")
Back to Journals » International Journal of Nanomedicine » Volume 15
The Impact of Surfactant Composition and Surface Charge of Niosomes on the Oral Absorption of Repaglinide as a BCS II Model Drug
Authors Yaghoobian M, Haeri A, Bolourchian N , Shahhosseni S , Dadashzadeh S
Received 13 May 2020
Accepted for publication 29 September 2020
Published 11 November 2020 Volume 2020:15 Pages 8767—8781
DOI https://doi.org/10.2147/IJN.S261932
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Morteza Yaghoobian,1 Azadeh Haeri,1 Noushin Bolourchian,1 Soraya Shahhosseni,2 Simin Dadashzadeh1,3
1Department of Pharmaceutics and Nanotechnology, School of Pharmacy, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 2Department of Pharmaceutical Chemistry, School of Pharmacy, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 3Pharmaceutical Sciences Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran
Correspondence: Simin Dadashzadeh
Department of Pharmaceutics and Nanotechnology, School of Pharmacy, Shahid Beheshti University of Medical Sciences, Tehran 14155-6153, Iran
Tel +98 21 88200070
Fax + 98 21 88209620
Email [email protected]
Background: Niosomes, bilayer vesicles formed by the self-assembly of nonionic surfactants, are receiving increasing attention as potential oral drug delivery systems but the impact of niosomal formulation parameters on their oral capability has not been studied systematically. The aim of this study was to investigate the impact of surfactant composition and surface charge of niosomes in enhancing oral bioavailability of repaglinide (REG) as a BCS II model drug.
Methods: Niosomes (13 formulations) from various nonionic surfactants having HLB in the range of 4– 28 (Tweens, Spans, Brijs, Myrj, poloxamer 188, TPGS and Labrasol) were prepared and characterized concerning their loading efficiency, hydrodynamic diameter, zeta potential, drug release profile, and stability. The oral pharmacokinetics of the selected formulations were studied in rats (8 in vivo groups).
Results: The results revealed that type of surfactant markedly affected the in vitro and in vivo potentials of niosomes. The Cmax and AUC values of REG after administration of the selected niosomes as well as the drug suspension (as control) were in the order of Tween 80> TPGS> Myrj 52> Brij 35> Span 60≈Suspension. Adding stearyl amine as a positive charge-inducing agent to the Tween 80-based niosomes, resulted in an additional increase in drug absorption and values of AUC and Cmax were 3.8- and 4.7-fold higher than the drug suspension, respectively.
Conclusion: Cationic Tween 80-based niosomes may represent a promising platform to develop oral delivery systems for BCS II drugs.
Keywords: repaglinide, niosome, oral bioavailability, surfactant type, surface charge, BCS II, HLB
Introduction
Oral administration of Biopharmaceutics Classification System (BCS) II drugs generally suffer from low and variable bioavailability owing to poor water solubility, efflux by gut wall transporters and first pass metabolism.1 To overcome these drawbacks, a variety of formulation strategies, such as polymer- and lipid-based nanoparticles,2,3 solid dispersions,4 nanoemulsions,5 and nanosuspentions6 have been developed.
Various studies have shown that the composition of nanocarriers is a key factor for their physicochemical characteristics and in vivo performance. For instance, as we have reported previously, the in vivo behavior of liposomes following peritoneal,7 intravenous (IV)8 or local intramural9 administration is influenced by both lipid composition and surface characteristics of the vesicles. For the oral route, multiple physiological barriers restrict the absorption of drugs, thus the role of carrier composition and surface characteristics is expected to be more critical.
Niosomes, bilayer vesicles formed by the self-assembly of nonionic surfactants, are biocompatible, non-immunogenic, flexible in their composition and have the potential to encapsulate both hydrophilic and lipophilic drugs. Niosomes are structurally similar to liposomes, however, in recent years, they have attracted high attention as alternative drug carriers to liposomes due to their higher chemical stability and lower production cost.10–12 Niosomes have shown promising results in improving the oral bioavailability of drugs.13–15 After oral administration, the small size of vesicles could provide a large interfacial surface area for intestinal absorption. Furthermore, the surfactant components of the formulations could improve drug solubility, may suppress P-glycoprotein (P-gp)-mediated efflux and inhibit activity of CYP enzymes which are responsible for first pass metabolism of drugs.16 The P-gp inhibition effects of nonionic surfactants have been attributed to the modulation of membrane fluidity. For instance, the inhibitory activity of three nonionic surfactants, Tween 80, Cremophor EL and vitamin E TPGS on P-glycoprotein in Caco-2 cell monolayers was demonstrated by Rege et al.17 Their further mechanistic studies revealed that Tween 80 and Cremophor EL fluidized cell lipid bilayers, while vitamin E TPGS rigidized lipid bilayers.17
Nonionic surfactants can also affect the metabolism of active drugs. CYP3A is a major subfamily of CYP enzymes responsible for drug first pass metabolism and bioavailability. Si and co-workers evaluated the effects of four common pharmaceutical surfactants on the metabolic activity of CYP3A in both in vitro and in vivo systems by using midazolam as a metabolic probe.18 Their results revealed that nonionic surfactants influence CYP3A activity with a mixed-type inhibitory pattern. They suggested that nonionic surfactants could bind to both the active and allosteric sites on CYP. Interfering with the active site on CYP3A results in decreased maximal velocity (Vmax) and binding to an allosteric site on CYP3A could change the conformation of CYP3A leading to a decrease in drug binding and its metabolism. Moreover, the ability of these excipients to alter the conformation of the microsomal monooxygenase’s components and consequently destroying their function19 may also contribute to nonionic surfactants mixed type inhibition on CYP3A.
A number of nonionic surfactants, such as polysorbates, alkyl esters and alkyl ethers are typically used for preparation of niosomes in drug delivery. The nature of the surfactant is an important factor affecting physicochemical characteristics of niosomes, namely, bilayer rigidity, particle size, stability and drug release rate, therefore, it is expected to play an important role in in vivo behavior of these drug delivery systems. Motivated by the advantages associated with niosomal drug delivery systems and also our previous results on the desirable influence of positively charged liposomes in improving oral drug absorption of a BCS III drug,20 the aim of this study was to investigate the impact of surfactant composition of niosomal formulations as well as positive surface charge of vesicles on the oral absorption of repaglinide (REG) as a BCS class II model drug. REG is an effective orally administered anti-hyperglycemic drug and is widely used in the treatment of type II diabetes mellitus.21–23 In spite of having excellent anti-diabetic activity and several advantages, REG suffers from low and variable oral bioavailability. REG has a low water solubility (approximately 15 μg/mL) and high lipophilicity (log P=3.97).23,24 Moreover, it undergoes significant first pass metabolism by the cytochrome P450 system.25
REG-loaded niosomes with various nonionic surfactants having hydrophilic lipophilic balance (HLB) in the range of 4–28 (Tween 20, Tween 60, Tween 80, Span 20, Span 40, Span 60, Brij 35, Brij 72, Brij 78, Myrj 52, TPGS, Labrasol or poloxamer 188) were prepared and thoroughly characterized for their in vitro properties. Subsequently, the pharmacokinetics of the selected formulations and controls were studied in eight groups of rats (n=6 in each group). MTT assay and tissue histology studies were performed to study possible cellular toxicity or tissue inflammation. To date, as far as the literature shows, the impact of the surfactant composition as well as positive surface charge of niosomes on the oral absorption of drugs has not yet been reported.
Materials and Methods
Materials
Brij 35, Brij 72, Brij 78, Span 20, Span 40, Span 60, Tween 20, Tween 60, Tween 80, D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS), Myrj 52, poloxamer 188, stearyl amine (STA), cholesterol (Chol), sodium taurocholate and 3-(4,5- Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma Chemical Co. (Germany). Labrasol was kindly gifted by GATTEFOSSE Company (France). REG was a kind gift from Darupakhsh Pharmaceutical Company (Iran). All organic solvents and the other chemicals were analytical grade and obtained from Merck (Germany).
Preparation of Niosomes
Different niosomal formulations (Table 1) composed of various nonionic surfactants were prepared using thin film hydration technique.26 The drug (1 mg/mL) and appropriate amount of surfactants and Chol (65:35, mol ratio) were dissolved in a mixture of methanol and chloroform (1:1 v/v) in a round bottom flask. In the case of positively charged niosomes, STA was also added to the related niosome composition. The organic phase was removed at 60°C under vacuum by rotary evaporator (Heidolph, Germany), yielding a thin film on the wall of the flask. The film was maintained under vacuum for at least 2 hours for removing the last traces of the organic solvent. The dried thin film was hydrated with 5 mL of deionized water at 65°C and was simultaneously bath sonicated (405, Hwashin Liarre, Korea) for 30 minutes (3 cycles of 10 minutes) to obtain niosomal dispersion.
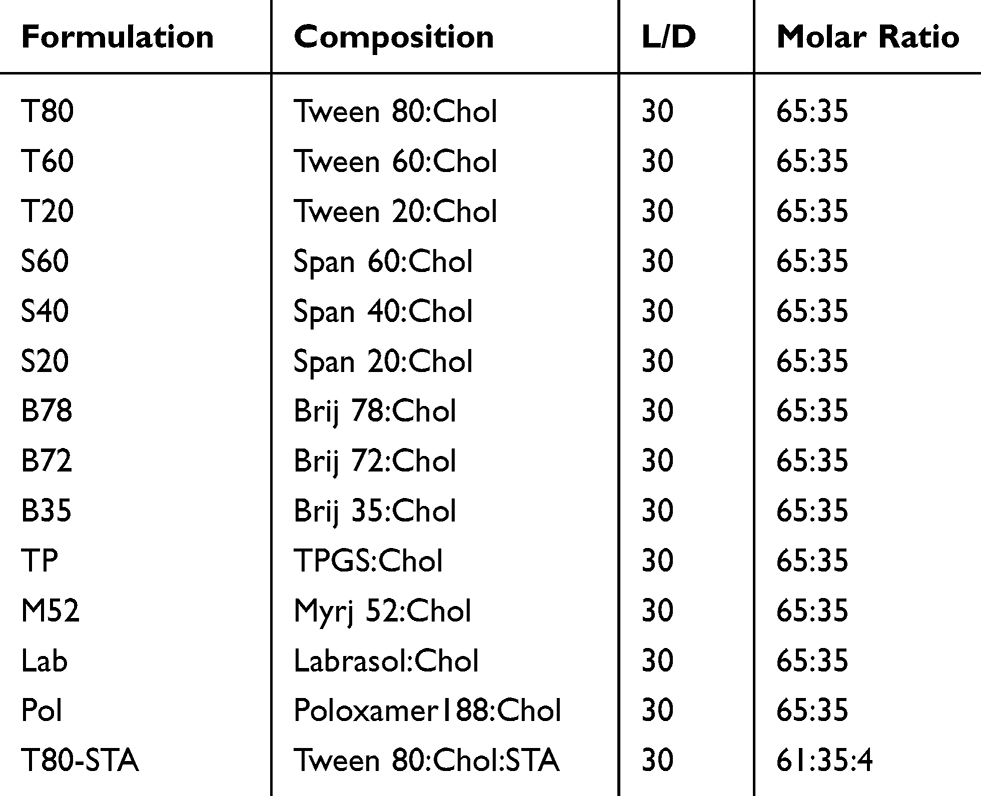 |
Table 1 Composition of the Prepared Niosomes (Mean ± SD, n=3) |
Drug Entrapment Efficiency
To determine entrapment efficiency (EE), separation of free drug from niosomal formulation was performed by the dialysis method. Samples of niosomal dispersion (equivalent to 1 mg/mL of REG) were placed in a dialysis bag (cut-off of 12,000 Da) and extensively dialyzed against 500 mL of deionized water at 4°C for 8 hours. At various time intervals (2, 4, 6 and 8 hours), aliquot samples of dialysate were withdrawn and subjected to drug content analysis. A small fraction of the dialysate (100 μL) was completely disrupted using a mixture of methanol and isopropanol (50:50, v/v; 900 μL) and bath-sonicated for 10 minutes. The obtained solution was centrifuged at 3000 rpm for 5 minutes. The supernatant was decanted off, and the entrapped REG was determined spectrophotometrically at the wavelength of 242 nm. The dialysis continued until the drug content did not change (6–8 hours). The EE% was calculated by the following equation:

Hydrodynamic Diameter and Zeta Potential
The hydrodynamic diameter, size distribution and zeta potential (ZP) of the prepared niosomes were determined by Zetasizer (Malvern Instruments, UK). Before performing the measurements, the formulations were diluted 100 times with deionized water and mixed thoroughly, and then measurements were done in triplicate at 25 ± 1°C with a scattering light angle of 90°.
Drug Release Studies
The in vitro drug release was carried out by dialysis method in different biorelevant media including simulated gastric fluid (SGF, pH=1.2), simulated intestinal fluid (SIF, pH=6.8) containing 0.1% (w/v) Tween 80 and SIF containing 0.1% (w/v) Tween 80 and 10 mM sodium taurocholate (B-SIF, pH=6.8). Freshly made niosomes were put in the dialysis bags (molecular mass cut-off of 12,000 Da). The bags were suspended in glass beakers containing each release media stirring at 37°C and 100 rpm. The total amount of drug inside the suspended vesicles was less than 10% of its solubility limit in the respected medium to maintain the sink condition. At regular time intervals (0.25, 0.5, 0.75, 1, 1.5 and 2 hours for SGF medium; 0.25, 0.5, 0.75, 1, 1.5, 2, 4, 6, 8 and 10 hours for SIF and B-SIF media), 100 µL aliquots of release media were withdrawn and replaced by the same volume of a fresh medium. After sampling at 4 and 6 hours the release medium was completely replaced with a fresh one to maintain the perfect sink condition. The drug concentration in each sample was quantified by a validated HPLC method.
Stability of Niosomes in the Biorelevant Media
The physical stability of niosomes in SGF, SIF and B-SIF, was determined by measuring the particle size, PDI and ZP. For this, the prepared formulations were incubated with each release medium at 37°C and stirred at 100 rpm. Samples were withdrawn at times 0 and 2 hours (SGF medium) or 0 and 10 hours (SIF and B-SIF media) and analyzed in terms of the above-mentioned characteristics.
Morphology of Niosomes
Atomic force microscopy (AFM, Nanowizard II) was used for visualization of niosomes. The niosomes were diluted with deionized water, spread thinly onto the glass slide and air-dried. For imaging, the microscope was operated in contact mode under ambient conditions employing low stress silicon nitride cantilevers (AppNano, Mountain View, CA, USA).
In vivo Studies
Male Wistar rats weighing 200 to 230 g were obtained from the Pasteur Institute (Tehran, Iran) and kept under controlled temperature (22°C), humidity (55%) and 12 hour-light, 12 hour-dark cycles. Before the experiments, the animals were fasted overnight with free access to water. The animal protocols were approved by the Institutional Research Ethics Committee of School of Pharmacy, Shahid Beheshti University of Medical Sciences (Approval No. SBMU.PHARMACY.REC.1396.044) which is based on “Guideline to care and use of laboratory animals in scientific research” released by Iran Ministry of Health and Medical Education.27 The rats were randomly divided into eight groups (n=6 in each group) and received either the various drug-loaded niosomes (6 groups), or controls (the drug suspension or a mixture of drug suspension and empty niosomes of the optimum formulation (T80-STA)). REG suspension was prepared by using 0.5% w/v sodium carboxymethyl cellulose as vehicle. All formulations were administered as a single-dose equivalent to 1 mg/kg of drug by oral gavage. Immediately before dosing and then at specified time intervals (0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 10 and 12 hours) after drug administration, blood samples were withdrawn from the tail vein and immediately centrifuged at 10,000 rpm for 10 minutes. Plasma samples were then collected and stored at −20°C until HPLC assay.
Analysis of Drug in Plasma
The HPLC method was conducted using a Knauer HPLC system (Germany). Chromatographic separation was performed on a PerfectSil RP-18 (150 mm ×4.6 mm i.d., MZ-Analysentechnik GmbH, Germany) at ambient temperature. The optimal mobile phase was a mixture of acetonitrile and phosphate buffer 0.05 M (60:40 v/v) and delivered at a flow rate of 1 mL/min. The detection wavelength was set at 242 nm and the injection volume at 100 μL. For sample preparation an aliquot of 30 μL of naproxen methanolic solution (1 μg/mL) as internal standard, 1 mL of methanol: phosphate buffer (60:40, pH =4.0) and 50 µL of ethyl acetate was added to 100 μL of plasma sample and thoroughly vortex-mixed for 10 minutes. After centrifuging (10,000 rpm, 10 minutes), the supernatant was evaporated to dryness under a nitrogen stream, reconstituted in 100 μL of the mobile phase and analyzed by HPLC. The average absolute recovery of REG and naproxen (I.S.) from the spiked plasma samples was more than 90%. The calibration curves were linear over the concentration range of 100–1000 ng/mL (R2> 0.994), and the lower limit of quantification was 10 ng/mL. The intra-day and inter-day coefficients of the variation (CV) were all less than 5%.
Pharmacokinetic Analysis
Pharmacokinetic analysis was performed by using a noncompartmental method. All pharmacokinetic parameters including maximum plasma concentration (Cmax), time to reach maximum plasma concentration (Tmax), area under the drug concentration-time curve (AUC) and mean residence time (MRT) were calculated from the plasma concentration–time data.
Evaluation of Formulations Biocompatibility
In vitro Cytotoxicity Evaluation of Niosomes
The effect of the empty niosomal formulations on cell growth was measured using the MTT assay. Caco-2 cells (obtained from Pasture Institute National Cell Bank of Iran in Tehran) were cultured in a medium consisting of RPMI 1640, Dulbecco’s modified Eagle medium (DMEM) and fetal bovine serum (FBS) (all from Gibco) in a ratio of 50:35:15 and maintained at 37°C at an atmosphere of 5% CO2 incubator. Cells were cultured for 48 hours and incubated with 500 µM and 1000 µM niosomal formulations for 3 hours at 37°C. Then, cells were washed twice with growth medium and the MTT solution (20 μL, 5 mg/mL) was added. Cells were maintained for another 4 hours at 37°C followed by addition of DMSO. UV absorbance of each well was measured on an Elisa Plate reader at a wavelength of 570 nm. Cell viability was calculated as a percentage of the optical density values relative to control cells treated with growth medium.
Histological Evaluation of Small Intestine Segments
Histological studies were conducted to evaluate any possible morphological damages caused by the optimal niosome formulation in the small intestine. For this purpose, rats were sacrificed 4 hours after administration of the niosomes. Tissue samples were taken from different parts of small intestine, fixed in buffered formalin, embedded in paraffin, sectioned at 5 μm and stained with hematoxylin and eosin (H&E) according to standard methods.
Statistical Analysis
All statistical analyses were performed using analysis of variance (ANOVA) and post hoc Bonferroni testing with SPSS version 22 (IBM Corporation, Armonk, NY, USA). Statistical significance was set at p < 0.05. Data were expressed as the mean ± SD (for 3 or 6 independent samples).
Results
Niosome Characteristics
To evaluate the effect of surfactant composition on in vitro and in vivo characteristics of REG-loaded niosomes, at the first step a variety of commonly used surfactants having HLB in the range of 4.5–27.8 (Tween 20, Tween 60, Tween 80, Span 20, Span 40, Span 60, Brij 35, Brij 72, Brij 78, Myrj 52, poloxamer 188, TPGS and Labrasol) were investigated for preparing formulations (Table 1). Chol content of 35% and lipid-to-drug (L/D) molar ratio of 30 were kept constant in all formulations. The physicochemical properties of the prepared vesicles are shown in Table 2. Among the investigated surfactants, poloxamer 188 vesicles were not formed and the hydrodynamic diameter of the Brij 78 niosomes was much larger (around 892 nm) than the other niosomes. Therefore, these two formulations were excluded from further studies. Z-average size of the other niosomes was suitable for oral administration ranging from 119.1 (T20) to 236.9 nm (TP). In all niosomal formulations, with the exception of B78, the average PDI values were less than 0.5 and ranged between 0.18 ± 0.13 and 0.48 ± 0.20. The values less than 0.7 are considered as suitable measurements.28,29 The lower PDI values indicate more uniformity of the dispersion. With regard to the REG loading, the average EE values varied between 52.8% (S 40 formulation) and 77.1% (TP formulation) indicating good niosomal entrapment of the drug. The exceptions were Labrasol- and Span 20-based vesicles which showed obviously low EE values (less than 50%). ZP of all the niosomal formulations were also determined which varied from −8.13 mV to −34.05 mV (Table 2).
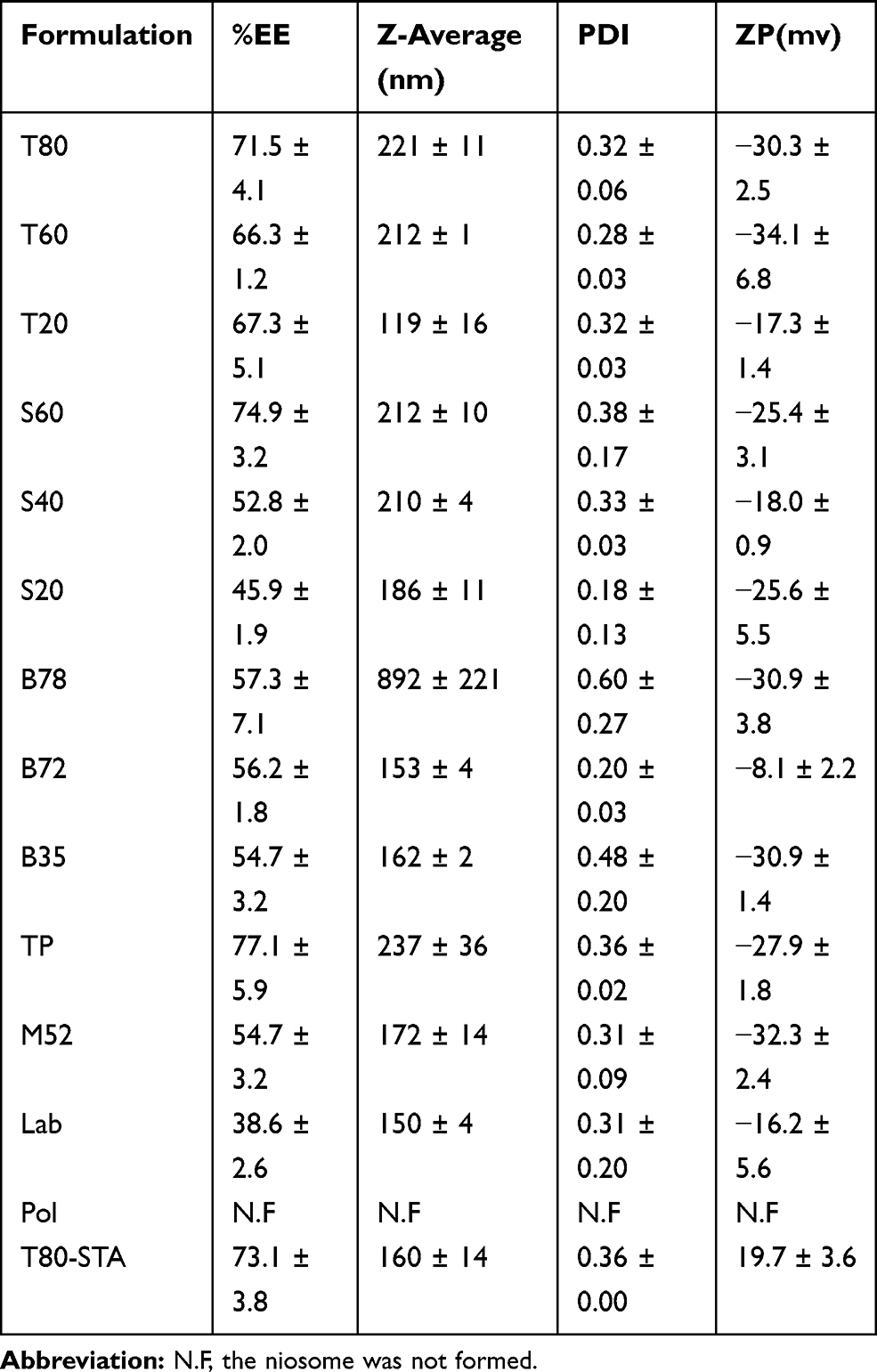 |
Table 2 %EE, Particle Size, Hydrodynamic Diameter, Polydispersity Index and Zeta Potential of the Prepared Niosomes (Mean ±SD; n =3) |
In vitro Drug Release
The niosomes with appropriate hydrodynamic diameter and EE higher than 50% underwent the drug release test (10 out of 13 formulations). The percentages of released drug in different simulated media (SGF, SIF and B-SIF) are shown in Figure 1. As illustrated, overall, niosomes showed markedly faster drug release than drug suspension. However, most of the formulations could retain more than 60% of their content after respectively 0.5 hours and 4 hours incubation in SGF (Figure 1A and B) and SIF (Figure 1C and D), respectively. Adding bile salt to simulated intestinal media (B-SIF), slightly increased drug release rate. Regarding Span surfactants, S60 showed significantly lower release rate compared to its class counterparts. Tween-based formulations showed nearly similar release rate and considering Brij class, the drug release from B72, particularly in SIF and B-SIF was obviously faster than B35. Both M52 and TP showed controlled release rates and were able to retain their encapsulated drug appropriately.
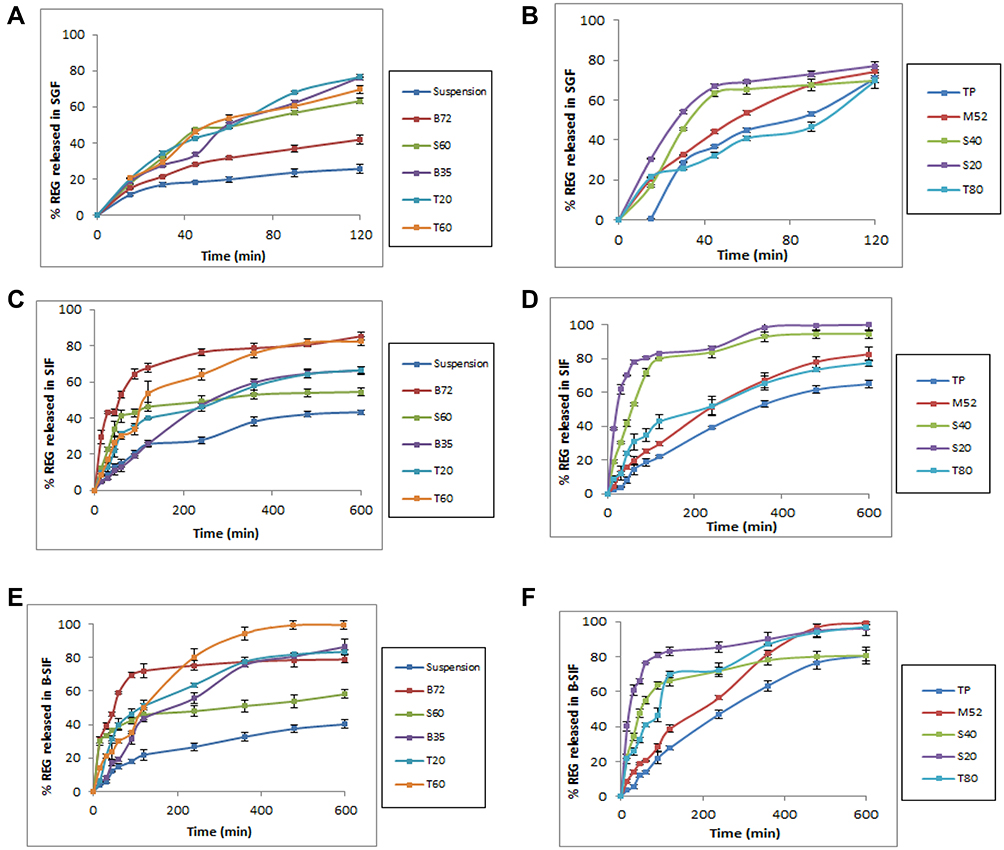 |
Figure 1 In vitro release profiles of REG from the various formulations in SGF (A and B), SIF (C and D) and B-SIF (E and F) at 37°C. Data represent mean ± SD (n=3). |
Stability of Niosomes in Gastrointestinal-Simulated Media
To investigate the potential of niosomal formulations in improving the oral bioavailability of REG as well as to evaluate the effect of surfactant type, as the main component of niosomes, the formulations containing one member of each surfactant class were chosen for in vivo studies. For this, T80, SP60, Bj35, Mj52 and TP formulations, which had more appropriate physicochemical characteristics (hydrodynamic diameter < 250 nm, PDI < 0.5 and EE > 50%) and almost sustained release profile, were chosen. Stability under gastrointestinal (GI) conditions is a major concern for the orally administered nanocarriers. Therefore, prior to in vivo studies, the stability of the selected REG niosomes was studied in the GI-simulated media. The size, PDI and ZP of the niosomes were measured before and after 2 hours and 10 hours incubation in SGF and B-SIF, respectively (Figure 2). There was no significant difference in size, PDI and ZP before and after incubation in the biorelevant media, implying that niosomes preserved their structure without any significant differences in their physical characteristics. Maximum increase in the size of the formulations after incubation time belonged to B35 formulation in SGF (16%) and S60 formulation in SIF-B (22%).
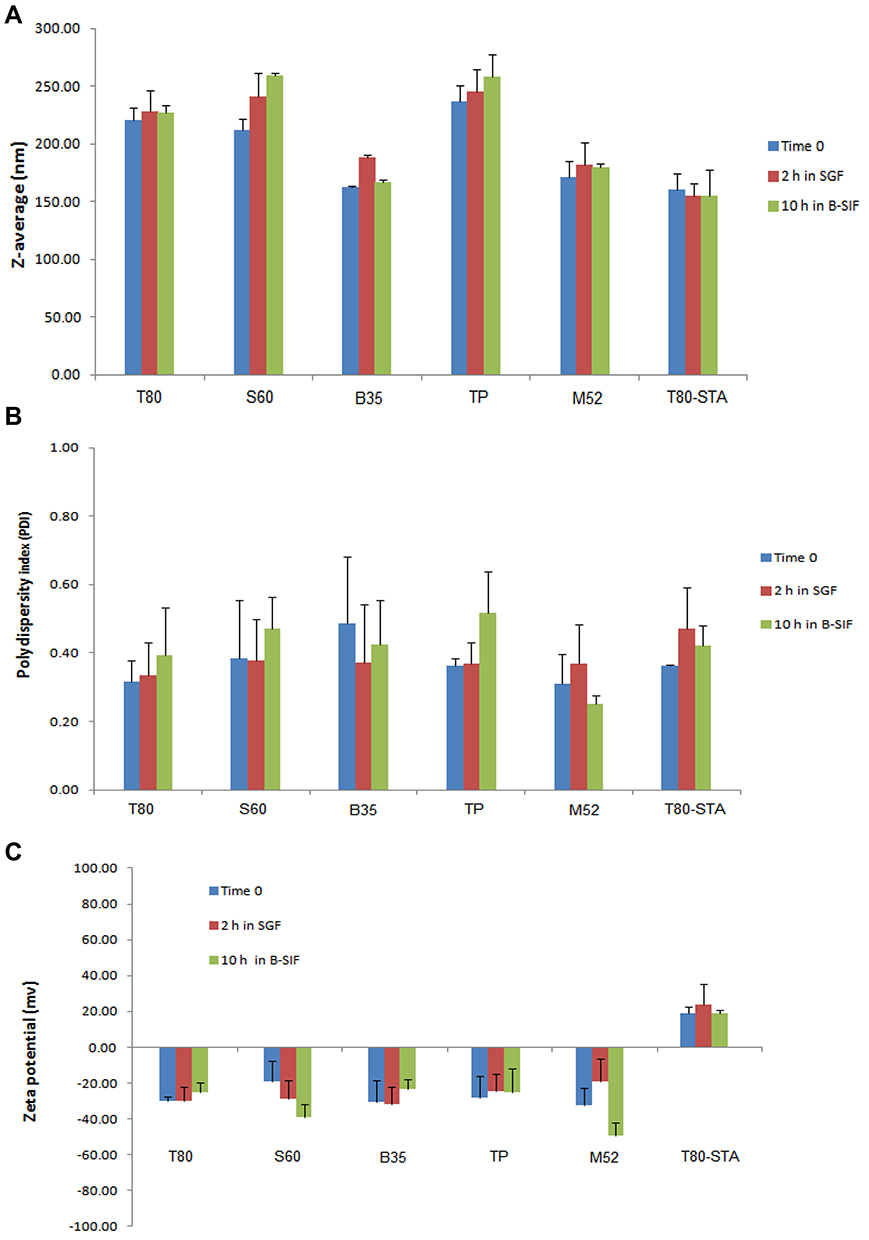 |
Figure 2 Hydrodynamic diameter (A), PDI (B) and ZP (C) of the selected niosomes immediately after preparation and after incubation in SGF (2 hours) and B-SIF (10 hours) at 37°C. |
In vivo Studies
As mentioned in previous sections, the formulations of T80, S60, B35, M52 and TP which had more desirable physicochemical characteristics and release profiles were chosen and their pharmacokinetics were evaluated following oral administration to rats. The plasma concentration vs time curves are shown in Figure 3 and the obtained pharmacokinetic parameters including Tmax, Cmax, MRT and AUC0→∞ are presented in Table 3.
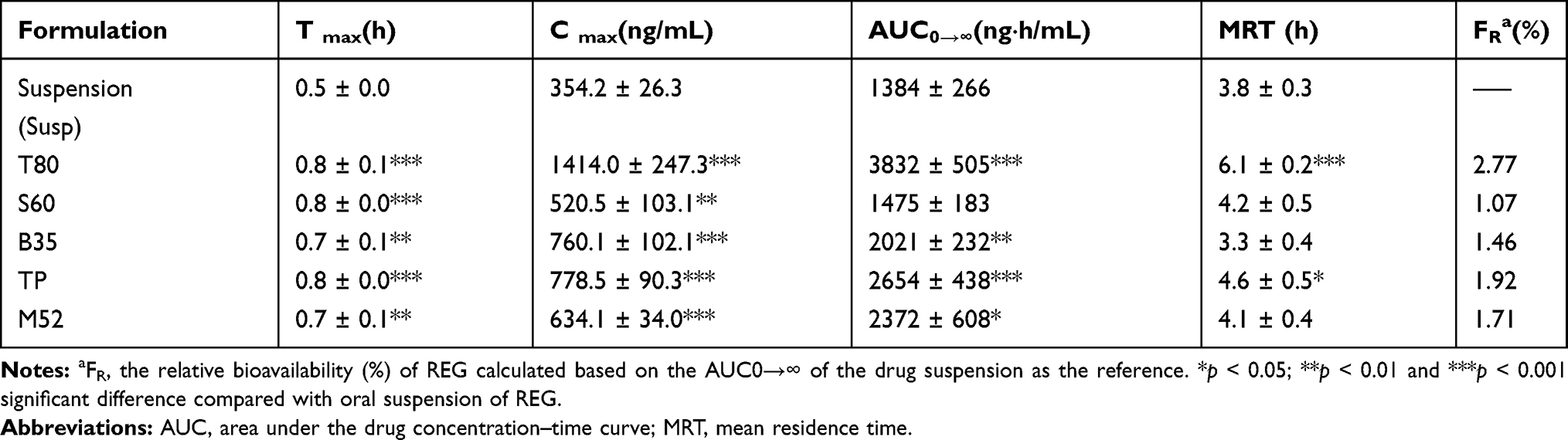 |
Table 3 Pharmacokinetic Parameters of REG After Oral Administration of Various Niosomal Formulations to Rats (n=6, Mean ± SD) |
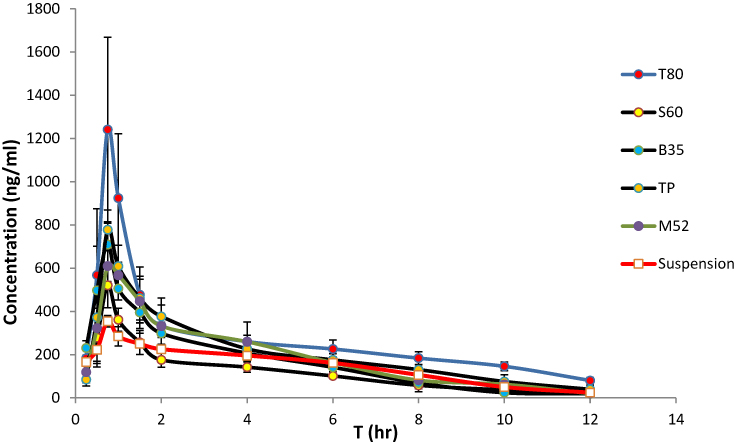 |
Figure 3 The mean plasma concentration of REG after a single oral dose (1 mg/kg) of drug suspension and the selected niosomal formulations to rats (n=6, mean ± SD). |
Significantly higher Cmax and AUC0→∞ values of REG were found in rats given drug-loaded niosomes than in rats given drug suspension, with the exception of S60 niosomes. The highest drug systemic exposure and maximum plasma concentration were achieved by T80 niosomes followed by TP vesicles. The Cmax and AUC0→∞ values of REG following oral administration of Tween 80-based niosomes (T80) were 1414 ± 247 ng/mL and 3832 ± 505 ng.h/mL, respectively which were about 4- and 2.8-fold more than the values calculated for the drug suspension (the respective values were 354.2 ± 26.3 ng/mL and 1384 ± 266 ng.h/mL). Statistical analysis through ANOVA and post hoc Bonferroni testing showed that T80 niosomes resulted in significantly higher Cmax and AUC values when compared with the other niosomes and control (p < 0.001 in all cases). Regarding TP vesicles, almost 2-fold enhancement was shown for both Cmax and AUC0→∞. However, for S60, there was no significant difference in the AUC0→∞ of REG compared to the drug suspension, indicating lack of the influence of this formulation on the extent of absorption of REG. For all preparations, the values of Tmax, which is a valid indicator of absorption rate, were significantly higher than those obtained with suspension (0.5 ± 0.0 h, p< 0.05).
Effect of Surface Positive Charge
According to our previous experiments and also the literature-based data,20 positively charged carriers may be more desirable for oral drug delivery. To take possible advantage of this issue, T80 formulation which showed the highest oral bioavailability was induced with some grades of positive charge by adding 4 mol% of STA. The positively charged niosome (T80-STA) was totally characterized in terms of in vitro tests and stability in the GI-simulated media. T80-STA showed about 73% drug entrapment, 51.2% drug release in 8 hours in SIF and was ≈160 nm in particle diameter and positively charged (ZP ≈ +18 mV). With regard to morphology as shown in Figure 4 the optimal formulation was found to be dispersed and have almost spherical particles. T80-STA vesicles were stable in the GI simulated media and there was no significant difference in size, PDI and ZP before and after incubation in the biorelevant media (Figure 2).
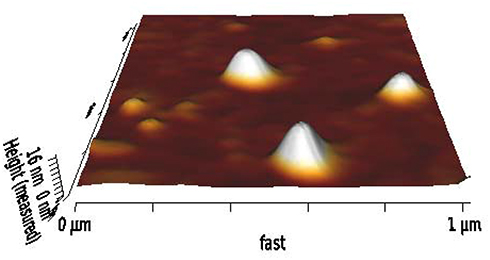 |
Figure 4 Morphology of the optimal REG-loaded niosomes (T80-STA) by AFM observation. |
REG release rate was influenced by adding STA. For instance in the SIF medium the drug release rate obviously decreased in comparison with the related negatively charged niosomes (Figure 5). In this medium, the cumulative drug released within 10 hours from T80-STA formulation was about 30% lower than that of T80 formulation (p < 0.0001). The membrane stabilizing ability of STA may be the cause of the slower drug release rate. Both Span 60 and STA are composed of stearyl chain in the molecule. Consequently, the closely packed bilayers could be achieved during the formation of vesicles30 leading to increase of membrane stability. Moreover, REG possesses one weakly acidic (pKa = 4.19) group,31 therefore could exhibit electrostatic affinity towards positively charged STA, resulting in a slower drug release rate.
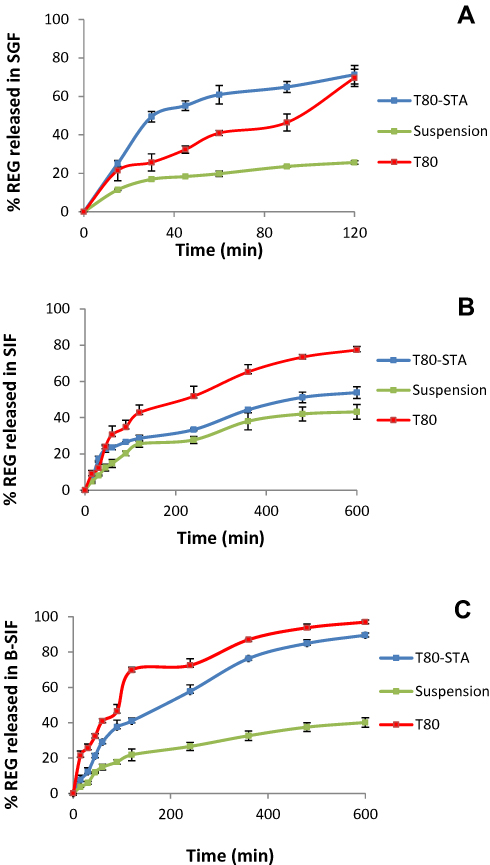 |
Figure 5 In vitro release profiles of REG from the T80-STA formulation in SGF (A), SIF (B) and B-SIF (C) at 37°C. Data represent mean ± SD (n=3). |
Subsequent to the in vitro tests, the T80-STA was orally administered to one group of Wistar rats (n=6).
The plasma concentration-time profiles of REG following oral administration of positively charged niosome and the relevant pharmacokinetic parameters are shown in Figure 6 and Table 4, respectively. When compared to drug suspension as well as plain niosome (niosomes without STA), the presence of positive charge in T80-STA resulted in significantly higher systemic availability and peak plasma concentration. For this formulation the AUC0→∞ and Cmax were respectively 3.8-fold and 4.7-fold higher than the drug suspension and statistical analysis revealed a very significant difference (p < 0.001).
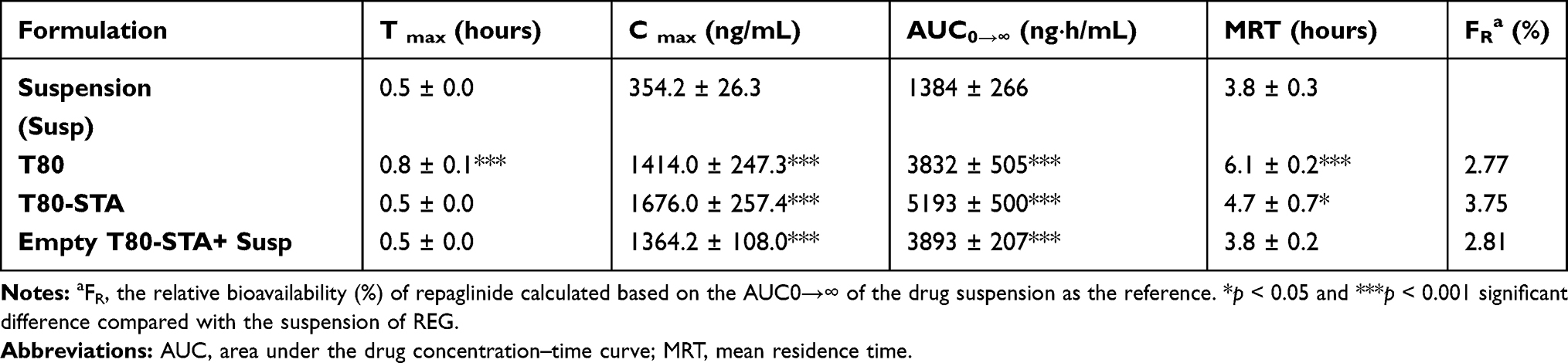 |
Table 4 Pharmacokinetic Parameters of REG After Oral Administration of Controls and T80-STA Niosomes to Rats (n=6, Mean ± SD) |
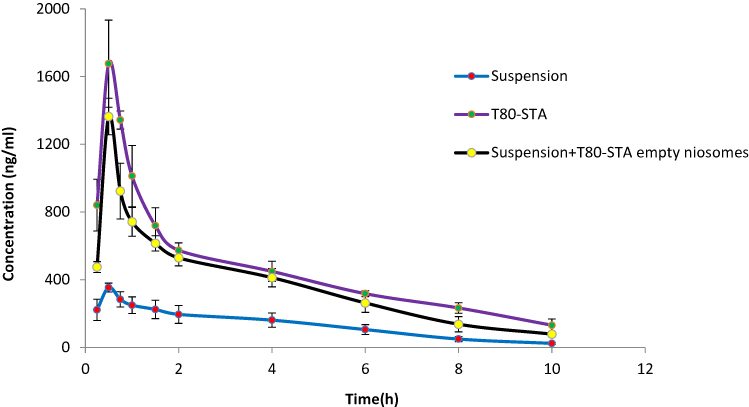 |
Figure 6 The mean plasma concentration of REG after a single oral dose of T80-STA and T80-STA empty niosomes with suspension of REG compared to suspension of REG after oral administration in rats (n=6, mean ± SD). |
Evaluation of Formulations Biocompatibility
Biocompatibility of the niosomal formulations was evaluated by in vitro toxicity (Figure 7) and tissue histological (Figure 8) analyses. In this study, the biocompatibility of Tween 80-based niosomes (non-enriched and STA-enriched) was assessed by the MTT viability test. The results indicate that neither the non-enriched formulation (T80) nor the positively charged niosomes (T80-STA) affected Caco-2 cell growth (Figure 7).
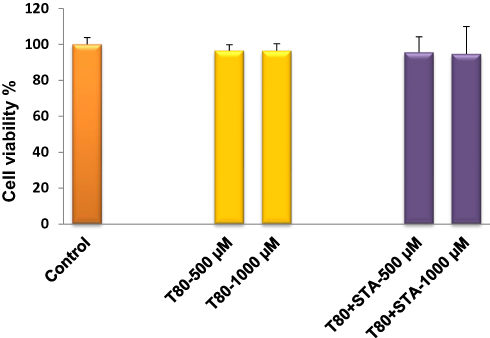 |
Figure 7 Cell viability of Caco-2 after 3 hours incubation with T80 and T80-STA niosomes (mean ± SD, n = 3). |
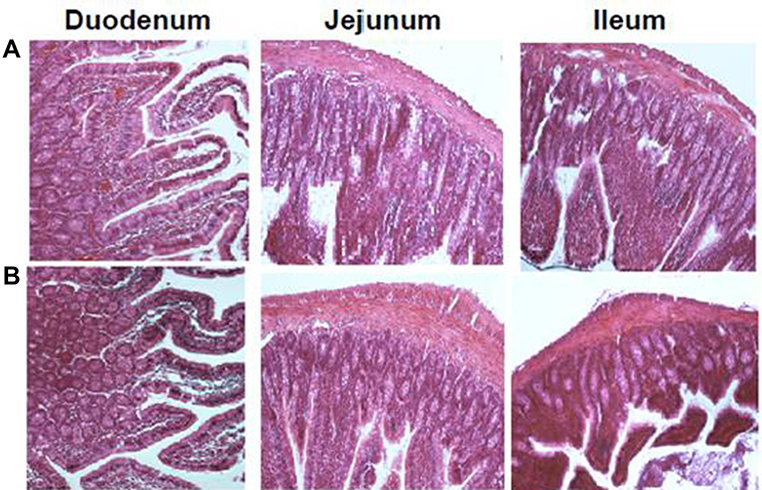 |
Figure 8 Histological sections of small intestinal segments from Wistar rats after oral administration of (A) Control and (B) T80-STA formulation. |
Tissue histological analysis of rat intestine was performed to assess possible pathological changes caused by the most in vivo effective niosomes in the intestinal epithelium in different segments (duodenum, ileum and jejunum) (Figure 8). After administration of T80-STA niosomal formulations, the pathophysiological analysis of rat intestine tissue did not show a clinically significant evidence of histological or morphological changes at the administered dose compared with the controls.
Discussion
Oral administration of BCS II drugs often suffers from low and variable bioavailability due to low water solubility, high first pass metabolism and being a substrate for efflux systems.1,32 To overcome this limitation, various drug delivery systems have been explored. Among them, niosomes, mainly consisting of nonionic surfactants, are becoming more attractive because of high chemical stability, biodegradability, biocompatibility, cost-effectiveness and easy storage and handling.10,11,33 A number of studies have shown the beneficial effect of niosomal vesicles in improving the oral bioavailability of BCS II drugs such as paclitaxel,13 glimepiride,14 carvedilol,15 telmisartan,34 and griseofulvin.35 Florence et al. 35 were the first to attempt applying niosomes in oral delivery. They found that the niosomal formulation significantly improved the bioavailability of methotrexate.36 Paclitaxel is an anticancer drug used to treat several types of cancers. Paclitaxel-loaded niosomes were prepared from Span 40 and coated with bioadhesive carbopol polymers. After oral administration of formulations to Wistar rats, higher drug plasma concentrations were observed for niosomes compared to paclitaxel suspension. The relative bioavailability of paclitaxel was increased 3.8- and 1.4-fold by uncoated and Carbopol 974P-coated niosomes emphasizing the ability of niosomes on improving the oral bioavailability of paclitaxel.13 Telmisartan-encapsulated niosomes have been investigated for improving bioavailability and extending the antihypertensive activity following oral administration.34 Telmisartan has poor bioavailability and approximately one half of an orally administered dose is absorbed. In vivo study with niosome formulation showed that the optimized formulation could reduce the systolic blood pressure in hypertensive rats and maintain it over an extended period.34 Niosomes encapsulating griseofulvin were prepared by using Span-based nonionic surfactants (span 20, span 40, and span 60). The niosomes prepared with span 60 which provided higher entrapment efficiency and indicated controlled in vitro release was tested for in vivo oral studies. The niosomal dispersion significantly improved the oral bioavailability of griseofulvin in albino rats after a single oral dose.35 Although niosomes have shown promise for enhancing oral absorption, but the impact of niosomal formulation parameters on their oral potential has not been studied systematically. In this regard, we attempted to explore the effects of surfactant composition and positive surface charge of niosomes on oral absorption of REG, as a model of poorly soluble therapeutic agents.
The niosomal formulations were prepared by the thin film hydration method and downsized by bath sonication. Chol was added to the formulations as a membrane stabilizing agent. To find an appropriate niosomal formulation with acceptable EE (higher than 50%), nearly similar average particle size (< 250 nm) and narrow distribution range, the effect of Chol content (from 0% to 50) and L/D molar ratio (from 10 to 30) were preliminarily evaluated (data not shown). The results showed that, for most of the surfactant compositions, a surfactant to Chol molar ratio of 65:35 and L/D of 30 can meet these criteria and therefore were chosen for preparation of all formulations. The in vitro properties of the formulations (hydrodynamic diameter, EE, PDI and ZP) are summarized in Table 2. Among the investigated surfactants, poloxamer 188, more likely due to its higher hydrophilicity, did not form vesicular structures, and Brij 78 resulted in formation of markedly large particles (around 892 nm), thus these two formulations were excluded from further studies. For surfactants with high HLB values, presence of adequate amounts of Chol is critical in the formation of bilayer vesicles. Poloxamers are nonionic triblock copolymers composed central lipophilic chains of propylene oxide (PO) and outer hydrophilic chains of ethylene oxide (EO).37 Poloxamer188 which possesses 27 PO chains and 80 EO chains at each end has a high HLB value of 27.837 and it seems that a higher amount of Chol is needed for niosome formation. Since in agreement with previous findings,21–24 our preliminary results indicated that niosomal size, regardless of the type of surfactant, increased with increasing Chol concentration, the surfactant to Chol molar ratio of 65:35 was kept constant for all formulations.
REG was successfully entrapped in all formulations; however, the EE value for Labrasol-based niosomes was significantly lower than that of other niosomes (≈39%), hence excluded from additional evaluation. The length of lipophilic alkyl chain of amphiphiles has been reported as one of the effective factors for drug loading.39 Labrasol is a PEG derivative of medium chain fatty acid triglyceride of capric and caprylic acid.40 The shorter lipophilic chain for Labrasol, when compared with other tested surfactants may explain the EE data. ZP is one of the indicators of the nanoparticles stability and the strong repellent forces of which could prevent aggregation of vesicles. The prepared vesicles had negative surface charge and the ZP values were sufficiently high for electrostatic stabilization of the niosomes. The ZP of vesicles revealed that the formulations can be suspended well in buffer, which is highly important for their administration.41,42 According to the size and EE data, ten out of the 13 niosomal formulations were selected for the drug release test.
As shown in Figure 1, when compared to all the prepared niosomes, Span 60 vesicles had the slowest drug release rate in SIF and B-SIF media. This result can be attributed to the lipophilic nature of this surfactant (HLB=4.7) and therefore the stronger drug-lipid chain interrelations of lipophilic REG in the lipid layers of niosomes. Overall, as the HLB of the surfactants increased (from 4.70 to 16.9) the REG release rate from niosomes, particularly in SIF, increased (from 54.4% for S60 formulation to 82.6% for M52 formulation), although it was not proportional to HLB values. Myrj 52 niosomes showed almost complete depletion of entrapped REG in B-SIF medium, which was significantly different from the behavior of Span 60 vesicles. When compared to the drug suspension (Figure 1) the rate of drug release across the dialysis membrane was faster for drug-loaded niosomes. For instance, the respective percentages of released drug in SIF during a period of 10 hours were approximately 40% and 80% for the suspension and T80 formulation. This was because the hydrodynamic diameter of niosomal formulations was much smaller than that of suspensions (mean hydrodynamic diameter ≈ 4 μm).32,43 Biphasic release was observed in all formulations, showing a primary and relatively fast release phase, which lasted for approximately 1.5 to 2 hours, followed by a slower release rate. The primary and secondary release phases might be due to differences in the diffusion distances for drug molecules in the outer and inner vesicle membranes.
To evaluate the effect of the main component of niosomes on the in vivo results, formulations containing one member of each surfactant class were chosen for in vivo studies. For this, T80, S60, B35, M52 and TP which had more appropriate physicochemical characteristics and release profiles were selected and their pharmacokinetics were investigated following oral administration to rats. According to our results (Figure 3 and Table 3), all the in vivo tested formulations caused a significant increase in oral bioavailability of REG when compared with the suspension. The only exception was S60 niosomes which resulted in similar AUC and Cmax values in comparison to the suspension. B35 caused a 47% increase in relative bioavailability (FR) of REG, but the difference was not statistically significant. As evident from the pharmacokinetics parameters, the most increase in AUC and Cmax values, as the major indicators of oral bioavailability, was related to T80 vesicles followed by TP. When compared to the suspension, T80 resulted in about 4-fold and 2.8-fold higher Cmax and AUC values, respectively. REG belongs to BCS class II drugs, and it has high permeability and poor aqueous solubility, so the rate-limiting step in the absorption of REG seems to be solubility. In our study, the size of REG niosomal formulations (162–237 nm) was considerably smaller than REG suspension (approximately 4 µm); a decrease in size and consequently increase in the surface area, along with solubility enhancement effect of surfactants might enhance dissolution rate and improve the drug absorption. As mentioned before, S60 niosomes, in spite of their small hydrodynamic diameter, did not improve the drug absorption. The obviously slower drug release rate from this formulation, as indicated in Figure 1, may describe this in vivo result. However, the enhancing effects of the niosomes could not be solely explained by the improvement of drug dissolution rate. The potency of some surfactants in inhibition of p-gp efflux system as well as cytochrome p450 enzymes and the significant difference in their potential should also be taken into account. BCS II drugs, in general, undergo significant first pass metabolism by cytochrome p450 enzymes.1 Close interplay between P-gp and CYP3A and other enterocytic drug metabolizing enzymes (eg CYP2C9, CYP2C19, CYP2C8, CYP2D6, esterases, N-acetyltransferases and glutathione-S-transferases) has been extensively reported. P-gp can work in alliance with CYP3A4 and other enzymes to increase first pass metabolism.44,45 In fact, inhibition of efflux transporter can cause a significant decrease in metabolism and subsequent increase of AUCs. Therefore, it is more likely that besides other mechanisms, some surfactants affect REG absorption via inhibition of the p-gp transporter. Lo studied the multidrug resistance (MDR) modulating effect of various excipients in Caco-2 cells and in rat intestine.46 The results showed that some excipients, such as Tween 20, Tween 80, Myrj 52 and Brij 30 not only significantly increased the absorption of epirubicin, as a known p-gp substrate, but also substantially reduced epirubicin efflux in the secretory direction. This indicates that some intestinal efflux pumps can be inhibited by these excipients. The enhancing effects of these excipients on the intestinal absorption and on absorptive transport of epirubicin in Caco-2 cells were in the order of Tween 80>Tween 20> Myrj 52> Brij 30. With regard to the modulating effect of the excipients on the basolateral to apical efflux (ie, secretory transport) of epirubicin across Caco-2 monolayers, Tween 80 was found to exhibit the most decreasing effect. Surfactants with a low HLB value, eg, Spans, had only moderate MDR modulating activity.46 The observed improvement effect of the different niosomal formulations on the oral absorption of REG (Table 3) is in agreement with the p-gp inhibitory potential of the employed surfactants. This implies that niosomes could suppress the efflux system leading to the decrease in drug first pass metabolism, and consequently increase of the drug oral absorption.
With regard to the effect of surface charge, as shown in Table 4, upon incorporation of STA in the vesicles and presence of positive charge (T80-STA) the oral absorption of REG was significantly increased compared with the suspension and the relevant plain niosomes (ie, T80). ZP of the plain T80 formulation was approximately −30 mV, however for T80-STA vesicles the ZP values increased to +18 mV. In comparison to the plain negatively charged niosomes, AUC0→∞ increased 1.36-fold for T80-STA (p<0.001). The positive charge of nanoparticles could aid mucus penetration, thus vesicles encapsulating the unreleased fraction of the drug could be internalized by either enterocytes or M-cells of the Peyer’s patches.47 The presence of positive charge could also facilitate nanoparticle adhesion onto the negatively charged epithelial cell surfaces and thereby promote cellular uptake of the particles. The uptake of positively charged nanoparticles by clathrin-mediated endocytosis has been reported,18,48,49 however, more studies are needed to understand the involved mechanisms. Furthermore, oral administration of a mixture of drug free T80-STA niosomes and REG suspension, and comparison of the results with REG suspension revealed a statistical increase in AUC0→∞ and Cmax values. However, this increase was not as large as the niosomal formulations containing REG, indicating the clear role of niosomes structure in improvement of drug absorption.
Overall, among all the evaluated surfactants, Tween 80 niosomes (T80 and T80-STA) had the highest enhancement effect on the oral bioavailability of REG and could be a promising platform to develop oral delivery systems for BCS II drugs.
Conclusion
In the present study, various nonionic surfactants having HLB in the range of 4–28 (Tweens, Spans, Brijs, Myrj, poloxamer 188, TPGS and Labrasol) were investigated for REG niosomes preparation. Drug loading efficiency, hydrodynamic diameter, ZP, PDI, in vitro release profile and stability were thoroughly characterized. Results revealed that the surfactant component of niosomes significantly affected physicochemical characteristics of vesicles. In vivo pharmacokinetics studies indicated that all the in vivo tested formulations, with the exception of Span 60-based niosomes, caused a significant increase in oral bioavailability of REG when compared with the suspension. However, the efficacy of oral niosomes was highly dependent on the surfactant composition (Tween 80> TPGS> Myrj 52> Brij 35> Span 60≈Suspension). Adding STA as a positive charge-inducing agent to Tween 80 niosomes resulted in an additional increase in drug absorption. For cationic Tween 80-based niosomal formulation, the AUC0→∞ and Cmax were 3.8- and 4.7-fold higher than the drug suspension (p < 0.001), respectively. Thus, Tween 80 niosomes with a positive surface charge could be considered as a potential carrier for oral administration of BCS II drugs.
Funding
This work was supported by a grant from Shahid Beheshti University of Medical Sciences.
Disclosure
The authors report no conflicts of interest.
References
1. Darwich AS, Neuhoff S, Jamei M, Rostami-Hodjegan A. Interplay of metabolism and transport in determining oral drug absorption and gut wall metabolism: a simulation assessment using the “Advanced Dissolution, Absorption, Metabolism (ADAM)” model. Curr Drug Metab. 2010;11(9):716–729. doi:10.2174/138920010794328913
2. Lu Y, Park K. Polymeric micelles and alternative nanonized delivery vehicles for poorly soluble drugs. Int J Pharm. 2013;453(1):198–214. doi:10.1016/j.ijpharm.2012.08.042
3. Luo Y, Chen D, Ren L, Zhao X, Qin J. Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J Control Release. 2006;114(1):53–59. doi:10.1016/j.jconrel.2006.05.010
4. Planinsek O, Kovacic B, Vrecer F. Carvedilol dissolution improvement by preparation of solid dispersions with porous silica. Int J Pharm. 2011;406(12):41–48. doi:10.1016/j.ijpharm.2010.12.035
5. McClements DJ. Nanoemulsion-based oral delivery systems for lipophilic bioactive components: nutraceuticals and pharmaceuticals. Ther Deliv. 2013;4(7):841–857.
6. Kesisoglou F, Mitra A. Crystalline nanosuspensions as potential toxicology and clinical oral formulations for BCS II/IV compounds. AAPS J. 2012;14(4):677–687.
7. Dadashzadeh S, Mirahmadi N, Babaei MH, Vali AM. Peritoneal retention of liposomes: effects of lipid composition, PEG coating and liposome charge. J Control Release. 2010;148(2):177–186.
8. Dadashzadeh S, Vali AM, Rezaie M. The effect of PEG coating on in vitro cytotoxicity and in vivo disposition of topotecan loaded liposomes in rats. Int J Pharm. 2008;353(12):251–259.
9. Haeri A, Sadeghian S, Rabbani S, Shirani S, Anvari MS, Dadashzadeh S. Physicochemical characteristics of liposomes are decisive for their antirestenosis efficacy following local delivery. Nanomedicine. 2017;12(2):131–145.
10. Thakkar M, Brijesh S. Opportunities and challenges for niosomes as drug delivery systems. Curr Drug Deliv. 2016;13(8):1275–1289. doi:10.2174/1567201813666160328113522
11. Chen S, Hanning S, Falconer J, Locke M, Wen J. Recent advances in non-ionic surfactant vesicles (niosomes): fabrication, characterization, pharmaceutical and cosmetic applications. Eur J Pharm Biopharm. 2019;144:18–39. doi:10.1016/j.ejpb.2019.08.015
12. Sohrabi S, Haeri A, Mahboubi A, Mortazavi A, Dadashzadeh S. Chitosan gel-embedded moxifloxacin niosomes: an efficient antimicrobial hybrid system for burn infection. Int J Biol Macromol. 2016;85:625–633. doi:10.1016/j.ijbiomac.2016.01.013
13. Sezgin-Bayindir Z, Onay-Besikci A, Vural N, Yuksel N. Niosomes encapsulating paclitaxel for oral bioavailability enhancement: preparation, characterization, pharmacokinetics and biodistribution. J Microencapsul. 2013;30(8):796–804.
14. Mohsen AM, AbouSamra MM, ElShebiney SA. Enhanced oral bioavailability and sustained delivery of glimepiride via niosomal encapsulation: in-vitro characterization and in-vivo evaluation. Drug Dev Ind Pharm. 2017;43(8):1254–1264. doi:10.1080/03639045.2017.1310224
15. Arzani G, Haeri A, Daeihamed M, Bakhtiari-Kaboutaraki H, Dadashzadeh S. Niosomal carriers enhance oral bioavailability of carvedilol: effects of bile salt-enriched vesicles and carrier surface charge. Int J Nanomedicine. 2015;10:4797–4813.
16. Christiansen A, Backensfeld T, Denner K, Weitschies W. Effects of non-ionic surfactants on cytochrome P450-mediated metabolism in vitro. Eur J Pharm Biopharm. 2011;78(1):166–172.
17. Rege BD, Kao JP, Polli JE. Effects of nonionic surfactants on membrane transporters in Caco-2 cell monolayers. Eur J Pharm Sci. 2002;16:237–246. doi:10.1016/S0928-0987(02)00055-6
18. Ren X, Mao X, Cao L, et al. (2009) Nonionic surfactants are strong inhibitors of cytochrome P450 3A biotransformation activity in vitro and in vivo. Eur J Pharm Sci. 2009;36(45):401–411. doi:10.1016/j.ejps.2008.11.002
19. da Silva ME, Meirelles NC. Interaction of non-ionic surfactants with hepatic CYP in Prochilodus scrofa. Toxicol in Vitro. 2004;18(6):859–867. doi:10.1016/j.tiv.2004.04.006
20. Daeihamed M, Haeri A, Ostad SN, Akhlaghi MF, Dadashzadeh S. Doxorubicin-loaded liposomes: enhancing the oral bioavailability by modulation of physicochemical characteristics. Nanomedicine. 2017;12(10):1187–1202. doi:10.2217/nnm-2017-0007
21. Tahrani, AA.; Barnet, AH.; bailey, CJ. pharmacology and therapeutic implications of current drugs for type 2 diabetes mellitus. Nat Rev Endocrinol. 2016, 12(10):566–592.
22. Repaglinide. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012.
23. Gao Y, Liao J, Qi X, Zhang J. Coamorphous repaglinide–saccharin with enhanced dissolution. Int J Pharm. 2013;450(12):290–295. doi:10.1016/j.ijpharm.2013.04.032
24. Mandic Z, Gabelica V. Ionization, lipophilicity and solubility properties of repaglinide. J Pharm Biomed Anal. 2006;41(3):866–871. doi:10.1016/j.jpba.2006.01.056
25. Hatorp V. Clinical pharmacokinetics and pharmacodynamics of repaglinide. Clin Pharmacokinet. 2002;41(7):471–483.
26. Tavano L, Muzzalupo R, Picci N, de Cindio B. Co-encapsulation of antioxidants into niosomal carriers: gastrointestinal release studies for nutraceutical applications. Colloids Surf B Biointerfaces. 2014;114(Supplement C):82–88. doi:10.1016/j.colsurfb.2013.09.058
27. https://ethics.research.ac.ir/docs/Ethics-Lab-Animal-Codes.pdf.
28. Yang DB, Zhu JB, Huang ZJ, Ren HX, Zheng ZJ. Synthesis and application of poly (ethylene glycol)-cholesterol (Chol-PEGm) conjugates in physicochemical characterization of nonionic surfactant vesicles. Colloids Surf B Biointerfaces. 2008;63(2):192–199.
29. Sezgin-Bayindir Z, Antep MN, Yuksel N. Development and characterization of mixed niosomes for oral delivery using candesartan cilexetil as a model poorly water-soluble drug. AAPS PharmSciTech. 2015;16(1):108–117. doi:10.1208/s12249-014-0213-9
30. Junyaprasert VB, Teeranachaideekul V, Supaperm T. Effect of charged and non-ionic membrane additives on physicochemical properties and stability of niosomes. AAPS PharmSciTech. 2008;9(3):851–859. doi:10.1208/s12249-008-9121-1
31. Purvis T, Mattucci ME, Crisp MT, Johnston KP, Williams RO. 3rd. Rapidly dissolving repaglinide powders produced by the ultra-rapid freezing process. AAPS PharmSciTech. 2007;8(3):E58. doi:10.1208/pt0803058
32. Charalabidis A, Sfouni M, Bergstrom C, Macheras P. The Biopharmaceutics Classification System (BCS) and the Biopharmaceutics Drug Disposition Classification System (BDDCS): beyond guidelines. Int J Pharm. 2019;566:264–281. doi:10.1016/j.ijpharm.2019.05.041
33. Azeem A, Anwer MK, Talegaonkar S. Niosomes in sustained and targeted drug delivery: some recent advances. J Drug Target. 2009;17(9):671–689.
34. Ahad A, Raish M, Al-Jenoobi FI, Al-Mohizea AM. Sorbitane monostearate and cholesterol based niosomes for oral delivery of telmisartan. Curr Drug Deliv. 2018;15(2):260–266. doi:10.2174/1567201814666170518131934
35. Jadon PS, Gajbhiye V, Jadon RS, Gajbhiye KR, Ganesh N. Enhanced oral bioavailability of griseofulvin via niosomes. AAPS PharmSciTech.
36. Azmin MN, Florence AT, Handjani-Vila RM, Stuart JF, Vanlerberghe G, Whittaker JS. The effect of non-ionic surfactant vesicle (niosome) entrapment on the absorption and distribution of methotrexate in mice. J Pharm Pharmacol. 1985;37(4):237–242. doi:10.1111/j.2042-7158.1985.tb05051.x
37. Kadam Y, Yerramilli U, Bahadur A, Bahadur P. Micelles from PEO–PPO–PEO block copolymers as nanocontainers for solubilization of a poorly water soluble drug hydrochlorothiazide. Colloids Surf B Biointerfaces. 2011;83(1):49–57. doi:10.1016/j.colsurfb.2010.10.041
38. Goliaei A, Lau EY, Adhikari U, Schwegler E, Berkowitz ML. Behavior of P85 and P188 poloxamer molecules: computer simulations using united-atom force-field. J Phys Chem B. 2016;120(33):8631–8641. doi:10.1021/acs.jpcb.6b03030
39. Mokhtar M, Sammour OA, Hammad MA, Megrab NA. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int J Pharm. 2008;361(12):104–111. doi:10.1016/j.ijpharm.2008.05.031
40. McCartney F, Jannin V, Chevrier S, et al. Labrasol® is an efficacious intestinal permeation enhancer across rat intestine: ex vivo and in vivo rat studies. J Control Release. 2019;310:115–126. doi:10.1016/j.jconrel.2019.08.008
41. Patrick JS. Martin’s Physical Pharmacy and Pharmaceutical Sciences: Physical Chemical and Biopharmaceutical Principles in the Pharmaceutical Sciences.
42. Kedar U, Phutane P, Shidhaye S, Kadam V. Advances in polymeric micelles for drug delivery and tumor targeting. Nanomedicine. 2010;6(6):714–729. doi:10.1016/j.nano.2010.05.005
43. Gaumet M, Vargas A, Gurny R, Delie F. Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur J Pharm Biopharm. 2008;69(1):1–9. doi:10.1016/j.ejpb.2007.08.001
44. Benet LZ. The drug transporter−metabolism alliance: uncovering and defining the interplay. Mol Pharm. 2009;6(6):1631–1643. doi:10.1021/mp900253n
45. Mudra DR, Desino KE, Desai PV. In silico, in vitro and in situ models to assess interplay between CYP3A and P-gp. Curr Drug Metab. 2011;12(8):750–773. doi:10.2174/138920011798356999
46. Lo Y-L. Relationships between the hydrophilic–lipophilic balance values of pharmaceutical excipients and their multidrug resistance modulating effect in Caco-2 cells and rat intestines. J Control Release. 2003;90(1):37–48. doi:10.1016/S0168-3659(03)00163-9
47. Roger E, Lagarce F, Garcion E, Benoit JP. Biopharmaceutical parameters to consider in order to alter the fate of nanocarriers after oral delivery. Nanomedicine. 2010;5(2):287–306.
48. Harush-Frenkel O, Rozentur E, Simon B, Altschuler Y. Surface charge of nanoparticles determines their endocytic and transcytotic pathway in polarized MDCK cells. Biomacromolecules. 2008;9(2):435–443.
49. Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61(2):158–171.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.