")
Back to Journals » Drug Design, Development and Therapy » Volume 10
The influence of dietary sodium content on the pharmacokinetics and pharmacodynamics of fimasartan
Authors Gu N, Cho J , Shin K, Jang I , Rhee M
Received 18 August 2015
Accepted for publication 17 February 2016
Published 19 April 2016 Volume 2016:10 Pages 1525—1531
DOI https://doi.org/10.2147/DDDT.S94694
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Namyi Gu,1,2 Joo-Youn Cho,3 Kwang-Hee Shin,4 In-Jin Jang,3 Moo-Yong Rhee2,5
1Department of Clinical Pharmacology and Therapeutics, 2Clinical Trial Center, Dongguk University College of Medicine and Ilsan Hospital, Goyang, 3Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Hospital, Seoul, 4Pharmacotherapy & Translational Research Lab., College of Pharmacy, Research Institute of Pharmaceutical Sciences, Kyungpook National University, Daegu, 5Cardiovascular Center, Dongguk University College of Medicine and Ilsan Hospital, Goyang, Republic of Korea
Abstract: A low sodium diet enhances the hemodynamic effect of renin–angiotensin system blockers. It was suggested that the substrates of P-glycoprotein or cytochrome P450 3A4 were reduced on a high sodium diet. This study aimed to investigate the influence of high sodium diet on the pharmacokinetics and pharmacodynamics of fimasartan, which is a substrate of cytochrome P450 3A4 but not P-glycoprotein. The study design was a two-diet, two-period, two-sequence, randomized, open-label, and crossover with 1-week washout for diet. Eligible subjects were fed with either low sodium (50 mEq/day) diet or high sodium diet (300 mEq/day) for 7 days in the first hospitalization period and the other diet in the second period. On the seventh morning of each period, subjects received a single dose of fimasartan 60 mg in a fasted state. The serial plasma concentrations of fimasartan, serum aldosterone concentration (SAC), and plasma renin activity (PRA) were measured for pharmacokinetic–pharmacodynamic analysis. Sixteen subjects completed the study satisfying the compliance test for diets. Although the mean systemic exposure of fimasartan is slightly (≈10%) decreased on a high sodium diet, the difference was not statistically or clinically significant (P>0.05). The SAC and PRA after fimasartan administration were highly dependent on their baseline levels. The dietary sodium content influenced the baseline of SAC and PRA, but did not influence the ratio change of SAC and PRA after fimasartan treatment. The ratio change of SAC after fimasartan treatment was correlated to the systemic exposure of fimasartan (P<0.05), while the correlation between the ratio change of PRA after fimasartan treatment and the individual systemic exposure of fimasartan was not significant (P>0.05). In conclusion, the pharmacokinetics of fimasartan and ratio changes of SAC and PRA after fimasartan treatment were not significantly influenced by dietary sodium content.
Keywords: aldosterone, renin activity, angiotensin receptor blocker, aldosterone, renin, cytochrome P450 3A4, P-glycoprotein, healthy, sodium diet
Introduction
The renin–angiotensin system (RAS) performs a critical role in the homeostatic regulation of body fluid volume, electrolyte balance, and blood pressure. A low sodium diet (LSD) is one of the stimulating factors of the RAS, and the RAS activation is an important physiological mechanism to guard against blood pressure lowering in a low sodium intake condition.1 Therefore, the RAS blockers have been known to enhance the blood pressure-lowering effect on a low sodium intake.2–4
On the other hand, the pharmacokinetic and pharmacodynamic properties of several drugs are also sometimes influenced by dietary sodium intake. High dietary sodium intake had been reported to be related to 20%–60% reduction of the oral bioavailability in some cardiovascular drugs, such as verapamil, atenolol, and quinidine, which are substrates of energy-dependent intestinal efflux transporter P-glycoprotein (P-gp) and intestinal metabolic enzyme cytochrome P450 3A (CYP3A) as reported from the previous clinical studies in humans.5–7 The prolonged hypertonic stress in the intestinal mucosal epithelial cells increases the expression of transporters or enzymes, including P-gp and CYP3A, which are involved in raising the intracellular level of compatible osmolytes in the previous in vitro and in vivo studies using human intestinal cell lines and rats.8,9 Therefore, modulation of intestinal P-gp and/or CYP3A activities has been suggested as the major mechanism to reduce the bioavailability of those drugs on a high sodium diet (HSD).6 Recently, Azizi et al10 reported that the systemic exposure of candersartan, which is a substrate of P-gp but not CYP3A, was ~30% lower on a HSD compared to on a LSD, while no such reduction was observed with valsartan and ramipril, which are neither substrate of P-gp nor CYP3A. These findings suggest that 1) the solitary modulation of intestinal P-gp activity by a high sodium intake could be a significant mechanism of the decreased bioavailability of a drug even without the modulation of CYP3A4 activity and that 2) some of the RAS blockers, such as candesartan, might have more prominent intra-subject variability in the clinical efficacy due to their pharmacokinetic change by dietary sodium content. If the bioavailability of a RAS blocker is reduced by a HSD, it might be a practically important issue. Because the reduction in the clinical efficacy of the RAS blockers when on a HSD is generally reported as a physiologic class even in the RAS blocker whose bioavailability is not affected by dietary sodium intake,11 a RAS blocker which has reduced bioavailability when on a HSD might have additionally lower, sometimes insufficient, clinical efficacy compared to other RAS blockers.
Fimasartan is a nonpeptide angiotensin II antagonist with an angiotensin II type 1 receptor selectivity. Although the pharmacologic mechanism is comparable to candersartan, fimasartan is known to be a substrate of CYP3A but not a substrate of P-gp unlike candersartan.12
Thus, in the present study, we aimed to investigate the influence of sodium intake on the pharmacokinetic and pharmacodynamic properties of fimasartan.
Materials and methods
Study population
The subjects eligible for this study were healthy normotensive Korean male volunteers, aged 20–45 years, weighing between 50 and 90 kg and with a body mass index between 19 and 27 kg/m2. Subjects were excluded if they had any clinically significant disease or had abnormal findings on physical examination, vital signs, 12-lead electrocardiography, serology, urinary drug screening test, and routine clinical laboratory tests (hematology, blood coagulation, clinical chemistry, and urinalysis), which were performed within 21 days prior to the first administration of the study drug.
Study design
The protocol was approved by the institutional review board of Dongguk University Ilsan Hospital. Written informed consent was obtained from every subject before screening. The study was conducted at the Clinical Trial Center of Dongguk University Ilsan Hospital in Goyang, Republic of Korea, in accordance with the Declaration of Helsinki for biomedical research involving human participants and the Guidelines for Good Clinical Practice (ClinicalTrials.gov identifier: NCT02147704).13,14
This study was designed as a two-diet, two-period, two-sequence, open-label, randomized, and crossover clinical trial. All the subjects experienced both 1 week of LSD and HSD periods with 1-week washout interval. The order of the periods (LSD → HSD or HSD → LSD) of each individual was randomly assigned with a ratio of 1:1. The identical sodium restricted diets (50 mEq/day) with fixed calories and composition of nutrients (2,300 kcal with 60%–65% carbohydrate, 15% protein, and 20%–25% fat) were served twice for subjects in the LSD and HSD periods. To induce HSD, subjects were additionally given sodium tablets (250 mmol/day) for 7 days in the HSD period. On the seventh morning of each period, subjects orally received a single dose of fimasartan 60 mg with ~240 mL of water in the fasting state. The excreted amounts of sodium, potassium, and creatinine in urine for 24 hours were measured on the sixth (baseline) and seventh day (fimasartan treatment) of each period to assess the compliance for having diets and sodium tablets. It was judged for subjects to be in a good compliance if their urinary excreted sodium amount is ≤100 mmol/day in the LSD period and ≥200 mmol/day in the HSD period. The pharmacokinetic blood samples to determine the plasma fimasartan concentrations were serially obtained before dose and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, and 24 hours after dose on the seventh day. The pharmacodynamic blood samples to determine the serum aldosterone concentration (SAC) and plasma renin activity (PRA) were obtained not only before dose and at 2, 4, 6, 8, 12, and 24 hours on the seventh day but also at the equivalently scheduled timepoints on the sixth day for the baseline assessment. Subjects were discharged after physician’s assessment of their health on the eighth morning of each period. The end visit was performed between the fourth and sixth day after the last discharge.
Pharmacokinetic assessment
A total of 8 mL blood was drawn and collected in a sodium heparin tube at each sampling timepoint to assess the pharmacokinetics of fimasartan. The plasma was separated by immediate centrifugation of the blood in the heparinized tube at 1,500 g for 10 minutes at 4°C. The separated plasma was immediately stored below −70°C until analysis.
The plasma concentrations of fimasartan were quantified with a validated method by using high performance liquid chromatography (Agilent 1200 series; Agilent Technologies, Santa Clara, CA, USA) coupled with mass spectrometry/mass spectrometry an API4000 QTrap hybrid triple-quadrupole linear ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) by the Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Hospital, Seoul, Republic of Korea. In brief, 50 μL of plasma samples was mixed with 250 μL of BR-A-563 (2.5 ng/mL in acetonitrile), an internal standard. After centrifugation at 19,320 g for 10 minutes at 4°C, the supernatant was separated on a Poroshell120 C18 column (2.7 μm, 50×3.0 mm inner diameter; Agilent Technologies). The mass transitions were m/z 502.277 → 207.400 for fimasartan and m/z 526.341 → 207.300 for internal standard. The standard curve interval for fimasartan was 0.5–500 μg/L. The within- and between-run accuracy of the analysis ranged from 89.30% to 103.2%, and precision was lower than 10.846%.
The pharmacokinetic parameters of fimasartan in individual subjects were analyzed with a noncompartmental method by using Phoenix® WinNonlin® software (v 1.3, Pharsight Corporation, St Louis, MO, USA). The maximal plasma concentration (Cmax) and the time to Cmax (Tmax) were obtained from the observed values. The terminal elimination rate constant (λz) was estimated by a log-linear regression of logarithmic-linear decline of the plasma concentration–time curve. The terminal elimination half-life (T1/2) was determined by the equation ln 2/λz. The area under the curve from before dose to 24 hours after dose (AUC0–24 h) was derived by using the log-linear trapezoidal rule, and the area under the curve from before administering a dose to infinite levels (AUC0–∞) was obtained by summation of AUC0–24 h and Clast/λz (Clast: the last measurable concentration).
Pharmacodynamic assessments
A total of 6 mL of blood was drawn in the upright position and collected in a serum separating tube and ethylenediaminetetraacetic acid tube for quantification of SAC and PRA, respectively. The blood in the serum separating tube was centrifuged after 30-minute standing, while the blood in the ethylenediaminetetraacetic acid tube was immediately centrifuged to separate serum and plasma. The centrifugation conditions of both tubes were 2,800 g for 10 minutes at 4°C. The separated serum and plasma were immediately stored below 70°C until analysis. The SAC and PRA were determined by the radioimmunoassay method using commercialized kits (SAC: Immunotech, Prague, Czech Republic, PRA: DiaSorin Inc., Stillwater, MN, USA) at Green Cross LabCell, Yongin, Gyeonggi, Republic of Korea. The log-linear trapezoidal rule was applied to calculate the AUC0–24 h of SAC and PRA for seventh day for the treatment as well as the sixth day for the baseline.
Tolerability assessment
Subjective symptoms were monitored throughout the study via investigator’s questionnaires or subjects’ spontaneous reports in verbal or by writing on the provided subject’s notebook. In addition, objective signs were monitored by conducting vital signs, physical examinations, clinical laboratory tests, and 12-lead electrocardiograms. Systolic and diastolic blood pressures were measured on the sitting position after at least 5-minute rest with an automatic device (WatchBP Office Twin200, Microlife Corp., Taipei, Taiwan).
Statistical analysis
The log-transformed Cmax, AUC0–24 h, and AUC0–∞ values were compared with the least-squares mean ratios and their 90% confidence intervals using a mixed model ANOVA, including sequence, period, and treatment as fixed effects and subjects nested within the sequence as random effects.15 Regression analysis was performed in baseline AUC0–24 h on the sixth day versus treatment AUC0–24 h on the seventh day of SAC and PRA by the least-squares method with log-transformed values. The pharmacokinetic–pharmacodynamic correlation was analyzed between the systemic exposure of fimasartan (logarithmic AUC0–24 h of fimasartan) and the changes of SAC and PRA (logarithmic treatment AUC0–24 h – logarithmic baseline AUC0–24 h). Other continuous parameters, such as Tmax, T1/2 of fimasartan, and changes of blood pressures, were compared by paired t-test. SAS® version 9.3 (SAS Institute Inc., Cary, NC, USA) was used for the statistical analysis, and the P-values <0.05 were considered significant.
Results
Subjects
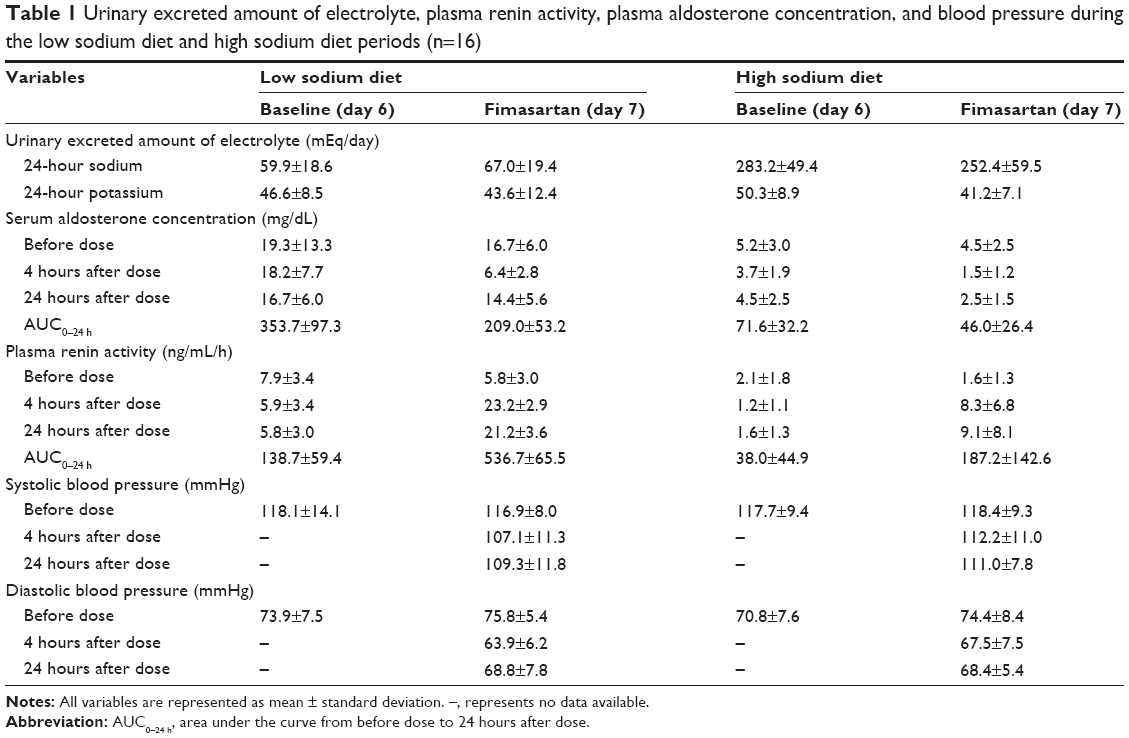
Thirty-three subjects were screened, and 27 subjects aged 20–44 years (mean: 28.8, standard deviation: 7.6) and weighing 54.5–91.0 kg (mean: 69.3, standard deviation: 8.8) were enrolled in this study. Seventeen subjects finally completed the study and one of them was unsatisfied in the compliance test; his excreted amounts of sodium, potassium, and creatinine in urine for 24 hours on sodium-restricted period were >100 mmol/day. Sixteen subjects who were satisfied in the compliance tests were included in the final pharmacokinetic and pharmacodynamics analysis. The urinary excretion of sodium on the sixth day (baseline) and seventh day (fimasartan treatment) represents good compliance of 16 subjects with the diets (Table 1).
 | Table 1 Urinary excreted amount of electrolyte, plasma renin activity, plasma aldosterone concentration, and blood pressure during the low sodium diet and high sodium diet periods (n=16) |
Pharmacokinetics
The time courses of mean plasma fimasartan concentrations were comparable in the LSD and HSD periods (Figure 1). There was no clear trend observed in the changes of the individual systemic exposures of fimasartan in LSD versus HSD periods (Figure 2). In the statistical analysis, the mean systemic exposure of fimasartan was slightly (≈10%) lower in the HSD period than in the LSD period, but the difference was not significant (P>0.05) (Table 2). The geometric mean ratios (HSD/LSD) and their 90% confidence intervals for Cmax, AUC0–24 h, and AUC0–∞ were 0.84 (0.60–1.18), 0.91 (0.78–1.05), and 0.90 (0.79–1.03), respectively. Tmax and T1/2 of fimasartan were also not significantly associated with the sodium status (P>0.05).
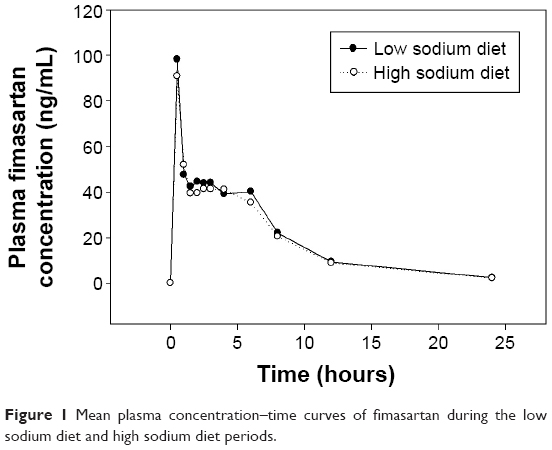 | Figure 1 Mean plasma concentration–time curves of fimasartan during the low sodium diet and high sodium diet periods. |
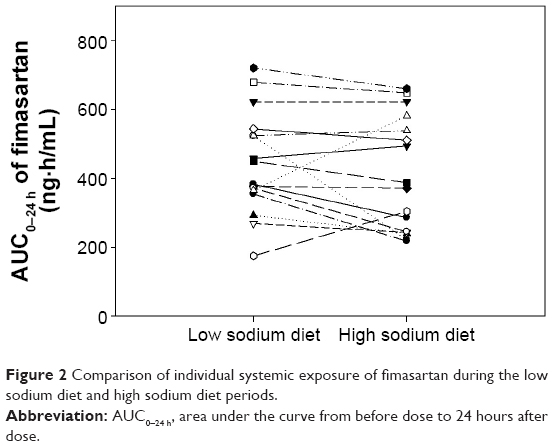 | Figure 2 Comparison of individual systemic exposure of fimasartan during the low sodium diet and high sodium diet periods. |
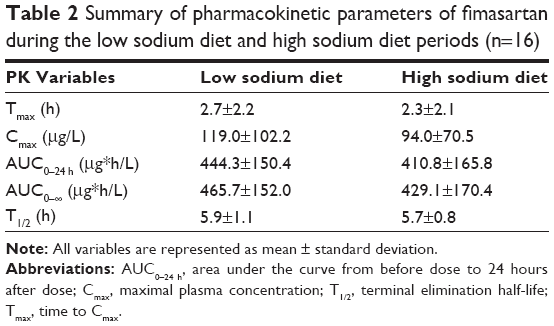 | Table 2 Summary of pharmacokinetic parameters of fimasartan during the low sodium diet and high sodium diet periods (n=16) |
Pharmacodynamics
The baseline levels of SAC were approximately fourfold higher in the LSD period compared to the HSD period (Table 1). The mean SAC were reduced to below a half of the baseline levels at 4 hours after the fimasartan administration and were incompletely recovered at 24 hours after treatment, on both LSD and HSD. The baseline levels of PRA were approximately threefold higher in the LSD period compared to the HSD period (Table 1). The fimasartan treatment increased mean PRA approximately fourfold of baseline levels, on both LSD and HSD. The elevated PRA continued over 24 hours after single administration of fimasartan with both diets.
In the comparison of regression slopes in the LSD versus HSD periods, the logarithmic linear regression line of the individual baseline AUC0–24 h versus treatment AUC0–24 h of SAC was not significantly different (P=0.2334), while the slope of the logarithmic regression curve of baseline AUC0–24 h versus treatment AUC0–24 h of PRA in the LSD period was significantly lower than that in the HSD period (P=0.0156).
In the combined analysis, the logarithmic linear regression line of the individual baseline AUC0–24 h versus treatment AUC0–24 h of SAC followed slightly transferred line of identity to the negative direction (r=0.9315, P<0.0001) (Figure 3A), while hyperbolic regression curve was generally well fitted to the relationship of baseline AUC0–24 h versus treatment AUC0–24 h of PRA (r=0.9058, P<0.0001) (Figure 3B).
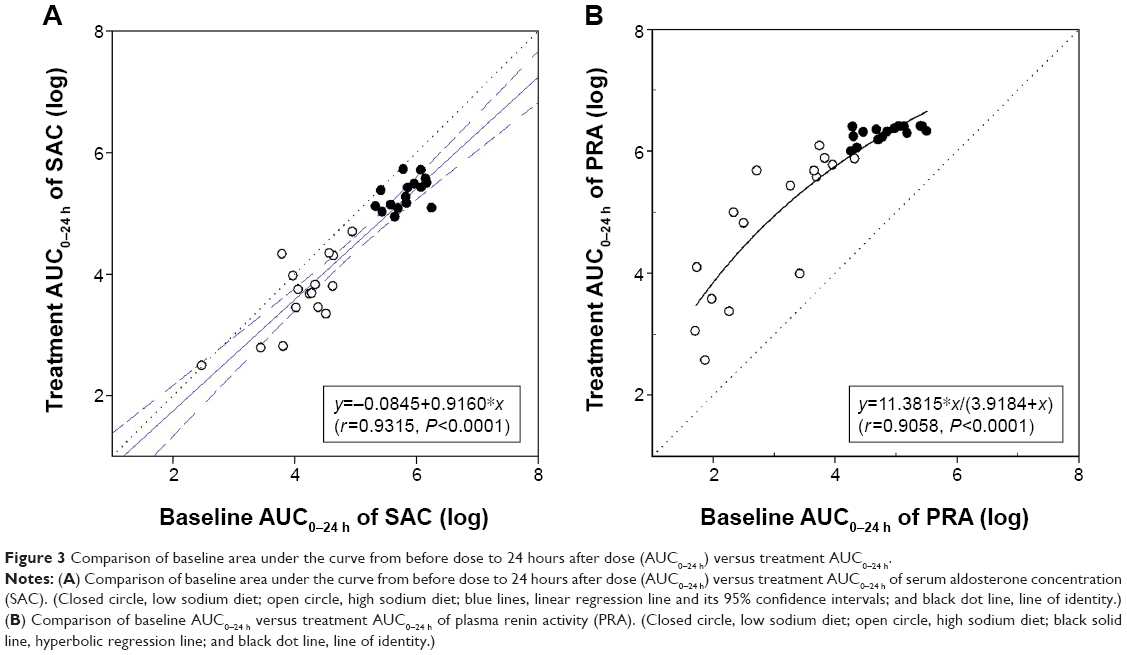 | Figure 3 Comparison of baseline area under the curve from before dose to 24 hours after dose (AUC0–24 h) versus treatment AUC0–24 h. |
Relationship between pharmacokinetics and pharmacodynamics
In the pharmacokinetic–pharmacodynamic correlation analyses, weak but significant correlation was found between the change in logarithmic AUC0–24 h of SAC after treatment and the logarithmic AUC0–24 h of fimasartan (r=−0.3595, P=0.0433), while the correlation between the PRA and systemic exposure of fimasartan was not significant (r=−0.0719, P=0.6959).
Tolerability
A single oral dose of fimasartan was well tolerated in the participants of this study. Sixteen cases (eight for each diet) of treatment-emergent adverse events were reported, including diarrhea, abdominal pain, dizziness, vasovagal syncope, headache, fatigue, increased serum creatine phosphokinase, and papule. All of them were in mild to moderate intensity and spontaneously resolved without any sequelae. There was no serious adverse event during the study.
Discussion
The present observation supported that the dietary sodium content rarely affects the pharmacokinetics of fimasartan. The mean systemic exposure of fimasartan is slightly (≈10%) decreased in the HSD period. The 90% confidence interval of the difference narrowly crossed the lower margin of the standard criteria for assuming bioequivalence with 0.78–1.05 for AUC0–24 h,16,17 and this difference was not statistically or clinically significant.
Fimasartan is reported to be metabolized primarily by CYP3A4 in the in vitro metabolism studies using human liver microsomes.18 However, the metabolism of CYP3A4 in the intestine has not been investigated yet. Fimasartan is reported to be transported by OAT1 (organic anion transporter 1), OATP1B1 (organic anion transporting polypeptides), P-gp (p-glycoprotein), MRP2 (multidrug resistance associated protein 2), BCRP (breast cancer resistance protein) in the in vitro studies using solute carrier overexpressed oocytes.18 The present study results suggest that the solitary modulation effect of intestinal CYP3A activity which is induced by dietary sodium intake influences little on the pharmacokinetics of fimasartan, supporting that the metabolism of intestinal CYP3A4 might be trivial on the elimination of fimasartan. It might be caused by the metabolically stable characteristics of fimasartan. Although the primarily responsible metabolic enzyme of fimasartan is CYP3A4, it is assumed to be involved to a minor extent because more than 90% of the fimasartan moiety in plasma is reported to be the parent form.19
The pharmacodynamics of the SAC and PRA after the fimasartan treatment showed their baseline-dependency with high correlation coefficients over 0.9. The logarithmic regression line of the baseline versus treatment AUC0–24 h of SAC was parallel to the line of identity (Figure 3A), suggesting that the ratio of SAC (fimasartan treatment/baseline) was constantly sustained regardless of their baseline SAC or sodium intake amounts. The pharmacokinetic–pharmacodynamic correlation analysis results indicated that the AUC0–24 h of SAC after fimasartan treatment was larger in the individuals with higher systemic exposure of fimasartan, and was not dependent on sodium intake.
The PRA level after the fimasartan treatment was also dependent on its baseline value. However, the relationship between the baseline versus after treatment was more fittable to the hyperbolic regression curve than the linear regression line, suggesting that PRA could be saturated after fimasartan treatment in some individuals whose PRA before treatment had been already highly enhanced by the LSD status. An explainable mechanism for this saturated phenomenon might be the negative feedback on the renin production by a direct action of increased angiotensin II level in response to the angiotensin II receptor blockade.20 No correlation between the AUC0–24 h ratios of PRA after fimasartan treatment and systemic exposure of fimasartan in this study seems to be due to this saturated phenomenon of PRA.
Limitations
The present study had some limitations due to its open-label, single-dose design, and small-sized population with younger healthy normotensive subjects. Although these experimental conditions of the study efficiently contributed to describe the effect of dietary sodium content on the pharmacokinetics and pharmacodynamics of fimasartan, the results of this study could not be directly generalized to the hypertensive patients with long-term treatment of fimasartan. The study results would be scientifically more robust, if a positive or negative control drug, such as candesartan or valsartan, was applied in the study. The effect of the body fluid status change, one of the confounding factors of dietary sodium intake, was not eliminated from the study results, because the observations of body fluid status change via repeated body weight measurements or water consumption restriction were not performed in this study.
Conclusion
The effect of solitary modulation of intestinal CYP3A4 by a high sodium intake did not significantly affect the pharmacokinetics of fimasartan. The pharmacodynamics of fimasartan was highly dependent on the baseline level of RAS activation, which is influenced by dietary sodium intake, and no additional reduction on the ratio change of SAC and PRA after fimasartan treatment was found when on a high sodium intake. Further investigations are needed to evaluate the effect of dietary sodium intake on the pharmacokinetics and pharmacodynamics of fimasartan in the hypertensive patients with long-term treatment.
Acknowledgments
This study was sponsored by Boryung Pharmaceutical Co., Ltd., Seoul, Republic of Korea. The authors would like to thank Ms. In-Ja Jung, Clinical Trial Center at Dongguk University Ilsan Hospital, who assisted in performing the clinical trial, and Ji Hyun Kim, Department of Pediatrics at Dongguk University Ilsan Hospital, who contributed as a researcher of the clinical trial. The authors do not have stock ownership or patents. There was no financial benefit from the sponsor, including the bonuses based on the success of this drug, and no payment to recruit and treat volunteers.
Disclosure
The authors report no conflicts of interest in this work.
References
Cappuccio FP, Markandu ND, Sagnella GA, MacGregor GA. Sodium restriction lowers high blood pressure through a decreased response of the renin system – direct evidence using saralasin. J Hypertens. 1985;3(3):243–247. | ||
MacGregor GA, Markandu ND, Singer DR, Cappuccio FP, Shore AC, Sagnella GA. Moderate sodium restriction with angiotensin converting enzyme inhibitor in essential hypertension: a double blind study. Br Med J. 1987;294(6571):531–534. | ||
MacGregor GA, Markandu ND, Sagnella GA, Singer DR, Cappuccio FP. Double-blind study of three sodium intakes and long-term effects of sodium restriction in essential hypertension. Lancet. 1989;2(8674):1244–1247. | ||
Singer DR, Markandu ND, Cappuccio FP, Miller MA, Sagnella GA, MacGregor GA. Reduction of salt intake during converting enzyme inhibitor treatment compared with addition of a thiazide. Hypertension. 1995;25(5):1042–1044. | ||
Fromm MF, Darbar D, Dell’Orto S, Roden DM. Modulation of effect of dietary salt on prehepatic first-pass metabolism: effects of beta-blockade and intravenous salt loading. J Pharmacol Exp Ther. 1999;290(1):253–258. | ||
Darbar D, Fromm MF, Dell’Orto S, et al. Modulation by dietary salt of verapamil disposition in humans. Circulation. 1998;98(24):2702–2708. | ||
Darbar D, Dell’Orto S, Morike K, Wilkinson GR, Roden DM. Dietary salt increases first-pass elimination of oral quinidine. Clin Pharmacol Ther. 1997;61(3):292–300. | ||
Kosuge K, Chuang AI, Uematsu S, et al. Discovery of osmosensitive transcriptional regulation of human cytochrome P450 3As by the tonicity-responsive enhancer binding protein (nuclear factor of activated T cells 5). Mol Pharmacol. 2007;72(4):826–837. | ||
Kang HJ, Song IS, Lee SS, Yoo MA, Shin JG. Effects of dietary salt on the expression of drug transporters, cytochrome P4503a, and nuclear receptors in rats. Xenobiotica. 2008;38(2):147–155. | ||
Azizi M, Blanchard A, Charbit B, et al. Effect of contrasted sodium diets on the pharmacokinetics and pharmacodynamic effects of renin-angiotensin system blockers. Hypertension. 2013;61(6):1239–1245. | ||
Charytan DM, Forman JP. You are what you eat: dietary salt intake and renin-angiotensin blockade in diabetic nephropathy. Kidney Int. 2012;82(3):257–259. | ||
Kim JW, Yi S, Kim TE, et al. Increased systemic exposure of fimasartan, an angiotensin II receptor antagonist, by ketoconazole and rifampicin. J Clin Pharmacol. 2013;53(1):75–81. | ||
World Medical Association Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects, 55th WMA General Assembly, Tokyo 2004. Available from: http://www.wma.net/en/30publications/10policies/b3/17c.pdf. Accessed April 4, 2015. | ||
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Guideline for Good Clinical Practice E6 (R1). Step 4 version, ICH Harmonised Tripartite Guideline. 1996. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf. Accessed April 2015. | ||
Bonate PL, Howard D. Pharmacokinetics in Drug Development. Volume 1: Clinical Study Design and Analysis. Arlington, VA: American Association of Pharmaceutical Scientists; 2004. | ||
Midha KK, Rawson MJ, Hubbard JW. The bioequivalence of highly variable drugs and drug products. Int J Clin Pharmacol Ther. 2005;43(10):485–498. | ||
Food and Drug Administration. Guidance for Industry: Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs – General Considerations. Rockville, MD: Food and Drug Administration; 2014. | ||
Yi S, Kim TE, Yoon SH, et al. Pharmacokinetic interaction of fimasartan, a new angiotensin II receptor antagonist, with amlodipine in healthy volunteers. J Cardiovasc Pharm. 2011;57(6):682–689. | ||
Chi YH, Lee H, Paik SH, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of fimasartan following single and repeated oral administration in the fasted and fed states in healthy subjects. Am J Cardiovasc Drugs. 2011;11(5):335–346. | ||
Atlas SA. The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J Manag Care Pharm. 2007;13(8 Suppl B):9–20. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.