")
Back to Journals » Drug Design, Development and Therapy » Volume 13
The MAP2K4/JNK/c-Jun Signaling Pathway Plays A Key Role In Dexmedetomidine Protection Against Acetaminophen-Induced Liver Toxicity
Authors Chou A-H, Liao CC , Lee HC , Liou J-T, Liu FC
Received 12 May 2019
Accepted for publication 2 October 2019
Published 14 November 2019 Volume 2019:13 Pages 3887—3898
DOI https://doi.org/10.2147/DDDT.S215473
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Manfred Ogris
An-Hsun Chou,1–3 Chia-Chih Liao,1,2 Hung-Chen Lee,1,4 Jiin-Tarng Liou,1,2 Fu-Chao Liu1,2
1Department of Anesthesiology, Chang Gung Memorial Hospital, Taoyuan, Taiwan; 2College of Medicine, Chang Gung University, Taoyuan, Taiwan; 3Department of Anesthesiology, Xiamen Chang Gung Hospital, Taoyuan, Taiwan; 4Graduate Institute of Clinical Medical Sciences, Chang Gung University, Taoyuan, Taiwan
Correspondence: Fu-Chao Liu
Department of Anesthesiology, Chang Gung Memorial Hospital and Chang Gung University College of Medicine, No. 5, Fusing Street, Gueishan District, Taoyuan City 33305, Taiwan
Tel +886-3-3281200 (ext. 3624)
Fax +886-3-3281200 (ext. 2787)
Email [email protected]
Purpose: Dexmedetomidine [DEX; (S)-4-[1-(2,3-dimethylphenyl)ethyl]-3H-imidazole] is a selective α2-adrenergic receptor (α2-AR) agonist that attenuates the liver damage associated with local or systemic inflammation. However, it remains unclear whether DEX has protective effects against acetaminophen (Paracetamol, PARA)-induced liver toxicity (PILT).
Methods: PILT mice were established by intraperitoneal administration of a hepatotoxic dose of acetaminophen (300 mg/kg). Thirty minutes later, the mice were treated with DEX at a concentration of 0, 5, 25, or 50 μg/kg. Blood and liver samples were obtained for further analysis.
Results: DEX treatment significantly attenuated PILT in mice, with the strongest beneficial effects at a dose of 25 μg/kg. The levels of hepatic cytokines, tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6), in addition to myeloperoxidase (MPO) activity, were significantly decreased following DEX treatment. Moreover, DEX treatment reduced macrophage recruitment around the area of hepatotoxicity and the expression levels of hepatic phosphorylated mitogen-activated protein kinase kinase 4 (MAP2K4), c-jun N-terminal kinase (JNK), and c-Jun expression induced by acetaminophen overdose.
Conclusion: The data suggest that DEX likely downregulates the JNK signaling pathway and its downstream effectors to promote its hepatoprotective effect, providing a clinical application of DEX for the attenuation of PILT.
Keywords: dexmedetomidine, acetaminophen-induced liver toxicity, MAP2K4/JNK/c-Jun
Introduction
Acetaminophen {N-acetyl-p-aminophenol, also known as paracetamol (PARA)} is an analgesic and antipyretic drug. Although safe and effective at recommended doses, overdose may lead to severe liver toxicity.1–3 At therapeutic doses, the majority of PARA is metabolized via glucuronidation and sulfation in the liver, and then excreted via the kidneys as water-soluble metabolites.4 Any remaining PARA is oxidized by the microsomal P-450 enzyme system to the reactive intermediate, N-acetyl-p-benzoquinone imine (NAPQI).5 Although NAPQI is rapidly detoxified via conjugation with glutathione (GSH), the availability of GSH is limited in the case of an overdose.6 The resulting depletion of GSH enables excessive NAPQI to covalently bind to cellular proteins, causing mitochondrial dysfunction, the formation of reactive oxygen species (ROS), and deoxyribonucleic acid (DNA) damage, which lead to hepatocyte necrosis and apoptosis.7–9 Although N-acetyl cysteine (NAC) is a known antioxidant for PARA-overdose, its use has certain limitations including adverse effects and a narrow therapeutic window.10 If the early treatment stage is missed, liver transplantation is the only option to improve survival in patients with acute liver failure;11 therefore, the development of novel drugs that are superior to NAC is clearly needed.
Dexmedetomidine [DEX; (S)-4-[1-(2,3-dimethylphenyl)ethyl]-3H-imidazole] is a selective and potent α2-adrenergic receptor (α2-AR) agonist, with sedative, anxiolytic, analgesic-sparing, sympatholytic, and hemodynamic stability characteristics.12 Previous studies have demonstrated that DEX exerts protective effects against ischemia and reperfusion (I/R) injury in several organs, including the kidneys, lungs, and liver.13–15 In addition, DEX has been shown to attenuate liver damage associated with sepsis, shock, and other diseases related to local or systemic inflammation.16 Moreover, recent studies have demonstrated that DEX reduces proinflammatory cytokine levels in experimental sepsis17 and in critically ill18,19 and postoperative patients.20 A significant decrease in leukocyte counts and the levels of CRP, IL-6, IL-8, and TNF-α in DEX-treated patients are indicative of its anti-inflammatory potential when used as a perioperative adjunct.18,19 Recent experimental study in rats demonstrated the beneficial effect of DEX in hepatotoxicity and nephrotoxicity after acetaminophen overdose by reducing the oxidative injury biomarker.21 However, detailed mechanisms to what role DEX plays in the hepatoprotective effects against PARA-induced liver toxicity (PILT) still need to evaluate. Therefore, the aim of the present study was to investigate the therapeutic effect and mechanism of DEX in a mouse model of PILT.
Methods
Animals And Experimental Protocol
Adult male C57BL/6J (B6) mice were used and purchased from BioLASCO Taiwan Co., Ltd. (Taipei, Taiwan). All the animal studies were conducted according to protocols that were approved by the Institutional Animal Care and Use Committee of Chang Gung Memorial Hospital (Approval number: 2017062001; date of approval August 18, 2017; Taiwan).
All animals were housed in a room with a controlled environment consisting of a 12 hrs light/dark cycle and were allowed free access to food and water. All experiments were carried out in 8- to 10-week-old male mice weighing 15 to 20 g. Mice received an intraperitoneal (i.p) injection of PARA (300 mg/kg; Sigma-Aldrich, St. Louis, MO), dissolved in sterile 0.9% saline at a concentration of 20 mg/mL, to create an acute liver injury model. For the treatment of the DEX group, mice were received i.p injection of DEX which was diluted in sterile 0.9% saline at a concentration of 10 ng/μL, 30 mins after injection of PARA 300 mg/kg. The mice were randomly assigned to 6 groups (n =6/group). Of the 6 groups of mice, 4 received an i.p injection of PARA (300 mg/kg) to induce liver toxicity and 30 mins later, the mice were intraperitoneally treated with DEX at a concentration of 0, 15, 25, or 50 μg/kg. The other 2 control groups were received an equal volume of sterile 0.9% saline and DEX (25 μg/kg) alone. Furthermore, in another experiment, mice were treated with intraperitoneal NAC 200 mg/kg after 30 mins of PARA administration for comparison with the effects of DEX (n = 6/group). Following 16 hrs of experiment, animals were anesthetized, blood was obtained by cardiac puncture and tissues were removed for various analyses.
Assessment Of The Liver Function
To determine the hepatic toxicity, serum levels of alanine transaminase (ALT) were measured using a Vitros DT60 II Chemistry System (Ortho-Clinical Diagnostics; Johnson & Johnson, New York, NY).
Glutathione (GSH) Assessment In Liver
The GSH level is measured by glutathione assay kit (Cayman Chemical) with absorbance amount read at 412 nm by spectrophotometer and GSH level was reported as μmol/g tissue.
Liver MPO Activity Assay
All activity assays were performed in triplicates on 96 well microtiter plates and analyzed with a Safire 2 microplate reader (Tecan, Durham, NC). Briefly, the liver tissues were submerged and homogenized in 10 volume of ice-cold 50 mM KPO4 buffer. The samples were centrifuged at 15,000 x g for 15 mins and MPO activity was measured spectrophotometrically using o-dianisidine (10 mg/mL) and 0.3% H2O2 with a maximum absorbance at 460 nm.
Histopathological Analysis
For histological analysis, liver tissues were obtained and fixed in 4% paraformaldehyde for 30 mins at room temperature and embedded in paraffin. The samples were subsequently transferred into 70% ethanol then embedded in paraffin. The paraffin blocks were sliced into 4-μm serial sections. The selected sections were stained with haematoxylin and eosin (H&E). The percentage of necrotic tissue estimated by evaluating the number of microscopic fields with necrosis compared to the entire histologic section. All histological evaluations were done in a blinded fashion by 2 investigators.
For immunohistochemical staining, the sections were deparaffinized in xylene and rehydrated in alcohol and distilled water. Antigen retrieval was performed and 3% H2O2 was used to eliminate endogenous peroxidase. Slides were washed with TBS-Triton three times and one time with TBS for 5 mins. For immunostaining of macrophages and neutrophils, tissue sections were incubated with a rat anti-mouse primary antibody (1 μg/mL) followed by a biotinylated goat anti-rat secondary antibody (Mac-2 for macrophages and Ly6G for neutrophils). Samples were incubated overnight with the specific primary antibody, followed by washing twice for 5 mins and incubation with the biotinylated secondary antibodies for 1 hr. The peroxidase reaction was performed following the manufacturer’s protocol. Proliferating cell nuclear antigen (PCNA)-positive stained hepatocytes were quantitated by randomly selecting five fields of views per section (five sections per animal for total six mice)
Measurement Of Cytokine Levels In Mouse Liver
The liver tissues were homogenized in digestion buffer (20 mM HEPES, 150 mM NaCl, 3 mM MgCl2, 1 mM 2-mercaptoethanol, 0.1 mM phenylmethane sulphonyl fluoride (PMSF), 0.01 μg aprotinin, 0.25 μg leupeptin, 0.05 μg pepstatin A, and 1 M DTT). The homogenate was centrifuged at 12,000 x g for 10 mins at 4°C and the supernatants were collected. The fluorescamine protein assay was used to determine total protein concentration. Subsequently, samples adjusted for protein concentration were analyzed for cytokine expression using the ELISA kit (San Diego, CA, USA) according to the supplier’s protocol. Standard curves were included for each experiment.
Western Blotting
The liver tissues were homogenized in protein extraction solution. Lysates were cleared by centrifugation at 12,000 x g for 10 mins at 4°C (two times). Proteins were quantified using the Bradford protein microassay (Bio-Rad, Hercules, USA). Equal amounts of protein from each sample were separated on 10% sodium dodecylsulphate polyacrylamide gels by electrophoresis. The proteins were transferred to polyvinylidene fluoride membranes, blocked in 5% fat-free milk solution for 1 hr, and rinsed 3 times in Tris buffer containing 1% Tween® 20. The membranes were incubated with 1:1000 dilution of primary antibodies against MAP2K4, ERK, JNK, c-Jun, phospho-MAP2K4 (p-MAP2K4), phospho-ERK (p-ERK), phospho-JNK (p-JNK), and phospho-c-Jun (p-c-Jun) (Cell Signaling Technology, MA, USA) overnight at 4°C. Bound antibody was detected with a horseradish peroxidase (HRP)-conjugated secondary antibody at room temperature for 1 hr (Cell Signaling Technology, MA, USA), and immunodetected proteins were visualized using the enhanced chemiluminescence (ECL) system. To confirm the equal amount of protein loaded, the membranes were stripped and reprobed with a monoclonal antibody against β-actin (Protein Tech Group, Chicago, IL, USA). Gel bands were quantitated by densitometry and normalised to β-actin.
Statistical Analysis
All results are expressed as the mean ± SEM (n = 6 mice/group). Calculations were performed with Prism statistical software version 5.0 (GraphPad Software Inc., San Diego, USA). Unpaired t-tests or one-way analysis of variance (ANOVA) with post-hoc Tukey multiple comparison tests were used to compare experimental groups with the control group. For all tests, P < 0.05 was considered statistically significant.
Results
Hepatoprotective Effects Of DEX On PILT
The effects of acute liver toxicity following increasing doses of PARA were directly compared. Sixteen hours of PARA 300 or 400 mg/kg administration, all mice developed hepatotoxicity. Both doses of PARA (300 and 400 mg/kg) significantly elevated the serum ALT levels compared to control group; however, ALT levels were not significantly different between the two treatment groups (Figure 1A). A time course study was performed following administration of 300 mg/kg PARA. Peaked serum ALT level at 16 and 24 hrs following administration of PARA was demonstrated as compared with the control group. However, there was no significant difference between these two time points (Figure 1B). Figure 1C shows the effects of DEX on PARA-induced increases in serum ALT levels. A single dose of PARA (300 mg/kg) significantly elevated serum ALT levels as compared with the control (6000 ± 500.2 vs 42.7 ± 4.8 U/L, p < 0.05). Treatment with different doses of DEX, after PARA administration markedly lowered serum ALT levels as compared with the PARA-only group (2045.2 ± 159.6 U/L, p < 0.05; 155.5 ± 16 U/L, p < 0.005; and 155.8 ± 14 U/L, p < 0.005 vs. 6000 ± 500.2 U/L, respectively). The effects of DEX were equivalent irrespective of the dose administered (25 or 50 μg/kg). We also compared the effects of DEX with NAC on PILT (Figure 1D). Serum ALT activity was significantly decreased in DEX (15ug/kg), NAC treatment, or combined therapy groups compared with the PARA-only group (1200 ± 150 vs. 6500 ± 500.5 U/L, p < 0.05; 2000.5 ± 50 vs. 6500 ± 500.5 U/L, p < 0.005; 500.8 ± 35 vs. 6500 ± 500.5 U/L, p < 0.05). NAC treatment 30 mins after PILT reduced elevated ALT levels, but there was no statistically significant difference when compared with the DEX treatment. When animals were treated with both DEX and NAC after PILT, the effect of protection was more significant than that after NAC treatment alone (500.8 ± 35 vs. 2000.5 ± 50 U/L, p <0.05).
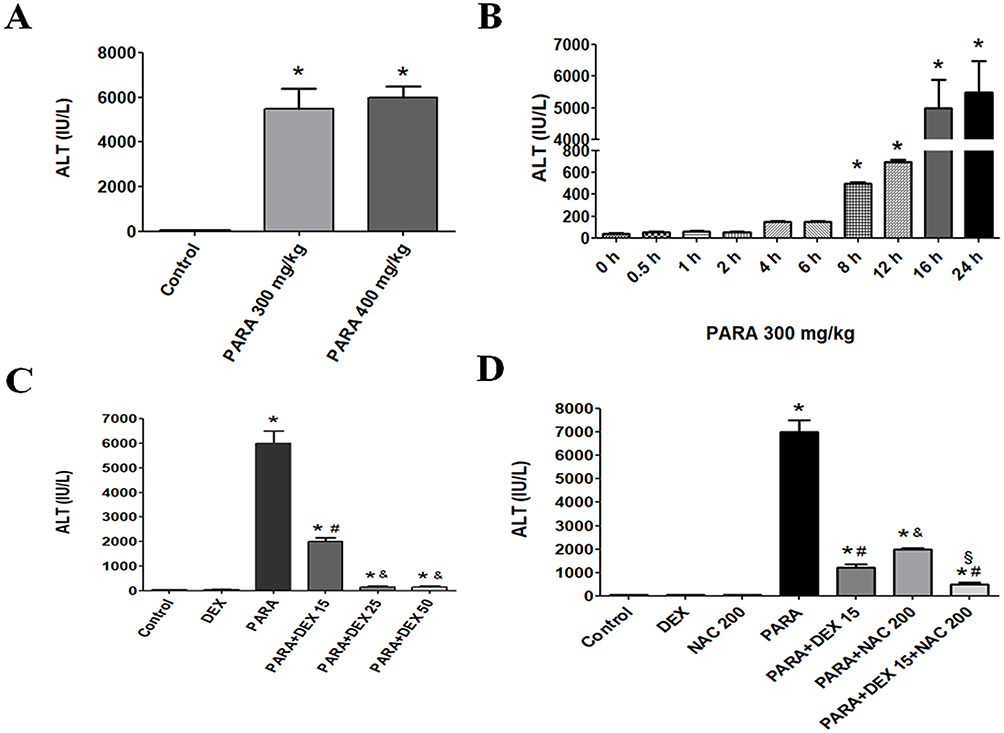 |
Figure 1 Serum ALT levels following PARA exposure. (A) ALT levels following PARA exposure at different doses. (B) Time courses of serum ALT levels following PARA exposure. (C) Dose-dependent protection effects of DEX against PARA-induced hepatotoxicity. Mice were intraperitoneally administered PARA (300 mg/kg) alone or various concentrations of DEX (15, 25, 50 μg/kg) 30 mins after injection of PARA and were sacrificed 16 hrs later for assessment of serum ALT levels. (D) Effects of DEX or NAC treatment on hepatic ALT levels. Mice were intraperitoneally administered an PARA dose (300 mg/kg) alone or treated with DEX (15 μg/kg), NAC 200mg/kg, or combination of both after injection of PARA, and were sacrificed 16 hrs after treatment for assay of serum ALT. Results are presented as the mean ± SEM; n = 6 mice per group. *p < 0.05 vs control; #p < 0.05 and &p < 0.005 vs PARA alone; §p < 0.05 vs. PARA + NAC 200. |
Supplemental Figure 1 shows the effects of DEX given 1 or 2 hrs after PILT. We demonstrated that treatment of DEX after 1 or 2 hrs after PARA (300 mg/kg) administration markedly lowered serum ALT levels as compared with the PARA only group (150 ± 30 vs. 6500 ± 500.2 U/L, p <0.05 for 1 h; 145 ± 35 vs. 6500 ± 500.2 U/L, p <0.05 for 2 hrs after PARA administration). These demonstrated that DEX also had protective effect on PILT if later given 1–2 hrs after PARA administration. In this study, we measured the total GSH at early time points of PARA and verified whether the DEX altered the PARA metabolism. Hepatic GSH was significantly lower in the 2 and 4 hrs after PARA 300 mg/kg administration when compared to the control group. There was no significant difference in hepatic GSH between in 2 and 4 hrs PARA and PARA+ DEX 25 μg/kg group (Figure 2).
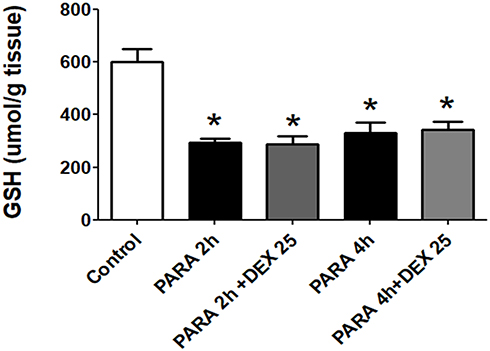 |
Figure 2 Effect of DEX on the GSH levels in the liver at early time points after PARA administration. Mice were intraperitoneally administered PARA (300 mg/kg) and DEX (25 μg/kg) was given 30 mins after PARA. Then, mice were sacrificed 2 and 4 hrs for assessment of GSH levels. Results are presented as the mean ± SEM; n = 6 mice per group. *p < 0.05 vs. control. |
Next, we determined the effect of DEX treatment on histopathology changes after PILT. H&E staining demonstrated severe sinusoidal swelling, centrilobular necrosis and destroyed endothelium of central vein in 16 hrs after PARA-treated mice. DEX treatment following PARA exposure, the animals showed well-preserved hepatocytes with less necrosis and less sinusoidal swelling (Figure 3A). Treatment with DEX after PARA administration markedly decreased the percent of necrosis as compared with the PARA-only group (20 ± 5% vs. 75 ± 10%, p <0.05) (Figure 3B). We also examined the early time course of PILT. There were no significant changes between control (0.9% saline-treated mice) and mice with 2 and 4 hrs after PARA administration in H&E staining (Supplemental Figure 2).
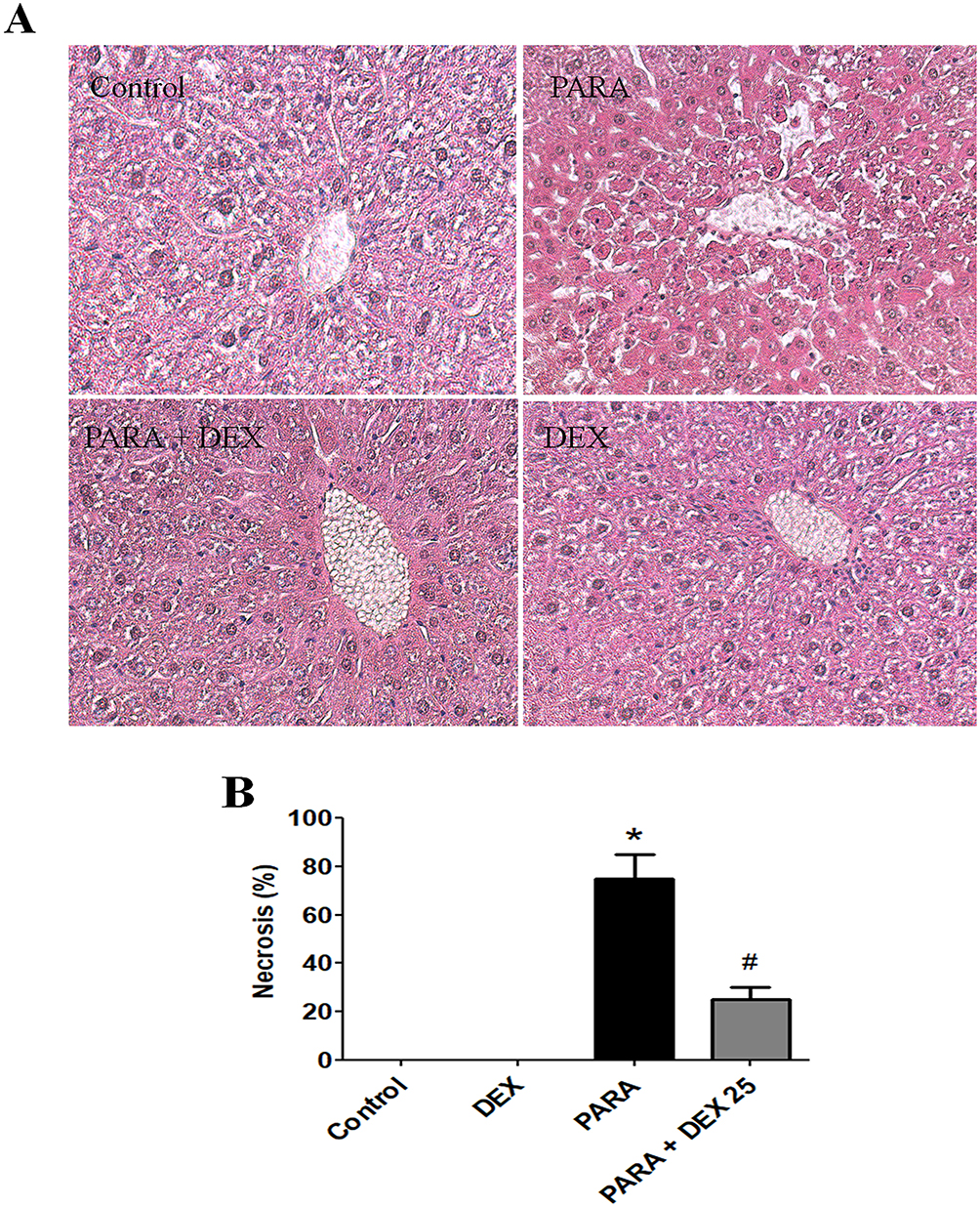 |
Figure 3 Effects of DEX on PARA-induced liver toxicity-related histology. (A) Mice were administered saline (control), PARA (300 mg/kg) alone, DEX (25 μg/kg) 30 mins after PARA injection, or DEX (25 μg/kg) alone, and were sacrificed 16 hrs later for H&E staining (200x). Typical images were chosen from each group. (B) Cell necrosis was evaluated in livers from controls, DEX alone, PARA (300 mg/kg) alone, and DEX (25 μg/kg) 30 mins after PARA injection. The percent of necrosis was estimated by evaluating the number of microscopic fields with necrosis compared to the entire histologic section. Data represent means ± SE of n=6 animals per group; *p < 0.05 vs. control; #p < 0.05 vs. PARA alone. |
Effects Of DEX Treatment On MPO Activity And Neutrophil Accumulation In PILT
Figure 4A shows the hepatic MPO expression levels. A single dose of PARA (300 mg/kg) significantly increased hepatic MPO activity as compared with the control (6.9 ± 0.6 vs. 2.3 ± 0.07 ΔOD460/g/min, p < 0.05). Treatment with DEX, after PARA administration lowered hepatic MPO levels, which were significantly decreased in the 25 μg/kg DEX group as compared with the PARA-only group (2.7 ± 0.17 vs. 6.9 ± 0.6 ΔOD460/g/min, p < 0.05). Figure 4B shows the immunohistochemical staining of LY6G, a granulocyte-specific marker, which is used for the evaluation of inflammatory infiltration of neutrophils in PILT. Animals treated with only PARA showed greater neutrophil infiltration in the liver parenchyma as compared with the control animals. DEX treatment after PARA injection significantly decreased neutrophil recruitment in the liver parenchyma.
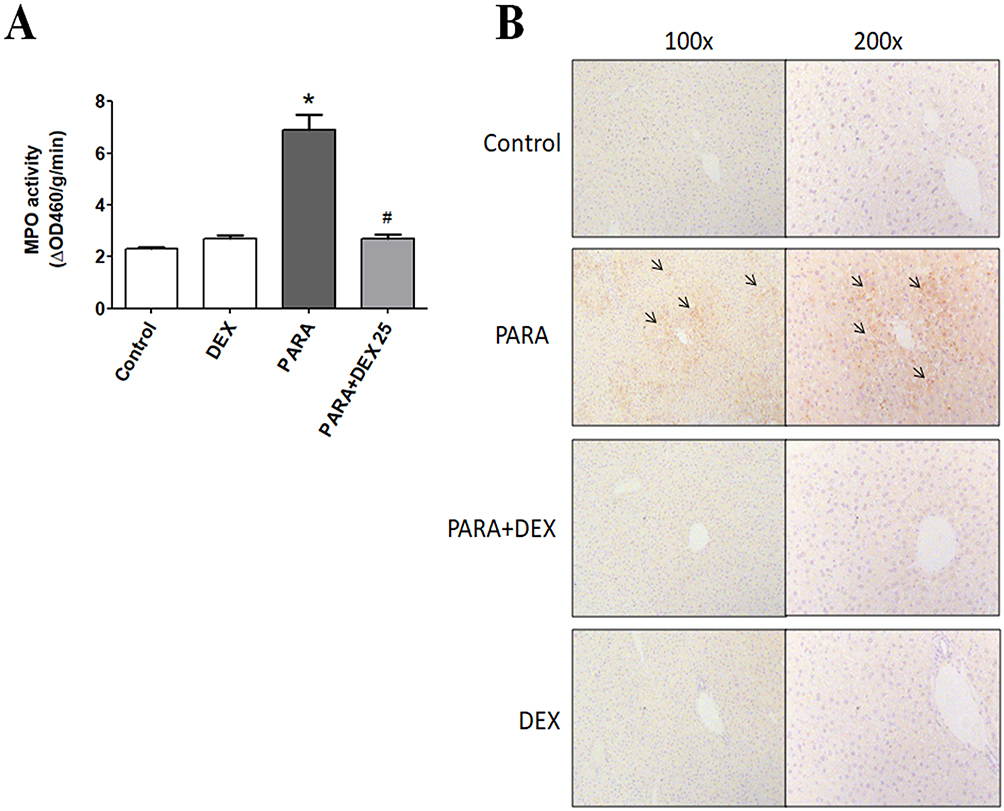 |
Figure 4 Effects of DEX treatment on hepatic MPO activity and immunohistochemical evidence of neutrophil accumulation following PARA-induced liver toxicity. (A) Mice were intraperitoneally administered PARA (300 mg/kg) alone or 25 μg/kg DEX 30 mins after injection of PARA and sacrificed 16 hrs later for the assessment of liver MPO activity. Results are presented as the mean ± SEM; n = 6 mice per group. *p < 0.05 vs control; #p < 0.05 vs PARA alone. (B) Mice were administered saline (control), PARA (300 mg/kg) alone, DEX (25 μg/kg) 30 mins after PARA injection, or DEX (25 μg/kg) alone, and were sacrificed 16 hrs later for immunohistochemical staining. Liver sections were immunostained for neutrophils (black arrows). Typical images were chosen from each group. |
Effects Of DEX On Immunohistochemical Staining For Macrophages
Immunohistochemical staining of Mac-2, a-specific marker for macrophages, was used for the evaluation of macrophage accumulation and infiltration in PARA-induced liver injury. Animals treated with only PARA showed greater macrophage infiltration in the liver parenchyma as compared with the control animals. DEX treatment after PARA injection significantly decreased macrophage recruitment around the area of hepatotoxicity (Figure 5).
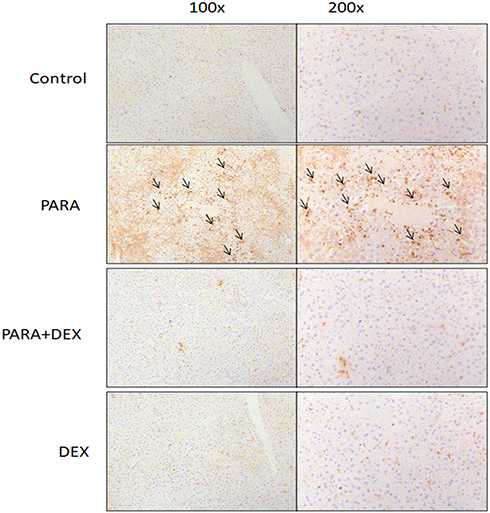 |
Figure 5 Effects of DEX on immunohistochemical evidence of macrophage accumulation following PARA-induced liver toxicity. Mice were administered saline (control), PARA (300 mg/kg) alone, DEX (25 μg/kg) 30 mins after PARA injection, or DEX (25 μg/kg) alone, and were sacrificed 16 hrs later for immunohistochemical staining. Liver sections were immunostained for macrophages (black; arrow). Typical images were chosen from each group. |
Effects Of DEX On Hepatic TNF-α And IL-6 Expression Levels
To determine the inflammatory response in PILT, we assessed the expression levels of inflammatory cytokines. There was no significant difference in hepatic TNF-α or IL-6 levels between the control and DEX-only treatment groups. After a single dose of PARA, the concentrations of TNF-α and IL-6 were significantly increased in the PARA-only group as compared with the control group (214.4 ± 15.0 vs. 118.7 ± 1.4 pg/100 μg protein, p < 0.05; 315.5 ± 15.5 vs. 170 ± 5.7 pg/100 μg protein, p < 0.05, respectively). However, DEX treatment (25 μg/kg) 30 mins after PARA administration significantly reduced the elevated hepatic TNF-α and IL-6 levels (106.3 ± 4.0 vs. 214.4 ± 15.0 pg/100 μg protein, p < 0.05; 169.4 ± 4.1 vs. 315.5 ± 15.5 pg/100 μg protein, p < 0.05, respectively) (Figure 6).
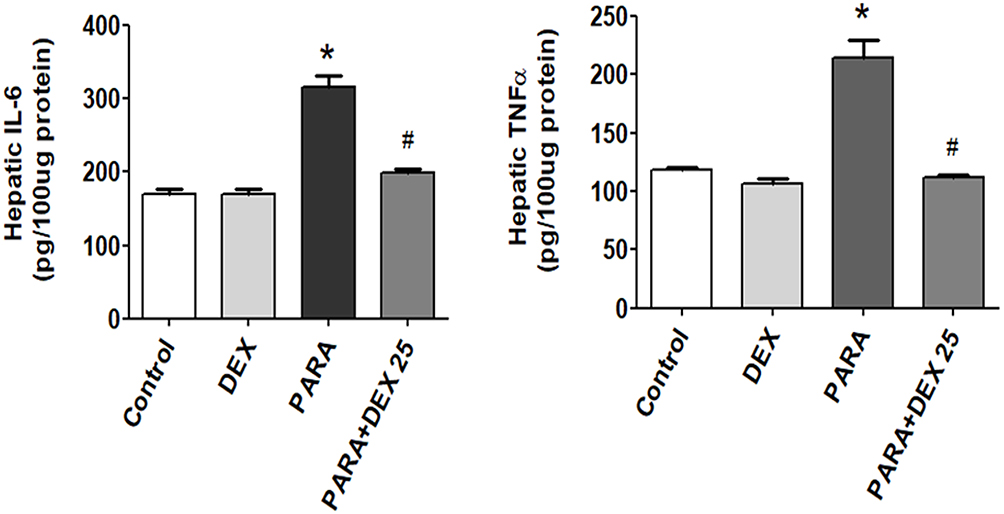 |
Figure 6 Effects of DEX treatment on hepatic TNF-α and IL-6 expression levels following PARA-induced liver toxicity. Mice were intraperitoneally administered PARA (300 mg/kg) alone, DEX (25 μg/kg) 30 mins after PARA injection, or DEX (25 μg/kg) alone, and were sacrificed 16 hrs later for assessment of hepatic TNF-α and IL-6 expression levels. Results are presented as the mean ± SEM; n = 6 mice per group. *p < 0.05 vs. control; #p < 0.05 vs. PARA alone. |
Effects Of DEX On Hepatic MAP2K4, ERK, JNK, And C-Jun Expression Levels
There were no significant differences in hepatic MAP2K4, JNK, or c-Jun protein expression levels between the control and DEX-only treatment groups. The levels of p-MAP2K4, p-JNK, and p-c-Jun were significantly increased following a single dose of PARA as compared with the control group. However, DEX treatment (25 μg/kg) 30 mins after PARA administration significantly decreased hepatic p-MAP2K4, p-JNK, and p-c-Jun expression levels (Figure 7). There was also no significant difference in hepatic ERK expression among the 4 groups (data not shown). In this study, the expression levels of hepatic JNK and c-Jun were also assessed in early time points and after DEX treatment. There were no significant differences in hepatic JNK, p-JNK protein expression levels between the control and mice with 2 and 4 hrs after PARA administration (Supplemental Figure 3).
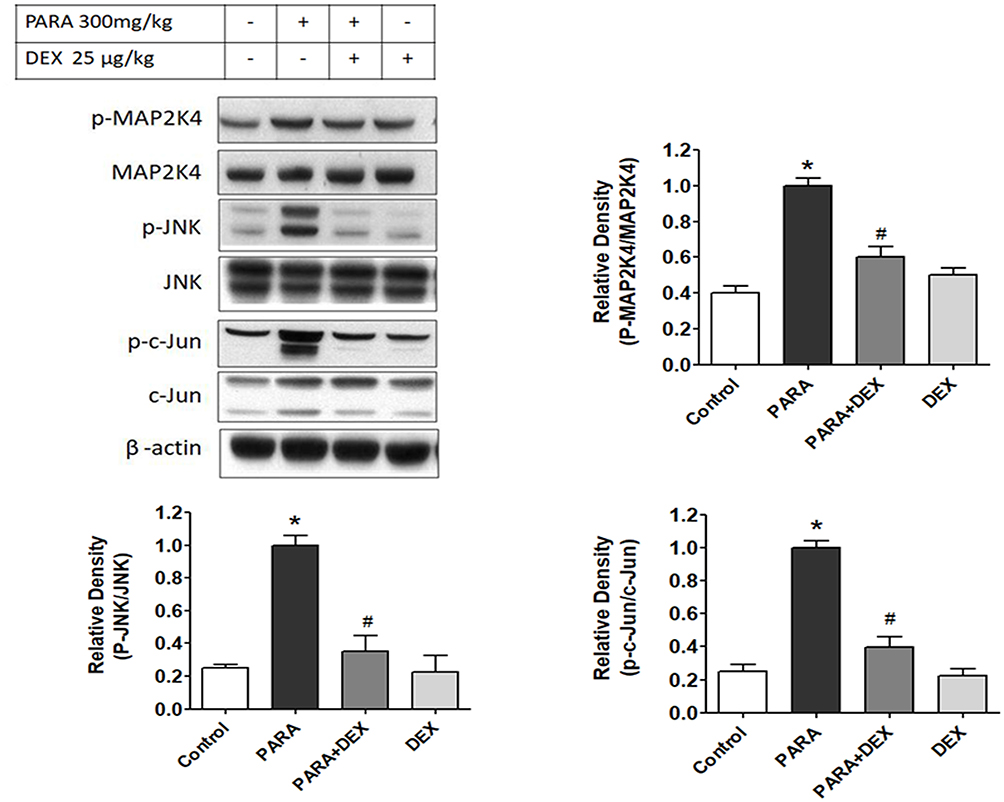 |
Figure 7 Effects of DEX on hepatic JNK protein expression and activity following PARA-induced liver toxicity. Hepatic p-MAP2K4, MAP2K4, p-JNK, JNK, p-c-Jun, and c-Jun protein expression levels were evaluated in mice administered normal saline (control; lane 1) or PARA (300 mg/kg) alone (PARA; lane 2), or after treatment with DEX (25 μg/kg) 30 mins after injection of PARA (PARA+ DEX; lane 3) or treatment with DEX (25 μg/kg) alone (DEX; lane 4). Blots were reprobed for β-actin to ensure equal protein loading in all lanes. The bands were analyzed using densitometry, and the values are presented as the mean ± SEM; n = 6 mice per group. *p < 0.05 vs. control; #p < 0.05 vs. PARA alone. |
Discussion
To examine the therapeutic effect of DEX on PILT, we prepared an PARA-induced hepatotoxic mouse model. Serum ALT, hepatic MPO, and hepatic TNF-α and IL-6 levels were markedly increased in hepatotoxic mice 16 hrs following PARA treatment. We found that treatment with 25 μg/kg DEX after PARA administration significantly improved hepatic injury. Serum ALT, hepatic MPO, and hepatic TNF-α and IL-6 levels were significantly decreased in DEX-treated mice as compared with PARA-only group.
DEX is a selective α2-adrenergic agonist with sedative, anxiolytic, analgesic-sparing, sympatholytic, and haemodynamic stability characteristics.12 A number of mechanisms of action have been postulated for DEX including modulation of cytokine production by macrophages/monocytes during the stress response, inhibition of apoptosis, and antinociceptive action involving interactions between pain and immune factors (proinflammatory cytokines).18,19 Animal and clinical trials12,13,22 have reported that DEX reduces the levels of TNF-α, Interleukin-1 β (IL-1β), and IL-6 stimulated by endotoxaemia. One such study demonstrated that DEX improves congestion and edema and decreases TNF-α, IL-1β, and IL-6 levels in lung tissues.13 In addition, the inhibitory effects of DEX on cytokines in polymicrobial septic mice model demonstrated that DEX could reduce the mortality rate and inhibit pro-inflammatory cytokine IL6 and TNF-α.23 The present study further reveals that DEX treatment was associated with decreased concentrations of TNF-α and IL-6 in hepatic tissue and attenuated the PILT.
Neutrophil and macrophage activation and infiltration into the hepatic vasculature are an essential component of PILT. The role of neutrophils has been shown in liver injury models such as ischemia-reperfusion24 and alcoholic hepatitis.25 Previous studies have indicated that the progression and severity of PILT are associated with increased neutrophil accumulation.26 Neutrophil infiltration and transmigration into the hepatic parenchyma are related to the increased release of cytokines and chemokines from injured hepatocytes.27 Increased tissue MPO levels are an indicator of neutrophil infiltration. In the present study, hepatic immunohistochemical staining of LY6G, a granulocyte-specific marker, and Mac-2, a-specific marker for macrophage infiltration, were markedly reduced following treatment with 25 μg/kg DEX after PARA administration.
Recent study had suggested that PILT is followed by sterile inflammation.28,29 The role of inflammation in the mechanism of PILT is highly controversial. In the sterile inflammation, initial necrotic cell death release damage-associated molecular patterns (DAMPs). The early release of DAMPs and the formation of cytokines during the sterile inflammatory response after PARA overdose lead to the recruitment of neutrophils and monocyte into the hepatic vasculature.30,31 Although the substantial sterile inflammatory response after the initial cell death, however, there is no convincing experimental evidence to support the hypothesis that Kupffer cells, neutrophils or monocytes directly cause cell injury by producing cytotoxic mediators. Despite the initial results arguing against an active role of neutrophils in PILT, previous studies using the identical experimental approach suggested that neutrophils are responsible in part for PILT.26,32 In contrast, study demonstrated that macrophages and neutrophils are instrumental in removing necrotic cell debris and promoting hepatocyte proliferation, ultimately resulting in tissue repair and resolution of the inflammatory response.33 Thus, the presence of an inflammatory infiltrate is obvious both histologically and biochemically, whether this infiltrate directly contributes to hepatocyte death remains controversial. Although the inflammatory infiltrate directly contributes to hepatocyte death remains controversial, our study demonstrated that DEX treatment decreased the concentration of cytokines and reduced infiltration of neutrophils and macrophages and PILT attenuated. Further studies are needed to evaluate the exact mechanisms of these inflammatory cells affect cell death or tissue repair. Also, further experimental designs are needed to evaluate whether DEX treatment was associated with attenuate the early hepatocyte injury or improve late tissue repair.
The MAPK pathways are activated by diverse extracellular and intracellular stimuli, including peptide growth factors, cytokines, hormones, and various cellular stressors such as oxidative and endoplasmic reticulum stress. These signaling pathways regulate a variety of cellular activities including proliferation, differentiation, survival, and death.34–36 MAP2K4, a member of the MAPK family, directly phosphorylates and activates the JNK in response to cellular stresses and proinflammatory cytokines.37 JNK activation in PARA toxicity has been studied previously. In both cultured hepatocytes and in vivo livers, treatment with PARA induces sustained activation of JNK, as reflected by increased phospho-c-Jun levels.38 Moreover, in a study in C6 glioma cells, PILT caused JNK activation.39 DEX has been shown to reduce the expression of p-JNK proteins in lung I/R injury via an anti-inflammatory mechanism. Administration of the α2-adrenoceptor antagonist, yohimbine, prior to DEX treatment failed to completely eliminate the effect of DEX on the phosphorylation of JNK and the production of inflammatory cytokines in lung ischemia and reperfusion injury, indicating that the anti-inflammatory mechanism of DEX may be associated with an α2-adrenoceptor-independent signaling pathway.40 A previous study has shown that activation of the ERK signaling pathway is also involved in inflammation, oxidative stress, and apoptosis. Inhibition of the ERK signaling pathway may protect against PILT;41 however, in the present study, DEX had no influence on the ERK signaling pathway (data not shown).
In the present study, p-MAP2K4 and p-JNK expression levels were significantly increased 16 hrs after PARA treatment. DEX treatment effectively inhibited this phosphorylation, consequently reducing protein expression. Up to now, the precise target of JNK in the pathogenesis of PILT remains unknown. Strong candidate targets of JNK are Bcl-2 family members; JNK can promote proapoptotic Bax translocation to mitochondria in COS cells;42 however, recent studies in Bax knockout mice showed a similar susceptibility to acetaminophen as wild-type mice,43 suggesting that JNK-induced Bax translocation alone cannot account for PILT. Previous study also demonstrated that JNK translocation to the mitochondria is required for the acetaminophen toxicity. Excessive NAPQI induces the ROS which phosphorylate MKK4 and JNK. Phosphorylated MKK4, JNK and Bax binding with Sab translocate to mitochondria through to increase the mitochondrial permeability transition, resulting in induction of massive hepatocyte death.44,45 In the present study, we demonstrated that JNK could phosphorylate c-Jun, and DEX treatment after PARA administration attenuated p-c-Jun expression. JNK can activate c-jun and activator protein 1 (AP-1) transcriptional activity or phosphorylate other targets not involved in transcription.46 Previous study stated that PARA-treated mice had elevated hepatic mRNA levels of inflammatory genes (NF-κB, TNF-α, IL1-β and IL-6) since 4 hrs after PARA administration. In mice, carbon monoxide releasing molecule A-1 attenuated the oxidative stress by induction of nuclear factor erythroid 2-related factor 2 (Nrf2) protein and anti-oxidant response element (ARE) genes, preventing GSH depletion and by reducing hepatocyte necrosis.47 However, a recent study in rats demonstrated that 24 hrs DEX administration had a beneficial effect on PARA-induced hepatotoxicity and nephrotoxicity through the antioxidant effect.21 In our study, hepatic GSH was lower in 2 and 4 hrs after PARA administration. However, we observed that DEX treatment had no effect on hepatic GSH in these time points. There were no significant histological changes and JNK phosphorylation in early stages of 2 and 4 hrs after PARA administration. According to our results, the possible mechanism of DEX protects against PILT was through the reduced phosphorylation of MAP2K4/JNK/c-Jun expression levels with reduced activation of downstream targets resulting in attenuated the hepatocyte death. However, the downstream targets of JNK that mediate PILT and the hepatoprotective effect of DEX are needed to evaluate in further study.
N-acetylcysteine (NAC), a glutathione precursor, reduces PILT by increasing glutathione synthesis, thereby helping in the detoxification of NAPOI and scavenging of the free radicals.48 In our study, DEX treatment (15 μg/kg) after PILT was not more effective than NAC (200 mg/kg), but the combination of DEX and NAC exhibited a higher efficacy than NAC treatment alone, suggesting that DEX and NAC may affect different pathways. Currently, NAC is the standard antidote for emergency treatment of PARA overdose. However, it is associated with a variety of side effects such as vomiting, diarrhea, and anaphylactic reaction.49 Therefore, anti-inflammatory properties of DEX with low toxicity are considered an alternative antidote for emergency treatment for PILT. Additionally, the additive effects of combined DEX and NAC treatment may increase the therapeutic effect or decrease the dosage of NAC required thus avoiding adverse effects but maintaining the same efficacy. Further studies are needed to compare DEX with NAC for its effectiveness as an antidote for PILT.
Conclusion
This study demonstrated that PARA toxicity increased the levels of TNF-α, IL-6, and MPO in liver tissue and induced sustained activation of MAP2K4, JNK, and c-Jun, contributing to hepatotoxicity. DEX treatment after administration of PARA decreased the levels of MPO and proinflammatory cytokines in the liver and reduced hepatic damage via the MAP2K4/JNK/c-Jun signaling pathway. The present work provides opportunities for the establishment of a potential clinical application of dexmedetomidine with the aim of reducing PILT.
Abbreviations
ALF, acute liver failure; ALT, alanine transaminase; ANOVA, one-way analysis of variance; AP-1, activator protein 1; α2-AR, α2-adrenergic receptor; BAX, BCL2-associated X protein; BCL2, B-cell lymphoma 2; CRP, C-reactive protein; DEX, dexmedetomidine; DNA, deoxyribonucleic acid; DTT, 1,4-dithiothreitol; ECL, enhanced chemiluminescence; ELISA, enzyme-linked immunosorbent assay; ERK, extracellular-signal-regulated kinase; GSH, glutathione; H&E, haematoxylin and eosin; HEPES, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid; HRP, horseradish peroxidase; IL-1β, interleukin-1 β; IL-6, interleukin-6; IL-8, interleukin-8; I/R, ischemia and reperfusion; JNK, C-jun N-terminal kinase; KPO4, potassium phosphate buffers; MAPK, mitogen-activated protein kinase; MAP2K4, mitogen-activated protein kinase kinase 4; MPO, myeloperoxidase; NAC, N-acetyl cysteine; NAPQI, N-acetyl-p-benzoquinone imine; NRf2, nuclear factor erythroid 2-related factor 2; PARA, acetaminophen; PBS, phosphate-buffered saline; PCNA, proliferating cell nuclear antigen; p-c-Jun, phospho-c-Jun; p-ERK, phospho-extracellular-signal-regulated kinase; p-JNK, phospho-c-jun N-terminal kinase; p-MAP2K4: phospho-mitogen-activated protein kinase kinase 4; PMSF, phenylmethane sulphonyl fluoride; ROS, reactive oxygen species; SEM, standard error of the mean; TNF-α, tumor necrosis factor-alpha.
Funding
The study was supported by a grant from the Chang Gung Medical Research Project (BMRPC19, CMRPG3D1471, CMRPG3D1472, CMRPG3D1473) and the Ministry of Science and Technology, Taiwan (MOST 106-2314-B-182A-061-MY2).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42(6):1364–1372. doi:10.1002/hep.20948
2. McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 2012;122(4):1574–1583. doi:10.1172/JCI59755
3. Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med. 2010;77(1):19–27. doi:10.3949/ccjm.77a.09084
4. Antoniades CG, Quaglia A, Taams LS, et al. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology. 2012;56(2):735–746. doi:10.1002/hep.25657
5. Lancaster EM, Hiatt JR, Zarrinpar A. Acetaminophen hepatotoxicity: an updated review. Arch Toxicol. 2015;89(2):193–199. doi:10.1007/s00204-014-1432-2
6. Bessems JG, Vermeulen NP. Paracetamol (acetaminophen)-induced toxicity: molecular and biochemical mechanisms, analogues, and protective approaches. Rev Toxicol. 2001;31(1):55–138. doi:10.1080/20014091111677
7. James LP, McCullough SS, Knight TR, Jaeschke H, Hinson JA. Acetaminophen toxicity in mice lacking NADPH oxidase activity: role of peroxynitrite formation and mitochondrial oxidant stress. Free Radic Res. 2003;37(12):1289–1297. doi:10.1080/10715760310001617776
8. Boulares AH, Ren T. Mechanism of acetaminophen-induced apoptosis in cultured cells: roles of caspase-3, DNA fragmentation factor, and the Ca2+ and Mg2+ endonuclease DNAS1L3. Basic Clin Pharmacol Toxicol. 2004;94(1):19–29.
9. Hinson JA, Roberts DW, James LP. Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol. 2010;196:369–405.
10. Du K, Ramachandran A, Jaeschke H. Oxidative stress during acetaminophen hepatotoxicity: sources, pathophysiological role and therapeutic potential. Redox Biol. 2016;10:148–156. doi:10.1016/j.redox.2016.10.001
11. Craig DG, Lee A, Hayes PC, Simpson KJ. Review article: the current management of acute liver failure. Aliment Pharmacol Ther. 2010;31(3):345–358. doi:10.1111/j.1365-2036.2009.04175.x
12. Cai Y, Xu H, Yan J, Zhang L, Lu Y. Molecular targets and mechanism of action of dexmedetomidine in treatment of ischemia/reperfusion injury. Mol Med Rep. 2014;9(5):1542–1550. doi:10.3892/mmr.2014.2034
13. Gu J, Sun P, Zhao H, et al. Dexmedetomidine provides renoprotection against ischemia-reperfusion injury in mice. Crit Care. 2011;15(3):R153. doi:10.1186/cc10324
14. Kocoglu H, Ozturk H, Ozturk H, Yilmaz F, Gulcu N. Effect of dexmedetomidine on ischemia-reperfusion injury in rat kidney: a histopathologic study. Ren Fail. 2009;31(1):70–74. doi:10.1080/08860220802546487
15. Geze S, Cekic B, Imamoğlu M, et al. Use of dexmedetomidine to prevent pulmonary injury after pneumoperitoneum in ventilated rats. Surg Laparosc Endosc Percutan Tech. 2012;22(5):447–453. doi:10.1097/SLE.0b013e31826183df
16. Sezer A, Memiş D, Usta U, Süt N. The effect of dexmedetomidine on liver histopathology in a rat sepsis model: an experimental pilot study. Ulus Travma Acil Cerrahi Derg. 2010;16(2):108–112.
17. Cavalcanti V, Santos CL, Samary CS, et al. Effects of short-term propofol and dexmedetomidine on pulmonary morphofunction and biological markers in experimental mild acute lung injury. Respir Physiol Neurobiol. 2014;203:45–50. doi:10.1016/j.resp.2014.08.008
18. Kurosawa S, Kato M. Anesthetics, immune cells, and immune responses. J Anesth. 2008;22(3):263–277. doi:10.1007/s00540-008-0626-2
19. Schneemilch CE, Schilling T, Bank U. Effects of general anaesthesia on inflammation. Best Pract Res Clin Anaesthesiol. 2004;18(3):493–507.
20. Kang SH, Kim YS, Hong TH, et al. Effects of dexmedetomidine on inflammatory responses in patients undergoing laparoscopic cholecystectomy. Acta Anaesthesiol Scand. 2013;57(4):480–487.
21. Tas N, Altinbas A, Noyan T, Kokturk S, Ayhan S, Canakci E. Acute acetaminophene-induced hepatotoxicity and nephrotoxicity; therapeutic effect of dexmedetomidine. Bratisl Lek Listy. 2019;120(4):270–276. doi:10.4149/BLL_2019_047
22. Paris A, Mantz J, Tonner PH, Hein L, Brede M, Gressens P. The effects of dexmedetomidine on perinatal excitotoxic brain injury are mediated by the alpha2A-adrenoceptor subtype. Anesth Analg. 2006;102(2):456–461. doi:10.1213/01.ane.0000194301.79118.e9
23. Xu L, Bao H, Si Y, Wang X. Effects of dexmedetomidine on early and late cytokines during polymicrobial sepsis in mice. Inflamm Res. 2013;62(5):507–514. doi:10.1007/s00011-013-0604-5
24. Mendes-Braz M, Elias-Miró M, Jiménez-Castro MB, Casillas-Ramóırez A, Ramalho FS, Peralta. C. The current state of knowledge of hepatic ischemia-reperfusion injury based on its study in experimental models. J Biomed Biotechnol. 2012;2012:298657. doi:10.1155/2012/298657
25. Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology. 2013;58(5):1814–1823. doi:10.1002/hep.26419
26. Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology. 2006;43(6):1220–1230. doi:10.1002/hep.21175
27. Williams CD, Bajt ML, Farhood A, Jaeschke H. Acetaminophen-induced hepatic neutrophil accumulation and inflammatory liver injury in CD18-deficient mice. Liver Int. 2010;30(9):1280–1292. doi:10.1111/j.1478-3231.2010.02284.x
28. Woolbright BL, Jaeschke H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J Hepatol. 2017;66(4):836–848. doi:10.1016/j.jhep.2016.11.017
29. Jaeschke H, Williams CD, Ramachandran A, Bajt ML. Acetaminophen hepatotoxicity and repair: the role of sterile inflammation and innate immunity. Liver Int. 2012;32(1):8–20. doi:10.1111/j.1478-3231.2011.02501.x
30. Lawson JA, Farhood A, Hopper RD, Bajt ML, Jaeschke H. The hepatic inflammatory response after acetaminophen overdose: role of neutrophils. Toxicol Sci. 2000;54(2):509–516. doi:10.1093/toxsci/54.2.509
31. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418(6894):191–195. doi:10.1038/nature00858
32. Ishida Y, Kondo T, Kimura A, Tsuneyama K, Takayasu T, Mukaida N. Opposite roles of neutrophils and macrophages in the pathogenesis of acetaminophen-induced acute liver injury. Eur J Immunol. 2006;36(4):1028–1038. doi:10.1002/eji.200535261
33. Holt MP, Cheng L, Ju C. Identification and characterization of infiltrating macrophages in acetaminophen induced liver injury. J Leukoc Biol. 2008;84(6):1410–1421. doi:10.1189/jlb.0308173
34. McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal. 2006;8(9–10):1775–1789. doi:10.1089/ars.2006.8.1775
35. Torii T, Yamamoto Y, Tsuchiya E, Nishida E. ERK MAP kinase in G cell cycle progression and cancer. Cancer Sci. 2006;97(8):697–702. doi:10.1111/j.1349-7006.2006.00244.x
36. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290. doi:10.1038/sj.onc.1210421
37. Cuenda A. Mitogen-activated protein kinase kinase 4 (MKK4). Int J Biochem Cell Biol. 2000;32(6):581–587. doi:10.1016/s1357-2725(00)00003-0
38. Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterol. 2006;131(1):165–178. doi:10.1053/j.gastro.2006.03.045
39. Bae MA, Pie JE, Song BJ. Acetaminophen induces apoptosis of C6 glioma cells by activating the c-Jun NH2-terminal protein kinase-related cell death pathway. Mol Pharmacol. 2001;60(4):847–856.
40. Jiang L, Li L, Shen J, Qi Z, Guo L. Effect of dexmedetomidine on lung ischemia-reperfusion injury. Mol Med Rep. 2014;9(2):419–426. doi:10.3892/mmr.2013.1867
41. Noh JR, Kim YH, Hwang JH, et al. Davallialactone protects against acetaminophen overdose-induced liver injuries in mice. Food Chem Toxicol. 2013;58:14–21. doi:10.1016/j.fct.2013.04.005
42. Tsuruta F, Sunayama J, Mori Y, et al. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. Embo J. 2004;23(8):1889–1899. doi:10.1038/sj.emboj.7600194
43. Bajt ML, Farhood A, Lemasters JJ, Jaeschke H. Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2008;324(1):8–14. doi:10.1124/jpet.107.129445
44. Win S, Than TA, Han D, Petrovic LM, Kaplowitz N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J Biol Chem. 2011;286:35071–35078. doi:10.1074/jbc.M111.276089
45. Win S, Than TA, Min RW, Aghajan M, Kaplowitz N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology. 2016;63(6):1987–2003. doi:10.1002/hep.28486
46. Schwabe RF, Uchiname H, Qian T, Bennett BL, Lemasters JJ, Brenner DA. Differential requirement for C-Jun NH2-terminal kinase in TNF-α and Fas-mediated apoptosis in hepatocytes. Faseb J. 2004;18(6):720–722. doi:10.1096/fj.03-0771fje
47. Upadhyay KK, Jadeja RN, Thadani JM, et al. Carbon monoxide releasing molecule A-1 attenuates acetaminophen-mediated hepatotoxicity and improves survival of mice by induction of Nrf2 and related genes. Toxicol Appl Pharmacol. 2018;360(1):99–108. doi:10.1016/j.taap.2018.09.034
48. Saito C, Zwingmann C, Jaeschke H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology. 2010;51:246–254. doi:10.1002/hep.23267
49. Kao LW, Kirk MA, Furbee RB, Mehta NH, Skinner JR, Brizendine EJ. What is the rate of adverse events after oral N-acetylcysteine administered by the intravenous route to patients with suspected acetaminophen poisoning? Ann Emerg Med. 2003;42:741–750. doi:10.1016/s0196-0644(03)00508-0
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.