents the formation of the NLRP3 oligomer and has been shown to effectively ameliorate cardiac fibrosis in mice with myocardial infarction.54 CY-09 and OLT1177 prevent ASC polymerization and inflammasome assembly.94,95 Tranilast binds directly to the NACHT domain of NLRP3, preventing the interaction between NLRP3 and ASC.96–98 Tranilast also reduces collagen synthesis, but the mechanism is unclear.99 Hydrogen gas reduces inflammasome levels and has anti-inflammatory effects.100 A study by Nie et al observed that hydrogen inhalation reduced the levels of pyroptosis-related proteins after NLRP3 activation and also reduced collagen synthesis in rats with myocardial infarction.101 Allopurinol is a xanthine oxidase inhibitor used primarily for treating gout. Kang et al reported that allopurinol alleviated cardiac fibrosis and inflammation induced by fructose by counteracting CD36-mediated TLR4/6-IRAK4/1 signaling to prevent NLRP3 inflammasome activation.102 Colchicine is an alkaloid that is also used for treating gout and familial Mediterranean fever due to its potent anti-inflammatory effects.103,104 Colchicine can prevent P2X7-mediated pyroptosis and inhibit NLRP3 inflammasome activation.105,106 Vincristine reduced the colocalization of NLRP3 and ASC, and directly inhibited NLRP3 inflammasome activation and cardiac fibrosis in LPS-ATP-stimulated cardiac fibroblasts.107 Further research is required to verify the clinical applicability of these NLRP3 inflammasome-targeting compounds.
Inhibition of Caspase-1, IL-1β, and IL-18
Considering that the NLRP3 inflammasome is a multiprotein complex, targeting other components of the complex or the products of the inflammasome may also be a useful treatment for cardiac fibrosis. VX-765 (belnacasan) and VX-740 (pralnacasan) are selective caspase-1 inhibitors.108 VX-765 and VX-740 are structurally similar and are both precursor drugs that are converted in the presence of plasma esterase to the active metabolites VRT-043198 and VRT-18858, which selectively inhibit caspase-1.109–112 However, clinical trials of VX-740 and VX-765 were discontinued due to hepatotoxicity observed in animal toxicity studies.109,113 AC-AC-YVAD-CMK is also an irreversible caspase-1 inhibitor that increases cell permeability.114 A study by Lu et al showed that AC-YVAD-CMK has an antifibrotic role through its inhibition of NLRP3 inflammasome signaling in obese hypertensive rats.115 In addition, it was also found that AC-YVAD-CMK was able to downregulate pressure overload-induced cardiomyocyte pyroptosis and improve cardiac function at both in vivo and in vitro levels.108
IL-1β is a key factor in the innate immune system.26 IL-1β expression is significantly elevated in cardiovascular disease and cardiac fibrosis.107,116 The human monoclonal antibody canakinumab binds and inhibits IL-1β.117 In a study involving 10061 patients with preexisting myocardial infarction, canakinumab was found to significantly reduce the inflammatory response.118 In addition to monoclonal antibodies, IL-1 receptor antagonists can also block IL-1 signaling. Anakinra, a human IL-1 receptor antagonist produced by genetic recombination technology, is the only FDA-approved drug used in neonatal-onset multisystem inflammatory disease(NOMID).119,120 A retrospective analysis found that anakinra treatment reduced the levels of high-sensitivity C-reactive protein and increased potassium and calcium fluxes in 29 COVID-induced ARDS patients, leading to a significant increase in 21-day overall survival (90% vs 56%).121 However, due to the small patient base and single-center nature of the study, the results need to be verified by a multicenter prospective study design.
Other Anti-Fibrotic Drugs
Metformin is used for treating type 2 diabetes either alone or in combination with other hypoglycemic agents.122 Yang et al reported that metformin blocks the NLRP3 inflammasome through inhibition of the AMPK/mTOR pathway, and improves cardiac fibrosis in mice with diabetic cardiomyopathy.123 Empaglifozin (EMPA) selectively inhibits the sodium-glucose co-transporter 2. Recent clinical trials have shown that EMPA can reduce the risk of hospitalization for heart failure and cardiovascular death in patients with type 2 diabetes.124,125 Moreover, EMPA has also been shown to be effective in reducing cardiac fibrosis and inflammation by preventing NLRP3 inflammasome activation in two rodent models of heart failure.126
Various Chinese medicinal herbs have also been shown to block both NLRP3 inflammasome activation and the development of cardiac fibrosis. Triptolide, a compound isolated from a Chinese medicinal herb, has demonstrated anticancer activities against a variety of tumor types, including leukemia, breast cancer, and lung cancer.127,128 Pan et al reported that triptolide prevented NLRP3 inflammasome assembly and activation of the NLRP3-TGF-β1-Smad signaling pathway, reducing myocardial hypertrophy and fibrosis caused by pressure overload.57 Huoxue Qianyang Qutan recipe(HQQR) is a widely used Chinese herbal compound that can reduce blood pressure and improve lipid metabolism in obese hypertensive patients.115,129 Recent findings have reported that HQQR could reduce cardiac fibrosis by inhibiting the NLRP3 inflammasome signaling pathway in obese hypertensive rats.115 Emodin is a natural medicine that is effective for treating several chronic diseases; it has various pharmacological properties, including antioxidant, antimicrobial, antidiabetic, and immunosuppressive effects.130 A study by Xiao et al found that Emodin has anti-fibrotic action through the suppression of NLRP3 inflammasome activation induced by LPS.131 Overall, these findings suggest potential treatments for cardiac fibrosis by targeting the NLRP3 inflammasome.
Conclusion
The NLRP3 inflammasome is a signaling protein complex with wide spread expression in cardiovascular tissues and cells. As it is able to be activated by a wide variety of pathogens and danger signals, the NLRP3 inflammasome plays an important role in cardiac fibrosis associated with atherosclerosis, myocardial infarction, diabetic cardiomyopathy, atrial fibrillation, and many other cardiovascular diseases. Targeting NLRP3 inflammasome signaling may thus be a promising treatment for cardiac fibrosis, as inhibition of the NLRP3 inflammasome and its associated signaling may both improve cardiac function and reduce cardiac fibrosis. Furthermore, Chinese herbal extracts have recently attracted attention for their ability to alleviate cardiac fibrosis, although their underlying mechanisms of action have yet to be verified. Currently, most of the evidence is provided by animal and in vitro cell experiments, and the clinical efficacy of these potential treatments is still unclear. Thus, further investigations are required in the future.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358(13):1370–1380.
2. Pathak A, Del monte F, Zhao W, et al. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res. 2005;96(7):756–766.
3. Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. 2021;117(6):1450–1488.
4. Carrizales-Sepúlveda EF, Ordaz-Farías A, Vera-Pineda R, Flores-Ramírez R. Periodontal Disease, Systemic Inflammation and the Risk of Cardiovascular Disease. Heart Lung Circ. 2018;27(11):1327–1334.
5. Anzai T. Inflammatory Mechanisms of Cardiovascular Remodeling. Circ J. 2018;82(3):629–635.
6. Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016;13(2):148–159.
7. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240–273.
8. Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin D in pyroptosis. Acta Pharm Sin B. 2021;11(9):2768–2782.
9. Rathinam VAK, Zhao Y, Shao F. Innate immunity to intracellular LPS. Nat Immunol. 2019;20(5):527–533.
10. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15(9):505–522.
11. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: an Overview of Mechanisms of Activation and Regulation. Int J Mol Sci. 2019;20:13.
12. Wang Q, Wu J, Zeng Y, et al. Pyroptosis: a pro-inflammatory type of cell death in cardiovascular disease. Clin Chim Acta. 2020;510:62–72.
13. Yang CS, Shin DM, Jo EK. The Role of NLR-related Protein 3 Inflammasome in Host Defense and Inflammatory Diseases. Int Neurourol J. 2012;16(1):2–12.
14. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–420.
15. Kanneganti TD, Lamkanfi M, Núñez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27(4):549–559.
16. Fidler TP, Xue C, Yalcinkaya M, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592(7853):296–301.
17. Sharma D, Kanneganti TD. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213(6):617–629.
18. Xu H, Yang J, Gao W, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513(7517):237–241.
19. Zhen Y, Zhang H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front Immunol. 2019;10:276.
20. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411.
21. Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733.
22. Cornut M, Bourdonnay E, Henry T. Transcriptional Regulation of Inflammasomes. Int J Mol Sci. 2020;21(21):849.
23. Toldo S, Mezzaroma E, McGeough MD, et al. Independent roles of the priming and the triggering of the NLRP3 inflammasome in the heart. Cardiovasc Res. 2015;105(2):203–212.
24. El-Sharkawy LY, Brough D, Freeman S. Inhibiting the NLRP3 Inflammasome. Molecules. 2020;25(23):548.
25. Shokoples BG, Paradis P, Schiffrin EL. P2X7 Receptors: an Untapped Target for the Management of Cardiovascular Disease. Arterioscler Thromb Vasc Biol. 2021;41(1):186–199.
26. Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011;22(4):189–195.
27. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225.
28. Bracey NA, Gershkovich B, Chun J, et al. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J Biol Chem. 2014;289(28):19571–19584.
29. Chung C, Seo W, Silwal P, Jo EK. Crosstalks between inflammasome and autophagy in cancer. J Hematol Oncol. 2020;13(1):100.
30. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–1153.
31. Mathur A, Hayward JA, Man SM. Molecular mechanisms of inflammasome signaling. J Leukoc Biol. 2018;103(2):233–257.
32. Liu G, Chen X, Wang Q, Yuan L. NEK7: a potential therapy target for NLRP3-related diseases. Biosci Trends. 2020;14(2):74–82.
33. He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530(7590):354–357.
34. Tao Y, Yang Y, Zhou R, Gong T. Golgi Apparatus: an Emerging Platform for Innate Immunity. Trends Cell Biol. 2020;30(6):467–477.
35. Hong S, Hwang I, Gim E, et al. Brefeldin A-sensitive ER-Golgi vesicle trafficking contributes to NLRP3-dependent caspase-1 activation. FASEB j. 2019;33(3):4547–4558.
36. Wang Y, Liu X, Shi H, et al. NLRP3 inflammasome, an immune-inflammatory target in pathogenesis and treatment of cardiovascular diseases. Clin Transl Med. 2020;10(1):91–106.
37. Guo C, Chi Z, Jiang D, et al. Cholesterol Homeostatic Regulator SCAP-SREBP2 Integrates NLRP3 Inflammasome Activation and Cholesterol Biosynthetic Signaling in Macrophages. Immunity. 2018;49(5):842–856.e847.
38. Gong T, Jiang W, Zhou R. Control of Inflammasome Activation by Phosphorylation. Trends Biochem Sci. 2018;43(9):685–699.
39. Frangogiannis NG. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med. 2019;65:70–99.
40. Park S, Nguyen NB, Pezhouman A, Ardehali R. Cardiac fibrosis: potential therapeutic targets. Transl Res. 2019;209:121–137.
41. Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123(2):255–278.
42. Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. 2005;65(1):40–51.
43. Tallquist MD. Cardiac Fibroblast Diversity. Annu Rev Physiol. 2020;82:63–78.
44. Liu M, López de Juan Abad B, Cheng K. Cardiac fibrosis: myofibroblast-mediated pathological regulation and drug delivery strategies. Adv Drug Deliv Rev. 2021;173:504–519.
45. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71(4):549–574.
46. Khalil H, Kanisicak O, Prasad V, et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127(10):3770–3783.
47. Baci D, Bosi A, Parisi L, et al. Innate Immunity Effector Cells as Inflammatory Drivers of Cardiac Fibrosis. Int J Mol Sci. 2020;21:19.
48. Sandanger Ø, Gao E, Ranheim T, et al. NLRP3 inflammasome activation during myocardial ischemia reperfusion is cardioprotective. Biochem Biophys Res Commun. 2016;469(4):1012–1020.
49. Pinar AA, Scott TE, Huuskes BM, Tapia Cáceres FE, Kemp-Harper BK, Samuel CS. Targeting the NLRP3 inflammasome to treat cardiovascular fibrosis. Pharmacol Ther. 2020;209:107511.
50. Louwe MC, Olsen MB, Kaasbøll OJ, et al. Absence of NLRP3 Inflammasome in Hematopoietic Cells Reduces Adverse Remodeling After Experimental Myocardial Infarction. JACC Basic Transl Sci. 2020;5(12):1210–1224.
51. Díaz-Araya G, Vivar R, Humeres C, Boza P, Bolivar S, Muñoz C. Cardiac fibroblasts as sentinel cells in cardiac tissue: receptors, signaling pathways and cellular functions. Pharmacol Res. 2015;101:30–40.
52. Cáceres FT, Gaspari TA, Samuel CS, Pinar AA. Serelaxin inhibits the profibrotic TGF-β1/IL-1β axis by targeting TLR-4 and the NLRP3 inflammasome in cardiac myofibroblasts. FASEB j. 2019;33(12):14717–14733.
53. Wu D, Chen Y, Sun Y, et al. Target of MCC950 in Inhibition of NLRP3 Inflammasome Activation: a Literature Review. Inflammation. 2020;43(1):17–23.
54. Gao R, Shi H, Chang S, et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces myocardial fibrosis and improves cardiac remodeling in a mouse model of myocardial infarction. Int Immunopharmacol. 2019;74:105575.
55. Gan W, Ren J, Li T, et al. The SGK1 inhibitor EMD638683, prevents Angiotensin II-induced cardiac inflammation and fibrosis by blocking NLRP3 inflammasome activation. Biochim Biophys Acta Mol Basis Dis. 2018;1864(1):1–10.
56. Lv SL, Zeng ZF, Gan WQ, et al. Lp-PLA2 inhibition prevents Ang II-induced cardiac inflammation and fibrosis by blocking macrophage NLRP3 inflammasome activation. Acta Pharmacol Sin. 2021;42(12):2016–2032.
57. Pan XC, Liu Y, Cen YY, et al. Dual Role of Triptolide in Interrupting the NLRP3 Inflammasome Pathway to Attenuate Cardiac Fibrosis. Int J Mol Sci. 2019;20(2):849.
58. Kawaguchi M, Takahashi M, Hata T, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123(6):594–604.
59. Lu A, Magupalli VG, Ruan J, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156(6):1193–1206.
60. Sandanger Ø, Ranheim T, Vinge LE, et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2013;99(1):164–174.
61. Qin L, Zang M, Xu Y, et al. Chlorogenic Acid Alleviates Hyperglycemia-Induced Cardiac Fibrosis through Activation of the NO/cGMP/PKG Pathway in Cardiac Fibroblasts. Mol Nutr Food Res. 2021;65(2):e2000810.
62. Luo B, Li B, Wang W, et al. Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model. Cardiovasc Drugs Ther. 2014;28(1):33–43.
63. Yue Y, Meng K, Pu Y, Zhang X. Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res Clin Pract. 2017;133:124–130.
64. Nie C, Zou R, Pan S, et al. Hydrogen gas inhalation ameliorates cardiac remodelling and fibrosis by regulating NLRP3 inflammasome in myocardial infarction rats. J Cell Mol Med. 2021;25(18):8997–9010.
65. Sharma A, Tate M, Mathew G, Vince JE, Ritchie RH, de Haan JB. Oxidative Stress and NLRP3-Inflammasome Activity as Significant Drivers of Diabetic Cardiovascular Complications: therapeutic Implications. Front Physiol. 2018;9:114.
66. Wang BF, Yoshioka J. The Emerging Role of Thioredoxin-Interacting Protein in Myocardial Ischemia/Reperfusion Injury. J Cardiovasc Pharmacol Ther. 2017;22(3):219–229.
67. Zhang H, Chen X, Zong B, et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J Cell Mol Med. 2018;22(9):4437–4448.
68. Che H, Wang Y, Li H, et al. Melatonin alleviates cardiac fibrosis via inhibiting lncRNA MALAT1/miR-141-mediated NLRP3 inflammasome and TGF-β1/Smads signaling in diabetic cardiomyopathy. FASEB j. 2020;34(4):5282–5298.
69. Li S, Li Z, Yin R, Nie J, Fu Y, Ying R. Knockdown of dual oxidase 1 suppresses activin A-induced fibrosis in cardiomyocytes via the reactive oxygen species-dependent pyroptotic pathway. Int J Biochem Cell Biol. 2021;131:105902.
70. Zhang X, Fu Y, Li H, et al. H3 relaxin inhibits the collagen synthesis via ROS- and P2X7R-mediated NLRP3 inflammasome activation in cardiac fibroblasts under high glucose. J Cell Mol Med. 2018;22(3):1816–1825.
71. Yao J, Li Y, Jin Y, Chen Y, Tian L, He W. Synergistic cardioptotection by tilianin and syringin in diabetic cardiomyopathy involves interaction of TLR4/NF-κB/NLRP3 and PGC1a/SIRT3 pathways. Int Immunopharmacol. 2021;96:107728.
72. Baman JR, Passman RS. Atrial Fibrillation. JAMA. 2021;325(21):2218.
73. Hu YF, Chen YJ, Lin YJ, Chen SA. Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol. 2015;12(4):230–243.
74. Ihara K, Sasano T. Role of Inflammation in the Pathogenesis of Atrial Fibrillation. Front Physiol. 2022;13:862164.
75. Qiu H, Liu W, Lan T, et al. Salvianolate reduces atrial fibrillation through suppressing atrial interstitial fibrosis by inhibiting TGF-β1/Smad2/3 and TXNIP/NLRP3 inflammasome signaling pathways in post-MI rats. Phytomedicine. 2018;51:255–265.
76. Yao C, Veleva T, Scott L, et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation. 2018;138(20):2227–2242.
77. Cheng T, Wang XF, Hou YT, Zhang L. Correlation between atrial fibrillation, serum amyloid protein A and other inflammatory cytokines. Mol Med Rep. 2012;6(3):581–584.
78. Powell-Wiley TM, Poirier P, Burke LE, et al. Obesity and Cardiovascular Disease: a Scientific Statement From the American Heart Association. Circulation. 2021;143(21):e984–e1010.
79. Sala L, Pontiroli AE. Role of obesity and hypertension in the incidence of atrial fibrillation, ischaemic heart disease and heart failure in patients with diabetes. Cardiovasc Diabetol. 2021;20(1):162.
80. Scott L, Fender AC, Saljic A, et al. NLRP3 inflammasome is a key driver of obesity-induced atrial arrhythmias. Cardiovasc Res. 2021;117(7):1746–1759.
81. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741.
82. Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and Autophagy: interactions and Molecular Regulatory Mechanisms. Cell Mol Neurobiol. 2015;35(5):615–621.
83. Liu PP, Liu HH, Sun SH, et al. Aspirin alleviates cardiac fibrosis in mice by inhibiting autophagy. Acta Pharmacol Sin. 2017;38(4):488–497.
84. Kovacs SB, Miao EA. Gasdermins: effectors of Pyroptosis. Trends Cell Biol. 2017;27(9):673–684.
85. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6(1):128.
86. Miao EA, Leaf IA, Treuting PM, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11(12):1136–1142.
87. Baker PJ, Boucher D, Bierschenk D, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol. 2015;45(10):2918–2926.
88. Zhang X, Hu C, Yuan YP, et al. Endothelial ERG alleviates cardiac fibrosis via blocking endothelin-1-dependent paracrine mechanism. Cell Biol Toxicol. 2021;37(6):873–890.
89. Jia C, Chen H, Zhang J, et al. Role of pyroptosis in cardiovascular diseases. Int Immunopharmacol. 2019;67:311–318.
90. Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: from Inflammation to Fibrosis. Circ Res. 2016;119(1):91–112.
91. Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016;365(3):563–581.
92. Luo B, Li B, Wang W, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One. 2014;9(8):e104771.
93. Mastrocola R, Penna C, Tullio F, et al. Pharmacological Inhibition of NLRP3 Inflammasome Attenuates Myocardial Ischemia/Reperfusion Injury by Activation of RISK and Mitochondrial Pathways. Oxid Med Cell Longev. 2016;2016:5271251.
94. Gao RF, Li X, Xiang HY, et al. The covalent NLRP3-inflammasome inhibitor Oridonin relieves myocardial infarction induced myocardial fibrosis and cardiac remodeling in mice. Int Immunopharmacol. 2021;90:107133.
95. Toldo S, Mauro AG, Cutter Z, et al. The NLRP3 Inflammasome Inhibitor, OLT1177 (Dapansutrile), Reduces Infarct Size and Preserves Contractile Function After Ischemia Reperfusion Injury in the Mouse. J Cardiovasc Pharmacol. 2019;73(4):215–222.
96. Chen S, Wang Y, Pan Y, et al. Novel Role for Tranilast in Regulating NLRP3 Ubiquitination, Vascular Inflammation, and Atherosclerosis. J Am Heart Assoc. 2020;9(12):e015513.
97. See F, Watanabe M, Kompa AR, et al. Early and delayed tranilast treatment reduces pathological fibrosis following myocardial infarction. Heart Lung Circ. 2013;22(2):122–132.
98. Kelly DJ, Zhang Y, Connelly K, et al. Tranilast attenuates diastolic dysfunction and structural injury in experimental diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2007;293(5):H2860–2869.
99. Huang Y, Jiang H, Chen Y, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med. 2018;10:4.
100. Ohta S. Molecular hydrogen as a preventive and therapeutic medical gas: initiation, development and potential of hydrogen medicine. Pharmacol Ther. 2014;144(1):1–11.
101. Nie C, Ding X. Hydrogen gas inhalation alleviates myocardial ischemia-reperfusion injury by the inhibition of oxidative stress and NLRP3-mediated pyroptosis in rats. Life Sci. 2021;272:119248.
102. Kang LL, Zhang DM, Ma CH, et al. Cinnamaldehyde and allopurinol reduce fructose-induced cardiac inflammation and fibrosis by attenuating CD36-mediated TLR4/6-IRAK4/1 signaling to suppress NLRP3 inflammasome activation. Sci Rep. 2016;6:27460.
103. Pascart T, Richette P. Colchicine in Gout: an Update. Curr Pharm Des. 2018;24(6):684–689.
104. Leung YY, Yao Hui LL, Kraus VB. Colchicine–Update on mechanisms of action and therapeutic uses. Semin Arthritis Rheum. 2015;45(3):341–350.
105. Fujisue K, Sugamura K, Kurokawa H, et al. Colchicine Improves Survival, Left Ventricular Remodeling, and Chronic Cardiac Function After Acute Myocardial Infarction. Circ J. 2017;81(8):1174–1182.
106. Marques-da-Silva C, Chaves MM, Castro NG, Coutinho-Silva R, Guimaraes MZ. Colchicine inhibits cationic dye uptake induced by ATP in P2X2 and P2X7 receptor-expressing cells: implications for its therapeutic action. Br J Pharmacol. 2011;163(5):912–926.
107. Ge C, Cheng Y, Fan Y, He Y. Vincristine attenuates cardiac fibrosis through the inhibition of NLRP3 inflammasome activation. Clin Sci (Lond). 2021;135(11):1409–1426.
108. Yue R, Zheng Z, Luo Y, et al. NLRP3-mediated pyroptosis aggravates pressure overload-induced cardiac hypertrophy, fibrosis, and dysfunction in mice: cardioprotective role of irisin. Cell Death Discov. 2021;7(1):50.
109. Rudolphi K, Gerwin N, Verzijl N, van der Kraan P, van den Berg W. Pralnacasan, an inhibitor of interleukin-1beta converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthritis Cartilage. 2003;11(10):738–746.
110. Wannamaker W, Davies R, Namchuk M, et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J Pharmacol Exp Ther. 2007;321(2):509–516.
111. Ku GFP, Raybuck S, Harding M, Randle J. Selective Interleukin-1β Converting Enzyme (ICE/Caspase-1) Inhibition with Pralnacasan (HMR 3480/VX-740) Reduces Inflammation and Joint Destruction in Murine Type II Collagen-Induced Arthritis (CIA). Arthritis Rheumatism. 2001;44(9):S241.
112. Audia JP, Yang XM, Crockett ES, et al. Caspase-1 inhibition by VX-765 administered at reperfusion in P2Y(12) receptor antagonist-treated rats provides long-term reduction in myocardial infarct size and preservation of ventricular function. Basic Res Cardiol. 2018;113(5):32.
113. Kudelova J, Fleischmannova J, Adamova E, Matalova E. Pharmacological caspase inhibitors: research towards therapeutic perspectives. J Physiol Pharmacol. 2015;66(4):473–482.
114. Hisahara S, Takano R, Shoji S, Okano H, Miura M. Role of caspase-1 subfamily in cytotoxic cytokine-induced oligodendrocyte cell death. J Neural Transm Suppl. 2000;1(58):135–142.
115. Lu B, Xie J, Fu D, et al. Huoxue Qianyang Qutan recipe attenuates cardiac fibrosis by inhibiting the NLRP3 inflammasome signalling pathway in obese hypertensive rats. Pharm Biol. 2021;59(1):1045–1057.
116. Pfeiler S, Winkels H, Kelm M, Gerdes N. IL-1 family cytokines in cardiovascular disease. Cytokine. 2019;122:154215.
117. Dhimolea E. Canakinumab. MAbs. 2010;2(1):3–13.
118. Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377(12):1119–1131.
119. Ramírez J, Cañete JD. Anakinra for the treatment of rheumatoid arthritis: a safety evaluation. Expert Opin Drug Saf. 2018;17(7):727–732.
120. Vastert SJ, Jamilloux Y, Quartier P, et al. Anakinra in children and adults with Still’s disease. Rheumatology. 2019;58(Suppl6):vi9–vi22.
121. Cavalli G, De Luca G, Campochiaro C, et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2020;2(6):e325–e331.
122. Sanchez-Rangel E, Inzucchi SE. Metformin: clinical use in type 2 diabetes. Diabetologia. 2017;60(9):1586–1593.
123. Yang F, Qin Y, Wang Y, et al. Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy. Int J Biol Sci. 2019;15(5):1010–1019.
124. Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med. 2015;373(22):2117–2128.
125. Muscelli E, Astiarraga B, Barsotti E, et al. Metabolic consequences of acute and chronic empagliflozin administration in treatment-naive and metformin pretreated patients with type 2 diabetes. Diabetologia. 2016;59(4):700–708.
126. Byrne NJ, Matsumura N, Maayah ZH, et al. Empagliflozin Blunts Worsening Cardiac Dysfunction Associated With Reduced NLRP3 (Nucleotide-Binding Domain-Like Receptor Protein 3) Inflammasome Activation in Heart Failure. Circ Heart Fail. 2020;13(1):e006277.
127. Li JX, Shi JF, Wu YH, Xu HT, Fu CM, Zhang JM. [Mechanisms and application of triptolide against breast cancer]. Zhongguo Zhong Yao Za Zhi. 2021;46(13):3249–3256. Chinese.
128. Cai J, Yi M, Tan Y, et al. Natural product triptolide induces GSDME-mediated pyroptosis in head and neck cancer through suppressing mitochondrial hexokinase-ΙΙ. J Exp Clin Cancer Res. 2021;40(1):190.
129. Zhou X, Lu B, Fu D, Gui M, Yao L, Li J. Huoxue Qianyang decoction ameliorates cardiac remodeling in obese spontaneously hypertensive rats in association with ATF6-CHOP endoplasmic reticulum stress signaling pathway regulation. Biomed Pharmacother. 2020;121:109518.
130. Semwal RB, Semwal DK, Combrinck S, Viljoen A. Emodin - A natural anthraquinone derivative with diverse pharmacological activities. Phytochemistry. 2021;190:112854.
131. Dai S, Ye B, Chen L, Hong G, Zhao G, Lu Z. Emodin alleviates LPS-induced myocardial injury through inhibition of NLRP3 inflammasome activation. Phytother Res. 2021;35(9):5203–5213.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.