")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 16
The Neuronal and Non-Neuronal Pathways of Sodium-Glucose Cotransporter-2 Inhibitor on Body Weight-Loss and Insulin Resistance
Authors Dong M , Chen H , Wen S , Yuan Y, Yang L, Li Y, Yuan X, Xu D, Zhou L
Received 28 November 2022
Accepted for publication 8 February 2023
Published 14 February 2023 Volume 2023:16 Pages 425—435
DOI https://doi.org/10.2147/DMSO.S399367
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Meiyuan Dong,1,2 Huiling Chen,2 Song Wen,2 Yue Yuan,2 Liling Yang,2 Yanyan Li,2 Xinlu Yuan,2 Dongxiang Xu,2 Ligang Zhou1– 3
1Graduate School of Hebei Medical University, Shijiazhuang, People’s Republic of China; 2Department of Endocrinology, Shanghai Pudong Hospital, Fudan University, Shanghai, People’s Republic of China; 3Shanghai Key Laboratory of Vascular Lesions Regulation and Remodeling, Shanghai Pudong Hospital, Shanghai, People’s Republic of China
Correspondence: Ligang Zhou, Department of Endocrinology, Shanghai Pudong Hospital, Fudan University, Shanghai, 201399, People’s Republic of China, Tel +8613611927616, Email [email protected]
Abstract: With the emergence of sodium-glucose cotransporter 2 inhibitors (SGLT2i), the treatment of type 2 diabetes mellitus (T2DM) has achieved a new milestone, of which the insulin-independent mechanism could produce weight loss, improve insulin resistance (IR) and exert other protective effects. Besides the well-acknowledged biochemical processes, the dysregulated balance between sympathetic and parasympathetic activity may play a significant role in IR and obesity. Weight loss caused by SGLT-2i could be achieved via activating the liver–brain–adipose neural axis in adipocytes. We previously demonstrated that SGLT-2 are widely expressed in central nervous system (CNS) tissues, and SGLT-2i could inhibit central areas associated with autonomic control through unidentified pathways, indicating that the role of the central sympathetic inhibition of SGLT-2i on blood pressure and weight loss. However, the exact pathway of SGLT2i related to these effects and to what extent it depends on the neural system are not fully understood. The evidence of how SGLT-2i interacts with the nervous system is worth exploring. Therefore, in this review, we will illustrate the potential neurological processes by which SGLT2i improves IR in skeletal muscle, liver, adipose tissue, and other insulin-target organs via the CNS and sympathetic nervous system/parasympathetic nervous system (SNS/PNS).
Keywords: sodium-glucose cotransporter 2 inhibitors, central nervous system, autonomic nervous system, insulin resistance, weight loss
Introduction
T2DM is a metabolic condition characterized by chronic hyperglycemia and is considered to be one of the primary causes of mortality and morbidity.1 Obesity is a significant exogenous factor, and recent data suggest that patients with obesity are at high-risk not only for T2DM but also for its comorbidities.2 Both T2DM and obesity are associated with IR, as IR contributes majorly to the progression of T2DM. IR could be traced to higher levels of serum pro-inflammatory cytokines in obese who possess considerable visceral fat accumulation. The higher incidence of ischemic heart disease, arrhythmia, and sudden death reported in obese patients may be attributed to the long-term activation of the SNS by hyperinsulinemia.3 Ectopic fat deposition, including visceral fat, skeletal muscle, liver, pancreas, and other organs, has been shown to contribute to T2DM by inducing liver and peripheral IR and progressive β-cell function decline.4
Sodium-glucose cotransporter-2 inhibitor (SGLT-2i) is a novel family of medications used to treat T2DM by inhibiting SGLT-2 in the renal proximal tubules, which is responsible for 90% of renal glucose reabsorption.5 According to the physical and chemical properties, SGLT-2i is lipid-soluble drugs with low molecular weight, indicating its ability to cross the blood–brain barrier.6 This mechanism depends on the level of blood sugar and has nothing to do with the effect and availability of insulin. The increase in urine glucose caused by SGLT2I is accompanied by osmotic diuresis and a decrease in blood pressure.7 In the cardiovascular outcome trial (CVOTS), SGLT2i has been shown to reduce major adverse cardiovascular events (MACE) and heart failure hospitalization, and is associated with slow progression of kidney disease and reduced incidence of renal end points, and these effects appear to be independent of hypoglycemic effects.8,9 In addition to the anti-diabetic effects, weight loss has also been observed in patients receiving SGLT-2i therapy.10,11 Clinical research has proved the profound weight-loss advantage of SGLT2i compared with other traditional oral antidiabetic drugs (OADs), as both randomized controlled trials (RCT) and real-world studies have shown that individuals treated with SGLT2i could lose 1–3 kg of mean body weight.11–13 However, in both human and animal research, this effect appears to be far less than predicted, and the underlying mechanisms remain unclear. On the other hand, according to previous reports, SGLT2i could ameliorate IR via numerous discrete mechanisms. Among the accumulating data from animal and human studies, increased central sympathetic activity may play a pivotal role in the aetiology and complications of diabetes and obesity (Table 1). In recent years, the autonomic nervous system has emerged as a unique potential regulator of metabolic homeostasis.14,15 However, the precise mechanism that underpins the connection between SGLT2i and the neurological system is yet to be determined.
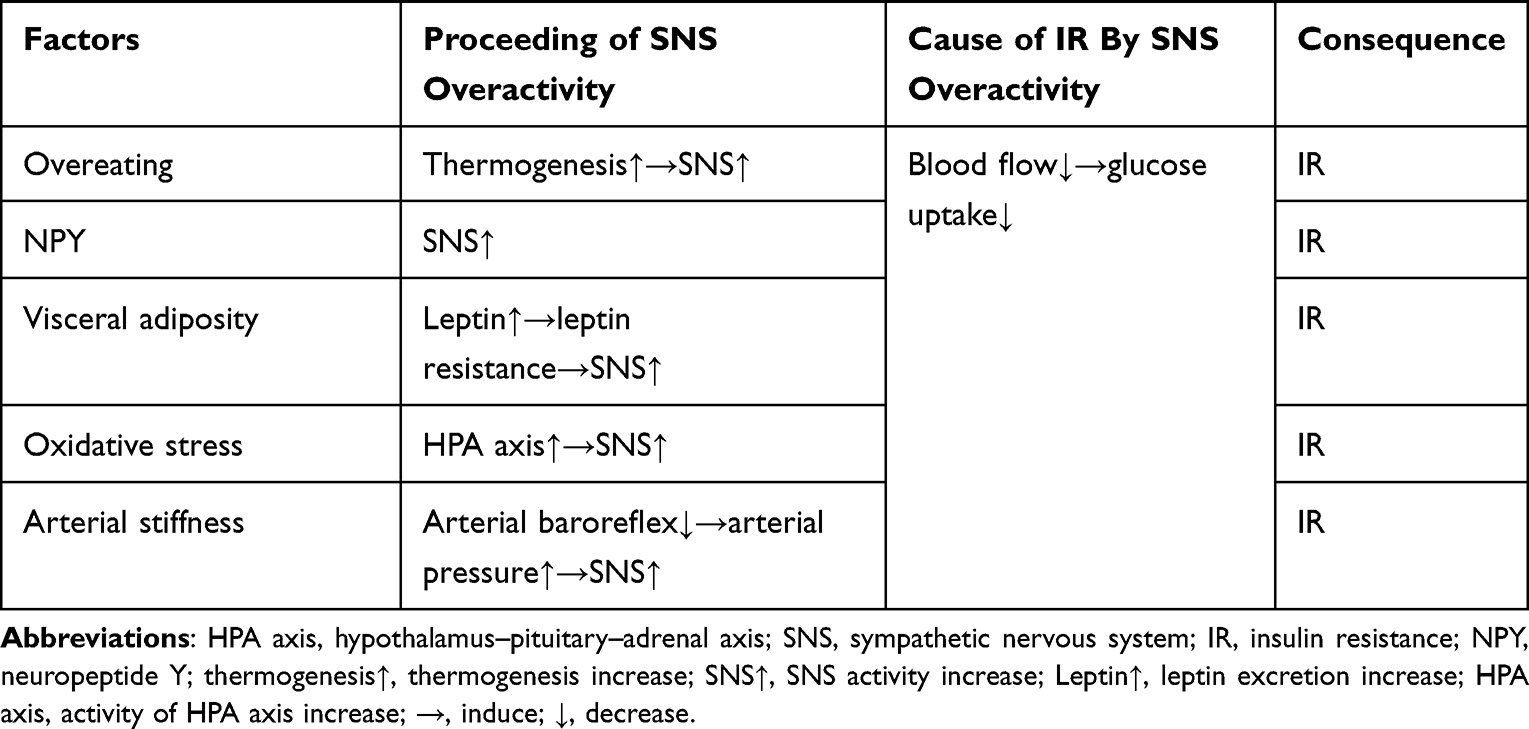 |
Table 1 Factors Induce Sympathetic Nervous System Overactivity and Insulin Resistance |
The purpose of this review is to exhibit an overview of the mechanisms of weight loss and IR improvement by SGLT-2i. This review will focus on the putative role of the nervous system in inducing IR, with particular emphasis on how SGLT2i ameliorates IR via central/peripheral neural pathways such as CNS, SNS, PNS.
The Effect and Mechanism of Body Weight Reduction by SGLT2i
According to a review study, diabetic patients treated with SGLT2i may achieve an average weight loss ranged from −0.591 kg (95% confidence interval [CI] −0.663, −0.519) to −2.1 kg (95% CI −2.3, −2.0).16 In a meta-analysis, data suggested that SGLT2i led a significantly greater decrease in the body weight (WMD, −2.01 kg; 95% CI, −2.18 to −1.83 kg, P<0.001, in random-effects).17 In a retrospective cohort analysis in Canada, 1052 T2DM patients treated with dapagliflozin for monotherapy for 3 to 6 months have lost weight about 2.2± 3.1 kg (P < 0.01).10 Furthermore, in a randomized to double-blind study (N=50, 25 dapagliflozin + exenatide and 25 placebo; aged 18–70 years; body mass index 30–45kg/m2), dapagliflozin, in combination with GLP-1 receptor agonist exenatide, resulted in a mean body weight loss of 4.5 kg and 5.7 kg at 24 weeks (95% confidence interval [CI] −6.09, −2.88) and 52 weeks (95% confidence interval [CI] −8.63, −2.75), respectively (P<0.005).18 According to another prior study conducted in Japanese patients with type 2 diabetes by a 24-week, ipragliflozin significantly decreased visceral and hepatic fat, along with body fat mass.19 In patients treated with SGLT2i displayed a tendency of weight loss in dose-dependent manner.20 Initial explanation for this effect with SGLT2i only relied on glycosuria, which results in energy deficit and water excretion via osmotic diuresis.21 Intriguingly, according to this mechanism, weight loss is predicted to reach 10 kg per year, which exceeds what has been reported in clinical research.22,23 Although fluid loss may have a little impact on weight loss, an observational research on body adipose composition indicated that fat mass reduction would be primarily responsible for weight loss, as significant reduction was observed in visceral adipose tissue after 16 weeks’ treatment with ipragliflozin (110±33 to 101±36 cm2, p = 0.005).24 In addition, glycosuria caused by SGLT2i might result in a decrease in serum insulin levels, accompanied by an elevation in glucagon concentration and in the ratio of glucagon to insulin, which accelerates lipolysis and lipid oxidation.25 In addition, the activity of brown adipose tissue (BAT) has anti-obesity and anti-diabetic properties, and SGLT2i could increase the browning of white adipose tissue by acting on the polarized adipose tissue macrophage M2.26 Additionally, mitochondrial function is linked to fat catabolism. Following treatment with SGLT2i, the activity of mammalian target of rapamycin complex 1 (mTORC1) is suppressed with induced autophagy and lysosomal degradation, resulting in a transformation of mitochondrial morphology and an improvement in its function.27 Multiple studies have demonstrated that weight loss may be associated with decreased SNS and increased PNS response in addition to the established non-neural mechanism.28,29
An Overview of Non-Neural Mechanisms SGLT2i Improving IR in Obesity and T2DM
Retrospective Review of Proposed Peripheral Mechanisms of IR
Mechanisms for generation of IR are complicated, which have not been elucidated thoroughly to date. Compiled evidence suggests that mitochondrial dysfunction driven by T2DM or obesity may accelerate IR progression through increased production of reactive oxygen species (ROS) and decreased synthesis rate of ATP.30 Glucotoxicity produced by chronic hyperglycemia inhibits the function of islet cells and causes IR in liver, muscle, and fat tissue.31 The decreased activation of the IRS-1/PI3K/Akt signaling pathway targeted by insulin may contribute to the development of IR in skeletal muscle.32 Non-esterified fatty acids (NEFAs) generated by adipose tissue are one critical resource of IR, and increased NEFAs release has been reported in T2DM and obesity highly associated with IR.33 NEFAs play a major role in reducing insulin clearance, and reduced insulin clearance in the liver can result in chronic hyperinsulinemia, which in turn causes downregulation of insulin receptor and IR.34 In patients with obesity and T2DM, it is well recognized that fatty acids (FAs) play a crucial role in impairment of insulin sensitivity.35 FAs inhibit the genetic expression of insulin receptor, through inducing phosphorylation of protein kinase Cε (PKCε), giving rise to an attenuation of insulin sensitivity.36 Diacylglycerol (DAG) and ceramides were positively correlated with the severity of IR, as DAG and ceramides can affect the insulin-mediated signal pathway of liver and muscle glucose metabolism. Ceramide mediates IR by inhibiting the phosphorylation and activation of protein kinase B (PKB), while DAG accumulation stimulates the activation of PKCθ, the phosphorylation of insulin receptor substrate-1 (IRS-1), ultimately generating IR37,38 In addition, latent inflammation is another risk factor for IR in adipose tissue, where inflammation-induced oxidative stress severely affect insulin signal transmission.39,40 Macrophages are the main types of immune cells that cause islet inflammation in obesity and T2DM. In normal adipose tissue, macrophages account for only 10% of the total local cells, while the proportion can be as high as 50% in obese people. The subtype that is increased is pro-inflammatory M1 macrophages, which have been prove to be the main source of inflammatory factors in adipose tissue. The study shows that deletion of M1 macrophages in mice can significantly improve IR.41
Improvement of IR Associated with SGLT2i Treatment
IR improvement is clinically significant following SGLT2i treatment. Notably, it is acknowledged that SGLT2i can increase whole-body insulin sensitivity, regardless this effect in individuals with T2DM is still debatable.42 Muscle glycogen synthesis is the main route for glucose disposal in normal and patients with T2DM and obesity, and muscle glycogen synthesis deficiency is predominantly in the occurrence of IR. However, SGLT2i could not directly affect the glucose metabolism in skeletal muscle since SGLT2 is merely expressed on skeletal muscle.43,44 One plausible explanation suggests that SGLT2i may minimize glucotoxicity by eliminating excessive glucose from tissue fluid and circulation.45 In T2DM patients treated with dapagliflozin, fasting plasma glucose decreased significantly, and dapagliflozin-induced glucosuria enhanced muscle insulin sensitivity.46 In a hyperinsulinemic-normoglycemic clamp study, tofogliflozin significantly improved insulin sensitivity and peripheral glucose uptake in patients with T2DM, and these improvements were transparently associated with reduced body fat mass.47 In addition, emerging evidence suggests that SGLT2i could improve redox state and oxidative stress, thereby reducing oxidative damages to ameliorate IR, by regulating the activity of the renin–angiotensin system (RAAS), down-regulating the pro-oxidant enzymes and enhancing mitochondrial function.48,49 Increased liver IR is related to decreased AMP kinase (AMPK) phosphorylation and the production of TNF and IL6 in hepatic tissue.50 Evidence suggested that empagliflozin could up-regulate AMPK activation to lower lipid deposition and the levels of FAs, serum TGs, and cholesterol.51 Tofogliflozin boosted glucose uptake in skeletal muscle and lipolysis in adipose tissue, leading to weight loss and a reduction in IR in male mice treated with tofogliflozin.52 Similarly, O’Brien et al conducted a study demonstrating that the insulin sensitivity of skeletal muscle improved due to the decreased intracellular lipid content and increased lipid oxidation, as well as the enhancement of skeletal muscle. In another previous study, Zucker diabetic fatty rats treated with SGLT2i were able to restore normal levels of whole-body IR with better glucose utilization in the liver and enhanced insulin sensitivity in the muscle.53 There are also hypotheses that SGLT2i can improve insulin sensitivity by reducing inflammation. For instance, empagliflozin can lower the proportions of T cells and M1 macrophages and raise that of M2 macrophages in obese patients, hence decreasing inflammation and IR.54 Most importantly, the proposed mechanism indicated that the infiltration of Th1 and CD8+ T cells precedes the recruitment of M1-polarized macrophages, and the interaction between T cells and macrophages constitutes a maladaptive feedforward loop, which lead to adipose inflammation and IR55 (Figure 1).
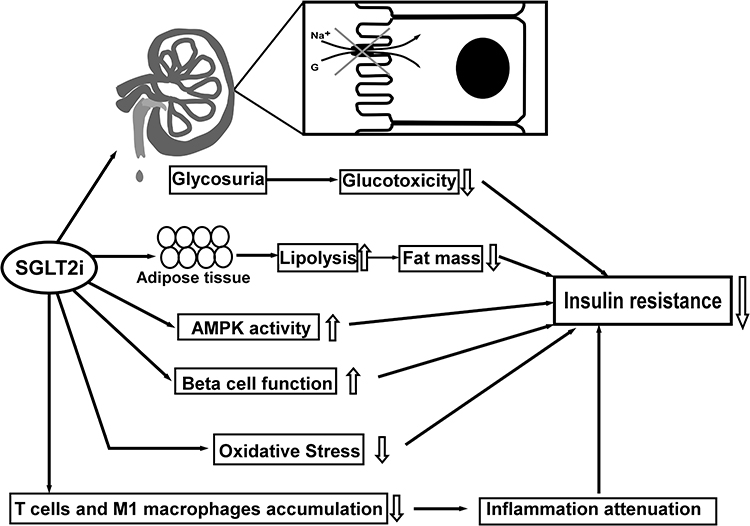 |
Figure 1 The Summaries on referred non-neural pathways regarding the mechanism of SGLT2i in improving IR: 1. Glucotoxicity alleviation via increasing glycosuria, 2. Lipolysis increase and lipid content reduction, 3. Up-regulation of AMPK activity, 4. β-cell function improvement, 5. Oxidative stress mitigation, 6. Inflammation attenuation by reducing T cells and M1 macrophages accumulation. Up arrows: increase; down arrows: decrease. |
Neural Pathways on Improvement of IR in Obesity and T2DM
It was proposed previously that overeating increases sympathetic activity in human, which can be measured with an increase in systemic norepinephrine spillover rate.56 Hyperinsulinemia caused by obesity produces long-term stimulation of the sympathetic nervous system and increased basal sympathetic activity has been reported and correlated with the degree of IR.57 Stina Lindmark et al demonstrated that the association between visceral adiposity and IR may be mediated in part by altered responsiveness of the sympathetic or parasympathetic activity.58 The link between the balance of sympathetic/parasympathetic activity and visceral abdominal fat suggest that large amounts of visceral fat may activate the sympathetic or suppress the PNS, which indicated the dysregulation of sympathetic/parasympathetic balance, and the outcome of IR.59 In addition, it is demonstrated that SGLT2i dapagliflozin may act on PVN, NTS, PAG and other nuclei, reducing the activity of SNS in organs such as kidney and heart to decrease the hypertension.60,61 SGLT-2 is expressed in the CNS of rats, including the brain and the blood–brain barrier, according to a previous anatomic localization study.62 In a recent work, we proposed that SGLT2 is highly expressed in the brain, primarily on the microvessels of the blood–brain barrier (BBB), as well as in the amygdala, hypothalamus, periaqueductal gray (PAG) and dorsomedial medulla-nucleus of solitary tract (NTS).63 In this research, after intra-gavage administration of SGLT-2i, c-Fos expression was widespread throughout the autonomic nerve region, extending from the telencephalon to the caudal brainstem. Furthermore, SGLT-2i may act on the rostral ventrolateral medulla (RVLM) and affect the sympathetic outflow of sympathetic preganglionic neurons to the intermediolateral nucleus of spinal cord (IML), thereby promoting parasympathetic activity.
SGLT2i May Decrease Liver IR Through the Parasympathetic Nervous System (PNS) and Sympathetic Nervous System (SNS)
Individuals with IR, especially those associated with central obesity, displayed a slow sympathetic response to physiological hyperinsulinemia, glucose consumption and changes in energy status. There is a great deal of evidence that the SNS is abnormally active in individuals with centripetal obesity and IR.64,65 Recently, the pathways such as the liver–brain–adipose axis, brain-liver circuit, gut-liver-kidney axis, and autonomic nervous system, have been proclaimed risingly in popularity. Numerous studies have demonstrated that circulating nutrients and peptides can affect food intake and alter hepatic glucose production via the vagus nerve efferent pathway in the dorsal motor nucleus of the vagus (DMV), which is the principal neural output of the parasympathetic nervous system (PNS).66 In addition, it has been suggested that attenuated parasympathetic activity may promote IR.67 The vagus nerve is thought to be a vital CNS communication pathway for regulating liver metabolism.68 Furthermore, insulin and leptin signals in the hypothalamic arcuate nucleus (ARC) can alter hepatic insulin sensitivity via the information transducted by vagus nerve.69 The glucose production and systemic insulin sensitivity are regulated via a circuit between the brain and liver.70 Liver glycogen depletion signals directly promote lipolysis in white adipose tissue by activating liver–brain–adipose neurocircuitry that is independent of blood glucose concentrations and insulin/glucagon levels.71
The ARC in hypothalamus is identified as one of the key sites where insulin increases sympathetic activity and sympathetic baroreflex.72 It is postulated that central hyperinsulinemia induced by overfeeding leads to a contradictory increase in the expression of neuropeptide Y (NPY) in the ARC, and then the activation of NPY neurons increases liver IR and endogenous glucose production by increasing sympathetic outflow to the liver.73 The action of NPY or agouti-related peptide (NPY/AgRP) neurons in the ARC mediates hepatic glucose production, inducing the IR of liver. For instance, intracerebroventricular injection of NPY in rats can acutely hinder the ability of insulin to inhibit glucose production by activating the sympathetic nerves which innervate the liver.74 Furthermore, resistin is a secreted protein produced by adipocytes, and studies show that central injection of resistin is related to the activation of neurons in ARC, paraventricular nucleus (PVN) and dorsomedial nucleus, increasing expression of NPY in hypothalamus, which impairs insulin sensitivity in liver.75 These results are in accord with previous reports showing that the ARC plays an important role in the control of the SNS. Intraventricular administration of SGLT2i can increase the expression of c-Fos in the PVN, ARC, and lateral hypothalamus nucleus (LH), elucidating that the excessive phagocytosis associated with SGLT2i increases food intake, at least in part via CNS.76 The ARC, PVN, and dorsomedial hypothalamic nucleus (DMH) all had their roles in glucose regulation. From the PVN and LHA, efferent projections synapse in the locus coeruleus (LC), which controls the SNS SGLT2i are supposed to activate the PVN of the hypothalamus, activating the parvocellular portion that could project to the brainstem to release factors, and regulate the autonomic neural efferent.77 Furthermore, it has been proposed that SGLT2i could down-regulate sympathetic activity as evidenced by decreases in markers of the SNS such as norepinephrine (NE) and NPY,78 which may be a pathway for SGLT2i to improve hepatic IR. In addition, activation of central melanocortin pathways enhances insulin sensitivity, and overexpression of pro-opiomelanocortin (POMC) attenuates IR.79 However, even though the mRNA levels of POMC and AgRP under treatment with an SGLT2i were observed significantly decreased and increased respectively, which is in contradict with the theory mentioned above, the glucose metabolism was observed to be improved, one possible explanation will be the compensatory response associated with glucose and energy deplete in SGLT-2i treatment.80 (Figure 2)
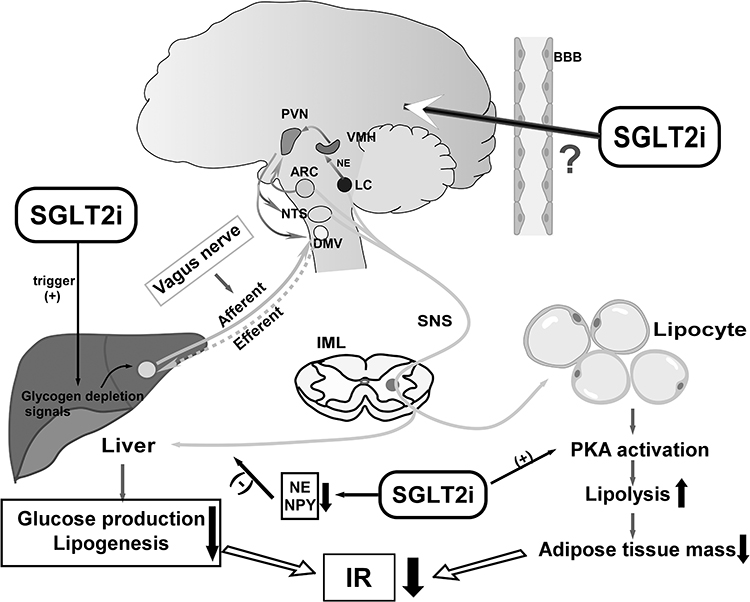 |
Figure 2 Scheme of the neural and metabolic mechanisms by which SGLT2i improves hepatic and adipose IR. SGLT2i triggers glycogen depletion signals in the liver, and liver may convey information to the CNS via the afferent vagus, activating efferent sympathetic nerves to adipose tissues, which promotes lipolysis leading to fat mass reduction. Meanwhile, activation of neurons in hypothalamus attenuates the glucose production and lipogenesis via efferent vagus to liver. SGLT2i could downregulate sympathetic activity innervating the liver by inhibiting expression of norepinephrine (NE) and NPY. SGLT2i activates the brain–adipose axis and induces fat mass loss. Both reduction of adipose tissue mass and hepatic glucose production could contribute to IR amelioration. Up arrows: increase; down arrows: decrease; (+): promote; (-): inhibit. |
SGLT2i May Improve the Adipose IR Through the Sympathetic Nervous System (SNS)
It is worth noting that adipose tissue is innervated only by sympathetic nerve, which makes it an important regulator of fat mobilization.81 Norepinephrine binds to β3 adrenergic receptors, stimulates hormone-sensitive lipase and promotes the decomposition of stored triglycerides into free fatty acids. Central sympathetic outflow directly stimulates lipolysis of adipocytes by binding to β-adrenergic receptors in white adipose tissue and activating cAMP-dependent pathways to translocate inactivated lipases, while activating α-adrenergic receptors will inhibit lipolysis.82 Dapagliflozin significantly inhibited the turnover of norepinephrine (NE) in brown adipose tissue and the expression of c-Fos in the raphe pallidus lateral nucleus (RRPA) of the thermogenic sympathetic premotor neurons, to regulate an interorgan neural network composed of common hepatic vagal branches and sympathetic nerves.83 These nuclei have been proposed by previous study in light of metabolic control and association with autonomic control. Canagliflozin increased sympathetic innervation and NE secretion in adipose tissue, via the cAMP-PKA signal pathway, consequently improving IR in mice fed a high-fat diet.84 SGLT2i induces the beiging of white adipose tissue by promoting sympathetic excitation in it, as well verified the existence of brain–adipose axis.85 In diet-induced obese mice, tofogliflozin activate liver–brain–adipose neurocircuitry by depleting hepatic glycogen, which leads to the activation of PKA (protein kinase A, thought to be an effector of the liver–brain–adipose axis that activates triglyceride lipase) in adipocytes and triggers fat decomposition in adipose tissue, leading to fat mass reduction and IR improvement.86 It is believed that a reduction in fat mass contributes to an improvement of IR.52,53 Therefore, it is plausible that SGLT2i activates the brain–adipose axis and induces fat mass loss, thereby ameliorating IR. (Figure 2)
SGLT2i Improve IR in Skeletal Muscle by Inhibiting SNS Directly and Indirectly
Skeletal muscle microvascular perfusion, hemodynamics and insulin permeability are the critical determinants of insulin action in skeletal muscle. The increase of sympathetic outflow to skeletal muscle plays an important role in glucose metabolism, mainly through the decrease in skeletal muscle blood flow.87 Chadderdon et al pointed out that in the early stage of obesity induced by high-fat diet in rhesus macaques, increasing basal and glucose-mediated capillary blood volume through endothelium-derived vasodilators may be a compensatory mechanism of IR.88 The enhanced activation of renin–angiotensin–aldosterone system (RAAS) in obesity/T2DM further leads to vascular IR and endothelial dysfunction.89,90 Aldosterone promotes IR by increasing the expression of insulin-like growth factor-1 (IGF-1) receptor and hybridization with IRS-1, and mediating the phosphorylation of ERK1/2 in vascular smooth muscle cells stimulated by angiotensin II (Ang II).91 The expression of angiotensin type 1 (AT1R) in the kidney of Otsuka Long-Evans Tokushima Fatty (OLETF) rats was increased, while the expression of AT1R was down-regulated after treatment with dapagliflozin, thus inhibiting the activation of RAAS.92 Leptin-induced sympathetic outflow from skeletal muscle vasculature mediates skeletal muscle vasoconstriction, reducing glucose transport and uptake in muscle, while the impairment of glucose uptake by skeletal muscle is a hallmark of IR syndrome.93,94 Considering a previous study in which treatment with SGLT2i led to attenuation of circulating leptin secretion and actions, IR in skeletal muscle could be improved with SGLT2i combination effects of suppressing SNS and decreasing leptin levels95 (Figure 3).
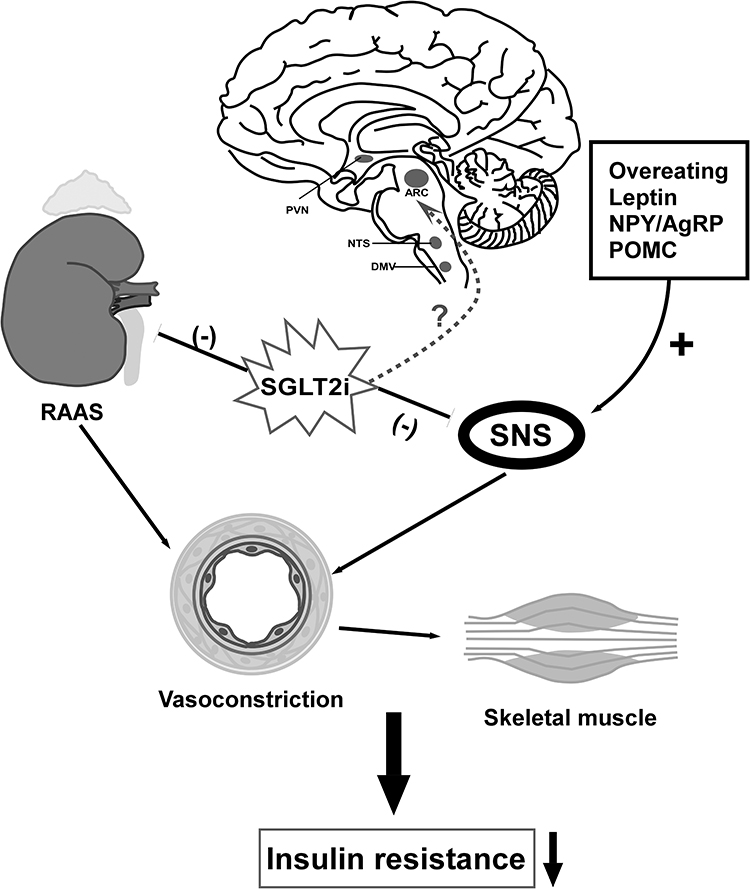 |
Figure 3 Regulation of neural pathway to attenuate muscular IR. Overeating and secretion of leptin increase sympathetic activity that induce IR of liver and skeletal muscle, so does overexpression of NPY. SGLT2i downregulates SNS by decreasing leptin, norepinephrine (NE) and NYP. SGLT-2i affect the sympathetic outflow of sympathetic preganglionic neurons to the intermediolateral nucleus of spinal cord (IML), thereby promoting parasympathetic activity of liver to improve hepatic glucose regulation and insulin sensitivity. Also, SGLT2i improves IR via suppression of renal RAAS component expression such as AT1R. Whether or to what extent SGLT2i act on neurons in the brain directly require further exploration. Down arrow: decrease; (-): inhibit; (+): promote. |
Conclusion
In this review, we discuss the existing evidence for the mechanisms of SGLT2i improving insulin resistance and reducing body weight. There are many factors that cause IR, such as superabundant visceral fat, oxidative stress, accumulation of inflammatory factors, excessive activation of sympathetic nervous system and so on. The neural, hormonal and nutritional mechanisms involved furtherly complicate this situation. As reported in this review, there are two avenues of communication between the brain and other tissues: humoral factors and neuronal pathways, via which SGLT2i could improve the IR of peripheral tissues. Even though a large number of studies have shown that SGLT2i can improve insulin resistance and weight loss, the specific mechanism is not clear, especially the neuroendocrine mechanism. SGLT2i improves IR and induces weight loss via glucotoxicity alleviation, inflammation attenuation, β-cell function improvement, lipid content reduction and oxidative stress mitigation. Of note, the activities of nervous system, including CNS, SNS, and PNS, play an important role in IR. Interestingly, SGLT2i activates efferent sympathetic nerves to adipose tissues and central efferent vagus nerve to liver via liver–brain–adipose axis, resulting in upregulation of lipolysis and reduction of hepatic glucose production, which attenuates IR. On top of that, through inhibiting activation of RAAS and excessive SNS, SGLT2i may improve muscle IR. Furthermore, it has been proposed that SGLT2i could downregulate sympathetic activity as evidenced by decreases in markers of the SNS such as norepinephrine (NE) and NPY. Overall, of potential major interest, a better understanding of the mechanisms linking SGLT2i and nervous system deserves further investigation.
Funding
This work was supported by the Project of Key Medical Discipline of Pudong Hospital of Fudan University (Zdxk2020-11), Project of Key Medical Specialty and Treatment Center of Pudong Hospital of Fudan University (Zdzk2020-24), Integrative Medicine special fund of Shanghai Municipal Health Planning Committee (ZHYY- ZXYJHZX-2-201712), Special Department Fund of the Pudong New Area Health Planning Commission (PWZzk2017-03), Outstanding Leaders Training Program of Pudong Health Bureau of Shanghai (PWR12014-06), Pudong New Area Clinical Plateau Discipline Project (PWYgy-2021-03), the Natural Science Foundation of China (21675034), National Natural Science Foundation of China (81370932), Shanghai Natural Science Foundation (19ZR1447500), Fudan Zhangjiang Clinical Medicine Innovation Fund Project (KP0202118), and Education Funding in Wenzhou Medical University (JG2021197).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14:88–98. doi:10.1038/nrendo.2017.151
2. Bluher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol. 2019;15:288–298. doi:10.1038/s41574-019-0176-8
3. Poon AK, Whitsel EA, Heiss G, et al. Insulin resistance and reduced cardiac autonomic function in older adults: the Atherosclerosis Risk in Communities study. BMC Cardiovasc Disord. 2020;20. doi:10.1186/s12872-020-01496-z
4. Skyler JS, Bakris GL, Bonifacio E, et al. Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes. 2017;66:241–255. doi:10.2337/db16-0806
5. Gallo LA, Wright EM, Vallon V. Probing SGLT2 as a therapeutic target for diabetes: basic physiology and consequences. Diab Vasc Dis Res. 2015;12:78–89. doi:10.1177/1479164114561992
6. Dong M, Wen S, Zhou L. The relationship between the blood-brain-barrier and the central effects of glucagon-like peptide-1 receptor agonists and sodium-glucose cotransporter-2 inhibitors. Diabetes Metabol Syndr Obes. 2022;15:2583–2597. doi:10.2147/DMSO.S375559
7. Ferrannini E, Solini A. SGLT2 inhibition in diabetes mellitus: rationale and clinical prospects. Nat Rev Endocrinol. 2012;8:495–502. doi:10.1038/nrendo.2011.243
8. Khat DZ, Husain M. Molecular mechanisms underlying the cardiovascular benefits of SGLT2i and GLP-1RA. Curr Diab Rep. 2018;18:45. doi:10.1007/s11892-018-1011-7
9. Zelniker TA, Wiviott SD, Raz I, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. 2019;393:31–39. doi:10.1016/S0140-6736(18)32590-X
10. Brown RE, Gupta N, Aronson R. Effect of dapagliflozin on glycemic control, weight, and blood pressure in patients with type 2 diabetes attending a specialist endocrinology practice in Canada: a retrospective cohort analysis. Diabetes Technol Ther. 2017;19:685–691. doi:10.1089/dia.2017.0134
11. Rosenstock J, Frias J, Pall D, et al. Effect of ertugliflozin on glucose control, body weight, blood pressure and bone density in type 2 diabetes mellitus inadequately controlled on metformin monotherapy (VERTIS MET). Diabetes Obes Metab. 2018;20:520–529. doi:10.1111/dom.13103
12. Bays HE, Weinstein R, Law G, Canovatchel W. Canagliflozin: effects in overweight and obese subjects without diabetes mellitus. Obesity. 2014;22:1042–1049. doi:10.1002/oby.20663
13. Wilding JP, Woo V, Soler NG, et al. Long-term efficacy of dapagliflozin in patients with type 2 diabetes mellitus receiving high doses of insulin: a randomized trial. Ann Intern Med. 2012;156:405–415. doi:10.7326/0003-4819-156-6-201203200-00003
14. Imbernon M, Beiroa D, Vazquez MJ, et al. Central melanin-concentrating hormone influences liver and adipose metabolism via specific hypothalamic nuclei and efferent autonomic/JNK1 pathways. Gastroenterology. 2013;144:636–649 e636. doi:10.1053/j.gastro.2012.10.051
15. Yahagi N. Hepatic control of energy metabolism via the autonomic nervous system. J Atheroscler Thromb. 2017;24:14–18. doi:10.5551/jat.RV16002
16. Lee PC, Ganguly S, Goh SY. Weight loss associated with sodium-glucose cotransporter-2 inhibition: a review of evidence and underlying mechanisms. Obes Rev. 2018;19:1630–1641. doi:10.1111/obr.12755
17. Cai X, Ji L, Chen Y, et al. Comparisons of weight changes between sodium-glucose cotransporter 2 inhibitors treatment and glucagon-like peptide-1 analogs treatment in type 2 diabetes patients: a meta-analysis. J Diabetes Investig. 2017;8:510–517. doi:10.1111/jdi.12625
18. Lundkvist P, Pereira MJ, Katsogiannos P, et al. Dapagliflozin once daily plus exenatide once weekly in obese adults without diabetes: s ustained reductions in body weight, glycaemia and blood pressure over 1 year. Diabetes Obes Metab. 2017;19:1276–1288. doi:10.1111/dom.12954
19. Ohta A, Kato H, Ishii S, et al. Ipragliflozin, a sodium glucose co-transporter 2 inhibitor, reduces intrahepatic lipid content and abdominal visceral fat volume in patients with type 2 diabetes. Expert Opin Pharmacother. 2017;18:1433–1438. doi:10.1080/14656566.2017.1363888
20. Cai X, Yang W, Gao X, et al. The association between the dosage of SGLT2 inhibitor and weight reduction in type 2 diabetes patients: a meta-analysis. Obesity. 2018;26:70–80. doi:10.1002/oby.22066
21. Komoroski B, Vachharajani N, Feng Y, et al. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin Pharmacol Ther. 2009;85(5):513–519. doi:10.1038/clpt.2008.250
22. Leiter LA, Yoon K-H, Arias P, et al. Canagliflozin provides durable glycemic improvements and body weight reduction over 104 weeks versus glimepiride in patients with type 2 diabetes on metformin: a randomized, double-blind, Phase 3 study. Diabetes Care. 2015;38:355–364. doi:10.2337/dc13-2762
23. Bolinder J, Ljunggren Ö, Johansson L, et al. Dapagliflozin maintains glycaemic control while reducing weight and body fat mass over 2 years in patients with type 2 diabetes mellitus inadequately controlled on metformin. Diabetes Obes Metab. 2014;16:159–169. doi:10.1111/dom.12189
24. Yamamoto C, Miyoshi H, Ono K, et al. Ipragliflozin effectively reduced visceral fat in Japanese patients with type 2 diabetes under adequate diet therapy. Endocr J. 2016;63:589–596. doi:10.1507/endocrj.EJ15-0749
25. Ferrannini E, Baldi S, Frascerra S, et al. Shift to fatty substrate utilization in response to sodium–glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes. 2016;65:1190–1195. doi:10.2337/db15-1356
26. Xu L, Ota T. Emerging roles of SGLT2 inhibitors in obesity and insulin resistance: focus on fat browning and macrophage polarization. Adipocyte. 2018;7:121–128. doi:10.1080/21623945.2017.1413516
27. Ost A, Svensson K, Ruishalme I, et al. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med. 2010;16:235–246. doi:10.2119/molmed.2010.00023
28. Lambert EA, Rice T, Eikelis N, et al. Sympathetic activity and markers of cardiovascular risk in nondiabetic severely obese patients: the effect of the initial 10% weight loss. Am J Hypertens. 2014;27:1308–1315. doi:10.1093/ajh/hpu050
29. Costa J, Moreira A, Moreira P, Delgado L, Silva D. Effects of weight changes in the autonomic nervous system: a systematic review and meta-analysis. Clin Nutr. 2019;38:110–126. doi:10.1016/j.clnu.2018.01.006
30. Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. 2011;8:92–103. doi:10.1038/nrendo.2011.138
31. Buren J, Lindmark S, Renstrom F, Eriksson JW. In vitro reversal of hyperglycemia normalizes insulin action in fat cells from type 2 diabetes patients: is cellular insulin resistance caused by glucotoxicity in vivo? Metabolism. 2003;52:239–245. doi:10.1053/meta.2003.50041
32. Zhang Y, Hai J, Cao M, et al. Silibinin ameliorates steatosis and insulin resistance during non-alcoholic fatty liver disease development partly through targeting IRS-1/PI3K/Akt pathway. Int Immunopharmacol. 2013;17:714–720. doi:10.1016/j.intimp.2013.08.019
33. Palomer X, Pizarro-Delgado J, Barroso E, Vazquez-Carrera M. Palmitic and oleic acid: the yin and yang of fatty acids in type 2 diabetes mellitus. Trends Endocrinol Metab. 2018;29:178–190. doi:10.1016/j.tem.2017.11.009
34. Ader M, Stefanovski D, Kim SP, et al. Hepatic insulin clearance is the primary determinant of insulin sensitivity in the normal dog. Obesity. 2014;22:1238–1245. doi:10.1002/oby.20625
35. Bakker LE, van Schinkel LD, Guigas B, et al. A 5-day high-fat, high-calorie diet impairs insulin sensitivity in healthy, young South Asian men but not in Caucasian men. Diabetes. 2014;63:248–258. doi:10.2337/db13-0696
36. Bhattacharya S, Dey D, Roy SS. Molecular mechanism of insulin resistance. J Biosci. 2007;32:405–413. doi:10.1007/s12038-007-0038-8
37. Coen PM, Goodpaster BH. Role of intramyocellular lipids in human health. Trends Endocrinol Metab. 2012;23:391–398. doi:10.1016/j.tem.2012.05.009
38. Lair B, Laurens C, Van Den Bosch B, Moro C. Novel insights and mechanisms of lipotoxicity-driven insulin resistance. Int J Mol Sci. 2020;22(1):21. doi:10.3390/ijms22010021
39. Vorotnikov AV, Stafeev IS, Menshikov MY, Shestakova MV, Parfyonova YV. Latent inflammation and defect in adipocyte renewal as a mechanism of obesity-associated insulin resistance. Biochemistry. 2019;84:1329–1345. doi:10.1134/S0006297919110099
40. Tangvarasittichai S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J Diabetes. 2015;6:456–480. doi:10.4239/wjd.v6.i3.456
41. Harford KA, Reynolds CM, McGillicuddy FC, Roche HM. Fats, inflammation and insulin resistance: insights to the role of macrophage and T-cell accumulation in adipose tissue. Proc Nutr Soc. 2011;70:408–417. doi:10.1017/S0029665111000565
42. So A, Sakaguchi K, Okada Y, et al. Relation between HOMA-IR and insulin sensitivity index determined by hyperinsulinemic-euglycemic clamp analysis during treatment with a sodium-glucose cotransporter 2 inhibitor. Endocr J. 2020;67:501–507. doi:10.1507/endocrj.EJ19-0445
43. Abdul-Ghani MA, Norton L, Defronzo RA. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev. 2011;32:515–531. doi:10.1210/er.2010-0029
44. Chen J, Williams S, Ho S, et al. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. 2010;1:57–92. doi:10.1007/s13300-010-0006-4
45. Joannides CN, Mangiafico SP, Waters MF, Lamont BJ, Andrikopoulos S. Dapagliflozin improves insulin resistance and glucose intolerance in a novel transgenic rat model of chronic glucose overproduction and glucose toxicity. Diabetes Obes Metab. 2017;19:1135–1146. doi:10.1111/dom.12923
46. Merovci A, Solis-Herrera C, Daniele G, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124:509–514. doi:10.1172/JCI70704
47. Matsuba R, Matsuba I, Shimokawa M, Nagai Y, Tanaka Y. Tofogliflozin decreases body fat mass and improves peripheral insulin resistance. Diabetes Obes Metab. 2018;20:1311–1315. doi:10.1111/dom.13211
48. Yaribeygi H, Atkin SL, Butler AE, Sahebkar A. Sodium-glucose cotransporter inhibitors and oxidative stress: an update. J Cell Physiol. 2019;234:3231–3237. doi:10.1002/jcp.26760
49. Osorio H, Coronel I, Arellano A, et al. Sodium-glucose cotransporter inhibition prevents oxidative stress in the kidney of diabetic rats. Oxid Med Cell Longev. 2012;(2012):542042. doi:10.1155/2012/542042
50. Saha AK, Xu XJ, Balon TW, et al. Insulin resistance due to nutrient excess: is it a consequence of AMPK downregulation? Cell Cycle. 2011;10(20):3447–3451. doi:10.4161/cc.10.20.17886
51. Zhang Z, Ni L, Zhang L, et al. Empagliflozin regulates the AdipoR1/p-AMPK/p-ACC pathway to alleviate lipid deposition in diabetic nephropathy. Diabetes Metab Syndr Obes. 2021;14:227–240. doi:10.2147/DMSO.S289712
52. Obata A, Kubota N, Kubota T, et al. Tofogliflozin improves insulin resistance in skeletal muscle and accelerates lipolysis in adipose tissue in male mice. Endocrinology. 2016;157:1029–1042. doi:10.1210/en.2015-1588
53. O’Brien TP, Jenkins EC, Estes SK, et al. Correcting postprandial hyperglycemia in Zucker diabetic fatty rats with an SGLT2 inhibitor restores glucose effectiveness in the liver and reduces insulin resistance in skeletal muscle. Diabetes. 2017;66:1172–1184. doi:10.2337/db16-1410
54. Xu L, Nagata N, Nagashimada M, et al. SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine. 2017;20:137–149. doi:10.1016/j.ebiom.2017.05.028
55. Sell H, Habich C, Eckel J. Adaptive immunity in obesity and insulin resistance. Nat Rev Endocrinol. 2012;8:709–716. doi:10.1038/nrendo.2012.114
56. O’Dea K, Esler M, Leonard P, Stockigt J, Nestel P. Noradrenaline turnover during under-and over-eating in normal weight subjects. Metabolism. 1982;31:896–899. doi:10.1016/0026-0495(82)90178-0
57. Feldstein C, Julius S. The complex interaction between overweight, hypertension, and sympathetic overactivity. J Am Soc Hypertens. 2009;3:353–365. doi:10.1016/j.jash.2009.10.001
58. Heisler LK, Jobst EE, Sutton GM, et al. Serotonin reciprocally regulates melanocortin neurons to modulate food intake. Neuron. 2006;51:239–249. doi:10.1016/j.neuron.2006.06.004
59. Lindmark S, Lönn L, Wiklund U, et al. Dysregulation of the autonomic nervous system can be a link between visceral adiposity and insulin resistance. Obes Res. 2005;13:717–728. doi:10.1038/oby.2005.81
60. Matthews VB, Elliot RH, Rudnicka C, et al. Role of the sympathetic nervous system in regulation of the sodium glucose cotransporter 2. J Hypertens. 2017;35:2059–2068.
61. Herat LY, Magno AL, Rudnicka C, et al. SGLT2 inhibitor-induced sympathoinhibition: a novel mechanism for cardiorenal protection. JACC Basic Transl Sci. 2020;5:169–179. doi:10.1016/j.jacbts.2019.11.007
62. Yu AS, Hirayama BA, Timbol G, et al. Functional expression of SGLTs in rat brain. Am J Physiol Cell Physiol. 2010;299:C1277–C1284. doi:10.1152/ajpcell.00296.2010
63. Nguyen T, Wen S, Gong M, et al. Dapagliflozin activates neurons in the central nervous system and regulates cardiovascular activity by inhibiting SGLT-2 in mice. Diabetes Metab Syndr Obes. 2020;13:2781–2799. doi:10.2147/DMSO.S258593
64. Grassi G, Dell’Oro R, Facchini A, et al. Effect of central and peripheral body fat distribution on sympathetic and baroreflex function in obese normotensives. J Hypertens. 2004;22(12):2363–2369. doi:10.1097/00004872-200412000-00019
65. Straznicky NE, Lambert GW, Masuo K, et al. Blunted sympathetic neural response to oral glucose in obese subjects with the insulin-resistant metabolic syndrome. Am J Clin Nutr. 2009;89:27–36. doi:10.3945/ajcn.2008.26299
66. Aviello G, Cristiano C, Luckman SM, D’Agostino G. Brain control of appetite during sickness. Br J Pharmacol. 2021;178:2096–2110. doi:10.1111/bph.15189
67. Takayama S, Sakura H, Katsumori K, Wasada T, Iwamoto Y. A possible involvement of parasympathetic neuropathy on insulin resistance in patients with type 2 diabetes. Diabetes Care. 2001;24:968–969. doi:10.2337/diacare.24.5.968
68. Kalsbeek A, Bruinstroop E, Yi CX, et al. Hypothalamic control of energy metabolism via the autonomic nervous system. Ann N Y Acad Sci. 2010;1212(1):114–129. doi:10.1111/j.1749-6632.2010.05800.x
69. German J, Kim F, Schwartz GJ, et al. Hypothalamic leptin signaling regulates hepatic insulin sensitivity via a neurocircuit involving the vagus nerve. Endocrinology. 2009;150:4502–4511. doi:10.1210/en.2009-0445
70. Uno K, Katagiri H, Yamada T, et al. Neuronal pathway from the liver modulates energy expenditure and systemic insulin sensitivity. Science. 2006;312:1656–1659. doi:10.1126/science.1126010
71. Izumida Y, Yahagi N, Takeuchi Y, et al. Glycogen shortage during fasting triggers liver-brain-adipose neurocircuitry to facilitate fat utilization. Nat Commun. 2013;4:2316. doi:10.1038/ncomms3316
72. Cassaglia PA, Hermes SM, Aicher SA, Brooks VL. Insulin acts in the arcuate nucleus to increase lumbar sympathetic nerve activity and baroreflex function in rats. J Physiol. 2011;589:1643–1662. doi:10.1113/jphysiol.2011.205575
73. Bruce KD, Zsombok A, Eckel RH. Lipid processing in the brain: a key regulator of systemic metabolism. Front Endocrinol. 2017;8:60. doi:10.3389/fendo.2017.00060
74. van den Hoek AM, van Heijningen C, Schroder-van der Elst JP, et al. Intracerebroventricular administration of neuropeptide Y induces hepatic insulin resistance via sympathetic innervation. Diabetes. 2008;57:2304–2310. doi:10.2337/db07-1658
75. Singhal NS, Lazar MA, Ahima RS. Central resistin induces hepatic insulin resistance via neuropeptide Y. J Neurosci. 2007;27:12924–12932. doi:10.1523/JNEUROSCI.2443-07.2007
76. Takeda K, Ono H, Ishikawa K, et al. Central administration of sodium-glucose cotransporter-2 inhibitors increases food intake involving adenosine monophosphate-activated protein kinase phosphorylation in the lateral hypothalamus in healthy rats. BMJ Open Diabetes Res Care. 2021;9(1):e002104. doi:10.1136/bmjdrc-2020-002104
77. Kenney M, Weiss M, Haywood J. The paraventricular nucleus: an important component of the central neurocircuitry regulating sympathetic nerve outflow. Acta Physiol Scand. 2003;177:7–15. doi:10.1046/j.1365-201X.2003.01042.x
78. Herat LY, Matthews J, Azzam O, Schlaich MP, Matthews VB. Targeting features of the metabolic syndrome through sympatholytic effects of SGLT2 inhibition. Curr Hypertens Rep. 2022;24:67–74. doi:10.1007/s11906-022-01170-z
79. Zhou L, Sutton GM, Rochford JJ, et al. Serotonin 2C receptor agonists improve type 2 diabetes via melanocortin-4 receptor signaling pathways. Cell Metab. 2007;6:398–405. doi:10.1016/j.cmet.2007.10.008
80. Yaginuma H, Banno R, Sun R, et al. Peripheral combination treatment of leptin and an SGLT2 inhibitor improved glucose metabolism in insulin-dependent diabetes mellitus mice. J Pharmacol Sci. 2021;147:340–347. doi:10.1016/j.jphs.2021.08.010
81. Bartness TJ, Shrestha YB, Vaughan CH, Schwartz GJ, Song CK. Sensory and sympathetic nervous system control of white adipose tissue lipolysis. Mol Cell Endocrinol. 2010;318:34–43. doi:10.1016/j.mce.2009.08.031
82. Cypess AM, Weiner LS, Roberts-Toler C, et al. Activation of human brown adipose tissue by a beta 3-adrenergic receptor agonist. Cell Metab. 2015;21:33–38. doi:10.1016/j.cmet.2014.12.009
83. Chiba Y, Yamada T, Tsukita S, et al. Dapagliflozin, a sodium-glucose co-transporter 2 inhibitor, acutely reduces energy expenditure in BAT via neural signals in mice. PLoS One. 2016;11:e0150756. doi:10.1371/journal.pone.0150756
84. Yang X, Liu Q, Li Y, et al. Inhibition of the sodium-glucose co-transporter SGLT2 by canagliflozin ameliorates diet-induced obesity by increasing intra-adipose sympathetic innervation. Br J Pharmacol. 2021;178:1756–1771. doi:10.1111/bph.15381
85. Matthews JR, Herat LY, Magno AL, et al. SGLT2 inhibitor-induced sympathoexcitation in white adipose tissue: a novel mechanism for beiging. Biomedicines. 2020;8(11):514. doi:10.3390/biomedicines8110514
86. Sawada Y, Izumida Y, Takeuchi Y, et al. Effect of sodium-glucose cotransporter 2 (SGLT2) inhibition on weight loss is partly mediated by liver-brain-adipose neurocircuitry. Biochem Biophys Res Commun. 2017;493:40–45. doi:10.1016/j.bbrc.2017.09.081
87. Jamerson KA, Julius S, Gudbrandsson T, Andersson O, Brant DO. Reflex sympathetic activation induces acute insulin resistance in the human forearm. Hypertension. 1993;21:618–623. doi:10.1161/01.HYP.21.5.618
88. Chadderdon SM, Belcik JT, Bader L, et al. Temporal changes in skeletal muscle capillary responses and endothelial-derived vasodilators in obesity-related insulin resistance. Diabetes. 2016;65:2249–2257. doi:10.2337/db15-1574
89. Wu H, Ballantyne CM. Skeletal muscle inflammation and insulin resistance in obesity. J Clin Invest. 2017;127:43–54.
90. Hitomi H, Kiyomoto H, Nishiyama A, et al. Aldosterone suppresses insulin signaling via the downregulation of insulin receptor substrate-1 in vascular smooth muscle cells. Hypertension. 2007;50:750–755. doi:10.1161/HYPERTENSIONAHA.107.093955
91. Mazak I, Fiebeler A, Muller DN, et al. Aldosterone potentiates angiotensin II-induced signaling in vascular smooth muscle cells. Circulation. 2004;109:2792–2800. doi:10.1161/01.CIR.0000131860.80444.AB
92. Shin SJ, Chung S, Kim SJ, et al. Effect of sodium-glucose co-transporter 2 inhibitor, dapagliflozin, on renal renin-angiotensin system in an animal model of type 2 diabetes. PLoS One. 2016;11:e0165703. doi:10.1371/journal.pone.0165703
93. Esler M, Rumantir M, Wiesner G, et al. Sympathetic nervous system and insulin resistance: from obesity to diabetes. Am J Hypertens. 2001;14:304S–309S. doi:10.1016/S0895-7061(01)02236-1
94. Russo B, Menduni M, Borboni P, Picconi F, Frontoni S. Autonomic nervous system in obesity and insulin-resistance-the complex interplay between leptin and central nervous system. Int J Mol Sci. 2021;23:22. doi:10.3390/ijms23010022
95. Packer M. Do sodium-glucose co-transporter-2 inhibitors prevent heart failure with a preserved ejection fraction by counterbalancing the effects of leptin? A novel hypothesis. Diabetes Obes Metab. 2018;20:1361–1366. doi:10.1111/dom.13229
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.