")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 11
The importance of the thick ascending limb of Henle’s loop in renal physiology and pathophysiology
Authors Zacchia M, Capolongo G, Rinaldi L, Capasso G
Received 13 October 2017
Accepted for publication 25 November 2017
Published 15 February 2018 Volume 2018:11 Pages 81—92
DOI https://doi.org/10.2147/IJNRD.S154000
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Pravin Singhal
Miriam Zacchia, Giovanna Capolongo, Luca Rinaldi, Giovambattista Capasso
Division of Nephrology, Department of Cardio-Thoracic and Respiratory Sciences, Università della Campania “Luigi Vanvitelli”, Naples, Italy
Abstract: The thick ascending limb (TAL) of Henle’s loop is a crucial segment for many tasks of the nephron. Indeed, the TAL is not only a mainstay for reabsorption of sodium (Na+), potassium (K+), and divalent cations such as calcium (Ca2+) and magnesium (Mg2+) from the luminal fluid, but also has an important role in urine concentration, overall acid–base homeostasis, and ammonia cycle. Transcellular Na+ transport along the TAL is a prerequisite for Na+, K+, Ca2+, Mg2+ homeostasis, and water reabsorption, the latter through its contribution in the generation of the cortico-medullar osmotic gradient. The role of this nephron site in acid–base balance, via bicarbonate reabsorption and acid secretion, is sometimes misunderstood by clinicians. This review describes in detail these functions, reporting in addition to the well-known molecular mechanisms, some novel findings from the current literature; moreover, the pathophysiology and the clinical relevance of primary or acquired conditions caused by TAL dysfunction are discussed. Knowing the physiology of the TAL is fundamental for clinicians, for a better understanding and management of rare and common conditions, such as tubulopathies, hypertension, and loop diuretics abuse.
Keywords: TAL, sodium handling, potassium handling, acid–base homeostasis, urine concentration
Physiology of the thick ascending limb (TAL)
Morphological features of TAL cells
The loop of Henle is a highly specialized nephron site, with two peculiar properties, its extreme heterogeneity and its anatomic configuration. It is composed of the pars recta of the proximal tubule (PT; thick descending limb), the thin descending and ascending limbs, the TAL, and the macula densa.1 The descending limb penetrates the medulla, where an increasing interstitial osmotic gradient is guaranteed by the noticeable addition of NaCl through the water impermeable TAL, amplified by the hairpin-shaped loop of Henle’s, with the so-called countercurrent multiplier system.2,3 A similar arrangement of the surrounding vessels prevents osmotic gradient dissipiation.4 Electron microscopy studies demonstrated the presence of two distinct cell types in the TAL, differing in the configuration of the apical membrane: rough-surface cells (R cells), characterized by abundant apical microvilli, and smooth-surface cells (S cells) with no microprojections, but with several subapical vesicles.5 The former is abundant in the cortex, and the latter in the medulla. Functional studies have demonstrated that R cells have high apical and low basolateral K+ conductance, while S cells have low apical and high basolateral K+ conductance, in hamster.6 The main differences in salt absorption between the two cell types are unclear. Both cell types show similar mechanisms of transport; however, Nielsen et al demonstrated that S cells showed an intense labeling of the Na-K-2Cl cotransporter (NKCC2) on subapical vesicles, that may serve as a reservoir that can be recruited based on homeostatic needs.7
Transcellular and paracellular salt absorption along the TAL
The loop of Henle is responsible for the reabsorption of ~40% of filtered Na+, mostly in the TAL.3 Here, Na+ entry from the lumen into the cells occurs mainly via the luminal electroneutral Na+,K+,2Cl− cotransporter, NKCC2, encoded by the SLC12A1 gene.8 This cotransporter mediates secondary active Na+, K+, and 2Cl− uptake across the apical membrane.9
The protein belongs to the family of solute carrier family 12, that includes two isoforms in humans encoded by two genes, SLC12A1 (NKCC2) and SLC12A2 (NKCC1).3 While NKCC1 is widely expressed in several organs and tissues, NKCC2 is exclusively localized along the TAL.10 Once reabsorbed from the lumen, Na+ exits the cell via the sodium pump; ClC-Ka and ClC-Kb channels, with their Barttin subunit, mediate Cl− exit from the basolateral side, participating in the electronegative blood side.11 The importance of ClC channels in human physiopathology is highlighted by the evidence that mutations in every channel lead to human disease.12 Finally, K+ ions are delivered back to the lumen through the apical renal outer medulla K+ channels (ROMK). These channels perform a dual crucial role in the TAL: first, they ensure K+ recycling to the lumen, essential for salt reabsorption; second, they set a positive transepithelial voltage, that drives paracellular reabsorption of cations.13,14
Additional Na+ reabsorption is driven by the generated electrical field through the paracellular way; moreover, other cations (Ca2+ and Mg2+) are reabsorbed by this route.
There is no doubt that the bulk of Ca2+ reabsorption along the TAL occurs through the paracellular pathway, following the electrochemical gradient. NKCC2 and in particular ROMK generate the “driving force” for paracellular cation transport. Interestingly, the evidence that paracellular transport is regulated by the calcium sensing receptor (CaSR) signaling in the absence of changes in transepithelial voltage suggested that paracellular permeability may vary, with consequent changes in divalent cations reabsorption.15 The CaSR, expressed on the basolateral membrane of TAL cells, has been shown to modulate Ca2+ absorption through two mechanisms: 1) by inhibiting ROMK activity, thus leading to a decreased lumen-positive voltage;16 and 2) by modulating tight junction (TJ) structure.17,18 The latter is the main determinant of paracellular permeability. Claudins are some of the main components of TJ. Along the TAL, several claudins are expressed, including claudin-16, 19, 10, 3, and 18.19 Their role in divalent cations absorption has been recently highlighted by the discovery of salt losing phenotype associated with their dysfunction due to genetic mutations.20 However, several evidences suggest that a significant component of Ca2+ reabsorption occurs also via transcellular pathway.21 The presence on the basolateral membrane of the Na+/Ca2+ exchanger (NCX) and the Ca2+ATPase (PMCA) confirms the presence of a basolateral exit, and suggests the possible presence of a luminal route for Ca2+entry.22
Micropuncture experiments have demonstrated that nearly 60% of the filtered Mg2+ is reabsorbed in the TAL.23 Passive paracellular transit is the main route, and it is driven by the lumen-positive voltage. The importance of claudin-16 (paracellin 1) and claudin-19 in Mg2+ homeostasis has been pointed out in the recent years, given the association of their mutation with familial hypomagnesemia.24
The synergic activity of the main transporters and channels involved in salt absorption (NKCC2, ROMK, the chloride channel Kb [also known as ClC-Kb], with the Barttin subunit) and the integrity of TJs are the prerequisite to prevent electrolytes imbalance (Figure 1, Table 1). The loss of function of any of those proteins leads to a salt losing phenotype. As salt absorption in the TAL is under the control of hormones, local mediators, and several intracellular signaling pathways, it is not uncommon that impaired salt absorption is secondary to these dysfunctions. As an example, activating mutations of CASR have been described to lead to salt losing nephropathy;25 similarly, dysfunction of uromodulin, Ste20-related proline–alanine-rich kinase (SPAK), and oxidative stress response kinase (OSR1) have been shown to influence NKCC2 activity and to determine fluid and electrolytes imbalance.26–28
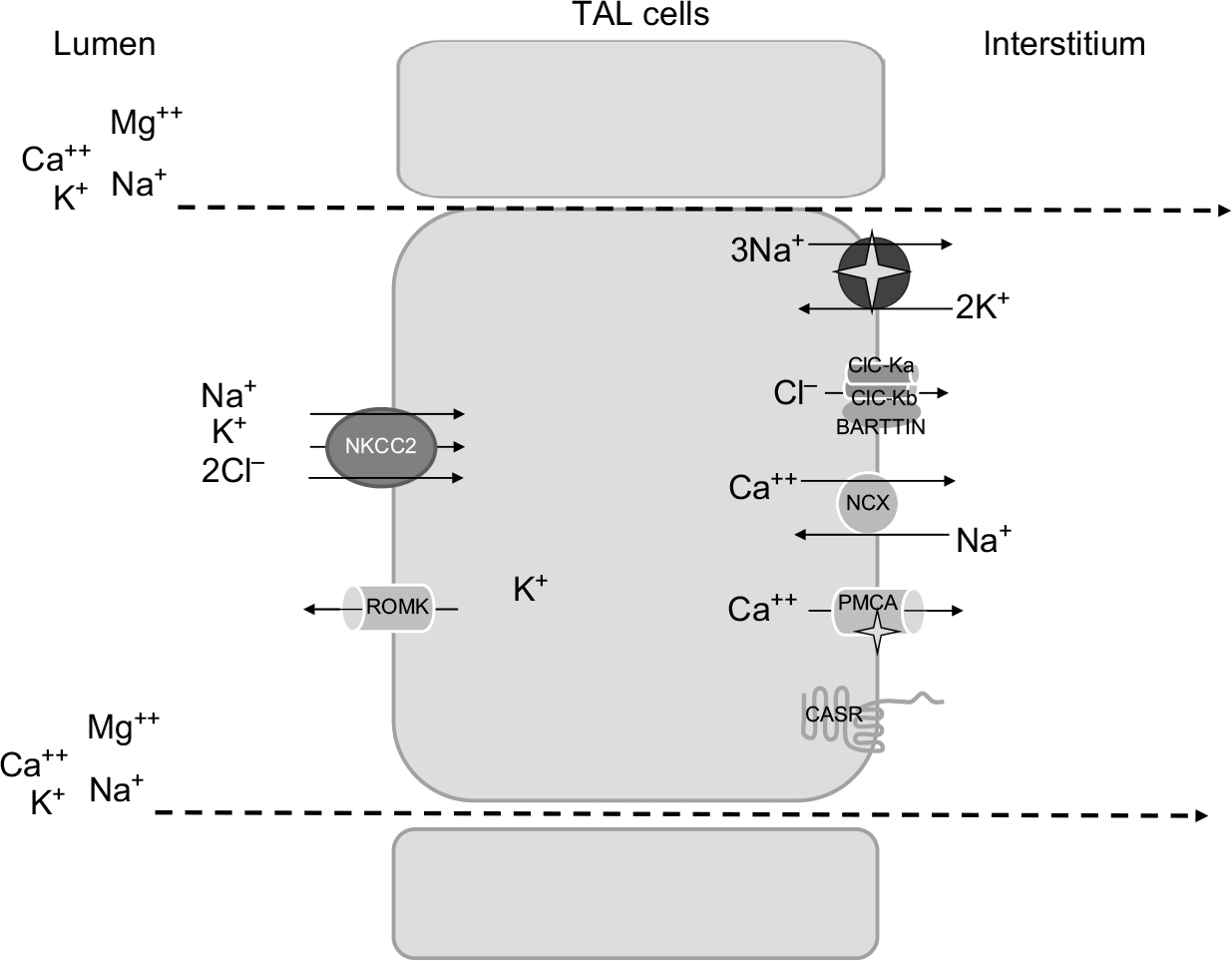 | Figure 1 Schematic representation of TAL cells, illustrating the major transcellular proteins involved in salt reabsorption. Abbreviations: TAL, thick ascending limb of the loop of Henle; ROMK, renal outer medulla K+ channels; CaSR, calcium sensing receptor; NCX, Na+/Ca2+ exchanger; PMCA, plasma membrane Ca2+ ATPase; NKCC2, Na-K-2Cl cotransporter. |
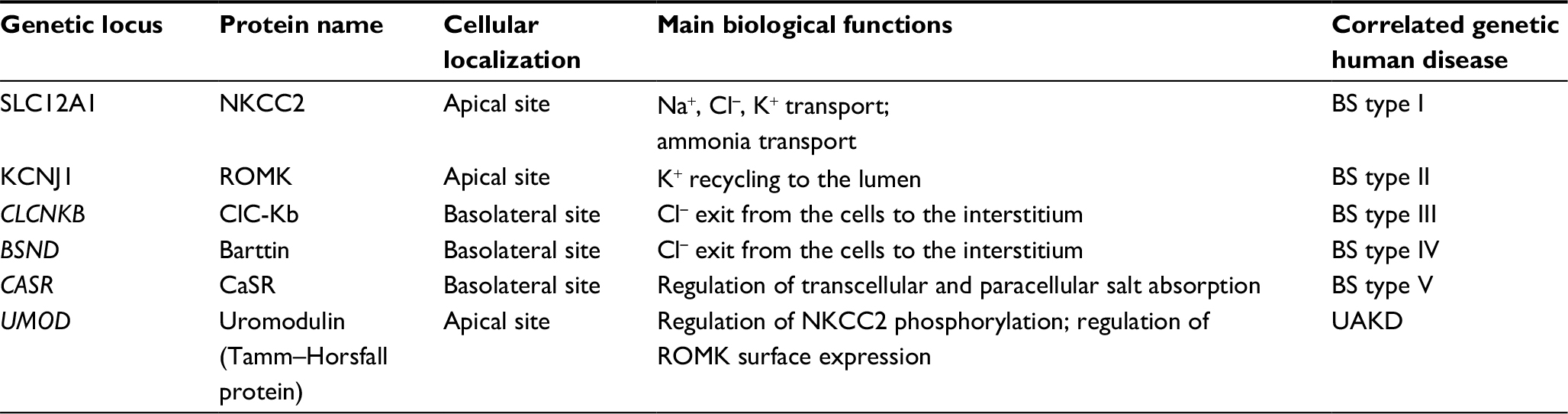 | Table 1 Main channels, transporters, and other proteins relevant in salt absorption along the TAL Abbreviations: UAKD, uromodulin-associated kidney disease; TAL, thick ascending limb of the loop of Henle; ROMK, renal outer medulla K+ channels; CaSR, calcium sensing receptor; NKCC2, Na-K-2Cl cotransporter; BS, Bartter syndrome. |
Generation of the cortico-medullar osmotic gradient
One of the major functions of TAL is the creation of the cortico-medullar osmotic gradient. In humans, interstitial osmolality progressively increases from the cortex, where it is ~290 mOsm/kg, to the tip of the medulla, where it reaches 1200 mOsm/kg.2 The U-shaped arrangement of the two branches of the loop of Henle, the different permeabilities of the descending and ascending limbs to Na+ and water, and the active Na+ reabsorption in TAL are fundamental factors in the creation of the medullary osmotic gradient.4
Fluid entering the descending limb from the PT is isotonic with the plasma and the surrounding interstitium. The renal epithelium along the descending limb is highly permeable to water but not to solutes.29 Here, the increasing interstitial osmolality from the cortex to the medulla provides the driving force for water reabsorption (Figure 2).30–32 Thus, water diffuses from the lumen to the interstitium along the descending limb and luminal osmolality rises progressively up to the tip of Henle’s loop. Active Na+ and Cl− reabsorption along the TAL serves two roles: 1) first, it dilutes the luminal fluid, as it is impermeable to water, so the solutes that accumulate into the interstitium are not followed by the osmotic flow of water; 2) second, by adding Na+ and Cl− to the interstitium, it provides a strong contribution in the generation of the interstitial osmotic gradient in the outer medulla.33
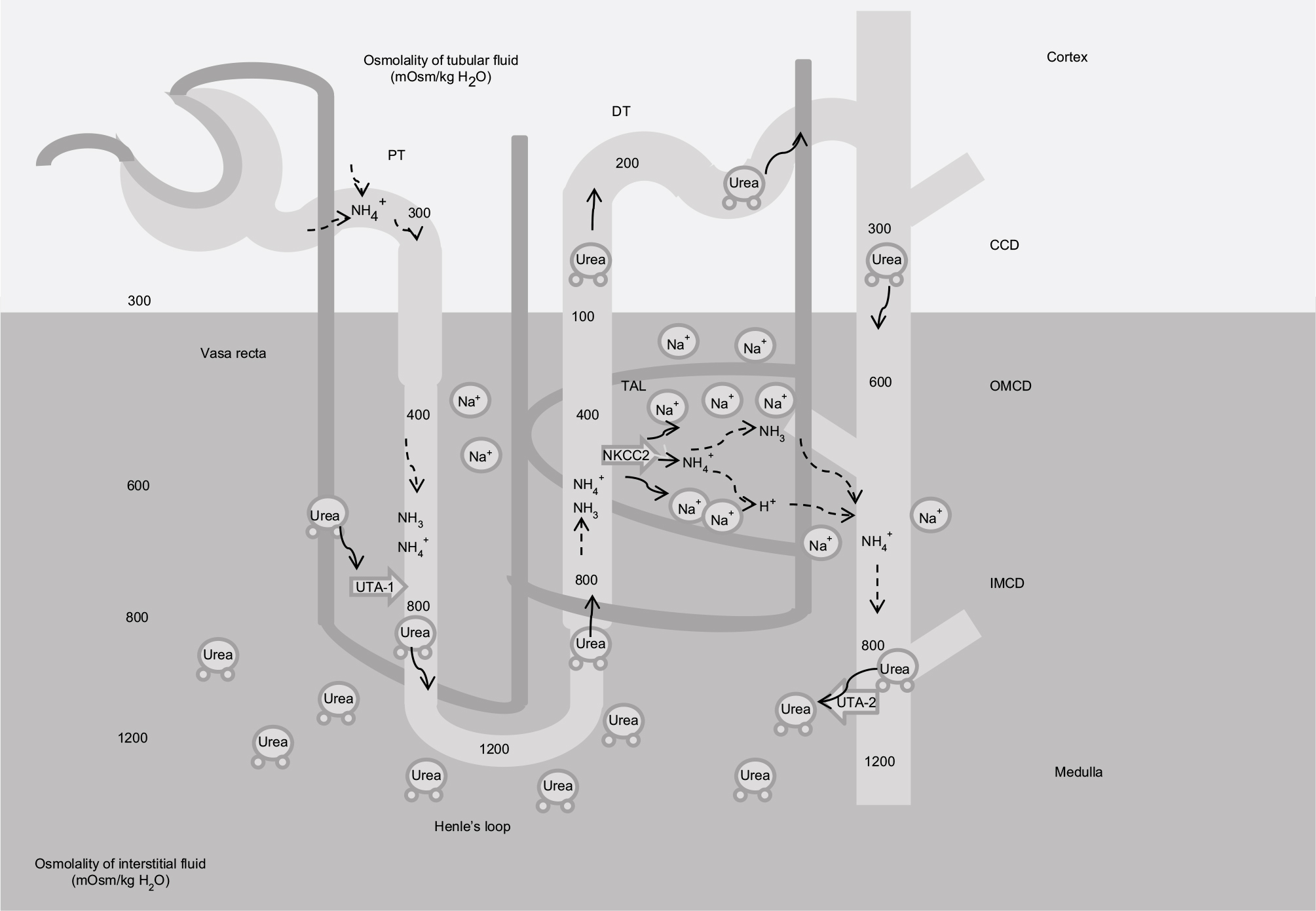 | Figure 2 Mechanism of urine concentration. Note: Contribution of Na+, urea, and NH4+/NH3 in the generation of the interstitial medullar hyperosmolality. Abbreviations: PT, proximal tubule; DT, distal tubule; UTA2, urea transporter; NKCC2, Na-K-2Cl cotransporter; TAL, thick ascending limb of the loop of Henle; CCD, cortical collecting duct; OMCD, outer medullar collecting duct; IMCD, inner medulla collecting duct. |
While Na+ is fundamental in the determination of the osmotic gradient in the outer medulla, in the inner medulla urea provides a strong contribution, thanks to its reabsorption from the lumen along the collecting duct (CD), and the recycling along the descending limb.2 Similarly to the two branches of Henle’s loop, the descending and ascending vasa recta are arranged in a specific anatomic relationship. The countercurrent flow configuration establishes an axial osmolality gradient that is distributed along the cortico-medullar axis.34 Osmotic equilibration is achieved through a combination of water absorption and solute secretion, as vasa recta are permeable to water, urea, and sodium. However, the descending vasa recta, penetrating in the medulla, lose water and gain solutes, while the ascending vasa recta show the opposite behavior, preventing dissipation of the osmotic gradient.35 Conditions that decrease medullary flow, such as dehydration, favor urine concentration as blood transit into the vasa recta has more time to achieve osmotic equilibration with the hypertonic interstitium. Conversely, an increased medullary flow decreases the urine concentrating ability.34,36
Acid–base homeostasis
The role of PT in bicarbonate reabsorption is well established.37–39 However, other downstream segments contribute to bicarbonate reabsorption. The loop of Henle reabsorbs a significant fraction (nearly 15%) of the filtered bicarbonate.40 In vivo studies have demonstrated that the descending limb of Henle’s loop has low bicarbonate permeability in rats. In contrast, the final segment of the PT (S3 portion) has been shown to be able to reabsorb bicarbonate. Under physiologic conditions, its contribution in bicarbonate reabsorption is modest, as it is actively reabsorbed along the early PT segments and its luminal concentration is low.
Micropuncture studies have shown that bicarbonate concentration increases significantly at the tip of Henle’s loop, most probably as an indirect effect resulting from luminal fluid concentration due to water reabsorption along the descending limb.41 In the TAL, bicarbonate is reabsorbed via the transcellular pathway; the main mechanism resembles bicarbonate reabsorption in the PT through Na+/H+ exchanger (NHE) activity.42 Both NHE2 and NHE3 isoforms have been localized to the luminal membrane.43 The contribution of NHE2 activity is considered low, as the addition of NHE2 knockdown in mice did not result in an overt acid–base disturbance compared with NHE3 knockout (KO) mice.44 Perfusions studies, in vivo and ex vivo, showed that bicarbonate reabsorption requires carbonic anhydrase, and is stimulated by bumetanide.41 Interestingly, de Bruijn et al have demonstrated that furosemide promotes luminal fluid acidification in the TAL by lowering the intracellular Na+ concentration, which in turn increases the driving force for NHE3 activity.45 Bicarbonate exit from the cells is mediated by the Cl−/HCO−3 exchanger 2 (AE2) (Figure 3).46 Interestingly, at the luminal site, the presence of the Cl−/HCO−3 exchanger 1 (AE1) has been shown in rats, that has been proposed to participate in Na+ reabsorption by coupling the Na+/H+ activity.47
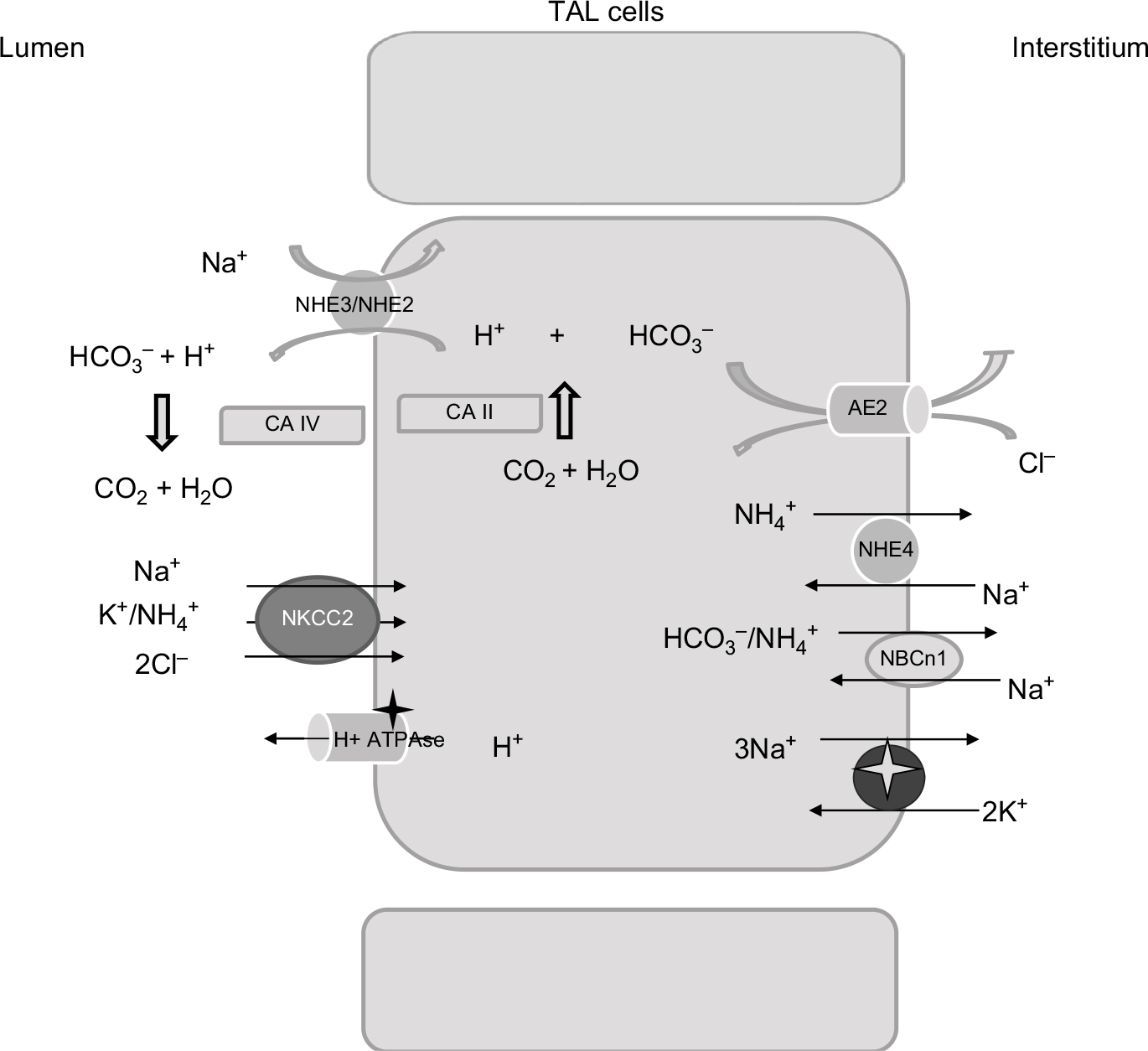 | Figure 3 Relevant proteins involved in acid–base homeostasis in the TAL. Abbreviations: NHE2/NHE3, isoforms 2 and 3 of the Na+, H+ exchangers; CA IV, isoform IV of carbonic anhydrase; CA II, isoform II of carbonic anhydrase; NKCC2, Na-K-2Cl cotransporter; AE2, Cl-HCO3– antiporter; TAL, thick ascending limb of the loop of Henle. |
Besides NHE3, additional molecules contribute to bicarbonate reabsorption in the TAL. Functional studies have suggested the presence of an active H+-ATPase, and immunohistochemical analysis has reported the presence of proton ATPase along the TAL.48 Its role in bicarbonate reabsorption is not considered as consistent as a Na+/H+ exchanger under physiological conditions.
Changes in acid–base balance modulate the rate of bicarbonate reabsorption along the TAL: both acute and chronic metabolic acidosis increase while metabolic alkalosis depresses bicarbonate reabsorption.49 Functional studies have demonstrated that both H+-ATPase and Na+/H+ exchanger are able to adapt to changes in the acid–base status, probably via hormonal stimulation by glucocorticoids and aldosterone.49
Ammonia handling is not of minor importance. Urine ammonia excretion derives mainly from renal ammoniagenesis, rather than glomerular filtration.50 It is produced from glutamine in the PT as ammonium ion (NH4+) and is released to the luminal fluid. TAL has a crucial role in ammonia reabsorption; this process occurs via NKCC2, at the K+ binding site.51 K+/NH4+ exchange and conductive K+ transport have also been described in the TAL, but their contribution is less significant compared with NKCC2.36 Basolateral exit is mediated by the Na+/NH4+ exchanged via NHE4, the Na+-bicarbonate NBCn1 cotransporter, and by a Cl−-dependent pathway.51–53 Interstitial ammonia is at least in part passively recycled into the thin descending limb of Henle’s loop, predominantly as ammonia (NH3). This recycling increases the axial ammonia gradient, paralleling the increasing tonicity of the interstitial fluid.54 Ammonia is then secreted into the luminal fluid along the CD, by the ammonia-specific transporters, Rhesus glycoproteins Rhbg and Rhcg.55
Pathophysiology of TAL dysfunction
Inherited and acquired conditions leading to an imbalance in salt reabsorption along the TAL have a significant impact on human pathophysiology.
Several water and electrolytes disturbances are attributed to TAL dysfunction. TAL hypofunction is responsible for water and electrolytes wasting, while increased salt absorption is considered a contributing factor in the pathogenesis of hypertension.
Bartter syndrome (BS)
BS is a rare disorder caused by genetic mutations leading to impaired salt reabsorption along the TAL. Five genetic BS subtypes have been described; BS type I is caused by mutations in SLCA12A1 (NKCC2) gene, while BS type II is associated to KCNJ1 (ROMK) mutations; type III and IV are caused by CLCNKB (ClC-Kb) and BSND (Barttin subunit) genetic aberrations; and BS type V results from activating mutation of the CASR (CaSR).56 Animal models of BS have provided a significant contribution in understanding the mechanism of the disease. Both NKCC2 and ROMK null mice are characterized by early lethality.57,58 The recessive mutant mouse line Slc12a1(I299F) resembled the features of patients suffering from type I BS, including polyuria, metabolic alkalosis, and hypercalciuria.59 Interestingly, ROMK null mice develop transient hyperkalemia after birth, consistent with ROMK hypofunction in the CD, where the channel mediates K+ secretion.60 Later, infants develop classic features of BS, including hypokalemia, sustained by K+ secretion along the CD mediated by the flow-dependent maxi-K+ channels, despite ROMK deficiency. All BS patients share similar clinical features and biochemical abnormalities, including fluid loss leading to low-normal blood pressure, hypokalemia, and metabolic alkalosis. Often, hypercalciuria occurs, while Mg2+ loss is generally balanced by increased reabsorption through downstream nephron sites, mainly the distal tubule (DT).61,62 Fluid wasting causes the activation of the renin–angiotensin–aldosterone system (RAAS) axis, while Ca2+ loss may lead to secondary hyperparathyroidism.63 Two major clinical forms of BS have been described, the antenatal (also known as hyperprostaglandin E syndrome) and the classic variant. The antenatal form is characterized by polyhydramnios and growth retardation in utero, with severe hypokalemia and fluid loss requiring intensive treatment at birth. This form is generally characteristic of type I, II, and IV. The latter is the only one that is associated with hearing loss, due to the presence of the Barttin subunit in the inner ear. The classic form tends to be less severe, with a late onset, generally in childhood.63 As a result of chronic volume depletion, increased PT urate reabsorption and consequent hyperuricemia may occur.64,65 Type III BS has a variable phenotype. It is linked to the CLCNKB gene, expressed on the basolateral site of both TAL and DT. The disease may have features of BS (either antenatal or classic) and/or Gitelman syndrome.66 ClC-K2 deficient mice exhibit salt losing phenotype, with compensatory increased RAAS axis, hypotension, and increased prostaglandin E2 generation; interestingly, the mice show a blunted response to both furosemide and thiazides, indicating a combined defective salt absorption along both TAL and DT.67 Activating mutations of the CaSR have been associated with hypocalcemia and hypercalciuria, and sometime hypomagnesemia.68 The pathogenesis of Ca2+ and Mg2+ wasting is multifactorial in this condition. CaSR is a well-known modulator of parathyroid hormone (PTH). In the presence of high plasma Ca2+ levels, it inhibits PTH secretion. Gain of function mutation in the CASR has been shown to determine autosomal dominant hypoparathyroidism, with consequent effects on serum levels of Ca2+, phosphate, and Mg2+.69 However, the activation of CaSR in the kidney is believed to contribute to Ca2+ wasting, as suggested by the evidence that patients carrying CASR mutations have more severe urine Ca2+ loss compared with patients with primary hypoparathyroidism.70
Recently, cases of some male infants showing features of transient antenatal BS, associated with mutations in the MAGE-D2 gene, have been reported.71 The gene product is expressed in the DT and in the TAL in both adult and fetal human kidney. Immunostaining studies revealed reduced NKCC2 and Na+-Cl− cotransporter (NCC) apical abundance in one of these patients, with increased NKCC2 cytoplasmic retention. In vitro, MAGE-D2 promotes trafficking to the apical membrane of both NCC and NKCC2.
Familial hypomagnesemia
Claudins are transmembrane proteins involved in intercellular adhesion and paracellular barrier formation.20 In mammals, over 24 claudin genes have been described; some genes exhibit peculiar tissue-specificity expression, others are expressed in several organs and tissues. In the kidney, several claudins have been mapped along the nephron.72 In 1999, Simon et al discovered a mutation in a gene encoding a protein belonging to this family, CLDN16 (encoding claudin-16, also named paracellin), causing familial hypercalciuric hypomagnesemia (FHHNC).73 Recessive mutations of CLDN16 and CLDN19 (encoding claudin-19) are the most common causes of FHHNC.20 The disease is believed to be the result of a deficient reabsorption of divalent cations in the TAL due to a defective paracellular barrier. This defect results in increased fractional excretion of Ca2+ and Mg2+, leading to hypocalcemia and secondary hyperparathyroidism and sometimes to hypomagnesemia.74 Nephrocalcinosis is the leading cause of renal failure. Patients with CLDN19 mutations will also exhibit severe ocular involvement.75 Interestingly, the role of claudin-10 in salt absorption along the TAL has been recently highlighted. Breiderhoff et al showed that claudin-10 deficiency affected paracellular permeability to monovalent and divalent cations in the TAL, causing nephrocalcinosis and hypermagnesemia.76 Its role in TAL salt absorption has been confirmed also in humans by the study of Bongers et al.77 They described two patients carrying CLDN10 mutations with a hypokalemic salt losing nephropathy, with hypermagnesemia. Accordingly, it has been recently shown that patients suffering from the so-called HELIX syndrome (characterized by hypohidrosis, salt losing nephropathy, lacrimal gland dysfunction, ichthyosis, and xerostomia) carried biallelic mutations of CLDN10 gene. Functional studies demonstrated that salt wasting depended on impaired salt absorption along the TAL.78 The role of claudins in salt homeostasis is further supported by a genome-wide association study demonstrating that claudin-14 variants are a risk factor for hypercalciuric nephrolithiasis.79 In addition, Corre et al have recently described a genome-wide association study demonstrating that claudin-14 variants influence differential urine excretion of Mg2+ and Ca2+, suggesting that there is still a lot to be discovered in paracellular electrolyte handling along the distal nephron.80
CNNM2 (hypomagnesemia with seizures and mental retardation)
The CNNM2 gene is mainly expressed in the kidney, brain, and lung in mammalian tissues and performs the function of a divalent cation transporter, including Mg2+.81 The protein has been localized on the basolateral membrane of both TAL and DT cells in human kidney sections. The presence along the DT is not surprising, as transcellular Mg2+ reabsorption from the lumen has been largely proven. The presence in the TAL may either indicate a role in the control of paracellular Mg2+ absorption, or a possible presence of apical Mg2+ uptake, as suggested by some authors.82
Mutations in this gene cause a rare disease characterized by cognitive impairment, seizure, and hypomagnesemia. The latter is believed to result from impaired luminal Mg2+ reabsorption. A genome-wide association study demonstrated a correlation between CNNM2 variants and plasma Mg2+ levels, supporting a significant role of CNNM2 in human Mg2+ homeostasis.83
The EAST syndrome
This is a rare recessive genetic disorder associated with mutations in the KCNJ10 gene, encoding the K+ channel Kir 4.1.84 The acronym EAST indicates the most important clinical features, including epilepsy, ataxia, senso-neural deafness, and tubulopathy. The Kir 4.1 channel is in fact expressed in several organs, including the central nervous system, the inner ear, and the nephron.85 In the latter, it has been shown on the basolateral membrane of the distal nephron, from the macula densa to the early CD and in cortical TAL.72 The salt losing phenotype resembles Gitelman syndrome, with hypocalciuria and hypomagnesemia, besides metabolic alkalosis and hypokalemia. Fan et al demonstrated that the vasopressin-dependent stimulation of the 80–150 pS basolateral K+ channel in Kcnj10 (Kir 4.1) KO mice counteracts the loss of function of Kir 4.1 in the TAL, preventing defective salt absorption at this nephron site.86
Mitochondrial diseases
Inherited mitochondrial diseases are a heterogeneous class of clinical conditions characterized by impaired function of the mitochondrial respiratory chain, due to genetic mutation of either genomic or mitochondrial DNA.87 The spectrum of clinical features is highly variable and the most common morbidities are neurologic and myopathic diseases. Some studies have described renal involvement, with a wide spectrum of tubular disorders, including Fanconi and Bartter-like syndrome; in addition, glomerular defects (focal and segmental glomerulosclerosis) and cystic diseases have been reported.88 The Kearns–Sayre syndrome is a rare mitochondrial disease caused by a large deletion of mitochondrial DNA.89 The clinical findings are Bartter-like syndrome, with hypokalemic metabolic alkalosis and nephrocalcinosis. Some reports described Fanconi-like phenotypes. Extrarenal features include ophthalmoplegia, cerebellar, heart, and several endocrine and metabolic dysfunctions.88
Increased salt absorption along the TAL: a potential role in salt-sensitive hypertension
Hypertension is a major public health problem, and it is considered to be of multifactorial origin. High dietary salt intake is considered one of the major environmental factors favoring its development. However, the effect of salt intake on blood pressure varies among individuals, a phenomenon called salt sensitivity.90 The molecular mechanism underlying salt sensitivity is only partially understood. A significant contribution is mainly attributable to an impaired ability to excrete Na+. Evidences demonstrate that increased Na+ reabsorption along the distal nephron plays a crucial role in the pathogenesis of hypertension.91,92 Monogenic forms of hypertension include clinical conditions characterized by increased salt absorption along the DT (Gordon syndrome) and the CD. The latter may be caused by gain of function mutations of the principal apical channel mediating Na+ entry, epithelial sodium channel (Liddle syndrome), or by the activation of mineralocorticoid receptors (autosomal dominant pseudoaldosteronism type I).93 Indirect data suggest that the TAL also has a crucial role in fluid balance and the pathogenesis of hypertension.94 TAL hypofunction, as in BS, clearly leads to fluid loss and low-normal blood pressure. Animal models of salt-sensitive hypertension show increased avidity in salt absorption along the TAL. Milan hypertensive strain rats displayed increased NKCC2 activity during the induction phase of hypertension.95 In Dahl salt-sensitive rats, increased TAL Cl− uptake has been demonstrated by in vivo micropuncture studies.96 In accordance with animal studies, several data suggest that also in humans TAL contributes to salt sensitivity and hypertension. African-American individuals show a much higher incidence of salt-sensitive hypertension than white people. In the first group of subjects, a lower plasma renin activity (PRA) has been shown, suggesting a primary increase in renal salt reabsorption and consequent compensatory PRA suppression.97 Results of Luft et al’s studies, showing a higher kaliuretic response after furosemide administration compared with white people, considered TAL as the major nephron site responsible for increased salt reabsorption in black individuals.98
In the Framingham heart study population, heterozygote genetic variants of NKCC2, ROMK, and NCC correlated with low blood pressure.99 In addition, a recent genome-wide association study indicated UMOD gene variants as risk factors for both salt-sensitive hypertension and kidney damage,27 further supporting the hypothesis of TAL contribution in salt-sensitive hypertension.100,101 In fact, besides other known functions, uromodulin (UMOD) has been proposed to modulate salt absorption in the TAL. Graham et al showed that UMOD−/− mice were resistant to salt-induced changes of blood pressure,102 while an independent study showed that UMOD overexpression increased blood pressure.102 In vitro studies suggest that UMOD increases either NKCC2 or ROMK activities.27 Moreover, the evidence that cyclosporine-induced hypertension is associated with NKCC2 upregulation further supports the hypothesis that increased avidity in salt absorption along the TAL contributes to the pathogenesis of hypertension.103,104
Acquired conditions resulting in TAL hypofunction: electrolytes disorders, loop diuretics, bilateral ureteral obstruction
Chronic electrolytes imbalance
Hypokalemia is known to inhibit urine concentrating ability and Na+ and Cl− absorption along the TAL.13 In vivo microperfusion studies have demonstrated that chronic hypokalemia reduced Na+ and fluid absorption in the TAL.105,106 In a study by Gutsche et al, impaired salt absorption along the TAL in rats under a low K+ diet was restored by acute K+ infusion; the evidence of impaired Na+ efflux across the peritubular membrane suggested hypokalemia-dependent inhibition of the basolateral Na+-K+-ATPase.107 The resulting increased Na+ and fluid loading to the distal K+-secretory segments of the nephrons, that is known to enhance K+ secretion, causes further K+ urinary excretion.105 Recent studies have addressed the role of the intracellular proteins SPAK and OSR1 in electrolytes homeostasis and salt absorption along the distal nephron. Both proteins have been shown to phosphorylate and activate NKCC2 and NCC in vitro.28 However, their function in vivo is controversial. In fact, SPAK null mice showed a Gitelman-like phenotype, suggesting a pivotal role of the protein in NCC regulation, rather than NKCC2, while kidney-specific OSR1 null mice had a Bartter-like phenotype, confirming its role in modulating NKCC2 function.108,109
Interestingly, total double KO mice is embryonic lethal, while constitutive SPAK KO mice with inducible OSR1 knockdown at steady state showed mild volume contraction and hypokalemia, a less severe phenotype than expected by combined NCC and NKCC2 hypofunction. Accordingly, phospho-NKCC2 abundance was unaffected despite the absence of SPAK and OSR1; conversely, phospho-NCC abundance in response to low K+ diet was reduced, suggesting a significant role of SPAK and OSR1 in NCC regulation by K+ plasma levels.28
Extracellular Ca2+ has a potent inhibitory effect on salt absorption. Chronic hypercalcemia leads to urinary salt loss and fluid wasting. By binding the CaSR on the basolateral membrane of the TAL, extracellular Ca2+ has been shown to inhibit ROMK activity via phospholipase A2, reducing the lumen-positive voltage and consequently paracellular cation reabsorption.16
As stated earlier, recent studies suggest that CaSR regulates paracellular calcium reabsorption along the TAL without affecting the transepithelial electrical gradient. CaSR activation is supposed to enhance CLD14 expression, leading to the downregulation of claudin-16/19, with consequent hypercalciuria. The requirement of calcineurin in the pathway regulating claudin-14 expression may provide an explanation for the cyclosporine-induced hypercalciuria.110
Loop diuretics
Diuretics are widely used in several clinical fields. Furosemide, torasemide, and bumetanide bind NKCC2 in a reversible fashion. The resulting NKCC2 inhibition leads to the reduction in Na+, K+, and Cl− absorption, an effect that overrules that the cortico-medullar osmotic gradient increases urine output and impairs paracellular cations reabsorption. This property has predictable beneficial effects in several conditions, and loop diuretics are the main therapy in fluid retentive states and hypercalcemic conditions.111 After 1 hour from the oral administration of furosemide, normal subjects experienced increased urine output and increased excretional fraction of Na+, K+, Cl−, Ca2+, and Mg2+ in a time-dependent manner, an effect that decreases after 4–6 hours. The effect is dose-dependent, and larger doses are required when the glomerular filtration rate (GFR) decreases.112 Low-dose loop diuretics rarely cause important side effects; conversely, high dosage or long-term treatments, as usually is needed in chronic conditions, result in electrolytes and acid–base imbalance. Hypokalemic metabolic alkalosis is a common side effect of loop diuretics treatment. The underlying mechanism is based on increased salt delivery to the distal nephron, that enhances K+ and H+ secretion along the CD.113 Extracellular volume contraction and the activation of the RAAS axis further contribute to metabolic alkalosis maintenance. Conversely, hyponatremia is uncommon in patients taking loop diuretics compared with patients under thiazides treatment, but still this complication can occur in the presence of concomitant pathologic conditions.114 By increased free water clearance, loop diuretics are expected to increase rather than decrease plasma Na+ concentration. However, high dose of loop diuretics, as often can be necessary in chronic heart failure or in advanced chronic kidney disease, can cause over-diuresis, intravascular volume depletion, and increased vasopressin incretion, worsening hyponatremia.115
Post-obstructive syndrome
Post-obstructive diuresis is a polyuric condition resulting after the relief of bilateral urinary tract obstruction. Diuresis is considered a physiological response to counteract water and salt retention during the obstruction. However, in some patients, diuresis does not cease when the excess of volume and salt is resolved, resulting in pathologic diuresis.116 Several factors have been implicated in the pathophysiology of this condition. Earlier studies from the 1950s suggested that increased urine and salt excretion was the result of reduced Na+ reabsorption, with some studies placing the site of reduced reabsorption in the PT, and others in the distal nephron.117
Micropuncture studies showed that post-obstructive diuresis resulted from impaired Na+ reabsorption at nephron sites beyond the PT in rats.118 McDougal and Wright showed that Na+ absorption was decreased either in PT, paralleling the reduction of the GFR, or in the TAL, leading to increased Na+ loading to the CD in rats.117 Subsequent studies have shown that polyuria following bilateral obstruction relief was associated with reduced expression of the major Na+ transporters, including NHE3, NKCC2, type II Na-Pi cotransporter, and NCC.101 Some authors have demonstrated the role of reduced responsivity of the CD to vasopressin, leading to a diabetes insipidus-like syndrome.119 Recent studies suggest that hydrogen sulphide (H2S), a gasotransmitter modulating a wide range of physiologic functions, may represent a novel therapeutic approach in this condition, given the evidence that increasing H2S levels may reduce renal fibrosis.120,121 It would be of interest to address its effect on salt absorption in this setting, considering that it has been shown to modulate urinary Na+ and K+ excretion, probably by affecting the function of both NKCC2 and Na-K ATPase.122
Conclusion
The TAL has a central role in water and electrolyte homeostasis and acid–base balance, and its dysfunction has a crucial role in human pathophysiology.
Increased awareness and understanding of its specific functions will enable physicians to better manage acquired and inherited conditions caused by TAL-defective salt absorption, and to reduce the incidence and the complications of potentially life-threatening conditions.
Disclosure
The authors report no conflicts of interest in this work.
References
Mount DB. Thick ascending limb of the loop of Henle.Clin J Am Soc Nephrol. 2014;9(11):1974–1986. | ||
Sands JM, Layton HE. The physiology of urinary concentration: an update.Semin Nephrol. 2009;29(3):178–195. | ||
Greger R. Ion transport mechanisms in thick ascending limb of Henle’s loop of mammalian nephron.Physiol Rev. 1985;65(3):760–795. | ||
Dantzler WH, Pannabecker TL, Layton AT, Layton HE. Urine concentrating mechanism in the inner medulla of the mammalian kidney: role of three-dimensional architecture.Acta Physiol (Oxf). 2011;202(3):361–378. | ||
Imai M, Tsuruoka S, Yoshitomi K, Junichi T, Suzuki M, Muto S. Morphological and functional heterogeneity of the thick ascending limb of Henle’s loop.Clin Exp Nephrol. 1999;3(1):9–17. | ||
Tsuruoka S, Koseki C, Muto S, Tabei K, Imai M. Axial heterogeneity of potassium transport across hamster thick ascending limb of Henle’s loop.Am J Physiol. 1994;267(1 Pt 2):F121–F129. | ||
Nielsen S, Maunsbach AB, Ecelbarger CA, Knepper MA. Ultrastructural localization of Na-K-2Cl cotransporter in thick ascending limb and macula densa of rat kidney.Am J Physiol. 1998;275(6 Pt 2):F885–F893. | ||
Mutig K. Trafficking and regulation of the NKCC2 cotransporter in the thick ascending limb.Curr Opin Nephrol Hypertens. 2017;26(5:392–397. | ||
Markadieu N, Delpire E. Physiology and pathophysiology of SLC12A1/2 transporters.Pflugers Arch. 2014;466(1):91–105. | ||
Ares GR, Caceres PS, Ortiz PA. Molecular regulation of NKCC2 in the thick ascending limb.Am J Physiol Renal Physiol. 2011;301(6:F1143–F1159. | ||
Hennings JC, Andrini O, Picard N, et al. The ClC-K2 chloride channel is critical for salt handling in the distal nephron.J Am Soc Nephrol. 2017;28(1):209–217. | ||
Stölting G, Fischer M, Fahlke C. CLC channel function and dysfunction in health and disease.Front Physiol. 2014;5:378. | ||
Trepiccione F, Zacchia M, Capasso G. Physiopathology of potassium deficiency. In: Alpern RJ, Caplan M, Moe OW, editors.Seldin and Giebisch’s The Kidney, Physiology and Pathophysiology. 5th ed. San Diego: Elsevier; 2013: 1713–1738. | ||
Zacchia M, Abategiovanni ML, Stratigis S, Capasso G. Potassium: from physiology to clinical implications.Kidney Dis (Basel). 2016;2(2):72–79. | ||
Loupy A, Ramakrishnan SK, Wootla B, et al. PTH-independent regulation of blood calcium concentration by the calcium-sensing receptor.J Clin Invest. 2012;122(9):3355–3367. | ||
Wang W, Lu M, Balazy M, Hebert SC. Phospholipase A2 is involved in mediating the effect of extracellular Ca2+ on apical K+ channels in rat TAL.Am J Physiol. 1997;273(3 Pt 2):F421–F429. | ||
Gong Y, Renigunta V, Himmerkus N, et al. Claudin-14 regulates renal Ca++ transport in response to CaSR signalling via a novel microRNA pathway.EMBO J. 2012;31(8):1999–2012. | ||
Riccardi D, Valenti G. Localization and function of the renal calcium-sensing receptor.Nat Rev Nephrol. 2016;12(7):414–425. | ||
Kiuchi-Saishin Y, Gotoh S, Furuse M, Takasuga A, Tano Y, Tsukita S. Differential expression patterns of claudins, tight junction membrane proteins, in mouse nephron segments. J Am Soc Nephrol. 2002;13(4):875–886. | ||
Gong Y, Hou J. Claudins in barrier and transport function-the kidney.Pflugers Arch. 2017;469(1):105–113. | ||
Blaine J, Chonchol M, Levi M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol. 2015;10(7):1257–1272. | ||
Hoenderop JG, Hartog A, Stuiver M, Doucet A, Willems PH, Bindels RJ. Localization of the epithelial Ca2+ channel in rabbit kidney and intestine. J Am Soc Nephrol. 2000;11(7):1171–1178. | ||
Ellison DH. Divalent cation transport by the distal nephron: insights from Bartter’s and Gitelman’s syndromes.Am J Physiol Renal Physiol. 2000;279(4):F616–F625. | ||
Godron A, Harambat J, Boccio V, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype–genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations. Clin J Am Soc Nephrol. 2012;7(5):801–809. | ||
Watanabe S, Fukumoto S, Chang H, et al. Association between activating mutations of calcium-sensing receptor and Bartter’s syndrome. Lancet. 2002;360(9334):692–694. | ||
Carmosino M, Procino G, Svelto M. Na+-K+-2Cl- cotransporter type 2 trafficking and activity: the role of interacting proteins. Biol Cell. 2012;104(4):201–212. | ||
Trudu M, Janas S, Lanzani C, et al. Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression. Nat Med. 2013;19(12):1655–1660. | ||
Ferdaus MZ, Barber KW, López-Cayuqueo KI, et al. SPAK and OSR1 play essential roles in potassium homeostasis through actions on the distal convoluted tubule. J Physiol. 2016;594(17):4945–4966. | ||
Kim WY, Lee HW, Han KH, et al. Descending thin limb of the intermediate loop expresses both aquaporin 1 and urea transporter A2 in the mouse kidney.Histochem Cell Biol. 2016;146(1):1–12. | ||
Yang B, Bankir L. Urea and urine concentrating ability: new insights from studies in mice. Am J Physiol Renal Physiol. 2005;288(5):F881–F896. | ||
Zacchia M, Di Iorio V, Trepiccione F, Caterino M, Capasso G. The kidney in Bardet–Biedl syndrome: possible pathogenesis of urine concentrating defect.Kidney Dis (Basel). 2017;3(2):57–65. | ||
Zona E, Zacchia M, Di Iorio V, Capolongo G, Rinaldi L, Capasso G. Il coinvolgimento renale nella sindrome di Bardet-Biedl [Patho-physiology of renal dysfunction in Bardet–Biedl syndrome].G Ital Nefrol. 2017;34(5):62–72. Italian. | ||
Fenton RA, Knepper MA. Urea and renal function in the 21st century: insights from knockout mice.J Am Soc Nephrol. 2007;18(3):679–688. | ||
Zimmerhackl BL, Robertson CR, Jamison RL. The medullary microcirculation.Kidney Int. 1987;31(2):641–647. | ||
Fenton RA. Urea transporters and renal function: lessons from knockout mice.Curr Opin Nephrol Hypertens. 2008;17(5):513–518. | ||
Zacchia M, Capasso G. Dehydration: a new modulator of klotho expression.Am J Physiol Renal Physiol. 2011;301(4):F743–F744. | ||
Ambühl PM, Amemiya M, Danczkay M, et al. Chronic metabolic acidosis increases NHE3 protein abundance in rat kidney.Am J Physiol. 1996;271(4 Pt 2):F917–F925. | ||
Preisig PA. The acid-activated signaling pathway: starting with Pyk2 and ending with increased NHE3 activity.Kidney Int. 2007;72(11:1324–1329. | ||
Liu L, Zacchia M, Tian X, et al. Acid regulation of NaDC-1 requires a functional endothelin B receptor.Kidney Int. 2010;78(9):895–904. | ||
Capasso G, Unwin R, Giebisch G. Role of the loop of Henle in urinary acidification.Kidney Int Suppl. 1991;33:S33–S35. | ||
Capasso G, Unwin R, Agulian S, Giebisch G. Bicarbonate transport along the loop of Henle. I. Microperfusion studies of load and inhibitor sensitivity. J Clin Invest. 1991;88(2):430–437. | ||
Good DW, Watts BA 3rd. Functional roles of apical membrane Na+/H+ exchange in rat medullary thick ascending limb.Am J Physiol. 1996;270(4 Pt 2):F691–F699. | ||
Paillard M. H+ and HCO3− transporters in the medullary thick ascending limb of the kidney: molecular mechanisms, function and regulation.Kidney Int Suppl. 1998;65:S36–S41. | ||
Ledoussal C, Lorenz JN, Nieman ML, Soleimani M, Schultheis PJ, Shull GE. Renal salt wasting in mice lacking NHE3 Na+/H+ exchanger but not in mice lacking NHE2. Am J Physiol Renal Physiol. 2001;281(4):F718–F727. | ||
de Bruijn PI, Larsen CK, Frische S, et al. Furosemide-induced urinary acidification is caused by pronounced H+ secretion in the thick ascending limb. Am J Physiol Renal Physiol. 2015;309(2):F146–F153. | ||
Houillier P, Bourgeois S. More actors in ammonia absorption by the thick ascending limb. Am J Physiol Renal Physiol. 2012;302(3:F293–F297. | ||
Eladari D, Blanchard A, Leviel F, et al. Functional and molecular characterization of luminal and basolateral Cl-/HCO-3 exchangers of rat thick limbs.Am J Physiol. 1998;275(3 Pt 2):F334–F342. | ||
Capasso G, Unwin R, Rizzo M, Pica A, Giebisch G. Bicarbonate transport along the loop of Henle: molecular mechanisms and regulation.J Nephrol. 2002;15 (Suppl 5):S88–S96. | ||
Capasso G, Unwin R, Ciani F, et al. Bicarbonate transport along the loop of Henle. II. Effects of acid-base, dietary, and neurohumoral determinants.J Clin Invest. 1994;94(2):830–838. | ||
Weiner ID, Verlander JW. Renal ammonia metabolism and transport.Compr Physiol. 2013;3(1):201–220. | ||
Amlal H, Paillard M, Bichara M. NH4+ transport pathways in cells of medullary thick ascending limb of rat kidney. NH4+ conductance and K+/NH4+(H+) antiport.J Biol Chem. 1994;269(35:21962–21971. | ||
Blanchard A, Eladari D, Leviel F, Tsimaratos M, Paillard M, Podevin RA. NH4+ as a substrate for apical and basolateral Na+-H+ exchangers of thick ascending limbs of rat kidney: evidence from isolated membranes.J Physiol. 1998;506(Pt 3):689–698. | ||
Lee S, Lee HJ, Yang HS, Thornell IM, Bevensee MO, Choi I. Sodium-bicarbonate cotransporter NBCn1 in the kidney medullary thick ascending limb cell line is upregulated under acidic conditions and enhances ammonium transport.Exp Physiol. 2010;95(9):926–937. | ||
Weiner ID, Mitch WE, Sands JM. Urea and ammonia metabolism and the control of renal nitrogen excretion.Clin J Am Soc Nephrol. 2015;10(8:1444–1458. | ||
Biver S, Belge H, Bourgeois, et al. A role for Rhesus factor Rhcg in renal ammonium excretion and male fertility.Nature. 2008;456(7220:339–343. | ||
Jain G, Ong S, Warnock DG. Genetic disorders of potassium homeostasis.Semin Nephrol. 2013;33(3):300–309. | ||
Takahashi N, Chernavvsky DR, Gomez RA, Igarashi P, Gitelman HJ, Smithies O. Uncompensated polyuria in a mouse model of Bartter’s syndrome.Proc Natl Acad Sci U S A. 2000;97(10):5434–5439. | ||
Lorenz JN, Baird NR, Judd LM, et al. Impaired renal NaCl absorption in mice lacking the ROMK potassium channel, a model for type II Bartter’s syndrome. J Biol Chem. 2002;277(40):37871–37880. | ||
Kemter E, Rathkolb B, Bankir L. Mutation of the Na(+)-K(+)-2Cl(−) cotransporter NKCC2 in mice is associated with severe polyuria and a urea-selective concentrating defect without hyperreninemia. Am J Physiol Renal Physiol. 2010;298(6):F1405–F1415. | ||
Bailey MA, Cantone A, Yan Q, et al. Maxi-K channels contribute to urinary potassium excretion in the ROMK-deficient mouse model of type II Bartter’s syndrome and in adaptation to a high-K diet. Kidney Int. 2006;70(1):51–59. | ||
McCormick JA, Ellison DH. Distal convoluted tubule.Compr Physiol. 2015;5(1):45–98. | ||
Zacchia M, Capasso G. Parvalbumin: a key protein in early distal tubule NaCl reabsorption.Nephrol Dial Transplant. 2008;23(4:1109–1111. | ||
Koulouridis E, Koulouridis I. Molecular pathophysiology of Bartter’s and Gitelman’s syndromes.World J Pediatr. 2015;11(2):113–125. | ||
Pressler CA, Heinzinger J, Jeck N, et al. Late-onset manifestation of antenatal Bartter syndrome as a result of residual function of the mutated renal Na+-K+-2Cl− co-transporter. J Am Soc Nephrol. 2006;17(8):2136–2142. | ||
Zacchia M, Capolongo G, Rinaldi L, Capasso G. Fisiopatologia dell’handling renale dell’acido urico [Renal handling of uric acid].G Ital Nefrol. 2015;32 (Suppl 62). Italian. | ||
Seys E, Andrini O, Keck M, et al. Clinical and genetic spectrum of Bartter syndrome type 3. J Am Soc Nephrol. 2017;28(8):2540–2552. | ||
Grill A, Schießl IM, Gess B, Fremter K, Hammer A, Castrop H. Salt-losing nephropathy in mice with a null mutation of the Clcnk2 gene. Acta Physiol (Oxf). 2016;218(3):198–211. | ||
Hannan FM, Thakker RV. Calcium-sensing receptor (CaSR) mutations and disorders of calcium, electrolyte and water metabolism.Best Pract Res Clin Endocrinol Metab. 2013;27(3):359–371. | ||
Riccardi D, Brown EM. Physiology and pathophysiology of the calcium sensing receptor in the kidney.Am J Physiol. 2010;298(3):F485–F499. | ||
Vargas-Poussou R, Huang C, Hulin P, et al. Functional characterization of a calcium-sensing receptor mutation in severe autosomal dominant hypocalcemia with a Bartter-like syndrome.J Am Soc Nephrol. 2002;13(9):2259–2266. | ||
Laghmani K, Beck BB, Yang SS, et al. Polyhydramnios, transient antenatal Bartter’s syndrome, and MAGED2 mutations.N Engl J Med. 2016;374(19):1853–1863. | ||
Hou J, Rajagopa M, Yu AS. Claudins and the kidney.Annu Rev Physiol. 2013;75:479–501. | ||
Simon DB, Lu Y, Choate KA, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption.Science. 1999;285(5424):103–106. | ||
Sikora P, Zaniew M, Haisch L, et al. Retrospective cohort study of familial hypomagnesaemia with hypercalciuria and nephrocalcinosis due to CLDN16 mutations.Nephrol Dial Transplant. 2015;30(4):636–644. | ||
Konrad M, Schaller A, Seelow D, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement.Am J Hum Genet. 2006;79(5):949–957. | ||
Breiderhoff T, Himmerkus N, Stuiver M, et al. Deletion of claudin-10 (Cldn10) in the thick ascending limb impairs paracellular sodium permeability and leads to hypermagnesemia and nephrocalcinosis. Proc Natl Acad Sci U S A. 2012;109(35):14241–14246. | ||
Bongers EMHF, Shelton LM, Milatz S, et al. A novel hypokalemic-alkalotic salt-losing tubulopathy in patients with CLDN10 mutations. J Am Soc Nephrol. 2017;28(10):3118–3128. | ||
Hadj-Rabia S, Brideau G, Al-Sarraj Y, et al. Multiplex epithelium dysfunction due to CLDN10 mutation: the HELIX syndrome. Genet Med. Epub 2017 Aug 3. | ||
Thorleifsson G, Holm H, Edvardsson V, et al. Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density.Nat Genet. 2009;41(8):926–930. | ||
Corre T, Olinger E, Harris SE, et al. Common variants in CLDN14 are associated with differential excretion of magnesium over calcium in urine. Pflugers Arch. 2017;469(1):91–103. | ||
Stuiver M, Lainez S, Will C, et al. CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia.Am J Hum Genet. 2011;88(3):333–343. | ||
Funato Y, Yamazaki D, Miki H. Renal function of cyclin M2 Mg2+ transporter maintains blood pressure.J Hypertens. 2017;35(3):585–592. | ||
Meyer TE, Verwoert GC, Hwang SJ, et al. Genome-wide association studies of serum magnesium, potassium, and sodium concentrations identify six loci influencing serum magnesium levels. PLoS Genet. 2010;6(8):e1001045. | ||
Abdelhadi O, Iancu D, Stanescu H, Kleta R, Bockenhauer D. EAST syndrome: clinical, pathophysiological, and genetic aspects of mutations in KCNJ10.Rare Dis. 2016;4(1):e1195043. | ||
Reichold M, Zdebik AA, Lieberer E, et al. CNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function.Proc Natl Acad Sci U S A. 2010;107(32):14490–14495. | ||
Fan L, Wang X, Zhang D, et al. Vasopressin-induced stimulation of the Na(+)-activated K(+) channels is responsible for maintaining the basolateral K(+) conductance of the thick ascending limb (TAL) in EAST/SeSAME syndrome. Biochim Biophys Acta. 2015;1852(11):2554–2562. | ||
Suomalainen A, Battersby BJ. Mitochondrial diseases: the contribution of organelle stress responses to pathology.Nat Rev Mol Cell Biol. Epub 2017 Aug 9. | ||
Emma F, Salviati L. Mitochondrial cytopathies and the kidney.Nephrol Ther. 2017;13 (Suppl 1):S23–S28. | ||
Emma F, Montini G, Parikh SM, Salviati L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury.Nat Rev Nephrol. 2016;12(5):267–280. | ||
Trepiccione F, Zacchia M, Capasso G. The role of the kidney in salt-sensitive hypertension.Clin Exp Nephrol. 2012;16(1):68–72. | ||
Frame AA, Wainford RD. Renal sodium handling and sodium sensitivity.Kidney Res Clin Pract. 2017;36(2):117–131. | ||
Petrazzuolo O, Trepiccione F, Zacchia M, Capasso G. Hypertension and renal calcium transport.J Nephrol. 2010;23(Suppl 16):S112–S117. | ||
Warnock DG. Genetic forms of human hypertension.Curr Opin Nephrol Hypertens. 2001;10(4):493–499. | ||
Graham LA, Dominiczak AF, Ferreri NR. Role of renal transporters and novel regulatory interactions in the TAL that control blood pressure.Physiol Genomics. 2017;49(5):261–276. | ||
Capasso G, Rizzo M, Garavaglia M. Upregulation of apical sodium-chloride cotransporter and basolateral chloride channels is responsible for the maintenance of salt-sensitive hypertension.Am J Physiol Renal Physiol. 2008;295(2):F556–F567. | ||
Kirchner KA. Increased loop chloride uptake precedes hypertension in Dahl salt-sensitive rats.Am J Physiol. 1992;262(2 Pt 2):R263–R268. | ||
Aviv A, Hollenberg NK, Weder A. Urinary potassium excretion and sodium sensitivity in blacks.Hypertension. 2004;43(4):707–713. | ||
Luft FC, Grim CE, Fineberg N, Weinberger MC. Effects of volume expansion and contraction in normotensive whites, blacks, and subjects of different ages.Circulation. 1979;59(4):643–650. | ||
Monette MY, Rinehart J, Lifton RP, Forbush B. Rare mutations in the human Na-K-Cl cotransporter (NKCC2) associated with lower blood pressure exhibit impaired processing and transport function.Am J Physiol Renal Physiol. 2011;300(4):F840–F847. | ||
Zacchia M, Capasso G. The importance of uromodulin as regulator of salt reabsorption along the thick ascending limb.Nephrol Dial Transplant. 2015;30(2):158–160. | ||
Zacchia M, Zacchia E, Zona E, et al. Renal phenotype in Bardet–Biedl syndrome: a combined defect of urinary concentration and dilution is associated with defective urinary AQP2 and UMOD excretion.Am J Physiol Renal Physiol. 2016;311(4):F686–F694. | ||
Graham LA, Padmanabhan S, Fraser NJ, et al. Validation of uromodulin as a candidate gene for human essential hypertension.Hypertension. 2014;63(3):551–558. | ||
Blankenstein KI, Borschewski A, Labes R, et al. Calcineurin inhibitor cyclosporine A activates renal Na-K-Cl cotransporters via local and systemic mechanisms. Am J Physiol Renal Physiol. 2017;312(3):F489–F501. | ||
Esteva-Font C, Guillén-Gómez E, Diaz JM. Renal sodium transporters are increased in urinary exosomes of cyclosporine-treated kidney transplant patients.Am J Nephrol. 2014;39(6):528–535. | ||
Unwin R, Capasso G, Giebisch G. Potassium and sodium transport along the loop of Henle: effects of altered dietary potassium intake.Kidney Int. 1994;46(4):1092–1099. | ||
McKay AJ, Peterson LN. K infusion corrects thick ascending limb Cl reabsorption in K-depleted rats by an aldosterone-independent mechanism.Am J Physiol. 1993;264(5 Pt 2):F792–F799. | ||
Gutsche HU, Peterson LN, Levine DZ. In vivo evidence of impaired solute transport by the thick ascending limb in potassium-depleted rats. J Clin Invest. 1984;73(4):908–916. | ||
Yang SS, Lo YF, Wu CC, et al. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol. 2010;21(11):1868–1877. | ||
Lin SH, Yu IS, Jiang ST, et al. Impaired phosphorylation of Na+-K+-2Cl− cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc Natl Acad Sci U S A. 2011;108(42):17538–17543. | ||
Chang CT, Hung CC, Tian YC, Yang CW, Wu MS. Ciclosporin reduces paracellin-1 expression and magnesium transport in thick ascending limb cells.Nephrol Dial Transplant. 2007;22(4):1033–1040. | ||
Hoorn EJ, Ellison DH. Diuretic resistance.Am J Kidney Dis. 2017;69(1:136–142. | ||
Bartoli E, Rossi L, Sola D, Castello L, Sainaghi PP, Smirne C. Use, misuse and abuse of diuretics.Eur J Intern Med. 2017;39:9–17. | ||
Kokot F, Hyla-Klekot L. Drug-induced abnormalities of potassium metabolism.Pol Arch Med Wewn. 2008;118(7–8):431–434. | ||
Sica DA, Carter B, Cushman W, Hamm L. Thiazide and loop diuretics.J Clin Hypertens (Greenwich). 2011;13(9):639–644. | ||
Lapsia V, Kazory A. Loop diuretics for heart failure-associated hyponatremia.Am J Med. 2010;123(8):e5–e6. | ||
Halbgewachs C, Domes T. Postobstructive diuresis: pay close attention to urinary retention.Can Fam Physician. 2015;61(2):137–142. | ||
McDougal WS, Wright FS. Defect in proximal and distal sodium transport in post-obstructive diuresis.Kidney Int. 1972;2(6):304–317. | ||
Yarger WE, Aynedjian HS, Bank N. A micropuncture study of postobstructive diuresis in the rat.J Clin Invest. 1972;51(3):625–637. | ||
Li C, Wang W, Kwon TH, Knepper MA, Nielsen S, Frøkiaer J. Altered expression of major renal Na transporters in rats with bilateral ureteral obstruction and release of obstruction.Am J Physiol Renal Physiol. 2003;285(5):F889–F901. | ||
Han SJ, Noh MR, Jung JM et al. Hydrogen sulfide-producing cystathionine γ-lyase is critical in the progression of kidney fibrosis.Free Radic Biol Med. 2017;112:423–432. | ||
Perna AF, Di Nunzio A, Amoresano A et al. Divergent behavior of hydrogen sulfide pools and of the sulfur metabolite lanthionine, a novel uremic toxin, in dialysis patients.Biochimie. 2016;126:97–107. | ||
Xia M, Chen L, Muh RW, Li PL, Li N. Production and actions of hydrogen sulfide, a novel gaseous bioactive substance, in the kidneys.J Pharmacol Exp Ther. 2009;329(3):1056–1062. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.