")
Back to Journals » Journal of Inflammation Research » Volume 15
The Key Molecular Mechanisms of Sini Decoction Plus Ginseng Soup to Rescue Acute Liver Failure: Regulating PPARα to Reduce Hepatocyte Necroptosis?
Authors He Y , Zhang Y , Zhang J , Hu X
Received 8 May 2022
Accepted for publication 9 August 2022
Published 22 August 2022 Volume 2022:15 Pages 4763—4784
DOI https://doi.org/10.2147/JIR.S373903
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Ying He,1,2,* Yang Zhang,3,* Junli Zhang,2 Xiaoyu Hu3
1Department of Gastroenterology, Clinical Medical College and the First Affiliated Hospital of Chengdu Medical College, Chengdu, People’s Republic of China; 2Department of College of Clinical Medicine, Chengdu University of Traditional Chinese Medicine, Chengdu, People’s Republic of China; 3Department of Infectious Disease, Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaoyu Hu, Department of Infectious Disease, Hospital of Chengdu University of Traditional Chinese Medicine, No. 39 Shi-er-qiao Road, Chengdu, Sichuan Province, People’s Republic of China, Tel +86 18981883931, Fax +86 28-87766041, Email [email protected]
Purpose: This study aimed to investigate the improvement effect of Sini Decoction plus Ginseng Soup (SNRS) on the LPS/D-GalN-induced acute liver failure (ALF) mouse model and the molecular mechanism of the SNRS effect.
Methods: To study the protective effect of SNRS on ALF mice, the ICR mice were firstly divided into 4 groups: Control group (vehicle-treated), Model group (LPS/D-GalN), SNRS group (LPS/D-GalN+SNRS), and Silymarin group (LPS/D-GalN+Silymarin), the therapeutic drug was administered by gavage 48h, 24h before, and 10 min after LPS/D-GalN injection. On this basis, the peroxisome proliferator-activated receptor (PPAR) α agonist (WY14643) and inhibitor (GW6471) were added to verify whether the therapeutic mechanism of SNRS is related to its promoting effect on PPARα. The animals are grouped as follows: Control group (vehicle-treated), Model group (LPS/D-GalN+DMSO), SNRS group (LPS/D-GalN+SNRS+DMSO), Inhibitor group (LPS/D-GalN+GW6471), Agonist group (LPS/D-GalN+WY14643), and Inhibitor+SNRS group (LPS/D-GalN+GW6471+SNRS).
Results: The protective effect of SNRS on the ALF model is mainly reflected in the reduction of serum alanine aminotransaminase (ALT) and aspartate aminotransaminase (AST) as well as the ameliorated pathology of the liver tissue. The survival rate of ALF mice treated with SNRS was significantly increased. Further mechanism studies showed that SNRS significantly promoted the protein expression of PPARα and decreased the expression of necroptosis proteins (RIP3, MLKL, p-MLKL) in ALF mice. Reduced necroptosis resulted in decreased HMGB1 release, which in turn inhibited the activation of TLR4-JNK and NLRP3 inflammasome signaling pathways and the expression of NF-κB protein induced by LPS/D-GalN. The expression of CPT1A, a key enzyme involved in fatty acid β-oxidation, was found to be significantly up-regulated in the SNRS treated group, accompanied by an increased adenosine-triphosphate (ATP) level, which may be the relevant mechanism by which SNRS reduces necroptosis.
Conclusion: The potential therapeutic effect of SNRS on ALF may be through promoting the expression of PPARα and increasing the level of ATP in liver tissue, thereby inhibiting necroptosis of hepatocytes, reducing hepatocyte damage, and improving liver function.
Keywords: acute liver failure, Sini Decoction plus Ginseng Soup, PPARα, necroptosis, damage-associated molecular patterns, proinflammatory
Introduction
The concept of fulminant hepatitis and fulminant liver failure was introduced by Roger Williams et al in 1993 and is often used as a related surrogate term for ALF. ALF is defined as the rapid loss of liver function caused by severe acute liver injury in the absence of known underlying chronic liver disease. The main clinical manifestations of ALF are jaundice and coagulopathy (international normalized ratio ≥1.5), which rapidly progresses to hepatic encephalopathy.1 Hepatotropic viruses infection and drug-induced liver injury contribute to the majority of ALF cases.2 Clinical treatment of ALF mainly includes internal medical treatment, artificial liver support therapy, stem cell transplantation, etc. Despite the progress in treatment methods, ALF mortality has not been significantly improved for a long time, and studies have shown that ALF mortality without liver transplantation is still close to 90%.3 Scarcity of liver donors, concurrent systemic inflammatory response syndrome (SIRS), and sepsis often lead to premature death of ALF patients before liver transplantation.4 In the future, if a novel therapy can effectively reduce the hepatocyte damage and reduce the local and systemic inflammatory response of the liver, it will greatly reduce the fatality rate of ALF and buy time for liver transplantation.
Hepatocyte necrosis plays an important role in the progression of ALF. Necroptosis, one of the many types of regulated necroptotic cell death, is upregulated in various inflammation-related diseases, including ALF.5 Under the attack of pathogenic factors, a large number of hepatocytes die, resulting in massive hepatic necrosis (MHN), which is the core histological feature and the most serious lesion of ALF.6,7 The destruction of plasma membrane integrity is a feature of all necrotic cell death modes, which is significantly different from other non-necrotic cell death modes, such as apoptosis and autophagy. Membrane rupture results in the release of damage-associated molecular patterns (DAMPs), a group of molecules with strong immunogenicity, into the extracellular region. Therefore, necrosis itself is a highly immunogenic event.8 As a representative product of DAMPs, a high acetylation level of HMGB-1 has been confirmed to be associated with poor prognosis in ALF patients.9 HMGB-1 can synergize with other pro-inflammatory cytokines, such as IL-1β, interferon-γ, and TNF-α to further amplify the inflammatory cascade reaction.10 Amplifying inflammatory cascade reactions mediate SIRS with concomitant multiple organ dysfunction syndromes (MODS) and recurrent infections, which are often the ultimate causes of death in patients with ALF. Therefore, to improve the hyperactivated pro-inflammatory response in ALF, the focus should be shifted from how to reduce hepatocyte death to how to reduce the necrotic death mode of hepatocytes (such as necroptosis) and thus reduce the release of DAMPs, since apoptosis and autophagy have little contribution to inflammation compared with hepatocyte necrosis.
PPARα belongs to the PPARs family, which is known for regulating the expression of genes involved in mitochondrial β-oxidation and reducing inflammation.11,12 Down-regulation of PPARα protein has been repeatedly demonstrated in ALF, and lack of PPARα aggravates LPS-induced hepatotoxicity and increases oxidative/nitrosative stress in liver tissue.13 In the LPS/D-GalN-induced ALF mouse model, activation of PPARα can reduce inflammation and protect the liver from an acute injury.14 Necrosis is thought to be a consequence of metabolic disruption and ATP depletion. In some cases, necroptosis is considered to be transformed from apoptosis. The deprivation of energy supply in cells during the process of apoptosis can induce apoptosis to necroptosis, while restoration of ATP supply or delay ATP depletion, apoptotic signaling may persist.15 The liver consumes about 24% of the body’s energy, and when ALF occurs, the liver’s energy expenditure further increases by up to 30%.16 ALF is usually accompanied by mitochondrial dysfunction,17 which means the balance between liver energy consumption and supply will be further broken. Studies have shown that sepsis suppresses mitochondrial β-oxidation, which leads to an increase in the accumulation of fatty acids (FAs) attached to carnitine molecules in the blood, with a concomitant decrease in energy production. This notion is supported by the blood accumulation of fatty acylcarnitines found in ACLF.18
The down-regulated PPARα may be involved in the impaired mitochondrial β-oxidation in ALF, which leads to the reduction of the energy supply of liver tissue, thus inducing necroptosis in hepatocytes, and finally mediating the excessive immune-inflammatory reactions. Activation of PPARα may improve hepatocyte energy depletion and reduce hepatocyte necroptosis by increasing mitochondrial β-oxidation. In an animal model of sepsis-induced cardiac dysfunction constructed by cecal ligation and puncture, the use of an activator of the PPAR family gene PPAR-γ was shown to inhibit necroptosis by inhibiting RIP1, RIP3, and MLKL.19 However, the regulatory role of PPARα in ALF necroptosis is still unknown, and further experiments are needed to confirm it.
SNRS is from “Treatise on Febrile Diseases” written by Zhong-jing Zhang, which consists of four herbs: Fu Zi (aconite lateral root), Gan Jiang (dried ginger), Zhi Gan Cao (licorice), and Ren Shen (ginseng). The main function of SNRS is to restore Yang for solid doff and promote fluid production, and it is the classical formulae of TCM to rescue emergency and severe cases including MODS, which has been used continuously for more than 1000 years. Studies have shown20 that Gan Jiang Fu Zi Decoction can reduce ischemia-reperfusion-induced apoptosis, alleviate oxidative damage, reduce mitochondrial mPTP opening, and increase ATPase activity. Our previous study also confirmed that Modified Sini Decoction can reduce D-GalN-induced liver necrosis and prolong survival time in ALF rats by reducing the mRNA expression of HMGB1, TLR4, and NF-κB.21 Our further study confirmed that aconitum aqueous extract can alleviate D-GalN-induced liver injury in the ALF rat model by inhibiting apoptosis and promoting liver tissue regeneration.22 Aconite is proposed to enhance the activity of Na+-K+-ATPase in myocardial tissue, reduce the level of MDA in liver tissue, and improve the membrane fluidity of hepatocytes.23 Ginsenoside Rg1 (G-Rg1), the main component of ginseng, can effectively inhibit carbon tetrachloride (CCl4)-induced inflammatory response in ALF by reducing hepatocyte apoptosis and necrosis.24
Taken together, whether it is a multi-herbal formula of SNRS or a single herbal medicine, or even an active ingredient, can participate in the metabolism of ALF energy and the regulation of cell necrosis at multiple targets, thereby producing a therapeutic effect on ALF. Therefore, we believe that SNRS may increase hepatocyte energy supply by activating PPARα, thereby reducing cell necroptosis, inhibiting tissue inflammation, and rescuing impaired hepatocytes. To test this hypothesis, we will construct an ALF mouse model by intraperitoneal (i.p.) injection of LPS/D-GalN, observe the therapeutic effect of SNRS on ALF, and explore the molecular mechanism by which SNRS affects the biomarkers of necroptosis.
Method
Preparation of Formula SNRS
SNRS consists of 4 herbs (Table 1), all of which were from Cheng Du Ji An Kang Pharmaceutical Co., Ltd. (Chuan 20160177). For the modern dosage of Chinese herbal medicine, refer to Tong Xiaolin’s textual criticism of the dosage of prescriptions in the Eastern Han Dynasty and the relevant results of the National 973 Program25 “Relative basic research of classical famous formulas based on quantity-effect relationship”. One tael in the Han Dynasty is equivalent to contemporary 15.625 g, 1 liter is equivalent to 200mL of contemporary, and one aconite is equivalent to contemporary 12 g aconite. Therefore, we converted the dosage of SNRS according to the original document of “Treatise on Febrile Diseases” into the modern dosage: baked licorice 31g, aconite 12g, dried ginger 23g, and ginseng 16 g. A total of 82g of raw herbs of SNRS was added to 600mL distilled water and soaked for 2h. The soaked herbs were heated to boil and continued to be decocted for 1h. Among them, aconite needs to be boiled for 1h in advance as required. The herb residue was filtered using multi-layer gauze, and a freeze dryer was used to freeze-dry the obtained decoctions to obtain solid extracts of SNRS.26 The dosage per kilogram of body weight of mouse was calculated based on 9.01 times the dosage for adults (70Kg).27 The required SNRS freeze-dried powder was calculated according to the daily bodyweight of the mouse and was fully dissolved with saline, then administered intragastrically at 20mL/kg.
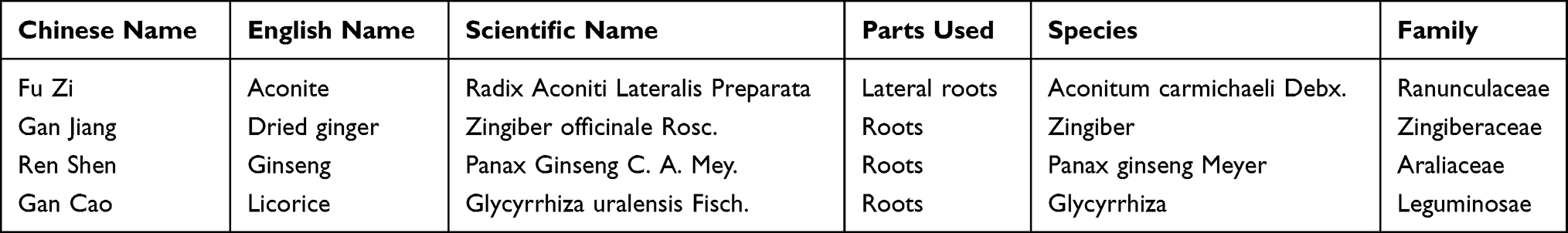 |
Table 1 Contents of SNRS |
Reagents
LPS (L2880-10MG) and D-GalN (G0500-5g) were purchased from Sigma-Aldrich. GW6471 (HY-15372) and WY14643 (HY-16995) were purchased from MedChemExpress. Legalon Silymarin Capsules (H20181067) were purchased from MADAUS GMBH. Sheep Anti-ASC antibody (ab175449), rabbit Anti-Casp1 antibody (A18646), rabbit Anti-JNK antibody (ab179461), mouse Anti-MLKL antibody (ab243142), mouse Anti-P38 MAPK antibody (ab31828), rabbit Anti-p-JNK antibody (ab124956), rabbit Anti-p-MLKL antibody (ab196436), rabbit Anti-PPARα antibody (ab126285), rabbit Anti-RIP3 antibody (ab62344), rabbit Anti-TLR4 antibody (ab13556), goat anti-mouse IgG (H+L) (ab6789), and rabbit anti-goat IgG (H+L) (ab97105) were purchased from Abcam. Rabbit Anti-β-actin antibody (AC026) purchased from Abclonal. Goat anti-rabbit IgG (H+L) (s0001) was purchased from Affinity. Mouse CPT1A ELISA KIT (ZC-54506), Mouse HMGB-1 ELISA KIT (ZC-38180), Mouse TNF-α ELISA KIT (ZC-39024), Mouse IL-1β ELISA KIT (ZC-37974), Mouse NLRP3 ELISA KIT (ZC-38967), Mouse IL-6 ELISA KIT (ZC-37988), and Mouse IL-10 ELISA KIT (ZC-37962) were purchased from Shanghai Zhuocai Biotechnology Co., Ltd. Mouse ATP ELISA KIT (ml981017) and Mouse ADP ELISA KIT (ml063251) were purchased from Shanghai Enzyme-linked Biotechnology Co., Ltd. Total protein (TP) determination kit (A045-2-2), total superoxide dismutase (T-SOD) determination kit (A001-1-2), Malondialdehyde (MDA) determination kit (A003-2-2), and reduced glutathione (GSH) determination kit (A006-2-1) were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Animals and Group Administration
Specific-pathogen-free (SPF) grade, 8–10-week-old, weighing about 25 ± 2 g, and male ICR mouse were purchased from Chengdu Da Shuo Experimental Animal Co., Ltd. (SCXK (Chuan) 2020–030, Chengdu, China). Adaptive feeding for 5–7 days in a barrier SPF environment with a temperature of 22°C, the humidity of 65%, and 12 h light/12 h dark cycle. We first divided the animals into 4 groups according to a random number table: Control group (vehicle-treated), Model group (LPS/D-GalN), SNRS group (LPS/D-GalN+SNRS), and Silymarin group (LPS/D-GalN+Silymarin), (n=25 each group, among them, 15 were used for survival observation and 10 for specimen acquisition). SNRS and Silymarin (100mg/kg) were given by gavage (0.2mL/kg) 48h, and 24h before LPS/D-GalN injection and 10 min after injection,28 respectively. The other groups were administered an equal volume of saline for control. The animals were fasted 12 hours before the experiment, and the drinking water was not restricted. The animals were euthanized 8 hours after the injection of LPS/D-GalN, and the blood and liver samples of the mouse were collected. On this basis, the further study added PPARα agonist (WY14643) and inhibitor (GW6471) to verify whether the mechanism of SNRS’s efficacy is related to its promoting effect on PPARα. We took another 60 mice and randomly divided them into 6 groups: Control group (vehicle-treated), Model group (LPS/D-GalN+DMSO), SNRS group (LPS/D-GalN+SNRS+DMSO), Inhibitor group (LPS/D-GalN+GW6471), Agonist group (LPS/D-GalN+WY14643), and Inhibitor+SNRS group (LPS/D-GalN+GW6471+SNRS) (n=10 each group). WY14643 (10mg/kg) and GW6471 (10mg/kg) were dissolved in DMSO solution (10% DMSO and 90% NS),29–31 and administered (i.p.) 2 h before the injection of LPS/D-GalN, respectively, and the other groups were injected with an equal volume of DMSO. The rest of the groups were treated in the same way as before. Grouping information from animal experiments is shown in Figure 1.
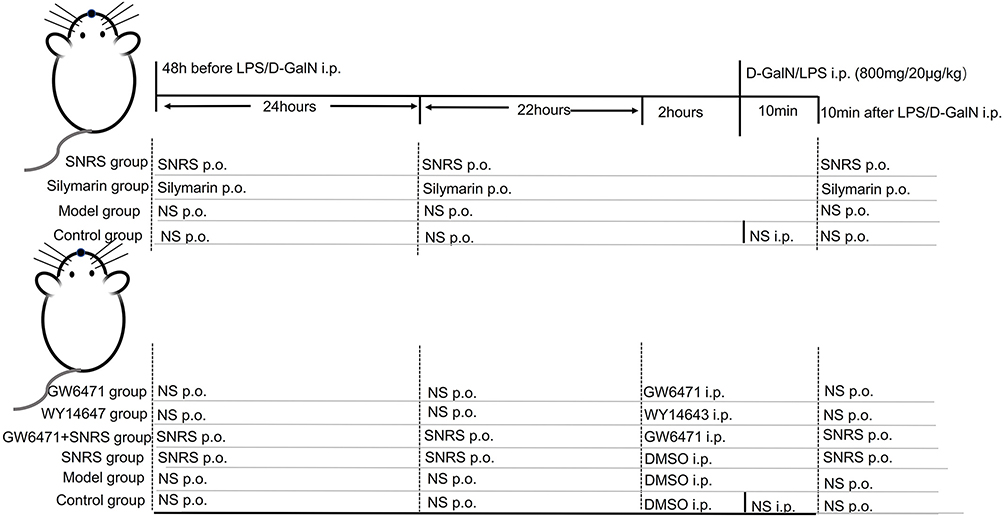 |
Figure 1 Animal grouping. |
Liver Injury Markers Test
Blood was collected and centrifuged for 15 minutes at 3000 rpm and separated the serum for the automatic blood biochemical analyzer to test the AST and ALT.
Pathological Examination
The fresh left lobe liver tissue was immersed in 10% formalin solution for fixation, embedded in paraffin, and cut into 5 μm-thick sections. The sections were subjected to hematoxylin/eosin staining (H&E) to evaluate the pathological changes in the liver. Lesion severity was graded and scored on a scale from 0 to 4 based on observations of liver tissue sections, with 0 being normal or minimal (<1%), 2 being mild (1–25%), and 3 being moderate (26–50%), 4 is moderate/severe (>50%).32
Survival Analysis
After administration of LPS/D-GalN, the death of mice in each group was observed and recorded every 1 h for 24 hours, and the mouse that did not die after the observation was euthanized. Survival rate was calculated according to the Kaplan–Meier method and compared by the Log rank test.
Ultrastructural Hepatocytes
Liver tissue samples were fixed with 3% glutaraldehyde and 1% osmium tetroxide successively, dehydrated by acetone and embedded, then cut into 60–70 nm thick ultra-thin sections, stained with uranium acetate for 10–15 min, and then with lead citrate for 1–2 min. Transmission electron microscopy (JEM-1400Flash, JEOL, Tokyo, Japan) was used to collect images from the copper mesh. Each copper mesh was observed 6000 times before, and the region to be observed was selected to collect images, which were further magnified to 20,000 times to observe specific lesions.
Real-Time Reverse-Transcriptase Polymerase Chain Reaction (RT-PCR)
An appropriate amount of mouse liver tissue was taken, and 1mL of pre-cooled Trizol was added, then ground at high speed and low temperature, stood for 5–15 min, centrifuged at 12000 rpm, and 2–8 °C for 10 min. Each 1mL supernatant with 0.2mL chloroform was added and centrifuged after standing (parameters were the same as before). The supernatant was continued to be collected, an equal amount of isopropanol was added, and after centrifugation again, the RNA precipitate at the bottom of the tube was retained and the supernatant was discarded. The RNA precipitate was washed and air-dried, dissolved in DEPC water, and placed at 4°C for later use. The RNA is then reverse transcribed into cDNA. The RT-PCR reaction parameters were set as 95°C denaturations for the 30s, followed by 40 cycles of denaturation at 95 °C for 5s, 55 °C annealing for 30s, and extension at 72 °C for 30s. The relative expression level of the gene to be tested was expressed by 2−ΔΔCT. PCR primers are shown in Table 2.
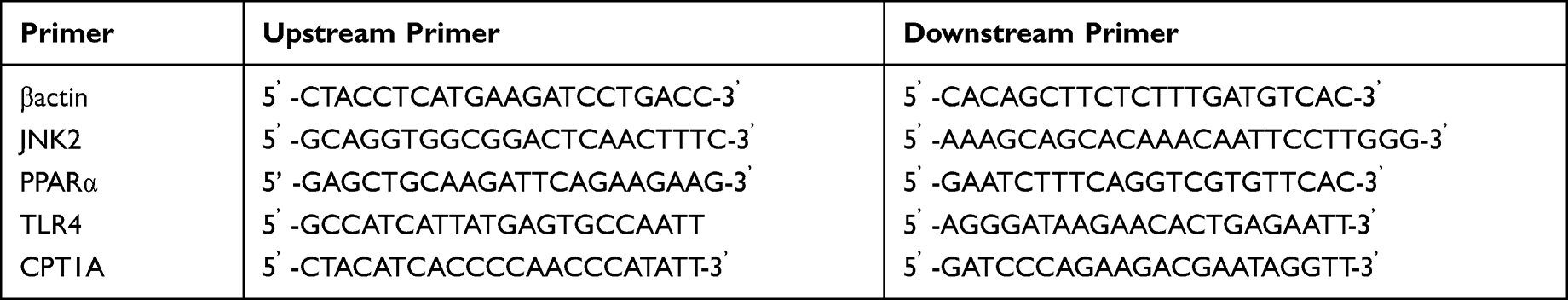 |
Table 2 Primer Design |
Western Blotting
A 100mg liver tissue sample was weighed and added with RIPA lysis solution (G2002-100mL, Servicebio) for high-speed and low-temperature grinding and lysis. After grinding, the sample was centrifuged at 12,000 rpm for 10 min at 4°C, and the supernatant was collected. The BCA kit (P0009, beyotime) was used to determine the protein concentration of the samples. The 32 μg sample was added to the well with a pipette gun, powered on, maintained at 100V voltage for 15 min, isolated in 10% SDS-PAGE gel, and then transferred to PVDF membrane (ISEQ00010, SigmaAldrich). The PVDF membrane was placed into the primary antibody, incubated at 4 °C overnight, and washed by TBST 3 times. Then the PVDF membrane was placed into the secondary antibody, incubated at room temperature for 2–3 hours, and washed with TBST 3 times. The PVDF membrane was tiled onto the exposure plate, and an equal volume of reagent A and B of an ECL luminescent kit was mixed and dropped uniformly onto the PVDF membrane to react for 1 minute. Then put the exposure plate with the film into the darkroom of chemiluminescence gel imager 5200 (Tanon Technology Co., Ltd., Shanghai, China), and adjust the exposure time appropriately according to the strength of the signal to expose (The exposure time was labeled on the original Western blot image). Tanon GIS software version 2.0 (Tanon, Shanghai, China) was used to scan the bands, and the results were normalized to β-actin. The raw data were generated with Photoshop software (Adobe Creative Suite 5). Photoshop software was used to process the original completely uncropped whole membranes to merge the ladder and protein bands without any modification or cutting.
Immunohistochemistry Analysis
Paraffin-embedded liver tissue sections were first dewaxed to water, then immersed in citrate buffer (PH 6.0) (ZLI-9065, Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China) for antigen repair, and then the sections were put in 3% hydrogen peroxide, incubate for 10 min at room temperature, and wash 3 times with PBS to block endogenous peroxidase. Sections were incubated with blocking solution (10% goat serum, 1% RIA-BSA, 0.3% Triton™ X-100 in PBS) for 20 min at room temperature, incubated with primary antibody (mouse Anti-NF-κB p65 antibody [E379], ab32536, Abcam) overnight at 4°C, and washed 3 times with PBS for 5 min each. Then, a secondary antibody (SP9001, Beijing Zhongshan Golden Bridge Biological Co., Ltd.) was added and incubated at 37°C for 30 min. DAB kit (ZLI-9018, Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China) was used for section color development. The sections were washed with distilled water to stop color development, counterstained with hematoxylin for 3 min, washed with water again, and finally sealed with neutral resin. The nuclei stained with hematoxylin are blue, and the positive expression of DAB is brown. A trinocular camera microscope (BA400Digital, McAudi Industrial Co., Ltd., Xiamen, China) was used for image acquisition. All tissues were observed at 100x magnification, and then three microscopic images were acquired at 400x magnification. The Halo data analysis system (Halo 101-WL-HALO-1, Indica labs) was used to calculate the percentage of positive area of each image (% DAB Positive Tissue), and the final result was displayed as the mean of three images.
Enzyme-Linked Immunosorbent Assay (ELISA)
ELISA was used to detect serum HMGB-1 and liver tissue NLRP3, IL-1β, IL-6, IL-10, TNF-α, CPT1A, ADP, and ATP levels. We weighed appropriate liver tissue sample, removed residual blood, added PBS at a mass-to-volume ratio of 1:9, and put it into a high throughput tissue lapping instrument (KZ-III-F, Wuhan Saiwell Biotechnology Co., Ltd., China) for tissue homogenization. Samples were centrifuged at 5000 × g for 10 min and supernatant was collected. According to the instructions of the ELISA kit, carry out the operation steps of sample addition, washing, color development, and termination, and finally, measure the absorbance (OD value) of each hole at the wavelength of 450 nm in sequence.
Bio-Indicators of Oxidative Stress in Liver Tissue
Liver tissue T-SOD, MDA, and GSH activities were detected using the T-SOD assay kit, MDA assay kit, and GSH assay kit, respectively, according to the manufacturer’s protocol.
Statistical Analysis
Statistical analysis was performed using IBM SPSS statistics software (SPSS 25.0. IBM Corp., Armonk, NY, USA) and graphs were drawn by GraphPad Prism (GraphPad Prism 5, Pugh computers, UK). Measurement data were expressed as mean±SD. One-way analysis of variance (ANOVA) was used to examine differences between multiple groups, and pairwise comparisons between groups were performed with LSD or Dunnett’s T3 test. P<0.05 considered the difference to be statistically significant.
Animal Ethics
This experimental protocol was approved by the Experimental Animal Ethics Committee of the Affiliated Hospital of Chengdu University of Traditional Chinese Medicine (2021DL-019). The animal experiments were following humane animal care standards, and all experimental procedures were strictly performed by the requirements of the “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health, USA.
Results
SNRS Attenuates Hepatocyte Injury Induced by LPS/D-GalN Attack in Mice
To evaluate the protective effect of SNRS on ALF mice, silymarin was selected as a positive control in this study. Silymarin, a natural product extracted from Milk thistle (Silybum marianum), has anti-inflammatory and antioxidant properties.33 In the past, several studies have used silymarin as a positive control to compare the efficacy of experimental drugs.34 In the liver failure of all causes, the majority of patients received medical therapy with silymarin or silibinin.35 Silibinin prevents D-GalN/LPS-induced liver injury.36 Compared with the blank control group, all LPS/D-GalN treated mice showed increased ALF and AST to varying degrees. SNRS and Silymarin can effectively reduce ALT and AST levels in the serum of ALF mice, and SNRS is more effective in reducing liver injury markers than Silymarin (Figure 2A and B).
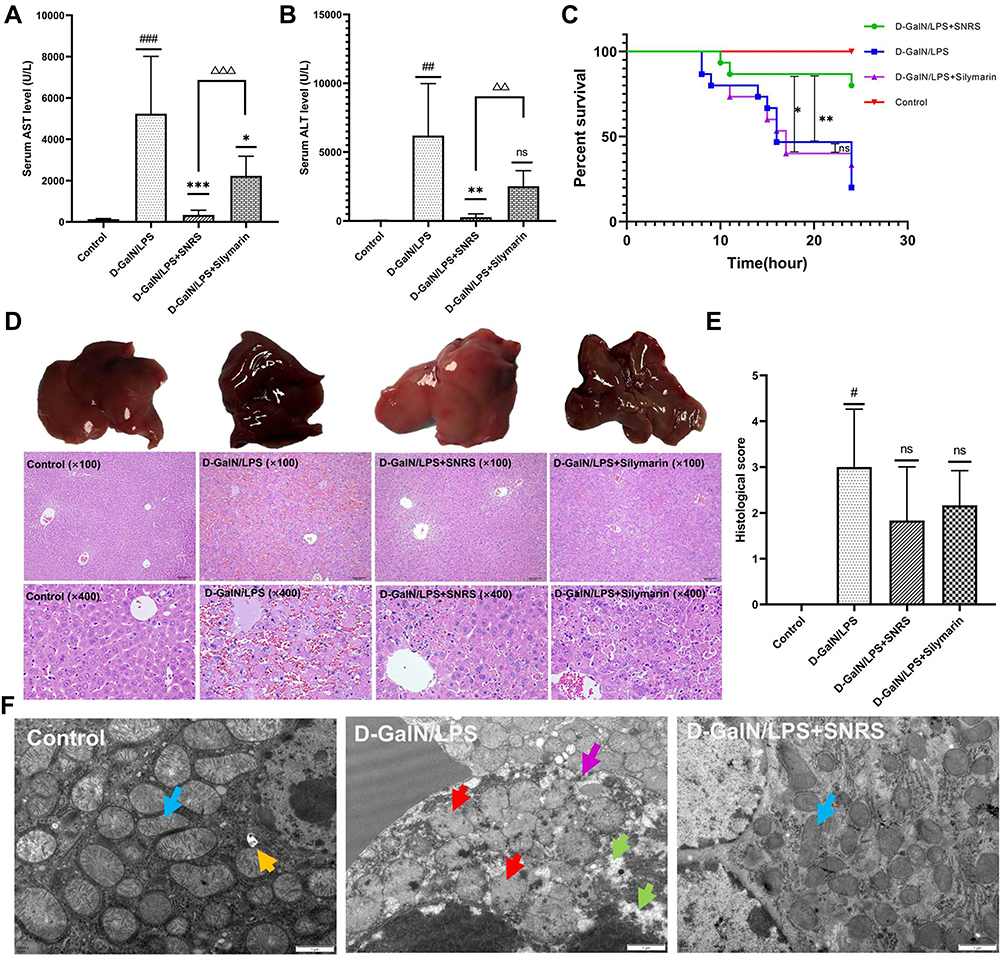 |
Figure 2 Protective effect of SNRS on LPS/D-GalN-induced ALF. Mouse serum was collected at 8 h after LPS/D-GalN injection, and the levels of ALF markers AST (A) and ALT (B) were assessed. (C) Effects of distinct treatments on the 24-hour survival rate of ALF mouse. (D) Hematoxylin and eosin (H & E) staining of mouse liver tissue in different treatment groups, the specimens were examined under microscope (×100 and ×400). (E) The severity of liver tissue sections was graded and scored from 0 to 4. (F) Microscopic changes of LPS/D-GalN attacked hepatocytes (blue arrow: normal mitochondrial structure, clearly visible internal ridge structure, intact mitochondrial membrane; red arrow: mitochondrial swelling and deformation, membrane continuity disruption, ridge disappearance; green arrow: intracellular cytoplasmic loss; purple arrow: discontinuous cell membrane structure; yellow arrow: autophagosome). #Model group compared with control group, #P<0.05, ##P<0.01, ###P<0.001. *Compared to model group, *P<0.05, **P<0.01, ***P<0.01. ΔSNRS group compared with silymarin group, ΔΔP<0.05, ΔΔΔP<0.05. |
Survival analysis of mouse within 24h after LPS/D-GalN injection showed that in the absence of other intervention, the mouse began to die at 8h after LPS/D-GalN attack, with a survival rate of 20% at 24h. Silymarin treatment can improve the 24-hour survival rate of the mouse (33.3%), but it cannot delay the time when mice start to die. The onset of death of SNRS-treated mice was delayed to 10h after LPS/D-GalN injection, and the survival rate increased to 80% (Figure 2C).
We further evaluated the ameliorating effect of SNRS on liver histopathology. After LPS/D-GalN treatment, the liver of the mouse was enlarged and purple. In the liver tissue of ALF mouse, the hepatic lobule structure disorder, hemorrhagic sheet necrosis of hepatocytes, hepatic sinusoidal congestion, and inflammatory cell infiltration in the necrotic area were observed under light microscopy. Silymarin and SNRS can alleviate the above pathological manifestations to varying degrees, and SNRS has significantly better improvement on liver histopathology than Silymarin (Figure 2D). After SNRS treatment, the appearance of the liver was not significantly different from that of the normal liver, the necrosis of hepatocytes was alleviated, and the pathological score was lower than that of the model group (Figure 2E).
Figure 2F shows the microscopic changes of mouse hepatocytes in different groups: The morphological structure of mitochondria in mice was severely damaged by LPS/D-GalN attack, resulting in swelling and deformation, and the disappearance of internal ridge structure, and destruction of mitochondrial membrane integrity. In addition, cells were also necrotic under LPS/D-GalN attack, showing loss of continuity of cell membrane structure and extravasation of cytoplasmic contents. Our research shows that SNRS can effectively improve the destruction of the mouse hepatocyte structure caused by LPS/D-GalN, SNRS was significantly better than silymarin group in improving survival rate, liver enzyme changes, and histopathology in mice.
SNRS Regulates PPARα Expression and Reduces Necroptosis of ALF Hepatocytes
The transcription and protein expression levels of PPARα in mouse liver tissue were significantly decreased after the LPS/D-GalN attack, and SNRS treatment reversed these trends and promoted the expression of PPARα (Figure 3A and B). The final execution of necroptosis is inseparable from the further activation of mixed lineage kinase domain-like protein (MLKL) by phosphorylated RIP3. Phosphorylated MLKL (p-MLKL) translocates to the plasma membrane, leading to membrane rupture, thus p-MLKL, as the final executor of necroptosis, can be used as a specific molecular marker to characterize necroptosis.37 We found that necroptosis-related proteins (RIP3, MLKL, and P-MLKL) were up-regulated in the liver of ALF mice, and the expressions of RIP3 and MLKL were significantly decreased in the SNRS-treated group. After SNRS treatment, the expression of p-MLKL showed a certain downward trend, however, this difference did not meet statistical significance (Figure 3C). The p-MLKL is the final effector molecule of necroptosis, and its phosphorylated structure can better characterize necroptosis than MLKL. In the present study, we observed a trend of inhibition of p-MLKL by SNRS, but since there was no statistical difference, it is too early to conclude that SNRS can inhibit necroptosis. In the following experiments, we will continue to observe the regulatory effect of SNRS on p-MLKL to clarify the effect of SNRS on necroptosis.
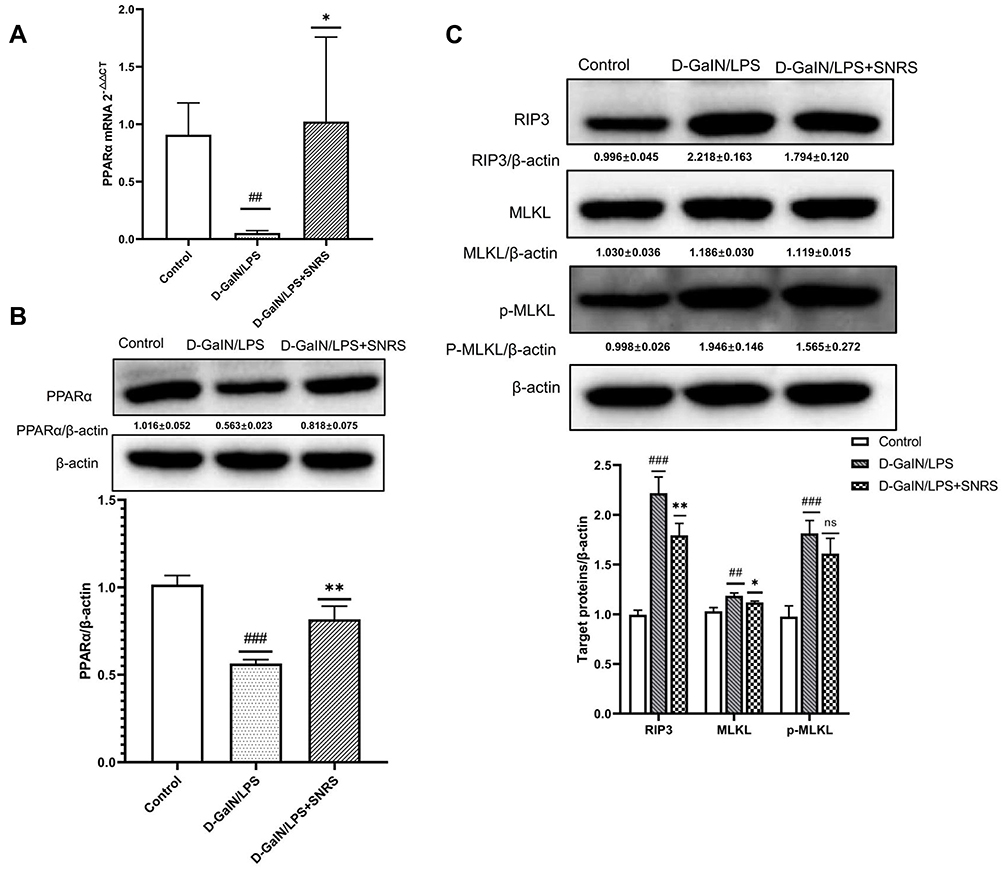 |
Figure 3 The effect of SNRS on PPARα and necroptosis protein (A) The expression of PPARα mRNA was detected by RT-PCR. (B) The effect of SNRS on PPARα protein expression was measured by Western blotting. (C) Changes in necroptosis-related protein RIP3, MLKL, and p-MLKL with SNRS treatment were measured by Western blotting. #Model group compared with control group, ##P<0.01, ###P<0.001. *SNRS group compared with model group, *P<0.05, **P<0.01. |
SNRS Reduces the Release of HMGB-1 and the Level of Inflammatory Cytokines in Liver Tissue
Plasma membrane rupture caused by necroptosis usually triggers the release of a large number of immunogenic DAMPs, and HMGB1 is one of the most well-studied components of DAMPs. We measured the serum HMGB1 level of an SNRS-treated mouse, and the results showed that LPS/D-GalN attack on liver cells resulted in increased HMGB1 release, and SNRS could improve the changing trend of HMGB1 (Figure 4A). HMGB1 is a biomarker and inflammatory mediator in numerous infectious and non-infectious injuries, and its release further promotes the release of inflammatory cytokines.38 We detected inflammatory cytokine levels in mouse liver tissue by ELISA. The results showed that the secretion of pro-inflammatory cytokines like TNF-α, TNF-β, IL-6, and IL-1β was significantly increased after LPS/D-GalN treatment (Figure 4B–E), while the secretion of IL-10, which has been proved to play an anti-inflammatory role in various inflammatory diseases, was decreased39 (Figure 4F). SNRS treatment reduced the secretion of TNF-α, TNF-β, IL-6, and IL-1β and promoted the secretion of IL-10, suggesting that SNRS has significant anti-inflammatory activity.
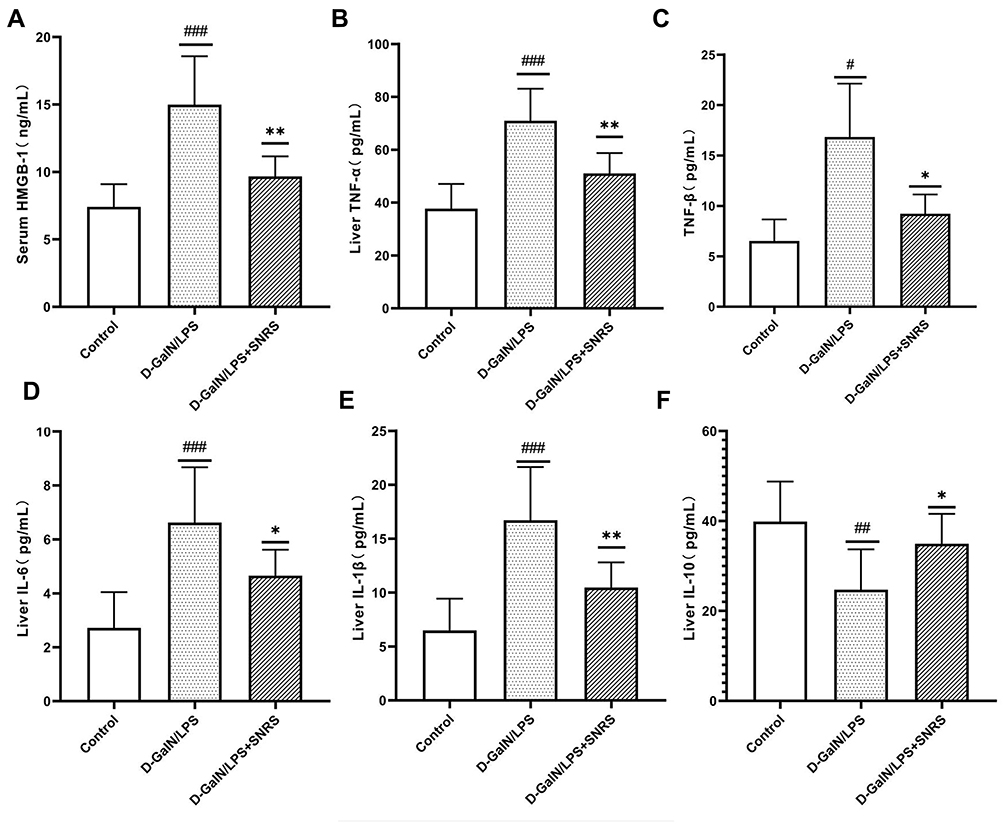 |
Figure 4 The effect of SNRS on cytokines in the liver tissue of ALF mouse. (A) Mouse serum was collected to detect the cell damage marker HMGB1 by ELISA. Mouse liver tissue was collected, and ELISA was used to evaluate the secretion of cytokines TNF-α (B), TNF-β (C), IL-6 (D), IL-1β (E), and IL-10 (F) in liver tissue. #Model group compared with control group, #P<0.05, ##P<0.01, ###P<0.001. *SNRS group compared with model group, *P<0.05, **P<0.01. |
SNRS Inhibited LPS/D-GalN-activated TLR4-JNK and NLRP3 Inflammasome Signaling Pathways and NF-κB Protein Expression
Toll-like receptor 4 (TLR4), as a pattern recognition receptor, can be activated by ligands of pathogen-associated molecular patterns (PAMPs) and DAMPs.40 Activated TLR4 further mediates the activation of MAPK and NF-κB signaling pathways, which normally act as pro-inflammatory signals to promote the production of inflammatory cytokines in response to LPS/D-GalN attack. SNRS treatment suppressed transcript levels of TLR4 and JNK, which were upregulated in ALF (Figure 5A and B). TLR4 protein expression was significantly up-regulated by LPS/D-GalN, and SNRS could inhibit this up-regulation trend of TLR4 (Figure 5C). There was no significant difference in the expression of P38 and JNK proteins between the treatment groups (Figure 5D–F), indicating that the mechanism of LPS/D-GalN and SNRS production was less related to the protein expression of these two molecules. Further studies showed that LPS/D-GalN treatment significantly increased the phosphorylation level of JNK (Figure 5G). Protein phosphorylation is one of the most common post-translational modifications (PTM), and SNRS can reduce the phosphorylation level of JKN, thereby inhibiting JNK activation. These results suggest that SNRS regulation of the JNK pathway may be related to the PTM of molecules in the pathway. NLRP3 inflammasome is composed of NLRP3, ASC, and pre-caspase-1, and can be activated by various membrane damaging agents and PAMPs,41 which can promote Casp1 maturation and further activate the cytokine IL-1β.42 NLRP3 was significantly upregulated after LPS/D-GalN treatment, and the other components of NLRP3 inflammasome, ASC and Casp1, were also upregulated. SNRS treatment significantly reduced the expression levels of NLRP3 and ASC in liver tissues but had little effect on Casp1 protein expression (Figure 5H–K). MAPK can be used as the upstream of NF-κB to activate and maintain NF-κB activity. Immunohistochemistry confirmed that NF-κB was significantly upregulated in ALF liver tissue, while SNRS inhibited the expression of NF-κB (Figure 5L). Activation of MAPK and NF-κB, known pro-inflammatory signaling pathways, usually implies increased secretion of inflammatory cytokines. These results suggest that SNRS inhibits inflammation by inhibiting the phosphorylation of key proteins in the TLR4-JNK pathway and NF-κB protein expression, as well as further inhibiting the NLRP3 and ASC protein expression in NLRP3 inflammasome.
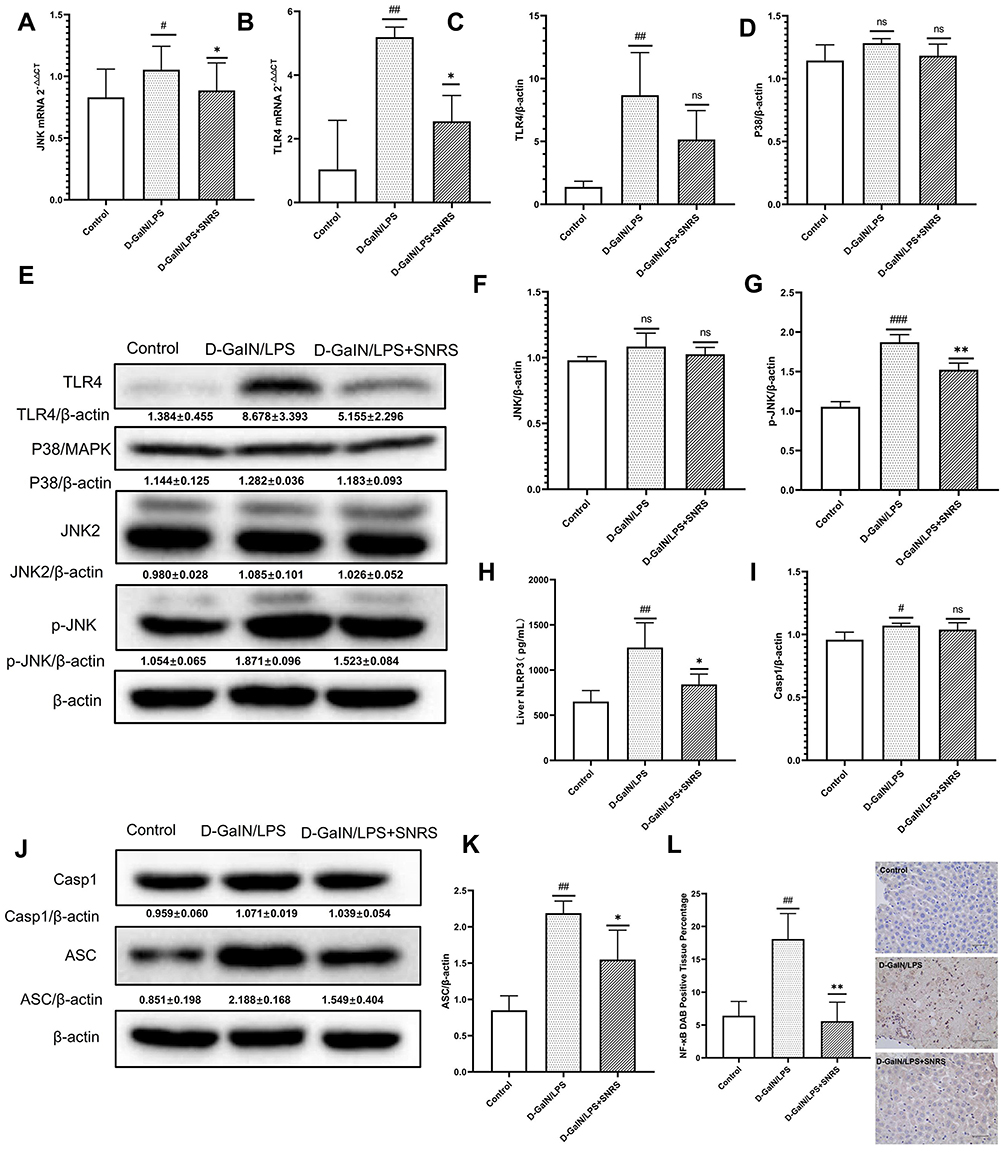 |
Figure 5 Effects of SNRS on inflammatory signaling pathways. Transcript levels of JNK (A) and TLR4 (B) were detected by RT-PCR. (C) Protein expression of TLR4; (D) Protein expression of p38; (E) Western blotting protein bands of TLR4, P38, JNK2, and P-JNK; (F) Protein expression of JNK2; (G) Protein expression of p-JNK2; (H) NLRP3 levels of liver tissue were determined by ELISA; (I) Protein expression of Casp1; (J) Western blotting protein bands of Casp1 and ASC; (K) Protein expression of ASC; (L) immunohistochemical detection of protein expression of NF-κB. #Model group compared with control group, #P<0.05, ##P<0.01, ###P<0.001. *SNRS group compared with model group, *P<0.05, **P<0.01. |
SNRS Protected ALF Mouse from LPS/D-GalN Attack by Regulating PPARα
PPARα has been proven to have protective effects on a variety of acute and chronic liver diseases including liver failure and drug-induced liver injury.43,44 In previous experimental steps, we confirmed that SNRS can promote the expression of PPARα, which was down-regulated by LPS/D-GalN (Figure 3A and B). Therefore, we believe that SNRS may improve the pathological damage of ALF by promoting the expression of PPARα. We pretreated the mouse with an agonist (WY14643) and inhibitor (GW6471) of PPARα to confirm our point. WY14643 can effectively promote the expression of PPARα in liver tissue of ALF mouse, while GW6471 has the opposite effect. Based on GW6471 treatment, superimposing SNRS can reverse the inhibitory effect of GW6471 on PPARα, which confirms that SNRS has a significant promoting effect on PPARα (Figure 6A). Subsequently, we confirmed that both SNRS and WY14643 treatments could effectively reduce the levels of serum liver injury markers (ALT and AST) in ALF mice, while the inhibition of PPARα (GW6471) increased the serum levels of ALT and AST to a certain extent, and SNRS reversed this effect (Figure 6B and C). Liver histopathological examination also suggested the protective effect of PPARα on ALF, promoting the expression of PPARα improved liver histopathology, while inhibiting the expression of PPARα aggravated the damage degree of liver cells. SNRS reversed the negative effect of PPARα inhibitors on the ALF mouse (Figure 6D). These results suggest that SNRS can protect hepatocytes from LPS/D-GalN attack by promoting PPARα.
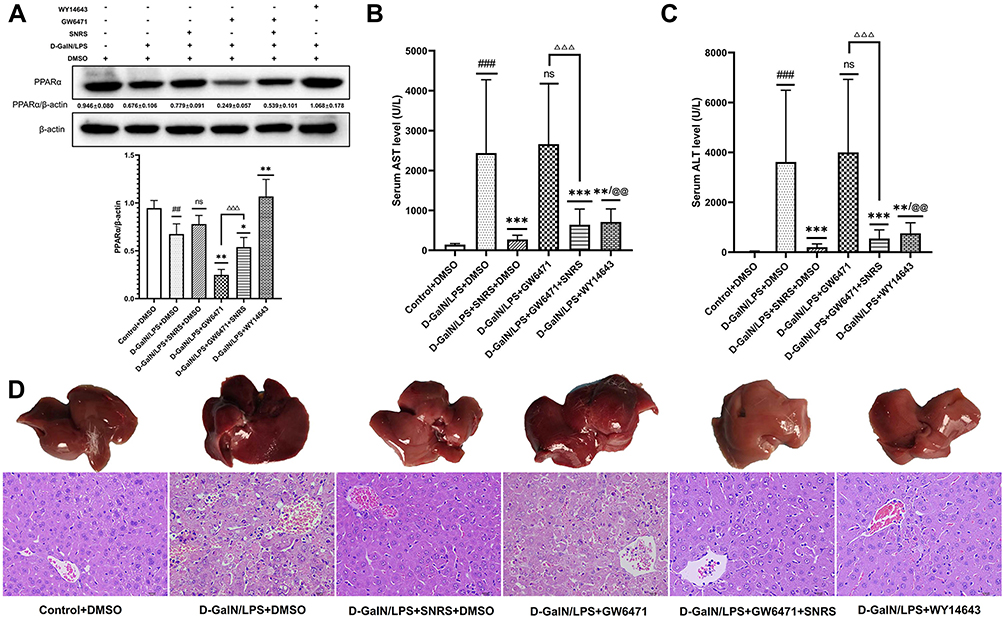 |
Figure 6 SNRS ameliorates liver injury by promoting the expression of PPARα. (A) The expression of PPARα protein in liver tissue of different treatment groups. (B) The serum level of AST. (C) The serum level of ALT. (D) Gross morphology and HE-stained histological section of the liver. #Model group compared with control group, ##P<0.01, ###P<0.001. *Compared to model group, *P<0.05, **P<0.01, ***P<0.001. ΔGW6471 group compared with GW6471+SNRS group, ΔΔΔP<0.05. @SNRS group compared with WY14643 group, @@P<0.01. |
SNRS Increases the Energy Supply of Hepatocytes in ALF Mice by Regulating PPARα
PPAR is best known for its role in regulating cellular lipid metabolism. Studies have shown that almost all downstream target genes of PPAR are involved in fatty acid metabolism and energy homeostasis in some way,45 and PPARα is believed to improve mitochondrial β-oxidation.46 Carnitine palmitoyltransferase (CPT) is a downstream target gene of PPARα.47 CPT1 is a key rate-limiting enzyme regulating β-oxidation of fatty acids, and CPT1A is a subtype of CPT1 that mainly regulates the transmembrane transport of acylcarnitines in mitochondria.48 SNRS can up-regulate the reduced CPT1A level in liver tissue under LPS/D-GalN attack (Figure 7A and B), and this effect may be related to the promotion of PPARα by SNRS (Figure 7C). The severe inflammatory response of the liver caused by ALF consumes a large amount of ATP, and the sharp decrease in ATP leads to more apoptosis transforming into necroptosis. The FAs β-oxidation is involved in the acute injury of liver tissue, and the energy generated by the complete decomposition of FAs through β-oxidation is several times that of ATP generated by carbohydrate metabolism. SNRS promotes the expression of the CPT1A upstream gene PPARα, which indirectly participates in the regulation of hepatocyte β-oxidation in ALF. We further studied the effect of SNRS on ATP, and the results showed that different treatments had little influence on the level of ADP in mouse liver tissue (Figure 7D), which mainly focused on the regulation of ATP. The ATP level in the liver tissue of mice treated with LPS/D-GalN was significantly decreased, and the inhibition of PPARα aggravated the decreasing trend of ATP. The use of SNRS reversed the down-regulation trend of ATP induced by PPARα inhibitor, and the use of PPARα agonists significantly upregulated the level of ATP in liver tissue of ALF mice (Figure 7E). At the same time, SNRS can also reduce the increased ADP/ATP ratio after LPS/D-GalN treatment by promoting PPARα (Figure 7F).
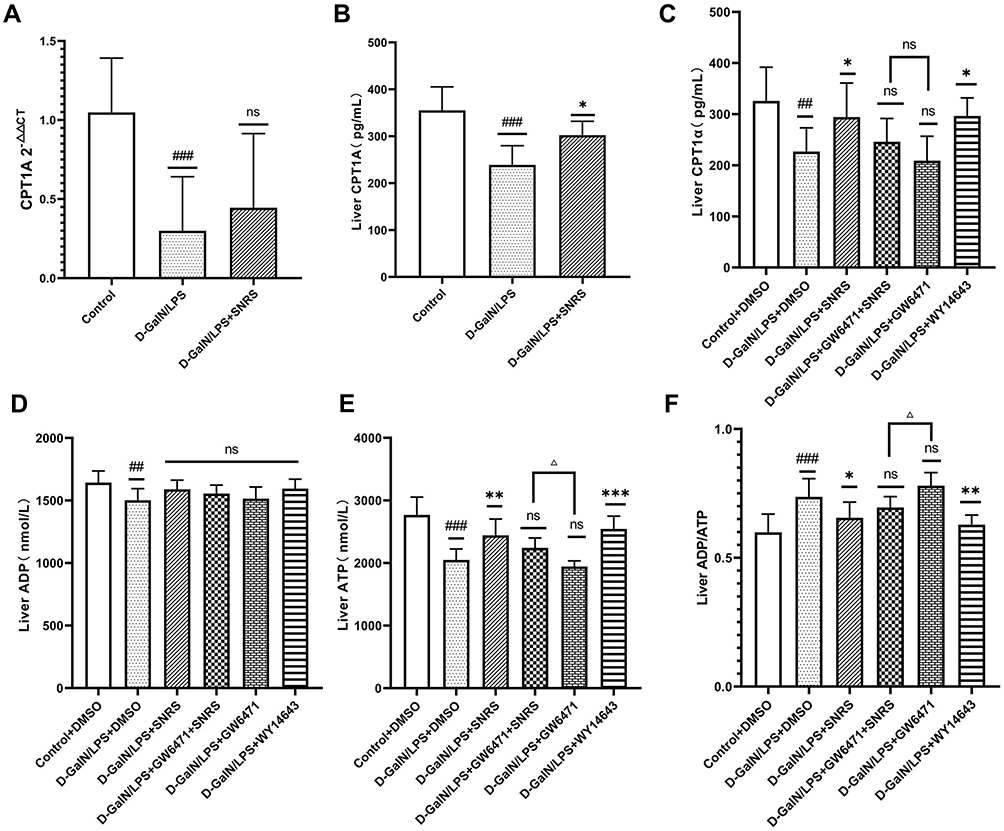 |
Figure 7 The effect of SNRS on energy metabolism in liver cells of ALF mice. (A) CPT1A mRNA expression; (B) Liver tissue CPT1A in 3 groups was detected by ELISA; (C) Liver tissue CPT1A in 6 groups was detected by ELISA; (D) ADP level in the liver of mice; (E) ATP level in the liver of mice; (F) ADP/ATP ratio in the liver of mice. #Model group compared with control group, #P<0.05, ##P<0.01, ###P<0.001. *Compared to model group, *P<0.05, **P<0.01, ***P<0.001. ΔGW6471 group compared with GW6471+SNRS group, ΔP<0.05. |
SNRS Inhibits Hepatocyte Necroptosis in ALF Mice by Regulating PPARα
We compared the changes in necroptosis after the addition of PPARα agonists and inhibitors. The results showed that for the expression of p-MLKL, the final effector protein of necroptosis, both PPARα agonists and SNRS significantly reduced its expression, while GW6471 had an insignificant effect on it. Based on GW6471 treatment, the add of SNRS can significantly reduce the expression of p-MLKL in liver tissue of ALF mice (Figure 8). The above results indicate that the regulation of PPARα can indirectly affect the necroptosis of ALF hepatocytes, and SNRS can reduce the necroptosis by promoting the expression of PPARα.
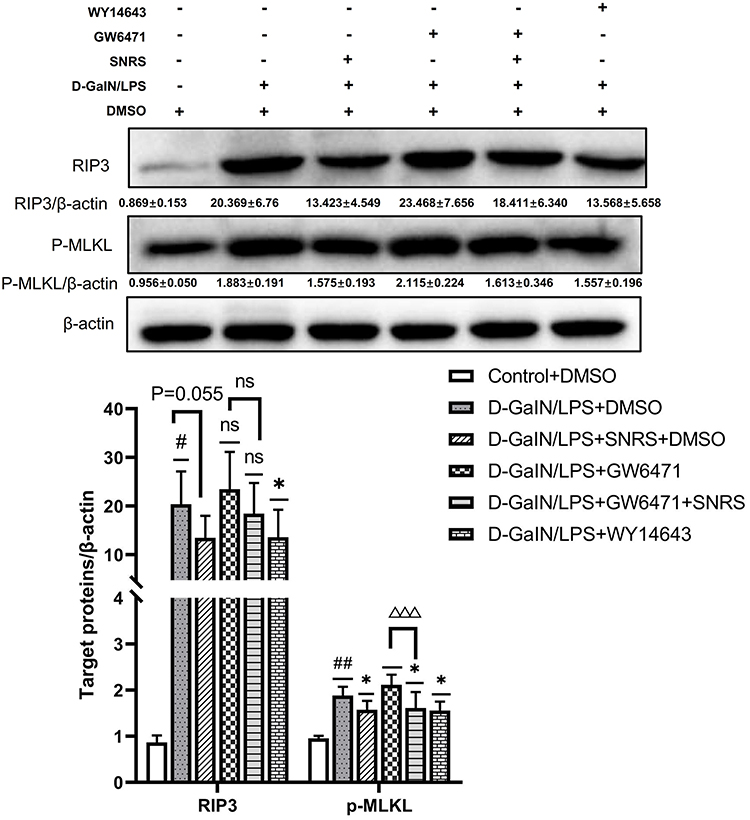 |
Figure 8 SNRS inhibits hepatocyte necroptosis by promoting PPARα. #Model group compared with control group, #P<0.05, ##P<0.01. *Compared to model group, *P<0.05. ΔGW6471 group compared with GW6471+SNRS group, ΔΔΔP<0. 001. |
SNRS Attenuates Oxidative Stress and Inflammation in Liver Tissue by Regulating PPARα
DAMPs released by hepatocyte necroptosis can induce a severe pro-inflammatory response. Up-regulated levels of inflammation and oxidative stress in liver tissue are characteristic pathological changes of ALF and can further exacerbate hepatocyte damage.49 We investigated whether SNRS could reduce the level of inflammation and oxidative stress upregulated in LPS/D-GalN-induced ALF by activating PPARα. The results showed that after inhibiting PPARα, the levels of pro-inflammatory cytokines (TNF-α, TNF-β, IL-1β, and IL-6) in liver tissue of ALF mouse were further increased compared with the model group. SNRS reversed the effect of GW6471, effectively reducing the levels of pro-inflammatory cytokines in ALF mouse liver tissue (Figure 9A–D), at the same time, SNRS also promoted the level of anti-inflammatory cytokine IL-10 (Figure 9E). MDA is the product of reactive oxygen species (ROS) lipid peroxidation, which leads to the lipid-free radical chain reaction to further damage the membrane structure organelles of cells.50 Both SOD and GSH are important antioxidant components of cells, which can neutralize the by-product ROS of oxidative phosphorylation. LPS/D-GalN treatment increased the production of MDA and decreased the levels of antioxidant SOD and GSH in cells. The use of PPARα agonists and SNRS can both reduce MDA and increase the levels of SOD and GSH in liver tissue. The effects of PPARα inhibitors on MDA, SOD, and GSH tended to be opposite to those of PPARα agonists, and SNRS could reverse the effects of PPARα inhibitors (Figure 9F–H). It indicated that SNRS could reduce the level of inflammation and oxidative stress in the liver tissue of ALF mice by promoting the expression of PPARα.
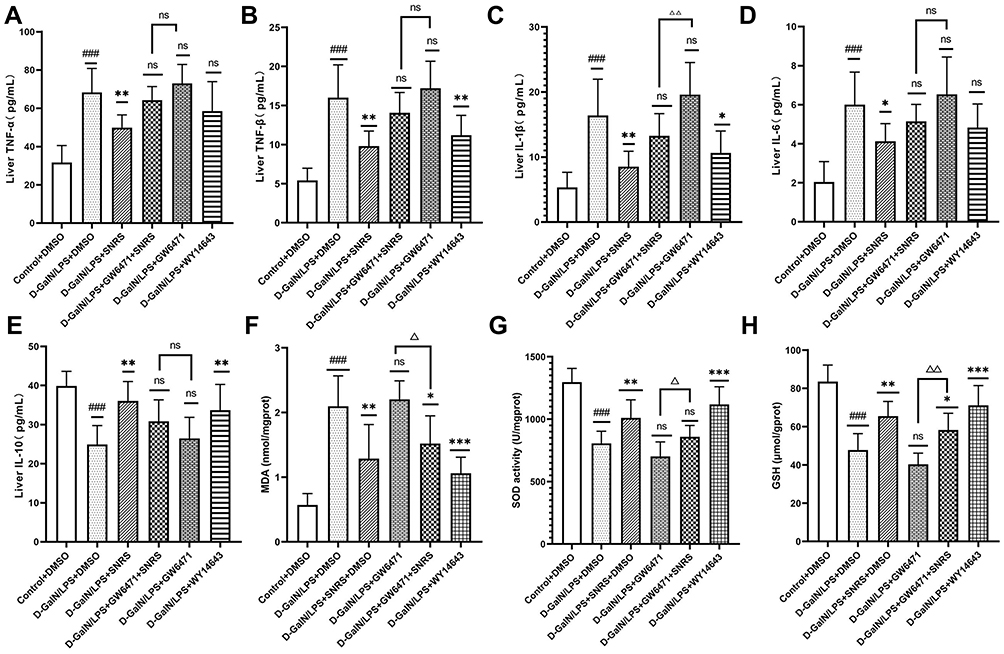 |
Figure 9 SNRS attenuates oxidative stress and inflammation in liver tissue. Changes in cytokines TNF-α (A), TNF-β (B), IL-1β (C), IL-6 (D), and IL-10 (E) in liver tissue with different drug treatments. Changes in oxidative stress-related indexes MDA (F), SOD (G), and GSH (H) in liver tissue with different drug treatments. #Model group compared with control group, ###P<0.001. *Compared to model group, *P<0.05, **P<0.01, ***P<0.001. ΔGW6471 group compared with GW6471+SNRS group, ΔP<0.05, ΔΔP<0.01. |
Discussion
ALF is a rare but fatal severe liver disease with a consistently high mortality rate compared to its low incidence. Immune inflammatory responses are involved in the key mechanisms that promote the progression of ALF. Various immune cells, including Kupffer cells, dendritic cells, and natural killers, are activated in ALF, and a large number of pro-inflammatory cytokines such as TNF-α, IL-1, and IFN-γ are generated subsequently.51 The pathological feature of ALF is massive liver cell death in a short period. For some cases of ALF, it is even difficult to find residual liver cells in liver tissues.56 The necrotic hepatocytes are not the perfect innocent victims. Instead, the damaged liver cells transform themselves from victim to perpetrator by releasing DAMPs to promote a more severe pro-inflammatory response. The MHN in ALF releases large amounts of DAMPs with pro-inflammatory effects, leading to SIRS and concomitant multi-organ failure. Therefore, finding a therapeutic method that can effectively reduce the necroptosis of hepatocytes is the key to improving the excessive immune response of ALF. In this study, we confirmed that SNRS can reduce the mortality of ALF mice and protect some liver cells from LPS/D-GalN attack. Further studies have found that SNRS can significantly reduce hepatocyte necroptosis by promoting the expression of PPARα, thus reducing the release of DAMPs, which may be the key mechanism of SNRS in the treatment of ALF.
The necrosomes formed by RIPK1 and RIPK3 mediate the phosphorylation of RIPK3 by RIPK1,52 and the phosphorylated RIPK3 further phosphorylates MLKL, resulting in the oligomerization of MLKL protein and its translocation to the membrane, which ultimately leads to the rupture of the plasma membrane and release the pro-inflammatory cell contents into the extracellular space.53 HMGB1, one of the most classical DAMPs, was used to characterize the release of cellular contents from necrotic hepatocytes in this experiment. HMGB-1 was identified as an alarm molecule involved in the pathogenesis of acute and chronic liver injury.54 A recent study showed that inhibition of the TNF-α/HMGB1 pathway reduced pyroptosis and tissue macrophage infiltration in LPS/D-GalN-induced mouse models of ALF and AKI.55 The use of SNRS reduced hepatocyte necroptosis (Figure 3C and Figure 8), decreased HMGB1 release, and decreased liver tissue proinflammatory cytokine levels. This may be an important mechanism for SNRS to reduce hepatocyte injury and play a therapeutic role in ALF.
Studies have shown that the inhibition of TLR4 signaling can significantly improve disease prognosis and reduce organ damage.56 PAMPs and DAMPs can act as ligands of TLR4 to activate TLR4 signaling pathways.40 The HMGB1-TLR4 pathway may induce inflammation through multiple mechanisms and is involved in the progression of diseases characterized by cell death and damage, such as ALF.57 The LPS-TLR4 axis has been shown to play an important role in liver failure, where it is involved in inflammatory processes leading to tissue damage.58 The LPS signaling cascade-induced cytokine production is thought to depend on the activation of mitogen-activated protein kinase (MAPK) family members, including p38, extracellular signal-regulated kinase (ERK), and JNK.59 The p38 MAPK can regulate the production of proinflammatory cytokines in response to environmental stress and has been implicated in the pathogenesis of many inflammation-driven conditions.60 TLR4 recognizes extracellular LPS and leads to the activation of myeloid differentiation primary response 88 (MyD88), which then activates NF-κB directly or indirectly by phosphorylating IκBα or by activating the MAPK pathway.61 NF-κB is a nuclear transcription factor that can regulate the expression of cytokines associated with inflammation, such as inducing the synthesis of IL-6, IL-1β, and TNF-a to mediate inflammation.62 Recognition of LPS and HMGB1 by TLR4 on the surface of macrophages leads to an increase in the release of TNF-α, which in turn maintains NF-κB activity by activating MAPK.60 Thus, MAPK and NF-κB play a huge role in cytokine production. In addition, cytokines that depend on MAPK and NF-κB production usually require further processing and maturation. The maturation and release of The pro-IL-1β and pro-IL-18 depend on the activation of the inflammatory complex. ASCs act as adaptor proteins to inflammasome sensor molecules, such as NLRP3 and AIM2, and then act on pro-caspase1 to mature, which in turn induces the processing and maturation of IL-18 and IL-1β.63 In this study, we confirmed that SNRS can inhibit the phosphorylation level of JNK in the TLR4-JNK pathway, thereby inhibiting the signaling of this pathway. At the same time, ALF mice treated with SNRS were observed to down-regulate the expression of NF-κB, These may be important mechanisms by which SNRS exerts its inhibition on the production of pro-inflammatory cytokines. SNRS also inhibited the NLRP3 inflammasome signaling pathway involved in cytokine maturation and release. It can be seen that SNRS can play a multi-channel and multi-target anti-inflammatory effect in the treatment of ALF.
After SNRS treatment, the expression of PPARα and CPT1A in liver tissue was up-regulated, and a marked increase in ATP levels in liver tissue was also observed. Manipulation of intracellular ATP levels can regulate the death mode of cells exposed to injury, and the classical apoptosis pathway can be converted to necroptosis when ATP level is reduced.64 As mentioned above, the intense inflammatory response in ALF is considered to be an energy-intensive biological process. The apparent ATP impairment observed in ALF suggests that the imbalance between ATP supply and demand is an important pathological link in ALF. Therefore, as we derive the mechanisms that influence cell death patterns upward, we turned our attention to the hepatocyte β-oxidation energy supply system. We believe that mitochondrial β oxidation, which is damaged in ALF, leads to the reduction of energy supply, which then leads to hepatocyte apoptosis turning to necroptosis.
PPARα is usually highly expressed in tissues with high fatty acid oxidation (FAO) rates such as liver and adipose tissue and is well known to function as a transcriptional regulator of genes related to FAs β-oxidation.65,66 In addition, pretreatment with PPARα can reduce the secretion of TNF, IL-1, and IL-6 induced by LPS to a certain extent.67 This study also observed that activation of PPARα could significantly inhibit the release of pro-inflammatory cytokines, thereby improving the pathological changes in ALF mice.
There are various explanations for why PPARα can exert anti-inflammatory effects, such as PPARα may mediate the up-regulation of certain anti-inflammatory genes.68 At the same time, PPARα was confirmed to have negative effects on the nuclear signaling pathways such as AP1 and NF-κB.69 PPARα can inhibit NF-κB-induced genes, such as VCAM-1, COX-2, and IL-6,70,71 thereby inhibiting the inflammatory responses mediated by the NF-κB pathway. This effect is consistent with our findings that downregulation of NF-κB was found in the PPARα agonist group. As mentioned above, PPARα activated by SNRS promotes FAs β-oxidation and reduces necroptosis by increasing ATP supply and reducing the immune inflammatory response of liver tissue. Of course, this is only one of the anti-inflammatory effects of PPARα.
The relationship between metabolism and inflammation is inseparable. Given the important role of PPARα in nutrient metabolism, it is reasonable to suspect that part of the anti-inflammatory effect of PPARα may be involved in the immunometabolism, which is also a key link in the anti-inflammatory effect of PPARα. The components of the immune system can play a vital role in regulating metabolism. Meanwhile, metabolism not only provides energy but also regulates the phenotype and function of macrophages, thus adversely affecting immunity.72,73 Adipose tissue, which plays an important role in metabolic diseases, has long been regarded as an active endocrine organ. Adipokines secreted by adipocytes include a variety of active substances related to immune inflammatory responses, such as leptin, TNFα, and IL-6.74 Therefore fat metabolism obstacle is usually associated with chronic low-level inflammation. PPARα plays an important role in hepatic FAs catabolism and clearance, and PPARα can reduce chronic liver inflammation caused by obesity. PPARα activation reduces hepatic triglyceride levels and alleviates steatohepatitis,43,75 while PPARα downregulation leads to hepatic triglyceride accumulation.45,76
Of course, the above content mainly reflects the contribution of PPARα-mediated fat metabolism in chronic persistent inflammation. For ALF, we pay more attention to the relationship between lipid metabolism and immune response. Infection and inflammation induce multiple alterations in lipid metabolism, usually manifested as lipolysis, increased de novo hepatic fatty acid synthesis, and suppression of fatty acid oxidation.77 Accumulation of lipid metabolites is associated with systemic inflammation.77,78 During acute inflammation, severe systemic inflammation inhibits mitochondrial β-oxidation, resulting in increased blood fatty acylcarnitine levels 60. Macrophages activated via the LPS/TLR4 pathway exhibited increased accumulation of succinate, leading to the induction of IL-1β.79 Increased levels of IL-1β in liver tissue of ALF mice were also observed in our study.
Mitochondrial dysfunction in ALF is an important factor leading to the reduction of β-oxidation levels. In this study, we characterized the damage of ALF to the mitochondrial morphological structure of hepatocytes by transmission electron microscopy, which resulted in the reduction of ATP levels (Figure 2). It has been suggested that inhibition of mitochondrial β-oxidation in ACLF may lead to a reduction in FAs-derived energy supply in peripheral organs.18 It has been reported in the past that impaired mitochondrial β -oxidation and ATP depletion lead to hepatocyte necrosis and ALF.80
LPS and cytokines are abundantly produced in ALF, which induces lipolysis by reducing the expression of acyl-CoA synthase (ACS) in adipose tissue.81 Mitochondrial ACS converts FAs to fatty acyl-CoA, and CPT1A catalyzes the transfer of acyl groups of long-chain FAs to carnitine to form acyl-carnitines, which is a key step in the entry of long-chain fatty acids into mitochondria.82 As a downstream target gene of PPARα, CPT1A has been confirmed to be down-regulated in the ALF mouse model in this study, while SNRS can significantly up-regulate the expression of CPT1A.
Severe inflammation-induced reduction in mitochondrial ACS prevents FAs from entering mitochondria for oxidation.77 This process is accompanied by downregulation of PPARα and decreased expression of CPT1A. FAs metabolism disorder will lead to increased production of free FAs (FFAs), which can induce ROS generation and endoplasmic reticulum stress by activating the unfolded protein response signaling pathway in adipocytes, thereby leading to aggravation of inflammation.83,84 In this study, we observed that the secretion of a large number of pro-inflammatory cytokines was inhibited by SNRS and WY14643. The mechanism may be partially explained by the regulation of immunometabolism after activation of PPARα. In the present study, we confirmed the exact regulatory effect of SNRS on PPARα (Figure 3) and confirmed that the use of SNRS can significantly increase ALF liver tissue ATP levels (Figure 7).
The regulation of PPARα by the constituent drug pairs in SNRS or a single herb has been reported for a long time. The combined application of aconite and dried ginger can significantly up-regulate the gene transcription and protein expression levels of PPARα, and improve ventricular muscle function.85 Ginsenoside Rg1, an important component of ginseng, can promote the expression of PPARα and CPT1A, thereby regulating lipid metabolism and effectively inhibiting liver damage caused by the release of IL-6 and TNF-α.86 The results of this study were largely consistent with the effects of SNRS on ALF that we observed (Figure 4). Nanoparticle conjugation of ginsenoside Rb3 can reshape cardiac energy metabolism by targeting the PPARα pathway to protect energy metabolism and mitochondrial function.87 6-gingerol, the active ingredient of ginger, can increase β-oxidation by activating PPARα and CPT1A. In addition, 6-gingerol can inhibit the decrease of ATP, alleviate the damage caused by ROS, and synergistically enhance mitochondrial function.88 The glycyrrhizic acid extracted from licorice can increase lipid metabolism by inducing the expression of PPARα and CPT1A, thereby exerting a protective effect on nonalcoholic steatohepatitis in mice.89 In conclusion, various active components in SNRS can increase mitochondrial function and reshape energy metabolism by promoting PPARα to regulate lipid metabolism.
However, it may be too early to conclude that PPARα can improve tissue ATP supply by regulating FAs β-oxidation and saving hepatocytes from inflammation, and a more rigorous and logical experimental design is still needed to confirm. In any case, we confirmed that SNRS can effectively promote the expression of PPARα, inhibiting the necroptosis of hepatocytes, reducing the level of oxidative stress and inflammation in liver tissue, thereby reducing the mortality of ALF mice and saving some hepatocyte immune cells from LPS/D-GalN attack.
Conclusion
SNRS can alleviate inflammation and oxidative stress injury by increasing hepatic ATP supply, reducing hepatocyte necroptosis, reducing DAMPs release, reducing TLR4-JNK and NLRP3 inflammasome pathway signal transduction, and inhibiting NF-κB protein expression. The mechanism may be related to the activation of PPARα by SNRS (Figure 10). This study involves part of the research on CPT1A, a key molecule of FAs β-oxidation, but further experiments are needed to confirm the regulatory effect of SNRS on FAs β-oxidation. Regardless, this study confirms the important potential of SNRS in the treatment of ALF, which will provide new insights into the application of traditional Chinese medicine in the field of liver disease.
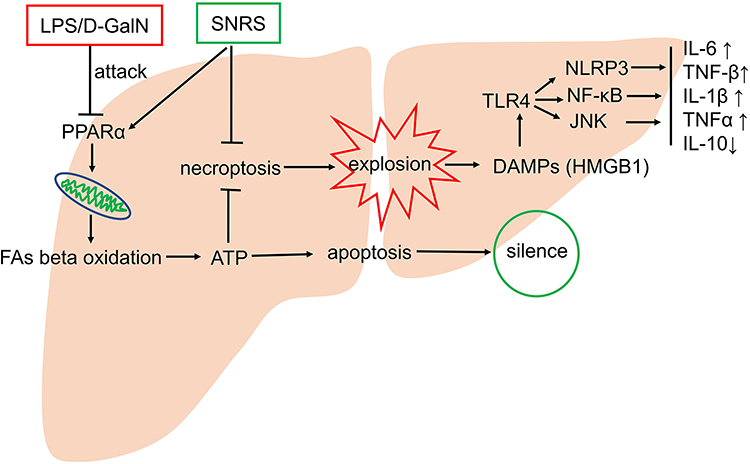 |
Figure 10 Possible mechanisms of SNRS in the treatment of ALF. |
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81973840).
Disclosure
The authors report no conflicts of interest in this work.
References
1. O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342(8866):273–275. doi:10.1016/0140-6736(93)91818-7
2. Lee WM. Acute liver failure. Semin Respir Crit Care Med. 2012;33(1):36–45. doi:10.1055/s-0032-1301733
3. Bernal W, Wong T, Wendon J. High-volume continuous veno-venous haemofiltration in hyper-acute liver failure: a pilot study. Critical Care. 1999;3(Suppl 1):P212. doi:10.1186/cc585
4. Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology. 2003;125(3):755–764. doi:10.1016/S0016-5085(03)01051-5
5. Seo M-J, Hong J-M, Kim S-J, Lee S-M. Genipin protects d-galactosamine and lipopolysaccharide-induced hepatic injury through suppression of the necroptosis-mediated inflammasome signaling. Eur J Pharmacol. 2017;812:128–137. doi:10.1016/j.ejphar.2017.07.024
6. Dong V, Nanchal R, Karvellas CJ. Pathophysiology of acute liver failure. Nutr Clin Pract. 2020;35(1):24–29. doi:10.1002/ncp.10459
7. Liang TJ, Hepatitis B. the virus and disease. Hepatology. 2009;49(5 Suppl):S13–21. doi:10.1002/hep.22881
8. Sarhan M, Land WG, Tonnus W, Hugo CP, Linkermann A. Origin and consequences of necroinflammation. Physiol Rev. 2018;98(2):727–780. doi:10.1152/physrev.00041.2016
9. Antoine DJ, Dear JW, Lewis PS, et al. Mechanistic biomarkers provide early and sensitive detection of Acetaminophen-induced acute liver injury at first presentation to hospital. Hepatology. 2013;58(2):777–787. doi:10.1002/hep.26294
10. Andersson U, Wang H, Palmblad K, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192(4):565–570. doi:10.1084/jem.192.4.565
11. Chinetti-Gbaguidi G, Fruchart J-C, Staels B. Role of the PPAR family of nuclear receptors in the regulation of metabolic and cardiovascular homeostasis: new approaches to therapy. Curr Opin Pharmacol. 2005;5(2):177–183. doi:10.1016/j.coph.2004.11.004
12. Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta. 2011;1812(8):1007–1022. doi:10.1016/j.bbadis.2011.02.014
13. Standage SW, Caldwell CC, Zingarelli B, Wong HR. Reduced peroxisome proliferator-activated receptor α expression is associated with decreased survival and increased tissue bacterial load in sepsis. Shock. 2012;37(2):164–169. doi:10.1097/SHK.0b013e31823f1a00
14. Jiao M, Ren F, Zhou L, et al. Peroxisome proliferator-activated receptor α activation attenuates the inflammatory response to protect the liver from acute failure by promoting the autophagy pathway. Cell Death Dis. 2014;5(8):e1397. doi:10.1038/cddis.2014.361
15. El-Gibaly AM, Scheuer C, Menger MD, Vollmar B. Improvement of rat liver graft quality by pifithrin-alpha-mediated inhibition of hepatocyte necrapoptosis. Hepatology. 2004;39(6):1553–1562. doi:10.1002/hep.20243
16. Schneeweiss B, Pammer J, Ratheiser K, et al. Energy metabolism in acute hepatic failure. Gastroenterology. 1993;105(5):1515–1521. doi:10.1016/0016-5085(93)90159-A
17. Xue R, Zhu X, Jia L, et al. Mitofusin2, a rising star in acute‐on‐chronic liver failure, triggers macroautophagy via the mTOR signalling pathway. J Cell Mol Med. 2019;23(11):7810–7818. doi:10.1111/jcmm.14658
18. Moreau R, Clària J, Aguilar F, et al. Blood metabolomics uncovers inflammation-associated mitochondrial dysfunction as a potential mechanism underlying ACLF. J Hepatol. 2020;72(4):688–701. doi:10.1016/j.jhep.2019.11.009
19. Peng S, Xu J, Ruan W, Li S, Xiao F. PPAR-γ activation prevents septic cardiac dysfunction via inhibition of apoptosis and necroptosis. Oxid Med Cell Longev. 2017;2017:8326749. doi:10.1155/2017/8326749
20. Xie F, Wu -Y-Y, Duan G-J, et al. Anti-myocardial ischemia reperfusion injury mechanism of dried ginger-aconite decoction based on network pharmacology. Front Pharmacol. 2021;12:609702. doi:10.3389/fphar.2021.609702
21. Luo J, Zhang Y, Hu X, et al. The effects of modified sini decoction on liver injury and regeneration in acute liver failure induced by D-galactosamine in rats. J Ethnopharmacol. 2015;161:53–59. doi:10.1016/j.jep.2014.12.003
22. Luo J-X, Zhang Y, Hu X-Y, et al. Aqueous extract from aconitum carmichaelii debeaux reduces liver injury in rats via regulation of HMGB1/TLR4/NF-ΚB/caspase-3 and PCNA signaling pathways. J Ethnopharmacol. 2016;183:187–192. doi:10.1016/j.jep.2016.01.020
23. Zhang T, Wang G, Bai S, Bai J, Tian L, Hu W. Experimental study on the effect of aconite on the antioxidant system of aged rats. Chin J Gerontol. 2001;2002(02):135–136.
24. Luo H, Huang W, Yang C, et al. Therapeutic efficacy and mechanism of action of ginsenosideRg1 in treating acute hepatic failure in mice. J Chin J Hepatol. 2017;25(03):217–222.
25. Tong X, Mu L, Ji H, Zhen Z, Chang B, Dong L. Textual research on dosage of Treatise on Exogenous Febrile Disease. J Tradit Chin Med. 2009;50(04):368–372.
26. Lin X, Shao T, Huang L, et al. Simiao decoction alleviates gouty arthritis by modulating proinflammatory cytokines and the gut ecosystem. Front Pharmacol. 2020;11:955. doi:10.3389/fphar.2020.00955
27. Du L, Ma Y, Liu M, Yan L, Tang H. Peroxisome Proliferators Activated Receptor (PPAR) agonists activate hepatitis B virus replication in vivo. Virol J. 2017;14(1):96. doi:10.1186/s12985-017-0765-x
28. Yamaura K, Shimada M, Nakayama N, Ueno K. Protective effects of goldenseal (Hydrastis canadensis L.) on Acetaminophen-induced hepatotoxicity through inhibition of CYP2E1 in rats. Pharmacognosy Res. 2011;3(4):250–255. doi:10.4103/0974-8490.89745
29. Rahmatollahi M, Baram SM, Rahimian R, Saeedi Saravi SS, Dehpour AR. Peroxisome proliferator-activated receptor-α inhibition protects against doxorubicin-induced cardiotoxicity in mice. Cardiovasc Toxicol. 2016;16(3):244–250. doi:10.1007/s12012-015-9332-0
30. Kong L, Ren W, Li W, et al. Activation of peroxisome proliferator activated receptor alpha ameliorates ethanol induced steatohepatitis in mice. Lipids Health Dis. 2011;10(1):246. doi:10.1186/1476-511X-10-246
31. Yang R, Wang P, Chen Z, et al. WY-14643, a selective agonist of peroxisome proliferator-activated receptor-α, ameliorates lipopolysaccharide-induced depressive-like behaviors by preventing neuroinflammation and oxido-nitrosative stress in mice. Pharmacol Biochem Behav. 2017;153:97–104. doi:10.1016/j.pbb.2016.12.010
32. Wu C-T, Deng J-S, Huang W-C, Shieh P-C, Chung M-I, Huang G-J. Salvianolic acid C against acetaminophen-induced acute liver injury by attenuating inflammation, oxidative stress, and apoptosis through inhibition of the Keap1/Nrf2/HO-1 signaling. Oxid Med Cell Longev. 2019;2019:9056845. doi:10.1155/2019/9056845
33. Giorgi VSI, Peracoli MTS, Peracoli JC, Witkin SS, Bannwart-Castro CF. Silibinin modulates the NF-κb pathway and pro-inflammatory cytokine production by mononuclear cells from preeclamptic women. J Reprod Immunol. 2012;95(1–2):67–72. doi:10.1016/j.jri.2012.06.004
34. Hong W, Hwang-Bo J, Jeon H, et al. A comparative study of the hepatoprotective effect of centella asiatica extract (CA-HE50) on lipopolysaccharide/d-galactosamine-induced acute liver injury in C57BL/6 mice. Nutrients. 2021;13(11):4090. doi:10.3390/nu13114090
35. Karvellas CJ, Tillman H, Leung AA, et al. Acute liver injury and acute liver failure from mushroom poisoning in North America. Liver Int. 2016;36(7):1043–1050. doi:10.1111/liv.13080
36. Hashem RM, Hassanin KM, Rashed LA, Mahmoud MO, Hassan MG. Effect of silibinin and vitamin E on the ASK1-p38 MAPK pathway in D-galactosamine/lipopolysaccharide induced hepatotoxicity. Exp Biol Med. 2016;241(11):1250–1257. doi:10.1177/1535370216636719
37. Chen X, Li W, Ren J, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24(1):105–121. doi:10.1038/cr.2013.171
38. Yamada S, Maruyama I. HMGB1, a novel inflammatory cytokine. Clin Chim Acta. 2007;375(1–2):36–42. doi:10.1016/j.cca.2006.07.019
39. Schetter AJ, Heegaard NHH, Harris CC. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis. 2010;31(1):37–49. doi:10.1093/carcin/bgp272
40. Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi:10.1126/science.282.5396.2085
41. Broz P, Cossart P, Roy CR, Sansonetti P. Recognition of intracellular bacteria by inflammasomes. Microbiol Spectr. 2019;7(2):2. doi:10.1128/microbiolspec.BAI-0003-2019
42. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254. doi:10.1016/j.tibs.2016.10.004
43. Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39(5):1286–1296. doi:10.1002/hep.20170
44. Patterson AD, Shah YM, Matsubara T, Krausz KW, Gonzalez FJ. Peroxisome proliferator-activated receptor alpha induction of uncoupling protein 2 protects against Acetaminophen-induced liver toxicity. Hepatology. 2012;56(1):281–290. doi:10.1002/hep.25645
45. Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103(11):1489–1498. doi:10.1172/JCI6223
46. Ashibe B, Motojima K. Fatty aldehyde dehydrogenase is up-regulated by polyunsaturated fatty acid via peroxisome proliferator-activated receptor alpha and suppresses polyunsaturated fatty acid-induced endoplasmic reticulum stress. FEBS J. 2009;276(23):6956–6970. doi:10.1111/j.1742-4658.2009.07404.x
47. Schoonjans K, Staels B, Auwerx J. Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J Lipid Res. 1996;37(5):907–925. doi:10.1016/S0022-2275(20)42003-6
48. Schlaepfer IR, Joshi M. CPT1A-mediated fat oxidation, mechanisms, and therapeutic potential. Endocrinology. 2020;161(2):bqz046. doi:10.1210/endocr/bqz046
49. Fakurazi S, Hairuszah I, Nanthini U. Moringa oleifera Lam prevents Acetaminophen induced liver injury through restoration of glutathione level. Food Chem Toxicol. 2008;46(8):2611–2615. doi:10.1016/j.fct.2008.04.018
50. Sharma D, Trivedi SS, Bhattacharjee J. Oxidative stress and eNOS (Glu298Asp) gene polymorphism in preeclampsia in Indian population. Mol Cell Biochem. 2011;353(1–2):189–193. doi:10.1007/s11010-011-0786-5
51. Wang X, Ning Q. Immune mediated liver failure. EXCLI J. 2014;13:1131–1144.
52. Cho Y, Challa S, Moquin D, et al. Phosphorylation-driven assembly of RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–1123. doi:10.1016/j.cell.2009.05.037
53. Wallach D, Kang T-B. Programmed cell death in immune defense: knowledge and presumptions. Immunity. 2018;49(1):19–32. doi:10.1016/j.immuni.2018.06.019
54. Wang H-L, Tsao S-M, Yeh C-B, Chou Y-E, Yang S-F. Circulating level of high mobility group box-1 predicts the severity of community-acquired pneumonia: regulation of inflammatory responses via the c-Jun N-terminal signaling pathway in macrophages. Mol Med Rep. 2017;16(3):2361–2366. doi:10.3892/mmr.2017.6892
55. Wang Y, Zhang H, Chen Q, et al. TNF-α/HMGB1 inflammation signalling pathway regulates pyroptosis during liver failure and acute kidney injury. Cell Prolif. 2020;53(6):e12829. doi:10.1111/cpr.12829
56. Mohammed S, Nicklas EH, Thadathil N, et al. Role of necroptosis in chronic hepatic inflammation and fibrosis in a mouse model of increased oxidative stress. Free Radic Biol Med. 2021;164:315–328. doi:10.1016/j.freeradbiomed.2020.12.449
57. Lu Y-N, Zhao X-D, Xu X, et al. Arctigenin exhibits hepatoprotective activity in Toxoplasma gondii-infected host through HMGB1/TLR4/NF-κB pathway. Int Immunopharmacol. 2020;84:106539. doi:10.1016/j.intimp.2020.106539
58. Shah N, Montes de Oca M, Jover-Cobos M, et al. Role of toll-like receptor 4 in mediating multiorgan dysfunction in mice with Acetaminophen induced acute liver failure. Liver Transplant. 2013;19(7):751–761. doi:10.1002/lt.23655
59. Lee J, Ha SJ, Lee HJ, et al. Protective effect of Tremella fuciformis Berk extract on LPS-induced acute inflammation via inhibition of the NF-κB and MAPK pathways. Food Funct. 2016;7(7):3263–3272. doi:10.1039/C6FO00540C
60. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–252. doi:10.1016/S0092-8674(00)00116-1
61. Yao H, Hu C, Yin L, et al. Dioscin reduces lipopolysaccharide-induced inflammatory liver injury via regulating TLR4/MyD88 signal pathway. Int Immunopharmacol. 2016;36:132–141. doi:10.1016/j.intimp.2016.04.023
62. Al-Harbi NO, Imam F, Al-Harbi MM, et al. Dexamethasone attenuates LPS-induced acute lung injury through inhibition of NF-κB, COX-2, and pro-inflammatory mediators. Immunol Invest. 2016;45(4):349–369. doi:10.3109/08820139.2016.1157814
63. von Moltke J, Ayres JS, Kofoed EM, Chavarría-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol. 2013;31(1):73–106. doi:10.1146/annurev-immunol-032712-095944
64. Nicotera P, Leist M, Ferrando-May E. Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol Lett. 1998;102–103:139–142. doi:10.1016/S0378-4274(98)00298-7
65. Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114(4):842–845. doi:10.1016/S0016-5085(98)70599-2
66. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2015;62(3):720–733. doi:10.1016/j.jhep.2014.10.039
67. Mansouri RM, Baugé E, Staels B, Gervois P. Systemic and distal repercussions of liver-specific peroxisome proliferator-activated receptor-alpha control of the acute-phase response. Endocrinology. 2008;149(6):3215–3223. doi:10.1210/en.2007-1339
68. Stienstra R, Mandard S, Tan NS, et al. The Interleukin-1 receptor antagonist is a direct target gene of PPARalpha in liver. J Hepatol. 2007;46(5):869–877. doi:10.1016/j.jhep.2006.11.019
69. Delerive P, Martin-Nizard F, Chinetti G, et al. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ Res. 1999;85(5):394–402. doi:10.1161/01.RES.85.5.394
70. Staels B, Koenig W, Habib A, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature. 1998;393(6687):790–793. doi:10.1038/31701
71. Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARα activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 1999;99(24):3125–3131. doi:10.1161/01.CIR.99.24.3125
72. Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. 2017;38(6):395–406. doi:10.1016/j.it.2017.03.001
73. Hotamisligil GS. Foundations of immunometabolism and implications for metabolic health and disease. Immunity. 2017;47(3):406–420. doi:10.1016/j.immuni.2017.08.009
74. Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89(6):2548–2556. doi:10.1210/jc.2004-0395
75. Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology. 2003;38(1):123–132. doi:10.1053/jhep.2003.50307
76. Stienstra R, Mandard S, Patsouris D, Maass C, Kersten S, Müller M. Peroxisome proliferator-activated receptor alpha protects against obesity-induced hepatic inflammation. Endocrinology. 2007;148(6):2753–2763. doi:10.1210/en.2007-0014
77. Khovidhunkit W, Kim M-S, Memon RA, et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res. 2004;45(7):1169–1196. doi:10.1194/jlr.R300019-JLR200
78. Feingold KR, Shigenaga JK, Kazemi MR, et al. Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol. 2012;92(4):829–839. doi:10.1189/jlb.1111537
79. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi:10.1038/nature11986
80. Liu H, Han T, Tian J, et al. Monitoring oxidative stress in acute-on-chronic liver failure by advanced oxidation protein products. Hepatol Res. 2012;42(2):171–180. doi:10.1111/j.1872-034X.2011.00911.x
81. Doerrler W, Feingold KR, Grunfeld C. Cytokines induce catabolic effects in cultured adipocytes by multiple mechanisms. Cytokine. 1994;6(5):478–484. doi:10.1016/1043-4666(94)90074-4
82. IJlst L, Mandel H, Oostheim W, Ruiter JP, Gutman A, Wanders RJ. Molecular basis of hepatic carnitine palmitoyltransferase I deficiency. J Clin Invest. 1998;102(3):527–531. doi:10.1172/JCI2927
83. Caputo T, Gilardi F, Desvergne B. From chronic overnutrition to metaflammation and insulin resistance: adipose tissue and liver contributions. FEBS Lett. 2017;591(19):3061–3088. doi:10.1002/1873-3468.12742
84. Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep. 2012;2(1):799. doi:10.1038/srep00799
85. Wen J, Zou W, Wang R, et al. Cardioprotective effects of aconiti lateralis radix praeparata combined with zingiberis rhizoma on doxorubicin-induced chronic heart failure in rats and potential mechanisms. J Ethnopharmacol. 2019;238:111880. doi:10.1016/j.jep.2019.111880
86. Hou Y, Gu D, Peng J, et al. Ginsenoside Rg1 regulates liver lipid factor metabolism in NAFLD model rats. ACS Omega. 2020;5(19):10878–10890. doi:10.1021/acsomega.0c00529
87. Zhang Y, Ji H, Qiao O, et al. Nanoparticle conjugation of ginsenoside Rb3 inhibits myocardial fibrosis by regulating PPARα pathway. Biomed Pharmacother. 2021;139:111630. doi:10.1016/j.biopha.2021.111630
88. Li J, Wang S, Yao L, et al. 6-gingerol ameliorates age-related hepatic steatosis: association with regulating lipogenesis, fatty acid oxidation, oxidative stress and mitochondrial dysfunction. Toxicol Appl Pharmacol. 2019;362:125–135. doi:10.1016/j.taap.2018.11.001
89. Wang C, Duan X, Sun X, et al. Protective effects of glycyrrhizic acid from edible botanical glycyrrhiza glabra against non-alcoholic steatohepatitis in mice. Food Funct. 2016;7(9):3716–3723. doi:10.1039/C6FO00773B
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.