")
Back to Journals » Journal of Inflammation Research » Volume 16
The Involvement of Glucose and Lipid Metabolism Alteration in Rheumatoid Arthritis and Its Clinical Implication
Authors Luo TT, Wu YJ , Yin Q, Chen WG, Zuo J
Received 25 November 2022
Accepted for publication 19 April 2023
Published 26 April 2023 Volume 2023:16 Pages 1837—1852
DOI https://doi.org/10.2147/JIR.S398291
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Ting-Ting Luo,1,2,* Yi-Jin Wu,1,2,* Qin Yin,1 Wen-Gang Chen,1 Jian Zuo2,3
1Department of Pharmacy, The Second Affiliated Hospital of Wannan Medical College, Wuhu, 241000, People’s Republic of China; 2Xin’an Medical Research Center, The First Affiliated Hospital of Wannan Medical College, Wuhu, 241000, People’s Republic of China; 3Research Center of Integration of Traditional Chinese and Western Medicine, Wannan Medical College, Wuhu, 241000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jian Zuo, Email [email protected]
Abstract: Obviously, immune cells like T cells and macrophages play a major role in rheumatoid arthritis (RA). On one hand, the breakdown of immune homeostasis directly induces systemic inflammation; on the other hand, these cells initiate and perpetuate synovitis and tissue damages through the interaction with fibroblast-like synoviocytes (FLS). In recent years, the pathological link between metabolic disorders and immune imbalance has received increasing attention. High energy demand of immune cells leads to the accumulation of metabolic byproducts and inflammatory mediators. They act on various metabolism-sensitive signal pathways as well as relevant transcription factors, such as HIF-1α, and STATs. These molecular events will impact RA-related effectors like circulating immune cells and joint-resident cells in return, allowing the continuous progression of systemic inflammation, arthritic manifestations, and life-threatening complications. In other words, metabolic complications are secondary pathological factors for the progression of RA. Therefore, the status of energy metabolism may be an important indicator to evaluate RA severity, and in-depth explorations of the mechanisms underlying the mystery of how RA-related metabolic disorders develop will provide useful clues to further clarify the etiology of RA, and inspire the discovery of new anti-rheumatic targets. This article reviews the latest research progress on the interactions between immune and metabolism systems in the context of RA. Great importance is attached to the changes in certain pathways controlling both immune and metabolism functions during RA progression.
Keywords: rheumatoid arthritis, metabolism, inflammation, immune cells
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease, characterized by chronic inflammation in synovial tissues and excessive production of autoantibodies.1 The pathogenesis of RA is related to many factors, typically immune dysfunction, epigenetic factors, environmental pollution, infections, lifestyle, and so on. Because of the complexity, it has not been thoroughly clarified yet.2 At present, the clinical treatment of RA mainly relies on conventional disease modifying anti-rheumatic drugs (cDMARDs) and biological agents. cDMARDs typically take weeks to months to take effect, and the related regimens may cause liver and bone marrow toxicity. Even though, they are still taken as first-line drugs because of their economic merits.3 Comparatively, biological agents produce more promising clinical efficacies, as they are designed to target specific molecules or cells that play key roles in RA. There are many commercially available products already, such as TNF inhibitors (etanercept, infliximab, and adalimumab), IL-6 receptor inhibitors (tocilizumab and sarilumab), and Janus kinase (JAK) inhibitors (tofacitinib, baricitinib). Basically, all biologics are more effective when used in combination with cDMARDs than prescribed alone.4–7 However, biologic agents are expensive, and not all patients respond well.8,9 Furthermore, current treatments cannot address all aspects of RA, especially the extra-articular manifestations like cardiovascular disease.10 In this context, a novel anti-rheumatic strategy is still in urgent need.
Recently, the pathological link between metabolic complications and immune imbalance in RA receives increasing attention. In fact, inflammation–metabolism interaction had been revealed decades ago.11,12 Metabolic diseases such as obesity and diabetes are always associated with chronic inflammation.13,14 Meanwhile, patients with inflammatory diseases tend to develop metabolic disorders. For example, the prevalence rates of hyperlipidemia were estimated as 42% and 31% in the patients enduring early and long-term RA, respectively.15 In addition, levels of metabolites related to glucose, amino acid, and lipid metabolism are all changed, and the metabolic alteration correlates to an increase in C-reactive protein (CRP) and many other inflammatory indicators.16 Insulin resistance (IR) is also very common in RA patients, which is believed to be caused by certain inflammatory cytokines, typically TNF-α and IL-6.17 As for the link between metabolic alteration and RA, the logic is simple. All cellular functions and physiological activities rely on energy supply. Given that RA is a condition characterized by elevated energy expenditure resulting from inflammation and associated symptoms, it is plausible that altered metabolic conditions may play a significant role in its pathological manifestations. That is, accelerated catabolism would sustain the proliferation, invasion, differentiation, and secretion of many RA-related cells such as fibroblast-like synoviocytes (FLSs), immune cells, and osteoclasts.18 Above changes are mediated by many signaling pathways, and the roles of those dominating both metabolism reprogramming and immune rebalancing are especially worthy of investigation. This review selectively focuses on the major energy metabolism changes occurred under RA conditions, and their impacts on immune system, based on currently known key signaling transduction mechanism. It is supposed to benefit a better understanding of the issue of how altered energy utilization and metabolism affect the course of RA. By discussing representative metabolism-targeting drugs, we highlight the prospect of this novel anti-rheumatic strategy.
Involvement of Energy Metabolism Alteration in RA
The majority of energy demands in mammals are met by glucose and fat catabolism. It is a priority for us to thoroughly clarify their metabolism profiles in RA. Meanwhile, we should realize that mitochondrion is the pivotal organelle responsible for energy regeneration. Any disturbance in glucose and fat metabolism will be reflected in functional changes in mitochondria, which will eventually impact metabolism status in return.19 In the following paragraphs, the relevance of mitochondrial dysfunction and RA-related inflammation is first discussed. Next, we review the current points about glucose and lipid metabolism alteration in RA. In these descriptions, FLSs and T cells are taken as representatives of effector cells, because they account for majority of RA-related clinical manifestations, and their functions are tightly controlled by their metabolic status.
Mitochondrial Dysfunction in Leukocytes Promotes Inflammation
Mitochondria serve as a major source of cellular energy by regulating multiple cellular signaling pathways; however, dysfunctional mitochondria generate reactive oxygen species (ROS), leading to oxidative stress and inflammation. Mitochondrial dysfunction has been shown to be common in T cell-mediated autoimmune diseases, including RA.20–22 Premature senescence of T lymphocytes in RA patients is a key characteristic differentiating them from normal cells, which is closely related to the acquired inflammatory phenotype.23–25 One reason for this is the decreased expression of MRE11A, the double-strand break repair nuclease. In healthy individuals, the expression of MRE11A protein in T cells decreases with age. However, in patients with RA, the majority of naive and memory CD4+ T cells exhibit low levels of MRE11A protein expression as early as middle age.26 The premature aging of T cells with damaged telomeres has been shown to be caused by a deficiency in MRE11A activity, and T cells with decreased expression of MRE11A differentiate into effector cells that are invasive to tissues, which contribute to the development of destructive synovitis.27 In fact, the abnormal MRE11A expression decrease directly leads to a series of unfavorable consequences, including chromosome disintegration and senescence markers up-regulation, in addition to telomeres damages.27 Accordingly, overexpressing MRE11A in RA T cells can reverse phenotype remodeling and ease synovial inflammation. It should be noted that MRE11A is also presented in cytoplasm and mitochondria.28 Loss of MRE11A functions will cause a significant decrease in oxygen consumption and ATP production within mitochondria.26 It implies the indispensable role of MRE11A in mitochondrial biogenesis. Indeed, MRE11A has an ability to bind to mitochondrial DNA (mtDNA). Inhibition on MRE11A expression will induce the leakage of mtDNA into cytoplasm, which is then recognized by NLRP3 and AIM2 inflammatory vesicles. This event is a corner stone of inflammatory cascades, and it triggers procaspase-1 cleavage, IL-1β release, and T cell pyroptosis.26 Some studies have reported that proteins encoded by nuclear DNA (nDNA) are also involved in mitochondrial dysfunction in RA patients. Specifically, these proteins participate in ROS generation, membrane potential maintenance, mitochondrial electron transport, energy metabolism, and intrinsic apoptosis.29 These properties partially explain the significant decreases in mitochondrial membrane potentials, superoxide, cellular ATP levels, and mitochondrial mass under RA circumstances when the expression of the relevant proteins is insufficient.30 Due to the aforementioned changes and many other mechanisms including these still unknown, mitochondrial structures in RA leukocytes are inevitably altered. In RA macrophages, mitochondria and endoplasmic reticulum together form an organelle named as “mitochondria-associated membranes” (MAMs). MAMs can inactivate glycogen synthase kinase-3β (GSK-3β), an upstream regulator of highly active mitochondria, probably through serine phosphorylation. In addition to its impact on mitochondrial biogenesis, the inactivation of GSK-3β should also account for the deteriorated RA severity directly, as it is related to the increased production of proteinase K, a collagenase implicated in joint tissue injuries.31
The consequences caused by mitochondrial structural changes are still largely unknown. However, current evidence suggests that mitochondrial dysfunction contributes significantly to RA pathology by increasing oxidative stress.32 Oxidative stress is a potent inducer of inflammation. Mitochondria are the main source of ROS. Many changes occurred in mitochondria including nutrient overloading, potential loss, and electron leakage can result in ROS accumulation, which are all evidenced in RA patients.33 It was observed that mitochondrial ROS levels in both whole blood and monocytes of RA patients increased by 5 times compared to the normal healthy controls.34 Because of its pro-inflammatory nature, ROS was found to be positively correlated with DAS 28 and RA diagnosis indicators CRP and anti-CCP (cyclic peptide containing citrulline).35,36 The pro-inflammatory properties of ROS rely on lipid peroxidation, and the products are deeply implicated in inflammatory arthritis and cartilage degeneration.37 Therefore, monitoring malondialdehyde and other similar derivatives becomes a routine when estimating RA severity and oxidative stress.37 It reminds us about the necessity of restoring antioxidant capacities during anti-RA therapies. Because of the excessive ROS production, reduced glutathione is consumed in a large amount in the cells enduring mitochondrial dysfunction. Replenishing reduced glutathione or any other approach that can potently ease oxidative stress will benefit the improvement of leukocytes-mediated inflammation in RA patients.38
Glycolysis Fuels FLSs-Mediated Pathological Changes
Glucose utilization includes three main approaches: glycolysis, aerobic oxidation, and pentose phosphate pathway (PPP). Both aerobic oxidation and glycolysis are vital for ATP production, and the ratio of glucose entering each metabolic pathway is determined by specific physiological and pathological conditions. For example, during intense exercises, glycolysis is the primary manner of glucose catabolism for ATP generation because of the speed merits and independence of oxygen supply. In contrast, during most of the time in daily life, aerobic oxidation is the primary way for glucose utilization because it produces more ATP per molecule of glucose and is more efficient and sustainable in the presence of oxygen.39 When oxidative stress is escalated, PPP becomes more active to generate NADPH, which is needed for scavenging ROS.40
According to the clues described above, we can conclude that cells tend to utilize glucose by aerobic oxidation under normal conditions. However, in many RA effector cells, like FLSs, ATP production is switched from oxidative phosphorylation to glycolytic pathway.41 It is known that RA is a wasting disease, and the resting metabolic rate is higher than the general population. It requires an efficient way to replenish energy.42 Under this circumstance, it is not surprising to reveal that glycolysis in RA patients is substantially up-regulated in certain hyper-activated cells. From the mechanism perspective, synovial hyperplasia directly creates a hypoxic environment. Even if oxygen supply is sufficient, glycolysis can still occur in FLSs because of persistent inflammation.43 Hence, abnormally activated glycolytic pathway in FLSs is an important factor implicated in the occurrence and development of articular manifestations in RA.
There are many convincing evidences about the up-regulation of glycolysis in FLSs and its involvement in RA.41 When activated by inflammatory CD4+ T cells, energy demand of FLSs is increased to sustain enhanced cellular behaviors, including proliferation, invasion, adhesion, and cytokines synthesis.44 To adapt to the metabolic requirement changes, some decisive enzymes should change accordingly. Indeed, the expression of two main rate-limiting glycolytic enzymes, glyceraldehyde 3-phosphate dehydrogenase (GAPD) and lactate dehydrogenase (LDH), are significantly up-regulated in the synovial tissue of RA patients.45 As a result, lactate production is increased.46 When glucose enters cells, it depends on glucose transporter (GLUT), the first step for glycolysis is catalyzed by hexokinases (HKs), which synthesizes glucose-6-phosphate (G-6-P) by using glucose.47 Therapeutic targeting of hexokinases (HKs) has the potential to significantly alleviate symptoms and slow the progression of RA.48,49 Glucose intake-related transporters are also important for glycolysis, as they control intracellular glucose availability. In most cases, cellular intake of glucose depends on GLUT.50 Interestingly, deletion of SLC2A3 the gene encoding GLUT3 can prevent RA-related joint injuries, indicating that as a glycolysis-related protein GLUT3 is a potential therapeutic target for RA.51 It is apparent that up-regulation of either HKs or GLUT would facilitate glycolysis. Indeed, HK2 and GLUT are highly expressed in RA synovium, which is believed to be a driving force for the enhanced migration and invasion ability of FLSs.48,52 The fore-mentioned clues basically explain the underlying mechanism, but meanwhile we should realize that both HK2 and GLUT are versatile molecules, which can induce the production of many pathological mediators such as MMPs and IL-6.41,48 HIF-1α is one of the most important regulator of glycolysis. When enriched in synovial fluid, it can promote the expression of many glycolysis-related genes/proteins, such as LDH, HK2, and GLUT1 in FLSs. In fact, HIF-1α increase is a driving force for many FLSs-related pathological changes, such as vascular proliferation, inflammation, and cartilage damages.53 Although FLS is the main source of HIF-1α in joints, the latter does not only solely impact FLSs but also greatly reshapes the microimmune environment. HIF-1α increases glycolytic flux in a variety of immune cells, including dendritic cells, classically activated M1 macrophages, neutrophils, lymphocytes, and B cells.54,55 This metabolic status confers inflammatory phenotypes of the cells, allowing immune responses to be persistent under hypoxic conditions.56 Taking T lymphocytes as an example, HIF1-α controls metabolic checkpoints and induces glycolysis there, a metabolic event favoring Th17 cells development but hampering Treg cell differentiation (Figure 1).57 Collectively, glycolytic status in synovial tissues thereby can be taken as a gauge of disease activity of RA.
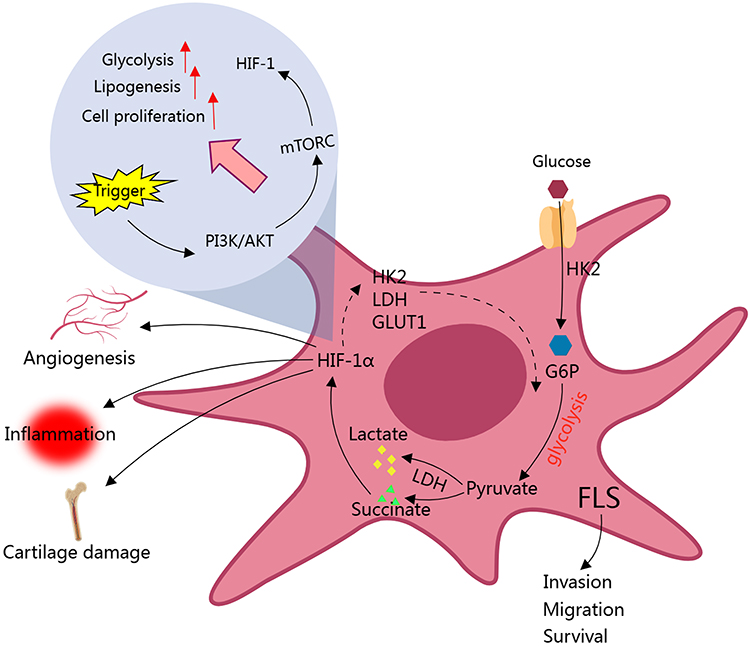 |
Figure 1 Glycolytic activity is enhanced in FLS of RA patients. Up-regulation of HK2 enhances the migration and invasion ability of FLS, which serves as an example how glycolysis drives RA. Many glycolytic intermediates like succinate up-regulate glycolysis-related genes via HIF-1α, further facilitate this metabolic process. Above changes eventually fuel vascular proliferation, inflammation, and cartilage damages. HIF-1α plays a key role in related signal transduction and pathological changes. Abbreviations: FLS, fibroblast-like synoviocyte; HK2, Hexokinase 2; G6P, glucose-6-phosphate; GLUT-1, Glucose transporter 1; LDH, lactate dehydrogenase; HIF-1α, Hypoxia-inducible factor 1-alpha; mTOCR, mammalian target of rapamycin complex; PI3K, Phosphatidylinositol 3-hydroxykinase. |
PPP Shunt Promotes the Development of Inflammatory CD4+ T Cells in RA
The properties of T cells, including lifespan, proliferative capacity, differentiation profile, and mobility ability are all greatly altered in RA patients.58,59 It is reasonable to conceive that they would prefer glycolysis due to the timely provision of energy, which indeed occurs under many pathological conditions like cancer.60 Interestingly, the metabolism of glucose in RA T cells is shifted to PPP, which is largely different from glycolysis, although they share some common steps.61,62 When studying CD4+CD45+ T cells in RA patients, it was found that 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) expression was decreased compared to normal healthy controls.63,64 This enzyme accounts for the production of fructose-2,6-diphosphate (F-2,6-P), a substrate and agonist of a rate-limiting glycolytic enzyme phosphofructokinase 1 (PFK1). Via this manner, PFKFB3 determines glycolytic flux and the downstream oxidative phosphorylation.65,66 In the context of aerobic oxidation inhibition, this finding raised a question, how glucose in these cells is utilized. Further studies revealed that T cells in RA patients tend to utilize glucose through PPP.67 When the T-cell receptor (TCR) is activated, it triggers a metabolic program that boosts mitochondrial function, increases glycolysis to produce ATP quickly and facilitates the generation of biosynthetic precursor molecules by shifting glucose to the (PPP).68 It distinguishes RA naïve T cells from their normal counterparts. A key molecule driving this change has been identified as glucose-6-phosphate dehydrogenase (G6PD), which is significantly up-regulated under RA conditions.69 G6PD is the rate-limiting enzyme of PPP and controls the production of nicotinamide adenine dinucleotide phosphate (NADPH).70 In CD4+ T cells of healthy people, expression of PFKFB3 and G6PD is finely tuned to balance the ratio of glycolysis/PPP, while the balance is tilted toward G6PD-controlled PPP in RA T cells.64 This metabolic phenotype is required by T cells to proliferate and differentiate into inflammatory subtypes.71 Hence, PPP at least partially accounts for the deterioration and recurrence of RA by affecting T cells.68
During inflammatory differentiation of T cells, PPP shunt provides large amounts of NADPH, which is the most abundant reducing molecule in cells and plays an important role in balancing redox/oxidation status. By donating electrons, NADPH does not only scavenge ROS,70 but also participates in the synthesis of many biomolecules. As a result, the over-production of NADPH promotes the replication of RA T cells.67 Meanwhile, NADPH derived from PPP protects inflammatory T cells from ROS injuries, and ensure their longevity. In fact, ROS has multiple facets about its impacts on T cells development.72 Insufficient intracellular ROS has a negative influence on ataxia telangiectasia mutation (ATM), a key kinase monitoring DNA damages and controlling cell cycle progression. When ATM levels in T cells are low, they bypass the G2/M cell cycle checkpoint, leading to abnormal proliferation, and predominantly differentiate into Th1 and Th17 cells.73 Via this mechanism, low ROS levels under PPP up-regulation status will induce the development of these inflammatory T cells. Meanwhile, NADPH is required for lipid synthesis. PPP up-regulation will accelerate lipogenesis, which provides the essential lipid components to build membranes, allowing the cells to replicate and migrate. With these changes, T cells are eventually activated in RA patients (Figure 2).74
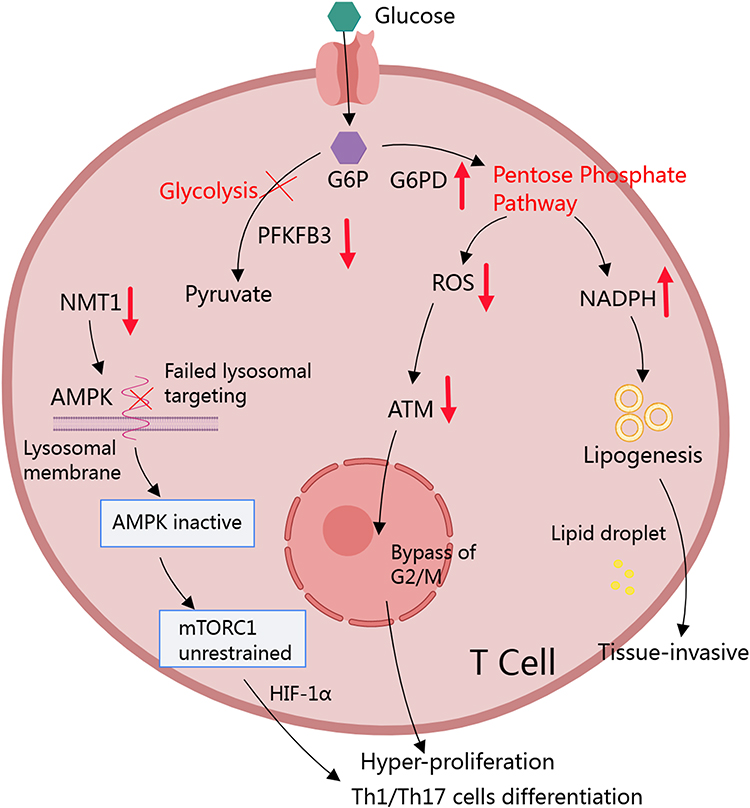 |
Figure 2 RA conditions favor PPP in CD4+ T cells. Increased PFKFB3/G6PD ratio serves as the fundament for the accelerated PPP in RA T cells. It leads to the production of large amounts of NADPH in cytoplasm. By supporting lipid synthesis, NADPH facilitates membranes construction, which makes T cells more reproductive and invasive. The increase in NADPH eventually depletes ROS. It directly prolongs the longevity of T cells, and induces them to rapidly proliferate by bypassing ATM-controlled G2/M cell cycle checkpoint. The low oxidative stress condition is also favorable for Th1 and Th17 differentiation. Concurrently, decreased NMT1 expression in T cells results in the inability to target AMPK to lysosomes, leading to elevated and unrestricted activation of mTORC1, which promotes Th1 and Th17 differentiation by mediating HIF-1α. Abbreviations: G6P, glucose 6-phosphate 6; G6PD, Glucose-6-phosphate Dehydrogenase; ROS, Reactive oxygen species; NADPH, Nicotinamide Adenine Dinucleotide Phosphate; ATM, ataxia telangiectasia mutated; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; NMT1, N-myristoyltransferase 1; AMPK, AMP-activated protein kinase; HIF-1α, Hypoxia inducible factor-1 alpha; Th, T helper. |
Inflammation Milieu in RA Promotes Lipid Catabolism
The risk of developing cardiovascular-related diseases (CVD) in RA patients is increased by 50% approximately, which is closely related to the increased mortality of RA.75,76 Apparently, chronic inflammation should account for this, in addition to some other well-known risk factors, such as smoking, obesity, hypertension, and diabetes.77 It also reminds us about the possibility of lipid metabolism alteration in RA, because high circulating lipid concentrations are always the main concern when evaluating CVD risks. Indeed, many RA patients are diagnosed with obesity and hyperlipidaemia.78–80 But the fact is much more complicated than one could assume. In fact, total cholesterol (TC), low-density lipoprotein cholesterol (LDL-c), and high-density lipoprotein cholesterol (HDL-c) are all decreased in most RA population, especially during the acute recurrence stages.81,82 Effective therapies will restore their levels. This situation together with the facts stated above lead to the conceptualization of RA lipid metabolism paradox.83 Many theories have been proposed in the attempts to clarify this mystery, but no convincing conclusion has been accepted yet. Herein we briefly discuss the relevance between cholesterol metabolism alteration and RA-related inflammation, because the implication of fat metabolism changes in RA had been reviewed well.
Studies have shown that lipids decline is closely related to CRP increase, while a decrease in CRP can be observed when LDL-c and HDL-c increase.84 Under this condition, it is not very difficult to understand the phenomenon that hypolipemia is closely related to the high incidence of CVD in RA patients. Increasing the potent inflammatory mediator CRP can induce a variety of inflammatory syndromes, including CVD.83,85 Inflammation affects the antioxidant and delivery capacity of HDL-c. When RA-related inflammation is exacerbated, HDL-c is detected with low contents of apolipoprotein A1 (apoA1) and paraoxonase-1 (an antioxidant enzyme).86 Hence, its capacity for transferring cholesterol and scavenging ROS is significantly impaired. Cholesterol will be accumulated in the vascular resident macrophages, endowing them with notable pro-atherosclerotic potentials.84,87 Based on these narrations, it can be easily noticed that inflammation-caused alteration of cholesterol metabolism is deeply implicated in RA-related extra-articular complications. A piece of convincing evidence is that high levels of TNF-α in RA patients induce the production of scavenger receptor class A (SR-A) and lectin-like oxidized low density lipoprotein receptor-1 (LOX-1), two proteins accounting for cholesterol catabolism and arteriosclerosis development.88 Furthermore, another pro-rheumatic cytokine IL-6 can stimulate the expression of LDL receptors.89 This change enhances the capacity of liver absorbing LDL, which in turn promotes the secretion of cholesterol by means of bile secretion. Cholesteryl ester catabolism is accelerated accordingly. Cholesteryl esters are carried by mature HDL particles and eventually transferred to LDL particles with the aid of cholesteryl ester transfer protein (CETP).90 According to this mechanism, the increase in cholesteryl ester catabolism in RA patients requires less HDL-c and LDL-c. These facts show that the shrink in cholesterol pool as well as HDL-c and LDL-c contents reflects the inflammatory conditions in vivo. The above evidence hints that improvements in immune environments will restore cholesterol metabolism alteration. Indeed, tofacitinib, a JAK1/JAK3 inhibitor and biological anti-rheumatic agent, can inhibit cholesteryl ester catabolism in RA patients, and therefore restore levels of HDL-c and LDL-c.90 Blocking IL-6 with tocilizumab will benefit the restoration of lipid levels too, especially for LDL-c.91 Although we have made great progress in elucidating the relationship between lipid levels and RA severity, our knowledge about the details of how cholesterol metabolism changes affect the immune environment is still poor.
Key Pathways Involved in RA Metabolism Alteration
Regardless of exact incentives, metabolism alteration in RA patients is controlled by certain signal transduction pathways. They respond to immune changes, and their changes directly create unfavorable metabolism environments. From this sense, a better understanding of their roles in RA would benefit the elucidation of many metabolism paradoxes, and inspire the introduction of novel anti-rheumatic strategies. More importantly, many pathways are evolved as dual functions roles. They control immune and metabolism systems simultaneously. As a result, they bridge the gap between inflammation and metabolic complications, and are therefore ideal therapeutic targets for RA-related CVD and other extra-articular syndromes.
PI3K/AKT
As discussed above, glycolysis is the predominant glucose utilization form for most immune cells in RA patients.92 As a key regulator of glycolysis, PI3K/AKT pathway induces the expression of GLUT1 and many other glycolytic enzymes, such as HK2 and PFK-2.93–96 The stimulation on TCR-CD3 and TLR will induce PI3K/AKT activation in lymphocytes and monocytes/macrophages, respectively. The activated AKT promotes glycolysis indirectly through its downstream GSK-3β, which can phosphorylate a panel of proteins/transcription factors related to metabolism.97–100 More importantly, PI3K/AKT activates mammalian target of rapamycin (mTOR). mTOR constructs two different complex isoforms, namely mTOR complex 1 and 2 (mTORC1/2). They are well known because of their ability to control cellular growth and proliferation. In fact, they are also indispensable for glucose metabolism reprogramming. AKT and mTORC2 form a positive feedback loop concerning their effects on glycolysis. On one hand, mTORC2 expression is up-regulated by AKT. On the other hand, mTORC2 activates AKT via phosphorylation at S473, and consequently induces the expression of GLUT1, HK2, and PFK-1.101 mTORC1 is also a direct downstream effector of PI3K/AKT that promotes protein/lipid synthesis and glycolysis.102 Up-regulation of mTORC1 will similarly induce the expression of GLUT1 and HK2, which leads to increased glucose intake and accelerated glycolysis, respectively.103 Besides the fore-mentioned impacts on AKT, mTOR can up-regulate glycolysis by promoting HIF-1 expression. As summarized above, HIF-1 initiates the expression of many glycolytic genes, such as GLUT1, HK2, PFKFB3, and LDHA by acting as a transcriptional factor (Figure 1). Meanwhile, it represses tricarboxylic acid cycle (TCA) by promoting the expression of its negative regulators like PDK.104–106 The net outcome from these changes is switching glucose from aerobic oxidation to glycolysis. Due to the abundance of TLR4 ligands, PI3K/AKT is always up-regulated in monocytes/monocytes in RA patients.107 This situation amplifies the hyper-activation of innate immune system. Interestingly, many cDMARDs and biological agents show the potential in regulating PI3K/AKT, and therefore their anti-rheumatic effects should be at least partially attributed to the inhibition of glycolysis-fueled inflammation and tissue injuries.108 Although T cells tend to utilize glucose through PPP in RA patients, regulating PI3K/AKT also affects the differentiation and development of this cell population, and affects the prognosis of RA. This implies that even for T lymphocytes, their functions are not thoroughly glycolysis-independent.
AMPK
Eukaryotes are endowed with the ability to rebalancing metabolism status according to the availability of nutrients, and AMP-activated kinase (AMPK) plays a key role in sensing environmental changes and reprogramming metabolism.109 AMPK controls many aspects of the fate of immune cells, like senescence, differentiation, apoptosis, and proliferation. We here discuss its role in immunometabolism by taking T cells as an example. Coordinating with mTOR, AMPK governs intracellular energy supply, which is decisive for memory and effector T cells development.110 Generally speaking, AMPK acts as a self-adapting buffer of energy generation in cells. It is activated when energy supply is insufficient, as indicated by the increased ratio of AMP/ATP or ADP/ATP. Once activated, AMPK phosphorylates a variety of metabolism regulators, and consequently stimulates mitochondrial biogenesis,111–114 affecting both lipid115–117 and glucose metabolism.118–121 In short, AMPK activation promotes catabolism of lipids and glucose, and hampers their anabolism, ultimately favoring ATP generation. When nutrients are sufficient, mTOR antagonizes the functions of AMPK mentioned above.122 In cells and tissues from RA patients, AMPK activation deficiency is extensively observed. This causes insufficient capacity of oxidative phosphorylation, and makes the cells depend on glycolysis for ATP replenishment. Interestingly, although the availability of ATP is reduced in RA T cells, AMPK activation does not occur there. To the contrary, its activity is down-regulated. It has been elucidated that this phenomenon is caused by dysfunction of signaling transduction within this pathway. Co-localization with mTORC1 in lysosome surface is required by AMPK to sense nutrient availability and ATP levels. RA conditions reduce the expression of protein-modifiable N-myristoyltransferase 1 (NMT1) in CD4+ T cells, which is necessary for AMPK to locate in lysosome and interact with mTORC1. Absence of NMT1 consequently causes unrestricted mTORC1 activation.123,124 As a result, HIF-1α is excessively produced, which then initiates the transcriptional expression of many inflammatory genes required by Th1 and Th17 differentiation (Figure 2).125 Another aspect of AMPK-related metabolism regulatory properties is mediated by its downstream target SIRT1. This deacetylase negatively regulates HIF-1α and NF-κB, and therefore inhibits glycolysis.126 Considering the pro-inflammatory impacts of glycolysis on both innate and adaptive immunity, deficiency of AMPK will contribute to RA-related inflammation. Chemical stimulus or genetic overexpression of AMPK would attenuate RA severity, which has been confirmed by many studies.127
JNK
Unlike the two fore-mentioned, JNK is usually categorized as an immune pathway rather than a metabolic regulator. In fact, many inflammatory pathways including JNK and NF-κB can also regulate metabolism.128,129 They exert metabolism regulatory effects through similar mechanisms. Here, we use JNK as an example to demonstrate how immune pathways change metabolic profiles of RA patients. JNK belongs to MAPK family, a group of pathways sensitive to both intracellular and extracellular stress. There are three isoforms of JNK, namely JNK1, JNK2, and JNK3. JNK1 and JNK2 are widely distributed, while JNK3 is selectively expressed in brain, heart, testis, and pancreatic β cells.130 Disturbance in JNK, especially the type 1 isoform, has been revealed to be implicated in many metabolic syndromes, such as diabetes, CVD, and fatty liver. Mice knocked out of JNK1 are protected from high-fat diet-induced obesity, insulin resistance (IR),131 oxidative damages to liver and adipose tissues.132,133
JNK regulates metabolism mainly through indirect manners. JNK can restrain the activity of peroxisome proliferator-activated receptor alpha (PPAR-α). This in turn inhibits the release of fibroblast growth factor-21 (FGF-21), which is involved in ketogenesis, insulin sensitivity, blood glucose maintenance, and obesity. Insulin receptor substrate 1 (IRS-1) is another key target for JNK to affect glucose metabolism. When activated by TNF-α, free fatty acids (FFAs), and some other inflammatory stimuli, JNK1 phosphorylates IRS-1 and prevents its interaction with insulin receptors.134,135 As a result, the potential for IRS-1 lowering blood glucose is impaired because of the altered gluconeogenesis and adipogenesis status. JNK also prevents insulin clearance in hepatocytes, which highly express insulin receptors.136 These factors cripple the physiological functions of insulin, but maintain it at high levels, which eventually results in the development of IR. Meanwhile, the role of JNK as an inflammatory cytokines inducer cannot be ignored, because these cytokines contribute to metabolic syndromes directly. For example, both of TNF-α and IL-1β can inhibit insulin signaling from activation by suppressing IRS-1 phosphorylation.137–139 Besides, they can regulate the expression and function of many metabolism-related signals such as PPAR-γ, suppressor of cytokine signaling (SOCS), and adiponectin.140,141 Based on this understanding, it was assumed that the anti-IR effects from JNK1 knockdown in adipose tissues may be caused by the decreased production of inflammatory mediators like IL-6. But this acclaim was questioned by a recent study, which showed that adipocytes-specific deletion of IL-6 gene had no effect on glucose tolerance in obese mice.142,143 Therefore, all aspects of JNK in metabolism regulation should be taken into consideration, and changes of cytokines cannot thoroughly explain all the consequences.
As a crucial pro-inflammatory signal, JNK is up-regulated in RA patients with no doubts. In addition to the direct consequence of inflammation, the metabolic complication is another contribution of JNK up-regulation to RA, which should be seriously treated as the secondary pathological factor. For example, hyperglycemia will cause nutrient overloading and toxicity to most cells.144 Consistently, high levels of ROS and mitochondrial dysfunction are observed in RA patients, and inhibition of JNK attenuates these situations, achieving similar metabolism-regulatory and anti-rheumatic effects to hypoglycemic therapies. Adipose tissues greatly account for the metabolic changes brought by JNK-targeting anti-RA regimens, which have been confirmed as the largest secretion organ and a governor of energy metabolism in mammals.145 Disruption of adipokine network is very common in RA patients, which serves as a foundation for developing metabolic complications.146 Under RA conditions, adipose tissues tend to secrete more inflammatory adipokines, and this process is controlled by JNK. Leptin is a good representative when elucidating the relevance between JNK-controlled adipokine secretion and RA pathology.147 Aside from effects of JNK on leptin production, leptin receptor sends signals through JNK.148 Because leptin levels in RA patients are elevated, it will amplify the unfavorable consequences from JNK up-regulation via positive feedback.149,150 In conformity to this, reducing leptin levels in RA patients by fasting improves overall clinical symptoms of RA.151 Perhaps due to the complicated metabolism regulatory properties of the adipokines, the availability of both FFA and glucose was observed to be reduced in RA animal models when JNK was suppressed. As a result, many metabolism-fueled pathological changes in peripheral tissues will be attenuated.152
Metabolism-Regulatory Anti-Rheumatic Therapies and the Prospect
Due to the importance of metabolic alterations in the occurrence and development of RA, regulating metabolism and related pathways could be a feasible anti-rheumatic strategy. In fact, many conventional drugs show impressive metabolism-regulating properties. Methotrexate (MTX), a first-line DMARD, can significantly down-regulate the expression of HK2 and SLC2A5 (a glucose/fructose carrier) in RA FLSs, and therefore restrain glycolysis.153 DMARDs and prednisolone combination therapy significantly elevate cholesterol levels when achieving notable improvement in disease activity in RA patients.154 Another report also revealed that this therapy would increase the levels of total cholesterol and HDL-c, which are inversely correlated to erythrocyte sedimentation rate (ESR) and CRP values.155 Likewise, the use of hydroxychloroquine (HCQ) in anti-RA regimens improved blood lipid composition profiles, indicated by the changes in TC, LDL-c, and HDL-c.156 Similar effects can also be achieved by biological anti-rheumatic therapies. Anti-TNF-α treatment was revealed to reduce the expression of GLUT1 and PKM2 in RA patients,157 and there is growing evidence that TNF inhibitors can reduce CVD risk.158,159 The inhibitor of JNK Tofacitinib can also inhibit LDH and HK2 expression in FLSs and inhibit their pathological activities by restraining glycolysis.160 No matter if it is monotherapy or in combination with cDMARDs, this drug can increase the levels of LDL-c and HDL-c by approximately 10–20%, which can be sustained for 3 months after treatments. This outcome is generally beneficial, as the incidence of CV did not increase with the increase in blood lipids.161–165 According to the current understanding, compounds with metabolism regulatory effects must have certain anti-rheumatic potentials. 2-Deoxyglucose (2-DG), a derivative of glucose, can inhibit downstream of HK2-catalyzing steps.166 Interestingly, 2-DG alleviated the severity of a spontaneous arthritis developed in mice. With a similar mechanism, it can also inhibit cancer cell proliferation. In fact, 2-DG has entered Phase I/II clinical trials for the treatment of advanced cancers. Cancer-related researches suggest that this compound would only induce mild side effects such as nausea and hypoglycemia. This safety merit motivates the evaluation of it as a therapeutic drug for many inflammatory diseases like RA.167,168 The hypoglycemic drug metformin partially acts as an agonist of AMPK. By affecting AMPK activity and downstream AKT/mTOR, metformin improved both metabolism and immune situations in animals with experimental autoimmune arthritis. FLSs-mediated arthritic manifestations were effectively contained.169 Metformin will bring extra benefits to patients with inflammatory diseases, who underwent glucocorticoids treatment, evidenced by improvements in metabolic conditions, disease activity, and infection risks.127 Many naturally occurring reagents possess dual pharmacological properties similar to metformin. A triterpenoid derivative lupeol can inhibit JNK-mediated neuroinflammation, and possesses anti-hyperglycemic and anti-dyslipidemic effects.170,171 Gingerol, a bioactive compound from common food ingredients, shows the potential in treating both obesity and c-Jun activation-caused inflammatory cytokine secretion.172 α-Mangostin, the main component identified in mangosteen, is well-known because of its impressive anti-inflammatory activity. However, increasing research has demonstrated that it is a promising reagent for treating obesity, diabetes, and CVD, and many metabolism pathways are involved in its therapeutic actions, like PPAR-γ.173 With these properties, α-mangostin can improve the immune and metabolism conditions in rheumatic subjects simultaneously.174
Considering these encouraging findings, more endeavors should be done to explore metabolism-related anti-rheumatic therapies from both synthetic and natural products. Traditional Chinese Medicine (TCM) usually emphasizes the importance of metabolism changes in diseases and will provide us useful clues to achieve this goal. A previous study demonstrated that Qingluoyin, a representative anti-rheumatic TCM decoction, eased RA severity mainly by affecting fatty acid and glucose metabolism.175 This paradigm encourages us to investigate the anti-rheumatic mechanism of TCM therapies from the mechanism perspective. Meanwhile, many conventional hypoglycemic and lipid-regulating drugs are also good candidates. We had known that metformin and rosiglitazone can help control RA progression,176 but more drugs are to be investigated.
Abbreviation
AMPK, Adenosine 5’-monophosphate (AMP)-activated protein kinase; apoA1, apolipoprotein A1; ATM, Ataxia Telangiectasia Mutated; CCP, cyclic peptide containing citrulline; CRP, cyclic peptide containing citrulline; CVD, cardiovascular-related diseases; CETP, cholesteryl ester transfer protein; DAS 28, Disease Activity Score 28; DMARDs, disease modifying anti-rheumatic drug; F-2,6-P, Fructose-2,6-diphosphate; FLS, Fibroblast-like synoviocytes; GAPD, Glyceraldehyde 3-Phosphate Dehydrogenase; G6PD, Glucose-6-Phosphate Dehydrogenase; G-6-P, Glucose-6-Phosphate; GSK-3β, Glycogen Synthase Kinase-3β; GLUT, Glucose Transporter; IR, Insulin Resistance; HDL-c, High-density lipoprotein cholesterol; HIF-1α, Hypoxia-inducible factor-1α; HKs, Hexokinases; JAK, Janus kinase; JNK, c-Jun N-terminal kinase; LDH, Lactate Dehydrogenase; LDL-c, Low-density Lipoprotein cholesterol; LOX-1; Lectin-like Oxidized Low density lipoprotein receptor-1; MAMs, Mitochondria-Associated Membranes, MAPK, Mitogen-Activated Protein Kinase; MetS, Metabolic Syndromes; MRE11A, Meiotic recombination 11 homolog A; mTOR, mammalian target of rapamycin; NADPH, Nicotinamide Adenine Dinucleotide Phosphate; NF-κB, Nuclear Factor kappa-B; NMT1, N-Myristoyltransferase 1; PDK, Pyruvate Dehydrogenase Kinase; PFK1, Phosphofructokinase 1; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; PPAR-α, Peroxisome Proliferator-activated Receptor alpha; PPP, Pentose Phosphate Pathways; RA, Rheumatoid arthritis; ROS, Reactive Oxygen Species; SIRT1, Silent Information Regulator 1; SR-A, Scavenger Receptor class A; TCA, Tricarboxylic Acid Cycle; TC, Total Cholesterol; Th, T helper; Treg, T regulatory cell.
Summary
In recent years, researches on metabolism-related inflammatory diseases have gained increasing knowledge about the complex interactions between metabolic pathways and immune system. Metabolic complications in inflammatory diseases like RA have been confirmed as an important part of the disease pathology itself, and provide an opportunity to introduce novel therapies. Metabolic pathway components, enzymes, and metabolites are emerging as potential biomarkers for the diagnosis of RA, which enable early intervention, personalized medication, and nutritional caring. Related researches will provide new directions for new anti-rheumatic drug discovery. More attentions are usually paid to signal transduction pathways, because they control every aspect of physiopathology functions. In fact, enzymes and metabolites are similarly important. In addition to the traditionally defined roles in metabolism, they usually also participate in signal transduction. That is, besides the biologics specifically targeting certain signaling pathways, agonists/antagonists of some enzymes and metabolic intermediates can all be used to rectify the abnormal metabolism alteration in RA as well as the immune consequences. However, our knowledge of the interplay between immune and metabolism systems especially under RA circumstances are still limited. Many immune/metabolism mediators have multiple facets in this feedback network. Targeting any of them will have a series of consequences. Even the so-called selective agonists or antagonists would cause certain unfavorable outcomes. Under these circumstances, thoroughly clarifying the signaling transduction mechanisms and identifying strictly target-specific reagents are the priority for developing novel anti-RA regimens.
Funding
This work was supported by National Natural Science Foundation of China (8197382), Anhui Natural Science Foundation Project (2108085QH386), Wannan Medical College Youth Research Fund (WK2022F36). Climbing Scientific Peak Project for Talents, the Second Affiliated Hospital of Wannan Medical College (DFJH2022002).
Disclosure
The authors declare that they have no conflict of interest.
References
1. Weyand CM, Goronzy JJ. The immunology of rheumatoid arthritis. Nat Immunol. 2021;22(1):10–18.
2. Bustamante MF, Garcia-Carbonell R, Whisenant KD, Guma M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res Ther. 2017;19(1):110.
3. Wang W, Zhou H, Liu L. Side effects of methotrexate therapy for rheumatoid arthritis: a systematic review. Eur J Med Chem. 2018;158:502–516.
4. Kaneko Y, Atsumi T, Tanaka Y, et al. Comparison of adding tocilizumab to methotrexate with switching to tocilizumab in patients with rheumatoid arthritis with inadequate response to methotrexate: 52-week results from a prospective, randomised, controlled study (Surprise study). Ann Rheum Dis. 2016;75(11):1917–1923.
5. Emery P, Burmester GR, Bykerk VP, et al. Evaluating drug-free remission with Abatacept in early rheumatoid arthritis: results from the phase 3b, multicentre, randomised, active-controlled AVERT study of 24 months, with a 12-month, double-blind treatment period. Ann Rheum Dis. 2015;74(1):19–26.
6. Burmester GR, Rigby WF, van Vollenhoven RF, et al. Tocilizumab in early progressive rheumatoid arthritis: FUNCTION, a randomised controlled trial. Ann Rheum Dis. 2016;75(6):1081–1091.
7. Fleischmann R, Mysler E, Hall S, et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and Adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet. 2017;390(10093):457–468.
8. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–2038.
9. Singh JA, Saag KG, Bridges SL
10. Panoulas V, Kitas GD. Pharmacological management of cardiovascular risk in chronic inflammatory rheumatic diseases. Expert Rev Clin Pharmacol. 2020;13(6):605–613.
11. Mehta S, Farmer JA. Obesity and inflammation: a new look at an old problem. Curr Atheroscler Rep. 2007;9(2):134–138.
12. Stavropoulos-Kalinoglou A, Metsios GS, Panoulas VF, et al. Associations of obesity with modifiable risk factors for the development of cardiovascular disease in patients with rheumatoid arthritis. Ann Rheum Dis. 2009;68(2):242–245.
13. Ali L, Schnitzler JG, Kroon J. Metabolism: the road to inflammation and atherosclerosis. Curr Opin Lipidol. 2018;29(6):474–480.
14. Rohm TV, Meier DT, Olefsky JM, Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity. 2022;55(1):31–55.
15. Chung CP, Oeser A, Solus JF, et al. Prevalence of the metabolic syndrome is increased in rheumatoid arthritis and is associated with coronary atherosclerosis. Atherosclerosis. 2008;196(2):756–763.
16. Young SP, Kapoor SR, Viant MR, et al. The impact of inflammation on metabolomic profiles in patients with arthritis. Arthritis Rheum. 2013;65(8):2015–2023.
17. Nicolau J, Lequerre T, Bacquet H, Vittecoq O. Rheumatoid arthritis, insulin resistance, and diabetes. Joint Bone Spine. 2017;84(4):411–416.
18. Pucino V, Certo M, Varricchi G, et al. Metabolic checkpoints in rheumatoid arthritis. Front Physiol. 2020;11:347.
19. Judge A, Dodd Michael S. Metabolism. Essays Biochem. 2020;64(4):607–647.
20. Kim EK, Kwon JE, Lee SY, et al. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis. 2017;8(1):e2565.
21. Alissafi T, Kalafati L, Lazari M, et al. Mitochondrial oxidative damage underlies regulatory T cell defects in autoimmunity. Cell Metab. 2020;32(4):591–604e597.
22. Wu B, Qiu J, Zhao TV, et al. Succinyl-CoA ligase deficiency in pro-inflammatory and tissue-invasive T cells. Cell Metab. 2020;32(6):967–980.
23. Weyand CM, Fujii H, Shao L, Goronzy JJ. Rejuvenating the immune system in rheumatoid arthritis. Nat Rev Rheumatol. 2009;5(10):583–588.
24. Goronzy JJ, Weyand CM. Aging, autoimmunity and arthritis: t-cell senescence and contraction of T-cell repertoire diversity - catalysts of autoimmunity and chronic inflammation. Arthritis Res Ther. 2003;5(5):225–234.
25. Koetz K, Bryl E, Spickschen K, O’Fallon WM, Goronzy JJ, Weyand CM. Cell homeostasis in patients with rheumatoid arthritis. P Natl Acad Sci USA. 2000;97(16):9203–9208.
26. Li Y, Shen Y, Jin K, et al. The DNA repair nuclease MRE11A functions as a mitochondrial protector and prevents T cell pyroptosis and tissue inflammation. Cell Metab. 2019;30(3):477–492.
27. Li Y, Shen Y, Hohensinner P, et al. Deficient activity of the nuclease MRE11A induces T cell aging and promotes arthritogenic effector functions in patients with rheumatoid arthritis. Immunity. 2016;45(4):903–916.
28. Syed A, Tainer JA. The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu Rev Biochem. 2018;87:263–294.
29. Panga V, Kallor AA, Nair A, Harshan S, Raghunathan S. Mitochondrial dysfunction in rheumatoid arthritis: a comprehensive analysis by integrating gene expression, protein-protein interactions and gene ontology data. PLoS One. 2019;14(11):e0224632.
30. Khanna S, Padhan P, Jaiswal KS, et al. Altered mitochondrial proteome and functional dynamics in patients with rheumatoid arthritis. Mitochondrion. 2020;54:8–14.
31. Zeisbrich M, Yanes RE, Zhang H, et al. Hypermetabolic macrophages in rheumatoid arthritis and coronary artery disease due to glycogen synthase kinase 3b inactivation. Ann Rheum Dis. 2018;77(7):1053–1062.
32. Lopez-Armada MJ, Fernandez-Rodriguez JA, Blanco FJ. Mitochondrial dysfunction and oxidative stress in rheumatoid arthritis. Antioxidants. 2022;11:6.
33. Ferraz-Amaro I, Gonzalez-Juanatey C, Lopez-Mejias R, Riancho-Zarrabeitia L, Gonzalez-Gay MA. Metabolic syndrome in rheumatoid arthritis. Mediators Inflamm. 2013;2013:710928.
34. Miesel R, Murphy MP, Kröger H. Enhanced mitochondrial radical production in patients with rheumatoid arthritis correlates with elevated levels of tumor necrosis factor alpha in plasma. Free Radic Res. 1996;25(2):161–169.
35. Kundu S, Ghosh P, Datta S, Ghosh A, Chattopadhyay S, Chatterjee M. Oxidative stress as a potential biomarker for determining disease activity in patients with rheumatoid arthritis. Free Radic Res. 2012;46(12):1482–1489.
36. Kundu S, Bala A, Ghosh P, et al. Attenuation of oxidative stress by allylpyrocatechol in synovial cellular infiltrate of patients with rheumatoid arthritis. Free Radic Res. 2011;45(5):518–526.
37. Biniecka M, Kennedy A, Fearon U, Ng CT, Veale DJ, O’Sullivan JN. Oxidative damage in synovial tissue is associated with in vivo hypoxic status in the arthritic joint. Ann Rheum Dis. 2010;69(6):1172–1178.
38. Smallwood MJ, Nissim A, Knight AR, Whiteman M, Haigh R, Winyard PG. Oxidative stress in autoimmune rheumatic diseases. Free Radic Biol Med. 2018;125:3–14.
39. Hargreaves M, Spriet LL. Exercise metabolism: fuels for the fire. Cold Spring Harb Perspect Med. 2018;8:8.
40. Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative stress in cancer. Cancer Cell. 2020;38(2):167–197.
41. Garcia-Carbonell R, Divakaruni AS, Lodi A, et al. Critical role of glucose metabolism in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol. 2016;68(7):1614–1626.
42. Rennie KL, Hughes J, Lang R, Jebb SA. Nutritional management of rheumatoid arthritis: a review of the evidence. J Hum Nutr Diet. 2003;16(2):97–109.
43. Masoumi M, Mehrabzadeh M, Mahmoudzehi S, et al. Role of glucose metabolism in aggressive phenotype of fibroblast-like synoviocytes: latest evidence and therapeutic approaches in rheumatoid arthritis. Int Immunopharmacol. 2020;89(Pt A):107064.
44. Petrasca A, Phelan JJ, Ansboro S, Veale DJ, Fearon U, Fletcher JM. Targeting bioenergetics prevents CD4 T cell–mediated activation of synovial fibroblasts in rheumatoid arthritis. Rheumatology. 2020;59(10):2816–2828.
45. Henderson B, Bitensky L, Chayen J. Glycolytic activity in human synovial lining cells in rheumatoid arthritis. Ann Rheum Dis. 1979;38(1):63–67.
46. Hakala M, Kroger H, Valleala H, et al. Once-monthly oral ibandronate provides significant improvement in bone mineral density in postmenopausal women treated with glucocorticoids for inflammatory rheumatic diseases: a 12-month, randomized, double-blind, placebo-controlled trial. Scand J Rheumatol. 2012;41(4):260–266.
47. Tan VP, Miyamoto S. HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy. 2015;11(6):963–964.
48. Bustamante MF, Oliveira PG, Garcia-Carbonell R, et al. Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann Rheum Dis. 2018;77(11):1636–1643.
49. Song G, Lu Q, Fan H, et al. Inhibition of hexokinases holds potential as treatment strategy for rheumatoid arthritis. Arthritis Res Ther. 2019;21:1.
50. Temre MK, Kumar A, Singh SM. An appraisal of the current status of inhibition of glucose transporters as an emerging antineoplastic approach: promising potential of new pan-GLUT inhibitors. Front Pharmacol. 2022;13:1035510.
51. Simpfendorfer KR, Li W, Shih A, et al. Influence of genetic copy number variants of the human GLUT3 glucose transporter gene SLC2A3 on protein expression, glycolysis and rheumatoid arthritis risk: a genetic replication study. Mol Genet Metab Rep. 2019;19:100470.
52. de Oliveira PG, Farinon M, Sanchez-Lopez E, Miyamoto S, Guma M. Fibroblast-like synoviocytes glucose metabolism as a therapeutic target in rheumatoid arthritis. Front Immunol. 2019;10:1743.
53. Hua S, Dias TH. Hypoxia-Inducible Factor (HIF) as a target for novel therapies in rheumatoid arthritis. Front Pharmacol. 2016;7:184.
54. Hu F, Liu H, Xu L, et al. Hypoxia-inducible factor-1alpha perpetuates synovial fibroblast interactions with T cells and B cells in rheumatoid arthritis. Eur J Immunol. 2016;46(3):742–751.
55. Hu F, Mu R, Zhu J, et al. Hypoxia and hypoxia-inducible factor-1alpha provoke toll-like receptor signalling-induced inflammation in rheumatoid arthritis. Ann Rheum Dis. 2014;73(5):928–936.
56. Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol. 2021;599(1):23–37.
57. Shi LZ, Wang R, Huang G, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208(7):1367–1376.
58. Goronzy JJ, Weyand CM. Mechanisms underlying T cell ageing. Nat Rev Immunol. 2019;19(9):573–583.
59. Vignali PDA, Barbi J, Pan F. Metabolic regulation of T cell immunity. Adv Exp Med Biol. 2017;1011:87–130.
60. Samuelson BT, Vesely SK, Chai-Adisaksopha C, Scott BL, Crowther M, Garcia D. The impact of ruxolitinib on thrombosis in patients with polycythemia vera and myelofibrosis: a meta-analysis. Blood Coagul Fibrinolysis. 2016;27(6):648–652.
61. Kerekes G, Nurmohamed MT, Gonzalez-Gay MA, et al. Rheumatoid arthritis and metabolic syndrome. Nat Rev Rheumatol. 2014;10(11):691–696.
62. Pi H, Zhou H, Jin H, Ning Y, Wang Y. Abnormal glucose metabolism in rheumatoid arthritis. Biomed Res Int. 2017;2017:9670434.
63. Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med. 2013;210(10):2119–2134.
64. Weyand CM, Shen Y, Goronzy JJ. Redox-sensitive signaling in inflammatory T cells and in autoimmune disease. Free Radic Biol Med. 2018;125:36–43.
65. Bartrons R, Rodriguez-Garcia A, Simon-Molas H, Castano E, Manzano A, Navarro-Sabate A. The potential utility of PFKFB3 as a therapeutic target. Expert Opin Ther Targets. 2018;22(8):659–674.
66. Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in cancer. Exp Mol Pathol. 2009;86(3):174–179.
67. Yang Z, Shen Y, Oishi H, et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med. 2016;8(331):331ra338–331ra338.
68. Weyand CM, Goronzy JJ. Immunometabolism in the development of rheumatoid arthritis. Immunol Rev. 2020;294(1):177–187.
69. Trefts E, Gannon M, Wasserman DH. The liver. Curr Biol. 2017;27(21):R1147–R1151.
70. Yang HC, Wu YH, Liu HY, Stern A, Chiu DT. What has passed is prolog: new cellular and physiological roles of G6PD. Free Radic Res. 2016;50(10):1047–1064.
71. Senolt L, Pavelka K, Housa D, Haluzik M. Increased adiponectin is negatively linked to the local inflammatory process in patients with rheumatoid arthritis. Cytokine. 2006;35(5–6):247–252.
72. Mittler R. ROS are good. Trends Plant Sci. 2017;22(1):11–19.
73. Jones JG. Hepatic glucose and lipid metabolism. Diabetologia. 2016;59(6):1098–1103.
74. Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019;20(3):137–155.
75. Peters MJ, Symmons DP, McCarey D, et al. EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann Rheum Dis. 2010;69(2):325–331.
76. Avina-Zubieta JA, Choi HK, Sadatsafavi M, Etminan M, Esdaile JM, Lacaille D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: a meta-analysis of observational studies. Arthritis Rheum. 2008;59(12):1690–1697.
77. van den Oever IA, van Sijl AM, Nurmohamed MT. Management of cardiovascular risk in patients with rheumatoid arthritis: evidence and expert opinion. Ther Adv Musculoskelet Dis. 2013;5(4):166–181.
78. Giles JT, Allison M, Blumenthal RS, et al. Abdominal adiposity in rheumatoid arthritis: association with cardiometabolic risk factors and disease characteristics. Arthritis Rheum. 2010;62(11):3173–3182.
79. Dougados M, Soubrier M, Antunez A, et al. Prevalence of comorbidities in rheumatoid arthritis and evaluation of their monitoring: results of an international, cross-sectional study (COMORA). Ann Rheum Dis. 2014;73(1):62–68.
80. Kruger K, Nusslein H. Kardiovaskuläre Komorbiditäten bei rheumatoider Arthritis[Cardiovascular comorbidities in rheumatoid arthritis]. Z Rheumatol. 2019;78(3):221–227. German.
81. Erum U, Ahsan T, Khowaja D. Lipid abnormalities in patients with rheumatoid arthritis. Pak J Med Sci. 2017;33:1.
82. Lazarevic MB, Vitic J, Mladenovic V, Myones BL, Skosey JL, Swedler WI. Dyslipoproteinemia in the course of active rheumatoid arthritis. Nat Libr Med. 1992;22(3):172–178.
83. Myasoedova E, Crowson CS, Kremers HM, et al. Lipid paradox in rheumatoid arthritis: the impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Ann Rheum Dis. 2011;70(3):482–487.
84. Liao KP, Playford MP, Frits M, et al. The association between reduction in inflammation and changes in lipoprotein levels and HDL cholesterol efflux capacity in rheumatoid arthritis. J Am Heart Assoc. 2015;4:2.
85. Zhang J, Chen L, Delzell E, et al. The association between inflammatory markers, serum lipids and the risk of cardiovascular events in patients with rheumatoid arthritis. Ann Rheum Dis. 2014;73(7):1301–1308.
86. Charles-Schoeman C, Watanabe J, Lee YY, et al. Abnormal function of high-density lipoprotein is associated with poor disease control and an altered protein cargo in rheumatoid arthritis. Arthritis Rheum. 2009;60(10):2870–2879.
87. Charles-Schoeman C, Lee YY, Grijalva V, et al. Cholesterol efflux by high density lipoproteins is impaired in patients with active rheumatoid arthritis. Ann Rheum Dis. 2012;71(7):1157–1162.
88. Hashizume M, Mihara M. Atherogenic effects of TNF-alpha and IL-6 via up-regulation of scavenger receptors. Cytokine. 2012;58(3):424–430.
89. Lubrano V, Gabriele M, Puntoni MR, Longo V, Pucci L. Relationship among IL-6, LDL cholesterol and lipid peroxidation. Cell Mol Biol Lett. 2015;20(2):310–322.
90. Charles-Schoeman C, Fleischmann R, Davignon J, et al. Potential mechanisms leading to the abnormal lipid profile in patients with rheumatoid arthritis versus healthy volunteers and reversal by tofacitinib. Arthritis Rheumatol. 2015;67(3):616–625.
91. Robertson J, Porter D, Sattar N, et al. Interleukin-6 blockade raises LDL via reduced catabolism rather than via increased synthesis: a cytokine-specific mechanism for cholesterol changes in rheumatoid arthritis. Ann Rheum Dis. 2017;76(11):1949–1952.
92. Weyand CM, Wu B, Goronzy JJ. The metabolic signature of T cells in rheumatoid arthritis. Curr Opin Rheumatol. 2020;32(2):159–167.
93. Chang M, Hamilton JA, Scholz GM, Elsegood CL. Glycolytic control of adjuvant-induced macrophage survival: role of PI3K, MEK1/2, and Bcl-2. J Leukoc Biol. 2009;85(6):947–956.
94. Cheng SC, Quintin J, Cramer RA, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345(6204):1250684.
95. Krawczyk CM, Holowka T, Sun J, et al. Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115(23):4742–4749.
96. Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015;36(4):257–264.
97. Inkster B, Zai G, Lewis G, Miskowiak KW. GSK3beta: a plausible mechanism of cognitive and hippocampal changes induced by erythropoietin treatment in mood disorders? Transl Psychiatry. 2018;8(1):216.
98. van der Vaart A, Meng X, Bowers MS, et al. Glycogen synthase kinase 3 beta regulates ethanol consumption and is a risk factor for alcohol dependence. Neuropsychopharmacology. 2018;43(13):2521–2531.
99. Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;59(1):1–16.
100. Sopjani M, Millaku L, Nebija D, Emini M, Rifati-Nixha A, Dermaku-Sopjani M. The glycogen synthase kinase-3 in the regulation of ion channels and cellular carriers. Curr Med Chem. 2019;26(37):6817–6829.
101. Sarbassov DD, Guertin DA, Ali SM, Sabatini DMJS. Phosphorylation and regulation of Akt/PKB by the Rictor-mTOR complex. Science. 2005;307(5712):1098–1101.
102. Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339(6125):1323–1328.
103. Maiese K, Chong ZZ, Shang YC, Wang S. mTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol Med. 2013;19(1):51–60.
104. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95.
105. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–732.
106. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21(3):297–308.
107. Li W, Wang K, Liu Y, et al. A novel drug combination of mangiferin and cinnamic acid alleviates rheumatoid arthritis by inhibiting TLR4/NFkappaB/NLRP3 activation-induced pyroptosis. Front Immunol. 2022;13:912933.
108. Liu S, Ma H, Zhang H, Deng C, Xin P. Recent advances on signaling pathways and their inhibitors in rheumatoid arthritis. Clin Immunol. 2021;230:108793.
109. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19(2):121–135.
110. Weyand CM, Goronzy JJ. Immunometabolism in early and late stages of rheumatoid arthritis. Nat Rev Rheumatol. 2017;13(5):291–301.
111. Toyama EQ, Herzig S, Courchet J, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351(6270):275–281.
112. Zong H, Ren JM, Young LH, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci. 2002;99(25):5983–5987.
113. Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci. 2007;104(29):12017–12022.
114. Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461.
115. Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223(2):217–222.
116. Munday MR, Campbell DG, Carling D, Hardie DG. Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur J Biochem. 1988;175(2):331–338.
117. Watt MJ, Holmes AG, Pinnamaneni SK, et al. Regulation of HSL serine phosphorylation in skeletal muscle and adipose tissue. Am J Physiol Endocrinol Metab. 2006;290(3):E500–508.
118. Marsin AS, Bertrand L, Rider MH, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10(20):1247–1255.
119. Bando H, Atsumi T, Nishio T, et al. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin Cancer Res. 2005;11(16):5784–5792.
120. Sakamoto K, Holman GD. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab. 2008;295(1):E29–37.
121. Wu N, Zheng B, Shaywitz A, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell. 2013;49(6):1167–1175.
122. Zhang C-S, Jiang B, Li M, et al. The lysosomal v-ATPase-ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014;20(3):526–540.
123. Liang J, Xu ZX, Ding Z, et al. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat Commun. 2015;6:7926.
124. Oakhill JS, Chen ZP, Scott JW, et al. beta-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc Natl Acad Sci USA. 2010;107(45):19237–19241.
125. Wen Z, Jin K, Shen Y, et al. N-myristoyltransferase deficiency impairs activation of kinase AMPK and promotes synovial tissue inflammation. Nat Immunol. 2019;20(3):313–325.
126. Canto C, Gerhart-Hines Z, Feige JN, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458(7241):1056–1060.
127. Pernicova I, Kelly S, Ajodha S, et al. Metformin to reduce metabolic complications and inflammation in patients on systemic glucocorticoid therapy: a randomised, double-blind, placebo-controlled, proof-of-concept, Phase 2 trial. Lancet Diabetes Endocrinol. 2020;8(4):278–291.
128. Hotamisligil GS, Davis RJ. Cell signaling and stress responses. Cold Spring Harb Perspect Biol. 2016;8(10):5.
129. Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25(10):1939–1948.
130. Nakano R, Nakayama T, Sugiya H. Biological properties of JNK3 and its function in neurons, astrocytes, pancreatic beta-cells and cardiovascular cells. Cells. 2020;9:8.
131. Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336.
132. Becattini B, Zani F, Breasson L, et al. JNK1 ablation in mice confers long-term metabolic protection from diet-induced obesity at the cost of moderate skin oxidative damage. FASEB J. 2016;30(9):3124–3132.
133. Yu XX, Murray SF, Watts L, et al. Reduction of JNK1 expression with antisense oligonucleotide improves adiposity in obese mice. Am J Physiol Endocrinol Metab. 2008;295(2):E436–445.
134. Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271(5249):665–668.
135. Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem. 2000;275(12):9047–9054.
136. Sabio G, Cavanagh-Kyros J, Ko HJ, et al. Prevention of steatosis by hepatic JNK1. Cell Metab. 2009;10(6):491–498.
137. Kang T, Huang H, Mandrup-Poulsen T, Larsen MR. Divalent metal transporter 1 knock-down modulates IL-1beta mediated pancreatic beta-cell pro-apoptotic signaling pathways through the autophagic machinery. Int J Mol Sci. 2021;22:15.
138. Aye IL, Jansson T, Powell TL. Interleukin-1beta inhibits insulin signaling and prevents insulin-stimulated system A amino acid transport in primary human trophoblasts. Mol Cell Endocrinol. 2013;381(1–2):46–55.
139. Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia. 2012;55(10):2565–2582.
140. Liu C, Feng X, Li Q, Wang Y, Li Q, Hua M. Adiponectin, TNF-alpha and inflammatory cytokines and risk of type 2 diabetes: a systematic review and meta-analysis. Cytokine. 2016;86:100–109.
141. Liang Y, Xu WD, Peng H, Pan HF, Ye DQ. SOCS signaling in autoimmune diseases: molecular mechanisms and therapeutic implications. Eur J Immunol. 2014;44(5):1265–1275.
142. Whitham M, Pal M, Petzold T, et al. Adipocyte-specific deletion of IL-6 does not attenuate obesity-induced weight gain or glucose intolerance in mice. Am J Physiol Endocrinol Metab. 2019;317(4):E597–E604.
143. Han MS, White A, Perry RJ, et al. Regulation of adipose tissue inflammation by interleukin 6. Proc Natl Acad Sci USA. 2020;117(6):2751–2760.
144. Runtuwene J, Cheng KC, Asakawa A, et al. Rosmarinic acid ameliorates hyperglycemia and insulin sensitivity in diabetic rats, potentially by modulating the expression of PEPCK and GLUT4. Drug Des Devel Ther. 2016;10:2193–2202.
145. Umar S, Palasiewicz K, Volin MV, et al. Metabolic regulation of RA macrophages is distinct from RA fibroblasts and blockade of glycolysis alleviates inflammatory phenotype in both cell types. Cell Mol Life Sci. 2021;78(23):7693–7707.
146. Kononoff A, Vuolteenaho K, Hamalainen M, et al. Metabolic syndrome, disease activity, and adipokines in patients with newly diagnosed inflammatory joint diseases. J Clin Rheumatol. 2021;27(8):e349–e356.
147. Stern JH, Rutkowski JM, Scherer PE. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 2016;23(5):770–784.
148. Obradovic M, Sudar-Milovanovic E, Soskic S, et al. Leptin and obesity: role and clinical implication. Front Endocrinol. 2021;12:585887.
149. Paquet J, Goebel JC, Delaunay C, et al. Cytokines profiling by multiplex analysis in experimental arthritis: which pathophysiological relevance for articular versus systemic mediators? Arthritis Res Ther. 2012;14(2):R60.
150. Tian G, Liang JN, Pan HF, Zhou D. Increased leptin levels in patients with rheumatoid arthritis: a meta-analysis. Ir J Med Sci. 2014;183(4):659–666.
151. Sugioka Y, Tada M, Okano T, et al. Acquired leptin resistance by high-fat feeding reduces inflammation from collagen antibody-induced arthritis in mice. Clin Exp Rheumatol. 2012;30:5.
152. Bastard J-P, Maachi M, Lagathu C, et al. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. 2006;17(1):4–12.
153. Shervington L, Darekar A, Shaikh M, Mathews R, Shervington A. Identifying reliable diagnostic/predictive biomarkers for rheumatoid arthritis. Biomark Insights. 2018;13:1177271918801005.
154. Boers M, Nurmohamed MT, Doelman CJA, et al. Influence of glucocorticoids and disease activity on total and high density lipoprotein cholesterol in patients with rheumatoid arthritis. Ann Rheum Dis. 2003;62(9):842–845.
155. Georgiadis AN, Papavasiliou EC, Lourida ES, et al. Atherogenic lipid profile is a feature characteristic of patients with early rheumatoid arthritis: effect of early treatment--a prospective, controlled study. Arthritis Res Ther. 2006;8(3):R82.
156. Morris SJ, Wasko MC, Antohe JL, et al. Hydroxychloroquine use associated with improvement in lipid profiles in rheumatoid arthritis patients. Arthritis Care Res. 2011;63(4):530–534.
157. Biniecka M, Canavan M, McGarry T, et al. Dysregulated bioenergetics: a key regulator of joint inflammation. Ann Rheum Dis. 2016;75(12):2192–2200.
158. Roubille C, Richer V, Starnino T, et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Ann Rheum Dis. 2015;74(3):480–489.
159. Greenberg JD, Kremer JM, Curtis JR, et al. Tumour necrosis factor antagonist use and associated risk reduction of cardiovascular events among patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70(4):576–582.
160. McGarry T, Orr C, Wade S, et al. JAK/STAT blockade alters synovial bioenergetics, mitochondrial function, and proinflammatory mediators in rheumatoid arthritis. Arthritis Rheumatol. 2018;70(12):1959–1970.
161. Lee EB, Fleischmann R, Hall S, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. 2014;370(25):2377–2386.
162. Fleischmann R, Kremer J, Cush J, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367(6):495–507.
163. van Vollenhoven RF, Fleischmann R, Cohen S, et al. Tofacitinib or Adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367(6):508–519.
164. van der Heijde D, Tanaka Y, Fleischmann R, et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month Phase III randomized radiographic study. Arthritis Rheum. 2013;65(3):559–570.
165. Kremer J, Li ZG, Hall S, Fleischmann R, Genovese M, Martin-Mola E. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2013;159(4):253–261.
166. Abboud G, Choi SC, Kanda N, Zeumer-Spataro L, Roopenian DC, Morel L. Inhibition of glycolysis reduces disease severity in an autoimmune model of rheumatoid arthritis. Front Immunol. 2018;9:1973.
167. Raez LE, Papadopoulos K, Ricart AD, et al. A Phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71(2):523–530.
168. Singh D, Banerji AK, Dwarakanath BS, et al. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther Onkol. 2005;181(8):507–514.
169. Wang T, Jiao Y, Zhang X. Immunometabolic pathways and its therapeutic implication in autoimmune diseases. Clin Rev Allergy Immunol. 2021;60(1):55–67.
170. Badshah H, Ali T, Shafiq-ur R, et al. Protective effect of lupeol against lipopolysaccharide-induced neuroinflammation via the p38/c-Jun N-terminal kinase pathway in the adult mouse brain. J Neuroimmune Pharmacol. 2016;11(1):48–60.
171. Papi Reddy K, Singh AB, Puri A, Srivastava AK, Narender T. Synthesis of novel triterpenoid (lupeol) derivatives and their in vivo antihyperglycemic and antidyslipidemic activity. Bioorg Med Chem Lett. 2009;19(15):4463–4466.
172. Beattie JH, Nicol F, Gordon MJ, et al. Ginger phytochemicals mitigate the obesogenic effects of a high-fat diet in mice: a proteomic and biomarker network analysis. Mol Nutr Food Res. 2011;55(Suppl 2):S203–213.
173. Hu YH, Han J, Wang L, et al. alpha-mangostin alleviated inflammation in rats with adjuvant-induced arthritis by disrupting adipocytes-mediated metabolism-immune feedback. Front Pharmacol. 2021;12:692806.
174. Jiang TT, Ji CF, Cheng XP, et al. alpha-mangostin alleviated HIF-1alpha-mediated angiogenesis in rats with adjuvant-induced arthritis by suppressing aerobic glycolysis. Front Pharmacol. 2021;12:785586.
175. Zuo J, Wang X, Liu Y, et al. Integrating network pharmacology and metabolomics study on anti-rheumatic mechanisms and antagonistic effects against methotrexate-induced toxicity of Qing-Luo-Yin. Front Pharmacol. 2018;9:1.
176. Byun SH, Lee JH, Jung NC, et al. Rosiglitazone-mediated dendritic cells ameliorate collagen-induced arthritis in mice. Biochem Pharmacol. 2016;115:85–93.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.