")
Back to Archived Journals » Oncolytic Virotherapy » Volume 7
The importance of imaging strategies for pre-clinical and clinical in vivo distribution of oncolytic viruses
Authors Pelin A , Wang J, Bell J, Le Boeuf F
Received 14 December 2017
Accepted for publication 31 January 2018
Published 28 March 2018 Volume 2018:7 Pages 25—35
DOI https://doi.org/10.2147/OV.S137159
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Athanasios Kyritsis
Adrian Pelin,1,2 Jiahu Wang,2,3 John Bell,1,2 Fabrice Le Boeuf2
1Department of Biochemistry, Faculty of Medicine, University of Ottawa, Ottawa, ON, Canada; 2Ottawa Hospital Research Institute, Center for Cancer Therapeutics, Ottawa, ON, Canada; 3Genvira Biosciences, Ottawa Hospital Research Institute, Ottawa, ON, Canada
Abstract: Oncolytic viruses (OVs) are an emergent and unique therapy for cancer patients. Similar to chemo- and radiation therapy, OV can lyse (kill) cancer cell directly. In general, the advantages of OVs over other treatments are primarily: a higher safety profile (as shown by less adverse effects), ability to replicate, transgene(s) delivery, and stimulation of a host’s immune system against cancer. The latter has prompted successful use of OVs with other immunotherapeutic strategies in a synergistic manner. In spite of extended testing in pre-clinical and clinical setting, using biologically derived therapeutics like virus always raises potential concerns about safety (replication at non-intended locations) and bio-availability of the product. Recent advent in in vivo imaging techniques dramatically improves the convenience of use, quality of pictures, and amount of information acquired. Easy assessing of safety/localization of the biotherapeutics like OVs became a new potential weapon in the physician’s arsenal to improve treatment outcome. Given that OVs are typically replicating, in vivo imaging can also track virus replication and persistence as well as precisely mapping tumor tissues presence. This review discusses the importance of imaging in vivo in evaluating OV efficacy, as well as currently available tools and techniques.
Keywords: oncolytic virus, mouse imaging, NIS virus, viral efficacy, clinical trial
Introduction
Oncolytic viruses (OVs) are an emerging therapy
Tumors arise through a combination of genetic and epigenetic changes that favor immortality. Malignant cells create a safety niche by evading host immunity.1 One of the major challenges in cancer treatment is overcoming 2-fold tumor heterogeneity, between individuals or within an individual. Deep sequencing of different cancer genomes has revealed differences in “driver” and “passenger” mutations between individuals, which contribute to tumorogenesis.2 Sequencing has also been used at the single cell level to show that within one individual breast cancer, high levels of variation are present between cells consisting of different mutations, gene copy numbers, and ploidy levels.3 These findings explain how genetically unique portions of a tumor can respond differently to selected treatments. Although a decrease in tumor burden is often observed following treatment in such cases, surviving clonal populations can be highly resistant to further attempts through already-utilized therapeutic interventions, which clinically present as aggressive treatment-refractory cancers.4 Despite recent therapeutic advances, novel and innovative approaches are required to move beyond the modest benefits achieved to date in cancer treatment. Some of the tools developed by “successful” tumors to help them to survive and expand within their host include mutations in genes involved in the interferon pathway, which is a fundamental component of the innate immune system. This contributes to tumors’ ability to evade host immune detection, however, it also leaves cancer cells vulnerable to infection by foreign pathogens.5 Oncolytic viral therapy uses engineered viruses to selectively infect and kill tumor cells and alert host’s immune system to target tumor.
Strategies for engineering successful OVs
OVs are most often engineered at the genomic level to increase their onco-selectivity and safety.6–8 The vaccinia virus (VacV) and herpes simplex virus (HSV) are DNA viruses, which are typically turned into oncolytics by the deletion of virally encoded metabolic genes.9 In this scenario, the virus is thought to target tumor cells preferentially since it is taking advantage of the higher metabolic rates associated with proliferating cancer cells. Rhabdoviruses do not encode metabolic genes so a different strategy is used to increase their tumor targeting. Since most tumors have lost their ability to mount an anti-viral response (defective interferon signaling), mutations are introduced to disable viruses’ ability to counter an anti-viral response. For instance, wild-type vesicular stomatitis virus (VSV) is able to block the translocation of interferon-related genes using its M protein. By deleting the fifty-first amino acid of this protein, the virus (VSVd51) is still able to infect cancer cells, but not normal cells.10
Another key feature of the tumor microenvironment is the presence of immunosuppressive factors. Typically, tumors have high expression of immunosuppressive cytokines such as IL-10 or TGF-beta.11 This is achieved either by recruitment of T-regs and/or by direct expression of immunosuppressive factors. Additional immune suppression can also come from expression of checkpoint blockade ligands such as PD-1, PD-L1 by the tumor. This typically further contributes to immune cell exhaustion and lack of anti-tumor immunity.12 The immunosuppressive properties of the tumor microenvironment is yet another property that favors OV replication. Successful viral replication in the tumor inadvertently renders the tumor more immunogenic and increases the chances of an anti-tumor immune response.13
Clinical trials achievements
The field of OV therapy is currently experiencing a strong momentum due to several developments in recent years. Mainly, HSV, VacV, and VSV are primary choices in the field of OVs. These include significant breakthroughs in developing the oncolytic VacV Pexa-Vec (formerly JX-594), which was tailored to express immunogenic transgenes and selectively infects cancer cells.14 Studies involving Pexa-Vec have demonstrated excellent safety profile of a VacV-based oncolytic, in addition to demonstrating a reduction in tumor burden.15 Capitalizing on Pexa-Vec success, another oncolytic VacV named VVdd concluded a Phase I trial in 2016 and reported high levels of safety and no dose-limiting toxicities.16
More recently, this success was followed by the 2015 US Food and Drug Administration approval of T-Vec–an oncolytic HSV-1-based therapeutic–for treatment in cases of advanced melanoma. This has paved the way for the approval of next generation OVs for clinical utility.17 Currently, ongoing trials are pairing T-Vec with the immune checkpoint inhibitor Ipilimumab (anti-CTLA-4 antibody).18 This approach to combination therapy has been previously demonstrated in a pre-clinical model by combining Newcastle Disease Virus (NDV) with CTLA-4 blockade. In a pre-clinical cancer model, the result of this approach was an increase in tumor-specific CD4+ and CD8+ T-cells in both NDV-injected and non-injected tumors.19 More recently, Reovirus has been shown to sensitize brain tumors to checkpoint blockade in a 9-person clinical trial.20 Thus far, over 1000 patients have been treated with OVs and the field is advancing rapidly.21
The role of imaging tools in OV engineering
Introduction
Strategies targeting high metabolism, high immune suppression, and lack of interferon signaling in tumors have shown promising results in current clinical trials. The main challenge of this approach is tumor heterogeneity.22,23 Indeed, interferon-expression can vary from one tumor cell to another in patients which in turn hinders the interferon sensitive OV’s ability to infect and replicate in certain tumor niches.10 This can result in areas of tumor to be shielded from OV replication and cancer cell lysis. This phenomenon is hard to address or study since there are limited tools to evaluate viral spread within a 3-D tumor model. Techniques such as immunohistochemistry require the scarification of an animal model in order to evaluate viral spread, making it difficult and costly to look at viral kinetics in vivo. Moreover, it is difficult to evaluate viral spread and viral loads simultaneously. Tittering of tumor organs is the gold standard to determine viral quantity yet lacks information on spread.24,25
The best tools to evaluate viral spread and replication are imaging technologies. This review will aim to address the advantages and shortcomings of available methods. We will touch upon tools available for research in in vivo animal models as well as techniques used to evaluate the efficacy of OVs during human clinical trials.
Imaging has become a key component in various diagnostic and therapeutic fields, notably in cancer.26 In pre-clinical models, early detection is central to achieve best treatment efficacy.27 These imaging tools allow the selection of a treatment regimen tailored to the size of the tumor, a key determinant in long-term cure.28 At various levels of research, imaging studies are still necessary as proof of concept before a new anti-cancer therapeutic can reach the milestone of early-stage clinical testing. Past decades witnessed an explosion of technologies able to detect and follow the evolution of tumors in small animals with high-resolution pictures (Figure 1). We will focus on these technologies and techniques currently used in pre-clinical and clinical cancer studies.
 | Figure 1 Decision paths for imaging tool technology. Notes: List of main imaging systems available for small animal research. Classification based on criteria that can help in the decision of the choice. Fluorescence and luminescence technologies are the most affordable and less invasive for animals, but less precise compared to MRI. Abbreviations: GFP, green fluorescent protein; RFP, red fluorescent protein; YFP, yellow fluorescent protein; MRI, magnetic resonance imaging; NIS, sodium iodide symporter; PET, positron emission tomography; SPECT, single-photon emission computed tomography; SSRT2, somatostatin receptor 2. |
Magnetic resonance imaging (MRI)
MRI, originally called nuclear MRI (NMRI), is widely used on patients for clinical diagnosis and also in the follow-up of disease progression due to its high resolution of anatomy representation. MRI is a non-invasive technology based on the manipulation of protons (hydrogen atoms in water) in tissue. After excitation, excited hydrogens relax and send a signal that is captured and measured. Contrast in pictures result from the rate at which excited protons come back to the original and non-excited state.
The use of MRI is particularly appealing in peritoneal tumors, allowing accurate size estimation, localization, and early detection. Despite some advantages, the associated high cost of use, the access to heavy machinery, and the necessary time to process single animal limit the use of this high-resolution technology at the pre-clinical level.29
Bioluminescent imaging (BLI)
BLI is an optical molecular imaging technology relying on the use of luciferase-based gene reporters (Figure 2). This technology has application in various fields of research, particularly in cancer research. While not in use clinically, it is widely used in pre-clinical studies with small animals and competes with or complements fluorescent imaging (FLI). The high sensitivity of this technique is based on the efficiency of photons released following the interaction between luciferase enzymes and luciferin substrates. This luminescence technology relies on the expressed luciferase gene using an in vivo imaging system (ex IVIS) and requires the administration of a luciferase substrate usually intraperitoneally. The most widely used substrate remains D-Luciferin that interacts with engineered Luciferase enzyme alongside other various substrates that have been developed to increase the spectrum use of BLI. Indeed, multiplex assays using various luciferase types results in the possibility to follow different biologic events simultaneously. One may use one luciferase for tracking the tumor growth and another for OV delivery and spread.30 Limitations have been observed, however, in imaging deep organs/tissues and remains an application for small animal models.31
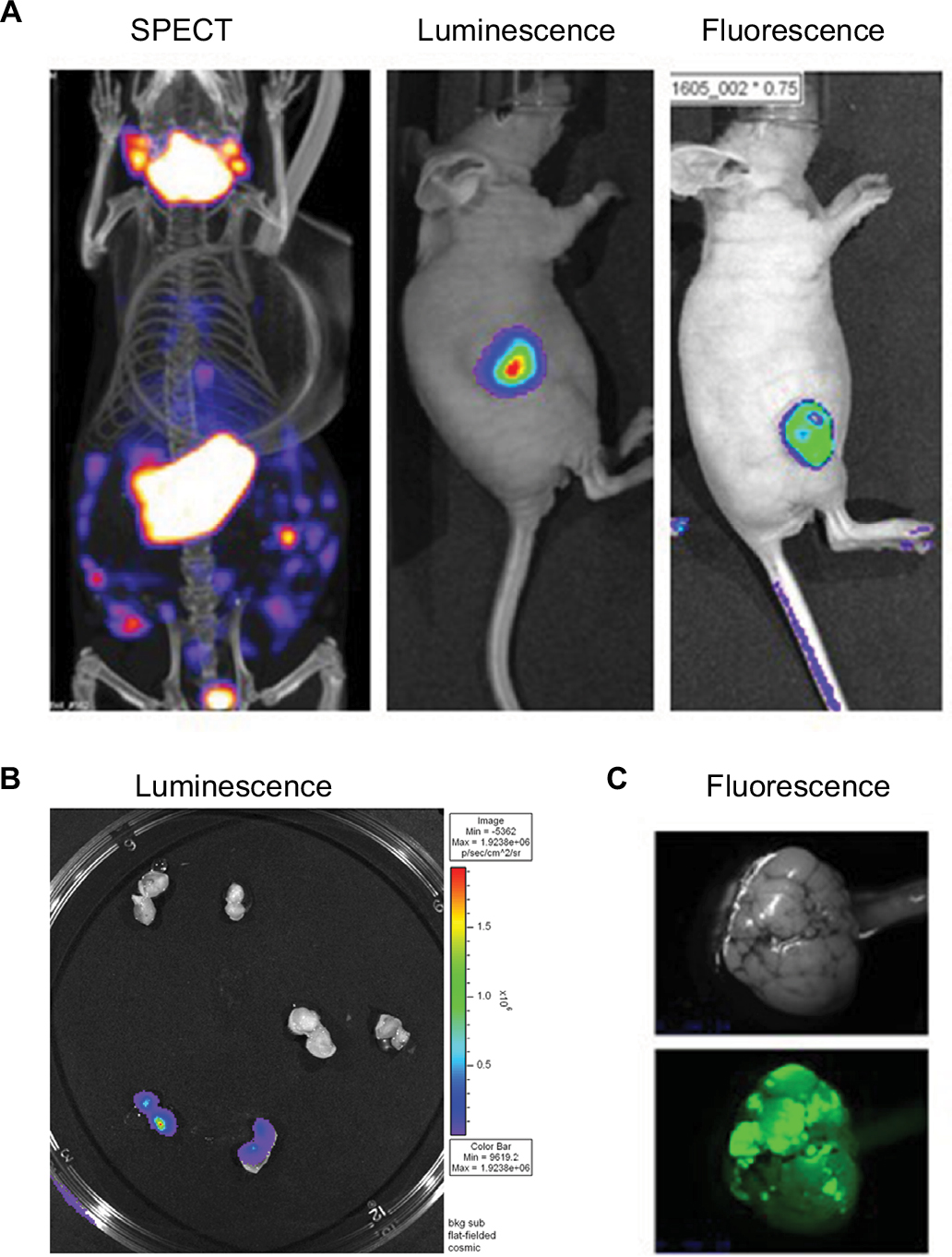 | Figure 2 PET CT; bioluminescence and fluorescence illustration for use in cancer field. Notes: (A) HT29 tumors were established subcutaneously in nude mice. Then vaccinia virus was injected intratumorally at 1×107 pfu as follow: vaccinia virus expressing NIS for SPECT image, vaccinia expressing firefly luciferase tag for bioluminescent image or vaccinia expressing eGFP tag for fluorescence. Four days after virus treatment, the mice were injected with 99Tc radioisotope for small-animal SPECT/CT imaging, D-luciferin (Molecular Imaging Products, Ann Arbor, MI, USA) for bioluminescent. Images were taken using the in vivo imaging system IVIS 200 Series Imaging System (Xenogen, Hopkinton, MA, USA). Luminescent and fluorescent images data acquisition and analysis were performed using Living Image v2.5 software. (B) Ht29 tumors were established subcutaneously in nude mice. Fourteen days after tumor seeding, the mice were injected with vaccinia virus expressing firefly luciferase tag (3), vaccinia virus with no imaging reporter (2) or not injected (1). Mice were sacrificed, tumor harvested and cut in half for imaging analysis. (C) Transgenic mice (90 days old) were treated with vaccinia-expressing fluorescent marker intravenously. Four days later, the mice were sacrificed, tumor harvested and processed for image testing. Top: brightness and contrast; Bottom: fluorescence. Abbreviations: PET, positron emission tomography; CT, computed tomography; NIS, sodium iodide symporter, SPECT, single-photon emission computed tomography. |
Fluorescence imaging
In vivo fluorescence imaging can detect the expression of fluorescent proteins across living animals (Figure 2). It is probably one of the technologies that has evolved the most in past years. Narrowly used in clinic, this technology is widely employed in pre-clinical studies at a relative affordable price. FLI uses a sensitive camera to detect fluorescence emission from various fluorophores in the entire body but has limitations of a reduced signal through the layers of organ tissues. In the cancer field, many examples of tumor expressing specific fluorophores are used in subcutaneous model where tumor evolution can be followed. On the other hand, specific tumor-targeted vectors expressing fluorophore genes can also be tracked through the body to detect primary or secondary tumor sites. As non-invasive technology and low cost, FLI does not require animal scarification, reducing the cost for experiments requiring imaging at multiple time points.32
Magnetic particle imaging (MPI)
MPI is a relatively new technology used in research that exploits the unique characteristics of superparamagnetic iron oxide nanoparticles. This technology can reconstruct images in 3D of the location where nanoparticles accumulate. In cancer field, it is used in small animal models, where tumors can be detected due to the accumulation of tracers.33 Great contrast images result from the high sensitivity of the tracer signal. Furthermore, safety, no radiation, no iodine, and the non-invasiveness are advantages of this technology, making it a hopeful modality for possible future use in the clinic.
Computed tomography (CT)
CT imagery or CT scan, is also a non-invasive imaging tool that is widely used clinically for various purposes, but mainly in preventive medicine to screen for diseases such as colon cancer. CT aids in patient comfort because of the reduced need for exploratory surgeries, particularly in reducing the length of hospitalization. Data acquisition is quick, typically within minutes. However, adverse effects have been documented; overusing this technology can significantly increase incidence of cancer. Mainly, CT scan is known as X-ray CT, but various other CTs exist such as single photon emission CT (SPECT) and positron emission tomography (PET).34
Single photon emission CT (SPECT)
SPECT imaging uses radioisotopes such as 99mTc, 111In, 123I and emits γ-rays that is captured by a gamma camera (Figures 2 and S1). Computer analysis of gamma signal provides a 3D spatial distribution of the target tissue where tracer accumulates. The advantage of radioisotopes used for SPECT is their relatively long half-life, which allows their usage for several hours after their production. This reduces the production cost, which, added to less expensive equipment, has allowed this technology to spread and be used widely in various fields of medicine like brain imaging or tumor imaging. SPECT is also widely used in pre-clinical studies in small animals.35
{kind=link}
Positron emission tomography (PET)
PET imaging detects positrons from radioisotopes like 11C, 13N, and 18F, and is the most clinically popular tool. With high sensitivity, this nuclear medicine imaging technology was adopted in oncology related research. PET allows the precise localization of tumor site(s), especially for lung cancers. The limitation of this technology is the short half-life of radioisotopes used, which drastically increase the cost of this technology. PET is appreciated by researchers who want to determine the pharmacokinetics of newly developed drugs labeled with radioisotopes, giving a clear picture of the biodistribution and clearance of the chemical in the body.36
Sodium iodide symporter (NIS)
A widely used protein for deep-tissue imaging is human NIS (hNIS). NIS is a 643 amino acid transmembrane glycoprotein of the plasma membrane regulating transport and concentration of iodide in the thyroid gland cells and some extra thyroidal tissues like the salivary gland, mammary gland, and gastric mucosa. After it was discovered that it mediated iodide uptake, hNIS has been extensively used for imaging in combination with radiolabeled 99Tc or 131I. The NIS symporter gene was first cloned from rat DNA in 1996,37 with the mouse version later in 2001.38 The human version was cloned in 1996.39 Following first cloning procedures, several in vivo experiments assessed NIS expression and its use in non-thyroid tissue, notably in tumor tissue.40
In vivo imaging used in OV field
Due to their tumor-tropic and directly lysing effect, OVs have shown great promise for cancer treatment. One additional advantage of using replication-enabled and tumor-targeted OVs is transgene delivery. For instance, in OVs based on engineered poxvirus, multiple transgenes can be inserted into a single virus, which not only provides the ability to enhance its therapeutic benefits, but also allows for visualization and quantification of OVs in vivo. One of the advantages of non-invasive detection of OVs directly is for safety or to determine the kinetics and dynamics of a viral platform. It permits the estimation of the abundance and the clearance of a biotherapeutics. Indeed, the high cancer selectivity aspect of OVs coupled with the high sensitivity of detection allows very precise tumor margin delimitation. Several methods have been investigated for OV in vivo imaging. These methods can be categorized broadly into optical imaging and deep-tissue imaging.
OV expressing fluorescent proteins
Fluorescence proteins are widely used for optical imaging.8,41–43 Authors reported that the location of tumors and metastases can be visualized in real time using a GFP or RFP encoding OV in mice.44,45 Fluorescent signal was detected using specially adapted charged-couple device cameras with high spatial resolution. A number of virus strains encoding fluorescent proteins, including GFP, enhanced GFP and RFP, have been developed and tested in different mouse xenograft models, and remain an important tool for researchers to follow the distribution of OVs.46–48 Given the large coding capacity of some OVs, some groups chose to encode both a fluorescent marker and a new transgene to monitor virus toxicity. In this case, tdTomato is encoded alongside an immunostimulatory gene CD40L to monitor virus distribution.49 Moreover, a GFP-encoding vaccina virus strain (GLV-1h68) is currently in Phase I and II clinical trials, in which GFP fluorescence is being used to confirm and monitor virus in tumor and metastases.50 The downside of using fluorescent proteins is auto-fluorescence coming from dead cells, an issue of particular importance when considering OVs, which lyse tumor cells.51
OV expressing luciferases
The other optical detection method utilizes Bioluminescence (BLI). Our results, as well as those reported by other groups,30,52 have suggested that using virus expressing luciferases genes allowed accurate tracking of tumor-specific viral replication in small animals.53 When using different luciferase types in tumor cells and OVs, one can follow the interaction of tumor and OV in vivo directly.8 This method supersedes fluorescence reporter genes for whole-body imaging due to higher sensitivity and lower background luminescence. The most commonly used luciferases in virus imaging are Firefly and Renilla with luciferin and coelenterazine as their respective substrates.54 More recently, Promega introduced Nanoluciferase, which uses a proprietary substrate and has a smaller coding sequence being the only option for viruses that cannot express longer transgenes.55 The downside of luciferases is their limited use in larger subjects due to their low spatial resolution.
Monitoring OV using MRI imaging
MRI is a powerful tool that can be used with photoacoustic technologies for detailed imaging. Currently, recombinant VacV has been engineered to produce tyrosinase protein.56 Since VacV-infected cells produce melanin, this opens the door for MRI and photoacoustic technology imaging, which can be combined with thermal therapy for cancer. Melanin’s production also allows for combination with near-infrared laser-induced thermal therapy against cancer.
OV expressing herpes virus thymidine kinase (TK)
HSV infection can be treated with acyclovir, a nucleoside analog, which when phosphorylated by the viral TK leads to DNA incorporation and damage, ultimately resulting in cell death. The oncolytic version of HSV provides a unique opportunity to increase the cancer-specific toxicity of the tumor-targeting virus in combination with acyclovir, but comes at the cost of viral replication.57
The mechanism of cell death when combing HSV-TK and drug is traceable by non-invasive PET imaging. A version of VSV has been engineered to express HSV-TK and has been used to assess viral biodistribution in hepatocellular carcinoma rat models.58 The effects of encoding HSV-TK can go beyond tumor toxicity and imaging. A group has engineered an adenoviral backbone to express HSV-TK and have shown both, ability to image virus localization using PET scanning and increased cancer cell death. They have also shown that cancer cell death induced by the combination of expressing HSV-TK and administering nucleoside analog ganciclovir, leads to the development of a stronger anti-tumor response.59
OV expressing human somatostatin receptor 2 (hSSRT2)
hSSRT2 is normally expressed in neuroendocrine tumors and is commonly used for imaging after being radiolabeled with indium-111 (111In).60 Gene therapy attempts to deliver SSRT2 to the tumor have mainly been successful when given intratumorally.61 OVs are more advantageous since systemic delivery will target OVs to all tumor sites and would be useful for both virus and tumor imaging.
It has been reported that tumors infected with hSSRT2-expressing OV accumulated higher radioactivity with 111In-pentetreotide-mediated SPECT, and could be visible up to 3 weeks with repeat radiotracer injection.62 Encoding hSSRT2 into vaccinia has shown therapeutic effect of radioactivity accumulation in tumor cells and has allowed long-term non-invasive virus spatial distribution in murine models.63
One disadvantage of using hSSRT2 for deep-tissue imaging is that radiotracers for hSSRT2 requires prior radiolabeling. Moreover, hSSRT2 usually has a 1:1 binding relationship with radiolabeled ligands, and therefore cannot provide significant signal amplification.
OV expressing human norepinephrine transporter (hNET)
hNET is a membrane transport protein of dopamine, norepinephrine, and epinephrine. Its use is appealing in the context of cancer therapy as the transporter functions across all cancer cell membranes. OVs can be engineered to express hNET and can then be visualized using PET or SPECT using the radiotracer meta-iodobenzylguanidine.64 Unlike some other transgenes, the advantage of hNET is its human origin, because of which, it is unlikely to induce an immunogenic response. These and other features of hNET have been first explored using a VacV Lister virus (GLV-1h99) expressing hNET in both in vitro cell lines and in vivo orthotopic murine pancreatic tumors.65
OV expressing hNIS
Accumulation of iodide in NIS-expressing tissues opened the doors to use and express the symporter through OVs. First, a non-replicating adenovirus (Ad) encoding NIS has been developed and tested in various types of cancer, including cervical cancer, breast cancer, and prostate cancer xenografts.66 Shortly after, a replication-competent Ad platform expressing NIS symporter (Ad5-yCD/mutTK[SR39]rep-hNIS), coupled with high-resolution picture has shown great promise in visualizing dog prostate cancer tissue.67 Following this great demonstration of NIS potential, the first RNA replicating OV platform expressing NIS appeared, which was the Edmonston-strain measles virus (MV).68 In their publication, authors provided in vitro and in vivo evidence of the positive observed therapeutic effect of NIS and radioisotopes in cancer models. These studies demonstrated positively the usefulness of NIS in cancer treatment and paved the path for use of the symporter in human.40
Amid other viruses is a VSV-expressing NIS that has been developed and tested in various mouse models. In fact, VSV remains an attractive platform as an OV: first, considering manufacture, VSV replicates well and can grow to high titers; second, virus lifecycle is kept in the cytoplasm and avoids host genome integration; third, there is almost no pre-existing immunity against VSV in the human population; fourth, VSV infections in humans result in very low manifestation symptoms. Notably, Russell’s group demonstrated that: first, a new VSVd51-expressing NIS is safe, and second, in immunocompetent mice with syngeneic 5TGM1 myeloma tumors, viral treatment showed significant benefits (tumor regression; prolonged survival), particularly when VSVd51-expressing NIS was combined with iodine radioisotopes and the use of 123I–γ-scintigraphy imaging.69 Subsequently, oncolytic VSV engineered to express interferon-beta (IFNβ) and NIS, VSV-IFNβ-NIS, has shown great promise as new potential therapeutic agent. Indeed, authors have shown a clear tumor remission following systemic therapy in various pre-clinical mouse models, including multiple myeloma.69 Interestingly, using the wild-type backbone of virus increased NIS expression and therefore iodine uptake when mice were treated with radioisotopes. Finally, potential concerns about the safety profile of this wild-type non-attenuated VSV platform were cleared by this study. VSV-IFNβ-NIS is now in early-stage clinical trials testing for patients with relapsed or refractory multiple myeloma, acute myeloid leukemia (AML), or T-cell lymphoma. On the other hand, a VSV-mIFNβ-NIS was applied in combination with anti-PD-L1 to treat mice bearing AML and resulted in the enhanced therapeutic outcome.70
VacV is another virus extensively developed into OVs. Indeed, a long history of use and knowledge has made this platform a good choice to express NIS. First vaccinia carrying NIS was in a Lister strain backbone, GLV-1h153.71,72 GLV-1h153, a replication-competent engineered VacV, carries transgenes encoding Renilla luciferase and GFP in F14.5L locus, beta-galactosidase in J2R locus (TK), and hNIS in the A56R locus under an early late promotor.73 This virus has shown a great safety profile and efficacy in various models, notably in pancreatic models or in malignant pleural mesotheliomas.74 Consistent with other reports, our group has demonstrated that hNIS-encoding virus preferentially replicates in various tumor sites in vivo and can be clearly visualized using SPECT/CT.40,75,76 Indeed, our group also developed a vaccinia (Wyeth backbone)-expressing hNIS. One difference was our usage of a late promoter to regulate transgenes, resulting in higher functional expression of the protein and much stronger used radioisotopes uptake in targeted tumor cells.40
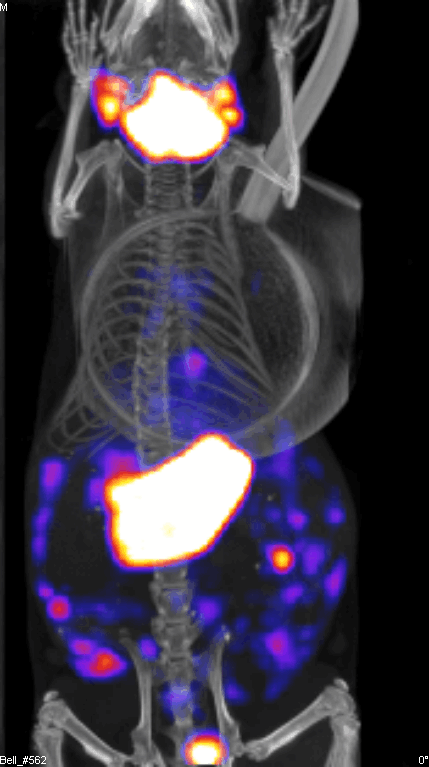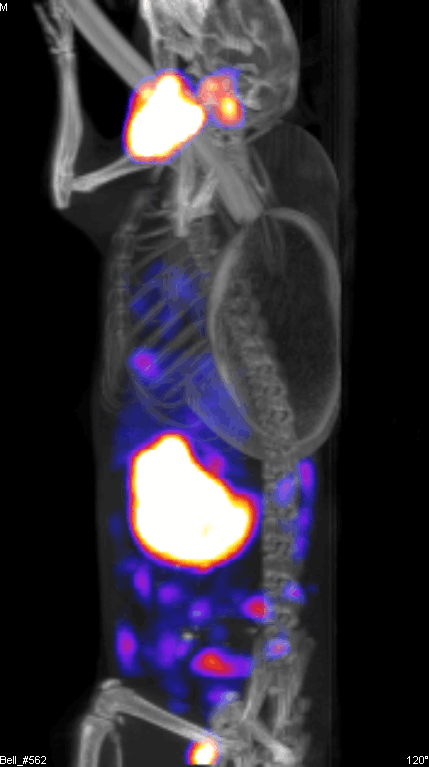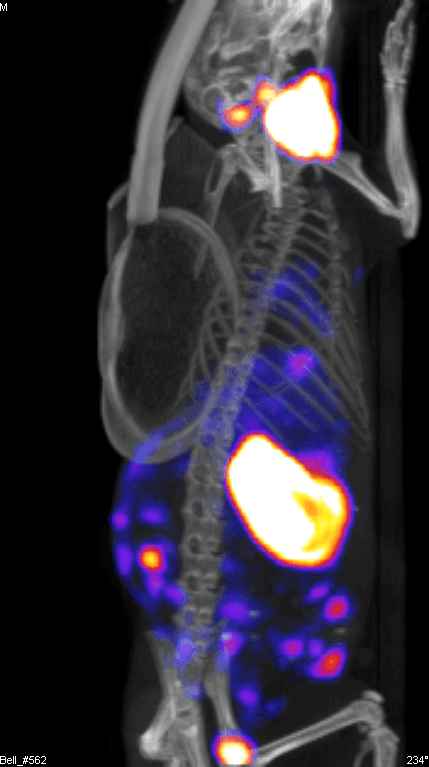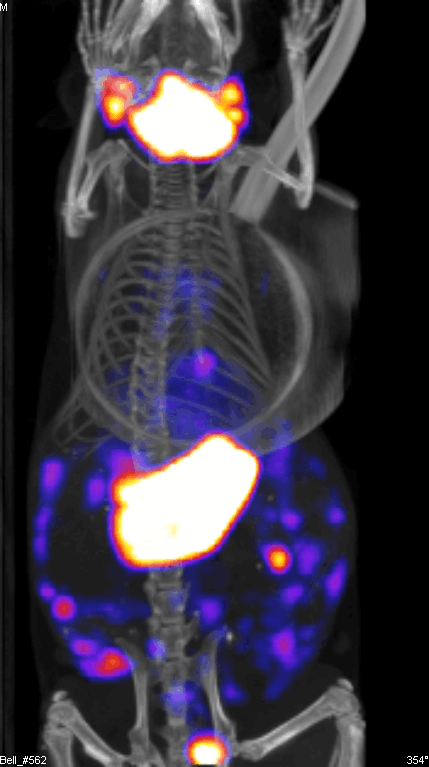
Figure 2 demonstrates the SPECT/CT imaging of a mouse bearing a transgenic ovarian tumor. In this case, CT was used to visualize the distribution of radioisotopes which localize in the tumor. The expression of NIS symporter was visualized through the uptake of 99Tc. Other than signals from thyroid, stomach due to endogenous NIS expression and excretion of radiotracer in bladder (and occasionally large intestinal), other signals result from NIS expression due to viral replication. Viral replication foci were evident. Further details of viral replication inside the tumor may be found in the 3D scan (Figure S1).
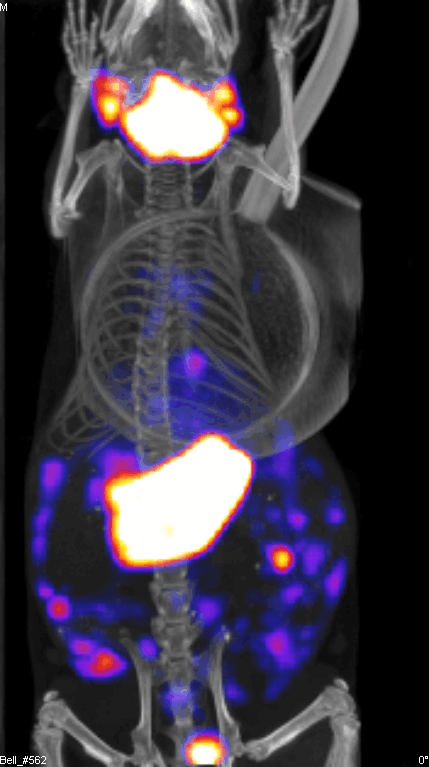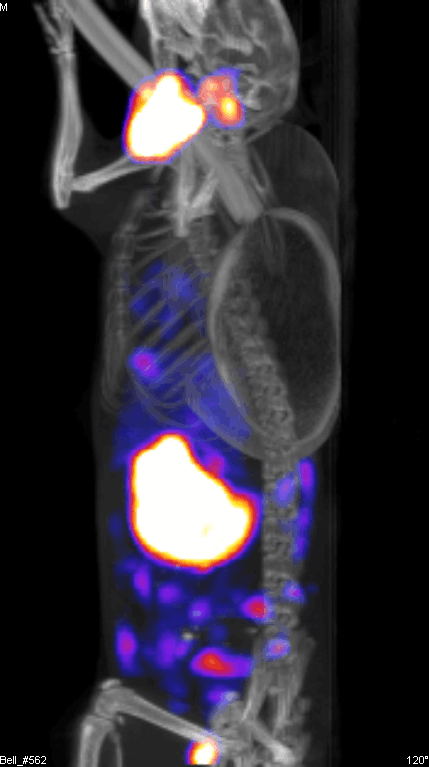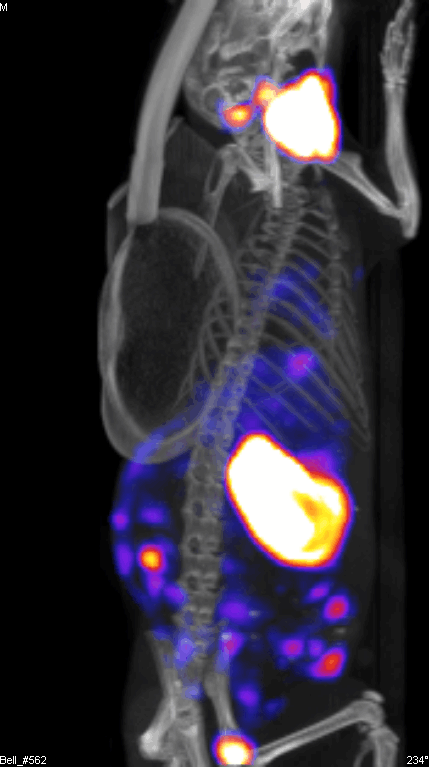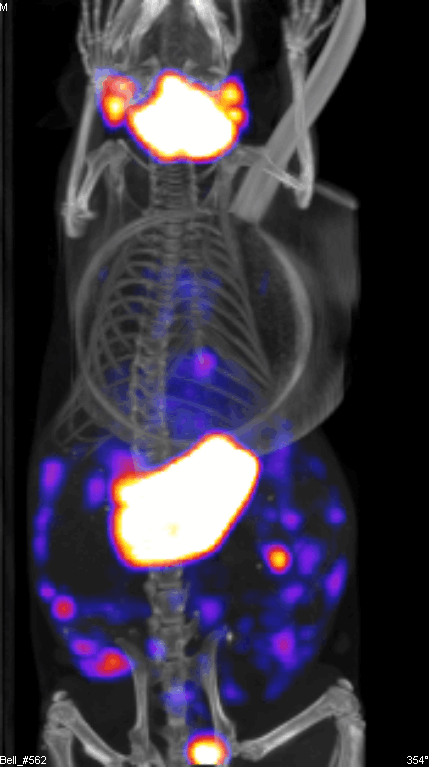{kind=link}
This non-invasive imaging technique allows for the monitoring of viral delivery and replication in vivo. We have also tested at different time points after each viral injection and assessed the effects of multiple injections based on NIS imaging. Besides monitoring viral spread within a tumor, NIS expression by OV can take up radioisotopes, leading to enhanced therapeutic benefit.40
OV clinical trial using expressing NIS marker
Pre-clinical studies using OV expressing reporter genes have also shown great hope.77 Several platforms used in various animal models and by different teams conclude the possible use of OV-expressing marker genes for early clinical testing. However, possible unexpected effects of marker gene expression in targeted cancer cells will need more investigations in future for safety, especially on the immunogenicity of these transgenes. We know now how important the activation of specific T-cells against the tumor is in leading to a complete cure. Potential modulation of any subset of immune cells by the presence of marker genes can have a dramatic effect on overall survival.78 Also, the route of administration of OVs, as well as the type of promoter used for the expression of these marker genes, will need more investigations. Indeed, a late versus an early expression of an imaging gene can impact which imaging strategies to use. Another very important consideration is the size of the subject. Since mice and other in vivo animals are much smaller than humans, humans typically get 100–10,000-fold more virus as treatment. To date, only a few clinical studies have been performed using OV expressing marker genes and they all use NIS symporter (Table 1).
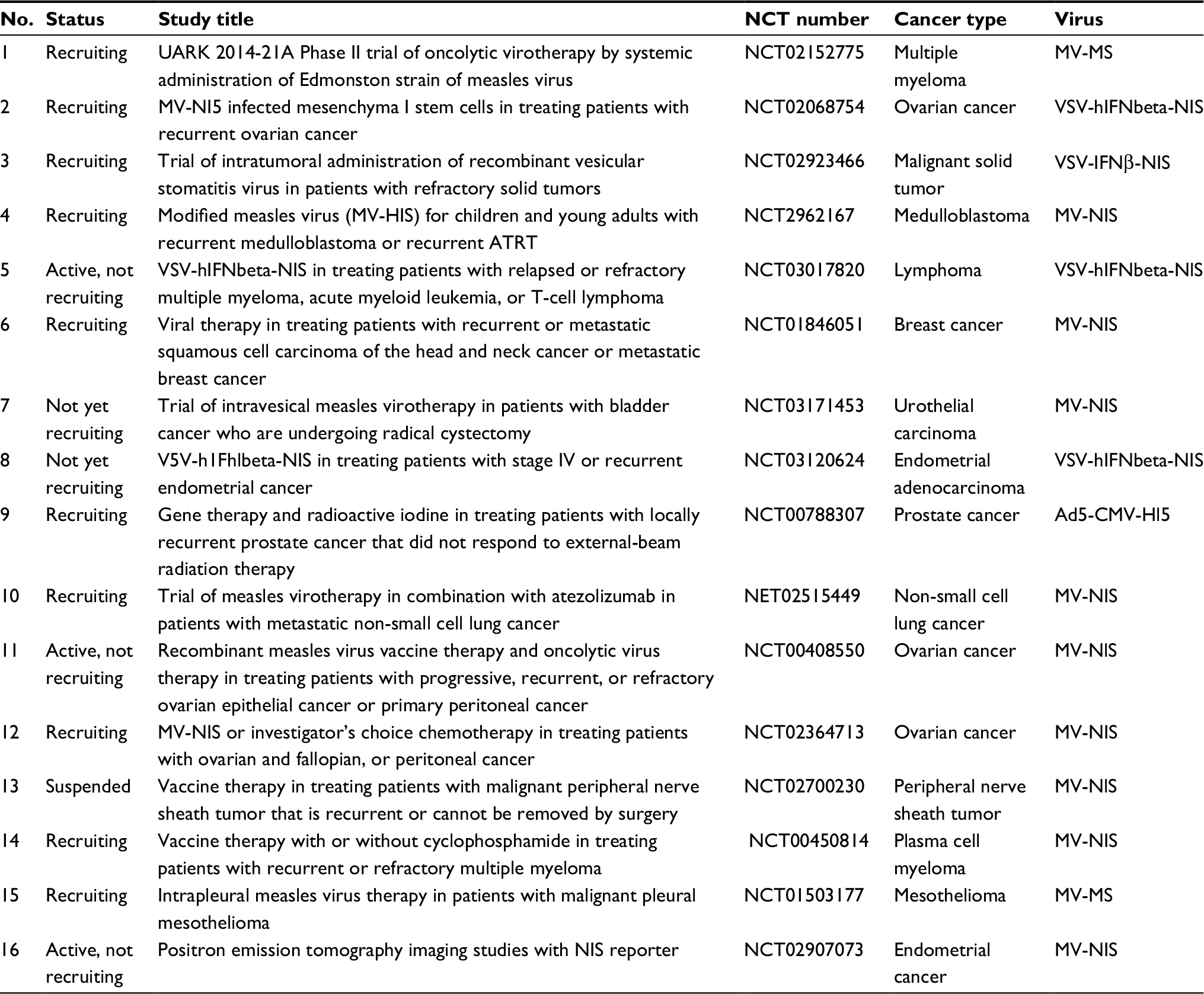 | Table 1 Clinical trials Note: List of main clinical trial using imaging reporter. Abbreviations: ATRT, atypical teratoid rhabdoid tumors; NCT, National Clinical Trial; NIS, sodium iodide symporter. |
NIS-based strategy has been successfully carried out in human early clinical trials. There are a number of viruses currently undergoing clinical translation, including Ad, HSV, MV, Rhabdoviruses (VSV, Maraba virus), and VacV.79 Interestingly enough, these platforms will not be ideal in expressing NIS symporter in targeted tumor cells. Indeed, pharmacodynamics of a fast and transient virus infection can be drastically different compared with a slower virus that might be persistent for a longer time after administration. Nonetheless, several platforms expressing NIS moved forward in the clinic, including replicative competent Ad5; MV and VSV.80,81 Ad5-yCD/utTKSR39rep-hNIS has been studied for prostate cancer.80 NIS gene expression was imaged non-invasively by the uptake of 99mTcO4 in infected cells using SPECT. Enhanced radioisotope-uptake was detected and found to increase in prostate following injection of virus, and no extraprostatic dissemination of the Ad was evident.
MV-NIS is a live attenuated MV (Edmonston lineage). This virus has been engineered to express the human NIS.82 MV targets tumor cells by entering through the CD46 receptor, a membrane regulator of complement activation that is known to be highly expressed in many human malignancies. MV-NIS biotherapeutic platform is in clinical trials for various tumor type, including multiple myeloma and urothelial carcinoma. A total of 32 patients with recurrent or refractory multiple myeloma were treated in the Phase I protocol. Patients received various doses of the therapeutic and the maximum tolerable dose has not been reached. Adverse grades 3–4 events have been noticed, including neutropenia or decreased leukocyte count. Interestingly, radioisotopes (I123)-injected patients’ scans were positive (8 patients). In terms of efficacy, 1 patient achieved a complete response.83
Mayo Clinic is at the forefront of pushing ahead the MV as a biotherapeutic against cancer.82 Indeed, several Phase I/II clinical trials are ongoing with the aim to treat various types of cancer, including: squamous cell cancer of the head and neck and breast cancer (NCT01846091), glioblastoma (NCT00390299), ovarian cancer (NCT02068794), mesothelioma (NCT01503177), and malignant peripheral nerve sheath tumor (NCT02700230). Also, a Phase II trial in patients with multiple myeloma (NCT00450814) is underway. Specifically, concerning the use of MV-NIS, a randomized Phase II trial is recruiting patients with ovarian tumors and fallopian or peritoneal tumors (NCT02364713). MV-NIS has been evaluated in combination with immune checkpoint inhibitors (antibody against PD-1; nivolumab) in a Phase I/II trial for patients with advanced non-small cell lung cancer (NCT02919449). Furthermore, spearheaded by a different team, a MV-NIS study in combination with cyclophosphamide is also in process in patients with multiple myeloma (NCT02192775).
A VSV harboring a mutation in the M protein at the amino-acid 51 (VSVd51)10 and expressing NIS symporter69 has been developed. Interestingly, VSVd51-NIS has been found to be a safe oncolytic agent with significant therapeutic potential in multiple myeloma. More recently, a newer version of VSV has been engineered by the same group at the Mayo Clinic based on a VSV wild-type backbone and expressing both IFNβ and NIS. In this platform, the expression of IFNβ is meant to slow down virus replication. Overall, this virus encoding a NIS symporter shows good safety profile, including the absence of cytokine storm and great efficacy in various murine models, including leukemia. A Phase I clinical trial started using rVSV-IFNβ-NIS in patients with refractory solid tumors (NCT02923466) and the outcome of this trial is highly anticipated.
Conclusion
The need for tumor tissue localization and precise size measurement is critical in devising a specific therapeutic regimen for a patient. The past decade has witnessed an increase in available non-invasive imaging tools. Notably, several groups published reports using various mouse models as proof of concept and specific reporter genes making clear picture of tumor evolution and dynamics. OVs have been often chosen as a vector to carry reporter genes into the tumor bed. Various type of virus has been assessed, including MV, VSV, and VacV, all showing great promise in different pre-clinical models.
To date, there are virtually no clinical trials that rely on imaging strategies discussed in this manuscript to increase efficacy or evaluate safety. This is perhaps due to the fact that OVs are new to the clinical field and will require time for implementing these tools as part of a study. The leading imaging tool to date is NIS. Although both MV- and VSV-expressing NIS have been in clinical trial for the past few years (Table 1), little is known about the effect of NIS on patients at the immune system level. This is not only because the stages of these trials are classified as early (i.e., Phase I or II), but also due to absence of data from these trials. In fact, only 2 trials are currently classified as “active” which means at least another few years before any conclusions can be drawn. Another factor currently not being tested for in these trials is the combination with radioiodine, which could increase the therapeutic efficacy of the OV expressing NIS as shown in animal models.40 Hopefully, future clinical trials can take full advantage of OV-driven NIS expression at tumor sites, for localizing tumor, tracking OV spread, and evaluating treatment efficacy, all through a non-invasive imaging technique. Altogether, we predict that current trials will demonstrate that OV encoding NIS are a promising platform to study the localization of both the virus and the tumor.
Disclosure
The authors report no conflicts of interest in this work.
References
Topalian SL, Drake CG, Pardoll, DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. | ||
Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–158. | ||
Navin N, Kendall J, Troge J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. | ||
Reardon DA, Wen PY. Glioma in 2014: unravelling tumour heterogeneity-implications for therapy. Nat Rev Clin Oncol. 2015;12(2):69–70. | ||
Russell SJ, Peng, KW, Bell, JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30(7):658–670. | ||
Le Boeuf F, Batenchuk C, Vähä-Koskela M, et al. Model-based rational design of an oncolytic virus with improved therapeutic potential. Nat Commun. 2013;4:1974. | ||
Le Boeuf, F, Bell, JC. United virus: the oncolytic tag-team against cancer! Cytokine Growth Factor Rev. 2010;21(2–3):205–211. | ||
Le Boeuf F, Diallo JS, McCart JA, et al. Synergistic interaction between oncolytic viruses augments tumor killing. Mol Ther. 2010;18(5):888–895. | ||
Yang S, Guo ZS, O’Malley ME, Yin X, Zeh HJ, Bartlett DL. A new recombinant vaccinia with targeted deletion of three viral genes: its safety and efficacy as an oncolytic virus. Gene Ther. 2007;14(8):638–647. | ||
Stojdl DF, Lichty B, Knowles S, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6(7):821–825. | ||
Massague J. TGFbeta in cancer. Cell. 2008;134(2):215–230. | ||
Kleinovink JW, van Hall T, Ossendorp F, Fransen MF. PD-L1 immune suppression in cancer: tumor cells or host cells? Oncoimmunology. 2017;6(7):e1325982. | ||
Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–662. | ||
Parato KA, Breitbach CJ, Le Boeuf F, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther. 2012;20(4):749–758. | ||
Breitbach CJ, Burke J, Jonker D, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477(7362):99–102. | ||
Downs-Canner S, Guo ZS, Ravindranathan R, et al. Phase 1 study of intravenous oncolytic poxvirus (vvDD) in patients with advanced solid cancers. Mol Ther. 2016;24(8):1492–1501. | ||
Johnson DB, Puzanov I, Kelley, MC. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy. 2015;7(6):611–619. | ||
Puzanov I, Milhem MM, Minor D, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol. 2016;34(22):2619–2626. | ||
Zamarin D, Holmgaard RB, Subudhi SK, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6(226):226ra32. | ||
Samson A, Scott KJ, Taggart D, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. 2018;10(422):eaam7577. | ||
Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14(8):559–567. | ||
Bhatia S, Frangioni JV, Hoffman RM, Iafrate AJ, Polyak K. The challenges posed by cancer heterogeneity. Nat Biotechnol. 2012;30(7):604–610. | ||
Fedele C, Tothill RW, McArthur, GA. Navigating the challenge of tumor heterogeneity in cancer therapy. Cancer Discov. 2014;4(2):146–148. | ||
Garcia V, Krishnan R, Davis C, et al. High-throughput titration of luciferase-expressing recombinant viruses. J Vis Exp. 2014(91):51890. | ||
Rodriguez-Brenes IA, Hofacre A, Fan H, Wodarz D. Complex dynamics of virus spread from low infection multiplicities: implications for the spread of oncolytic viruses. PLoS Comput Biol. 2017;13(1):e1005241. | ||
Fass L. Imaging and cancer: a review. Mol Oncol. 2008;2(2):115–152. | ||
Day CP, Merlino G, Van Dyke T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell. 2015;163(1):39–53. | ||
Lee WC, Chang C-H, Ho C-L, et al. Early detection of tumor response by FLT/microPET Imaging in a C26 murine colon carcinoma solid tumor animal model. J Biomed Biotechnol. 2011;2011:535902. | ||
Menezes GL, Knuttel FM, Stehouwer BL, Pijnappel RM, van den Bosch MA1. Magnetic resonance imaging in breast cancer: a literature review and future perspectives. World J Clin Oncol. 2014;5(2):61–70. | ||
Power AT, Wang J, Falls TJ, et al. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol Ther. 2007;15(1):123–130. | ||
de Jong M, Essers J, van Weerden, WM. Imaging preclinical tumour models: improving translational power. Nat Rev Cancer. 2014;14(7):481–493. | ||
Shin D, Vigneswaran N, Gillenwater A, Richards-Kortum R. Advances in fluorescence imaging techniques to detect oral cancer and its precursors. Future Oncol. 2010;6(7):1143–1154. | ||
Yu EY, Bishop M, Zheng B, et al. Magnetic particle imaging: a novel in vivo imaging platform for cancer detection. Nano Lett. 2017;17(3):1648–1654. | ||
Mansueti JR, Maillie S. A review of non-small cell lung cancer post-treatment follow-up imaging procedures with PET/CT scans versus CT scans and the effect on patient survival. Int J Radiat Oncol Biol Phys. 2017;98(1):231. | ||
Mariani G, Bruselli L, Kuwert T, et al. A review on the clinical uses of SPECT/CT. Eur J Nucl Med Mol Imaging. 2010;37(10):1959–1985. | ||
Farwell MD, Pryma DA, Mankoff, DA. PET/CT imaging in cancer: current applications and future directions. Cancer. 2014;120(22):3433–3445. | ||
Dai G, Levy O, Carrasco N. Cloning and characterization of the thyroid iodide transporter. Nature. 1996;379(6564):458–460. | ||
Pinke LA, Dean DS, Bergert ER, Spitzweg C, Dutton CM, Morris JC. Cloning of the mouse sodium iodide symporter. Thyroid. 2001;11(10):935–939. | ||
Smanik PA, Liu Q, Furminger TL, et al. Cloning of the human sodium lodide symporter. Biochem Biophys Res Commun. 1996;226(2):339–345. | ||
Wang J, Arulanandam R, Wassenaar R, et al. Enhancing expression of functional human sodium iodide symporter and somatostatin receptor in recombinant oncolytic vaccinia virus for in vivo imaging of tumors. J Nucl Med. 2017;58(2):221–227. | ||
Hoffman RM. Application of GFP imaging in cancer. Lab Invest. 2015;95(4):432–452. | ||
Ruotsalainen JJ, Kaikkonen MU, Niittykoski M, et al. Clonal variation in interferon response determines the outcome of oncolytic virotherapy in mouse CT26 colon carcinoma model. Gene Ther. 2015;22(1):65–75. | ||
Vaha-Koskela MJ, Le Boeuf F, Lemay C, et al. Resistance to two heterologous neurotropic oncolytic viruses, Semliki Forest virus and vaccinia virus, in experimental glioma. J Virol. 2013;87(4):2363–2366. | ||
Soboleski MR, Oaks J, Halford WP. Green fluorescent protein is a quantitative reporter of gene expression in individual eukaryotic cells. FASEB J. 2005;19(3):440–442. | ||
Yu YA, Shabahang S, Timiryasova TM, et al. Visualization of tumors and metastases in live animals with bacteria and vaccinia virus encoding light-emitting proteins. Nat Biotechnol. 2004;22(3):313–320. | ||
Alberti C. From molecular imaging in preclinical/clinical oncology to theranostic applications in targeted tumor therapy. Eur Rev Med Pharmacol Sci. 2012;16(14):1925–1933. | ||
Deng H, Tang N, Stief AE, et al. Oncolytic virotherapy for multiple myeloma using a tumour-specific double-deleted vaccinia virus. Leukemia. 2008;22(12):2261–2264. | ||
Zhang Q, Yu YA, Wang E, et al. Eradication of solid human breast tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus. Cancer Res. 2007;67(20):10038–10046. | ||
Parviainen S, Ahonen M, Diaconu I, et al. CD40 ligand and tdTomato-armed vaccinia virus for induction of antitumor immune response and tumor imaging. Gene Ther. 2014;21(2):195–204. | ||
Binz E, Berchtold S, Beil J, et al. Chemovirotherapy of pancreatic adenocarcinoma by combining oncolytic vaccinia virus GLV-1h68 with nab-paclitaxel plus gemcitabine. Mol Ther Oncolytics. 2017;6:10–21. | ||
Costantini LM, Snapp, EL. Going viral with fluorescent proteins. J Virol. 2015;89(19):9706–9708. | ||
Barry MA, May S, Weaver EA. Imaging luciferase-expressing viruses. Methods Mol Biol. 2012;797:79–87. | ||
Brun J, Mahoney DJ, Le Boeuf F, et al. Oncolytic Vaccinia virus safely and effectively treats skin tumors in mouse models of xeroderma pigmentosum. Int J Cancer. 2013;132(3):726–731. | ||
Coleman SM, McGregor A. A bright future for bioluminescent imaging in viral research. Future Virol. 2015;10(2):169–183. | ||
Zhang F, Perez-Martin E, Juleff N, Charleston B, Seago J. A replication-competent foot-and-mouth disease virus expressing a luciferase reporter. J Virol Methods. 2017;247:38–44. | ||
Stritzker J, Kirscher L, Scadenget M, et al. Vaccinia virus-mediated melanin production allows MR and optoacoustic deep tissue imaging and laser-induced thermotherapy of cancer. Proc Natl Acad Sci USA. 2013;110(9):3316–3320. | ||
Kaur B, Chiocca EA, Cripe TP. Oncolytic HSV-1 virotherapy: clinical experience and opportunities for progress. Curr Pharm Biotechnol. 2012;13(9):1842–1851. | ||
Munoz-Alvarez KA, Altomonte J, Laitinen I, et al. PET imaging of oncolytic VSV expressing the mutant HSV-1 thymidine kinase transgene in a preclinical HCC rat model. Mol Ther. 2015;23(4):728–7436. | ||
Abate-Daga D, Andreu N, Camacho-Sánchez J, et al. Oncolytic adenoviruses armed with thymidine kinase can be traced by PET imaging and show potent antitumoural effects by ganciclovir dosing. PLoS One. 2011;6(10):e26142. | ||
Slooter GD, Mearadji A, Breeman WA, et al. Somatostatin receptor imaging, therapy and new strategies in patients with neuroendocrine tumours. Br J Surg. 2001;88(1):31–40. | ||
Rogers BE, McLean SF, Kirkman RL, et al. In vivo localization of [(111)In]-DTPA-D-Phe1-octreotide to human ovarian tumor xenografts induced to express the somatostatin receptor subtype 2 using an adenoviral vector. Clin Cancer Res. 1999;5(2):383–393. | ||
Zinn KR, Buchsbaum DJ, Chaudhuri TR, Mountz JM, Grizzle WE, Rogers BE. Noninvasive monitoring of gene transfer using a reporter receptor imaged with a high-affinity peptide radiolabeled with 99mTc or 188Re. J Nucl Med. 2000;41(5):887–895. | ||
McCart JA, Mehta N, Scollard D, et al. Oncolytic vaccinia virus expressing the human somatostatin receptor SSTR2: molecular imaging after systemic delivery using 111In-pentetreotide. Mol Ther. 2004;10(3):553–561. | ||
Moroz MA, Serganova I, Zanzonico P, et al. Imaging hNET reporter gene expression with 124I-MIBG. J Nucl Med. 2007;48(5):827–836. | ||
Brader P, Kelly KJ, Chen, N, et al. Imaging a genetically engineered oncolytic vaccinia virus (GLV-1h99) using a human norepinephrine transporter reporter gene. Clin Cancer Res. 2009;15(11):3791–801. | ||
Boland A, Ricard M, Opolon P, et al. Adenovirus-mediated transfer of the thyroid sodium/iodide symporter gene into tumors for a targeted radiotherapy. Cancer Res. 2000;60(13):3484–3492. | ||
Barton KN, Tyson D, Stricker H, et al. GENIS: gene expression of sodium iodide symporter for noninvasive imaging of gene therapy vectors and quantification of gene expression in vivo. Mol Ther. 2003;8(3):508–518. | ||
Dingli D, Bergert ER, Bajzer Z, O’connor MK, Russell SJ, Morris JC. Dynamic iodide trapping by tumor cells expressing the thyroidal sodium iodide symporter. Biochem Biophys Res Commun. 2004;325(1):157–166. | ||
Goel A, Carlson SK, Classic KL, et al. Radioiodide imaging and radiovirotherapy of multiple myeloma using VSV(Delta51)-NIS, an attenuated vesicular stomatitis virus encoding the sodium iodide symporter gene. Blood. 2007;110(7):2342–2350. | ||
Shen W, Patnaik MM, Ruiz A, Russell SJ, Peng KW. Immunovirotherapy with vesicular stomatitis virus and PD-L1 blockade enhances therapeutic outcome in murine acute myeloid leukemia. Blood. 2016;127(11):1449–1458. | ||
Haddad D, Chen CH, Carlin S, et al. Imaging characteristics, tissue distribution, and spread of a novel oncolytic vaccinia virus carrying the human sodium iodide symporter. PLoS One. 2012;7(8):e41647. | ||
Haddad D, Chen NG, Zhang Q, et al. Insertion of the human sodium iodide symporter to facilitate deep tissue imaging does not alter oncolytic or replication capability of a novel vaccinia virus. J Transl Med. 2011;9:36. | ||
Gholami S, Chen CH, Lou E, et al. Vaccinia virus GLV-1h153 in combination with 131I shows increased efficiency in treating triple-negative breast cancer. FASEB J. 2014;28(2):676–682. | ||
Mansfield DC, Kyula JN, Rosenfelder N, et al. Oncolytic vaccinia virus as a vector for therapeutic sodium iodide symporter gene therapy in prostate cancer. Gene Ther. 2016;23(4):357–368. | ||
Hakkarainen T, Rajecki M, Sarparanta M, et al. Targeted radiotherapy for prostate cancer with an oncolytic adenovirus coding for human sodium iodide symporter. Clin Cancer Res. 2009;15(17):5396–5403. | ||
Merron A, Peerlinck I, Martin-Duque P, et al. SPECT/CT imaging of oncolytic adenovirus propagation in tumours in vivo using the Na/I symporter as a reporter gene. Gene Ther. 2007;14(24):1731–1738. | ||
Jhawar SR, Thandoni A, Bommareddy PK, et al. Oncolytic viruses-natural and genetically engineered cancer immunotherapies. Front Oncol. 2017;7:202. | ||
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | ||
Pol J, Buqué A, Aranda F, et al. Trial Watch-Oncolytic viruses and cancer therapy. Oncoimmunology. 2016;5(2):e1117740. | ||
Barton KN, Stricker H, Brown SL, et al. Phase I study of noninvasive imaging of adenovirus-mediated gene expression in the human prostate. Mol Ther. 2008;16(10):1761–1769. | ||
Msaouel P, Dispenzieri A, Galanis E. Clinical testing of engineered oncolytic measles virus strains in the treatment of cancer: an overview. Curr Opin Mol Ther. 2009;11(1):43–53. | ||
Msaouel P, Opyrchal M, Dispenzieri A, et al. Clinical trials with oncolytic measles virus: current status and future prospects. Curr Cancer Drug Targets. Epub 2017 Feb 22. | ||
Dispenzieri A, Tong C, LaPlant B, et al. Phase I trial of systemic administration of Edmonston strain of measles virus genetically engineered to express the sodium iodide symporter in patients with recurrent or refractory multiple myeloma. Leukemia. 2017;31(12):2791–2798. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.