")
Back to Journals » OncoTargets and Therapy » Volume 13
The Impact of Smoking on Pulmonary Metastasis in Colorectal Cancer
Authors Makino A, Tsuruta M, Okabayashi K, Ishida T, Shigeta K , Seishima R, Ikebata A, Koishikawa K, Hasegawa H, Shimoda M, Fukunaga K , Betsuyaku T, Kitagawa Y
Received 9 June 2020
Accepted for publication 1 September 2020
Published 30 September 2020 Volume 2020:13 Pages 9623—9629
DOI https://doi.org/10.2147/OTT.S263250
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Akitsugu Makino,1 Masashi Tsuruta,1 Koji Okabayashi,1 Takashi Ishida,1 Kohei Shigeta,1 Ryo Seishima,1 Akiyoshi Ikebata,1 Kaoru Koishikawa,1 Hirotoshi Hasegawa,2 Masayuki Shimoda,3 Koichi Fukunaga,4 Tomoko Betsuyaku4 ,* Yuko Kitagawa1
1Department of Surgery, Keio University School of Medicine, Tokyo, Japan; 2Department of Surgery, Tokyo Dental College Ichikawa General Hospital, Chiba, Japan; 3Department of Pathology, Keio University School of Medicine, Tokyo, Japan; 4Division of Pulmonary Medicine, Department of Medicine, Keio University School of Medicine, Tokyo, Japan
†Professor Tomoko Betsuyaku passed away on September 1, 2018.
Correspondence: Masashi Tsuruta Department of Surgery
Keio University School of Medicine, 35 Shinanomachi Shinjyuku-Ku, Tokyo 160-8582, Japan
Tel +81-3-3353-1211
Fax +81-3-3355-4707
Email [email protected]
Introduction: Recently, clinical studies have revealed that smoking can contribute to the poor prognosis of colorectal cancer (CRC) and, additionally, can be a risk factor for pulmonary metastasis of CRC. However, there has been no basic research regarding the underlying molecular mechanism. The purpose of this study was to clarify the mechanism by which smoking causes pulmonary metastasis of CRC.
Methods: First, pulmonary metastasis model mice inhaled cigarette smoke or air (control) for 1 h once a day for 3 weeks. We attempted to clarify the effect of smoking on the incidence of pulmonary metastasis. On the 15th day, CMT-93 cells were injected into the tail vein. At 6 and 8 weeks following injection, the extent of pulmonary metastasis was evaluated using in vivo micro CT. After the last CT examination, the mice were sacrificed, and the lungs were extracted for pathological examination.
Results: The number of mice with pulmonary metastases in the smoking group was significantly higher than in the control group. Three weeks of smoking induced mild inflammation in the lungs, as evidenced by increases in the levels of IL-6 and TNF-α in bronchoalveolar lavage. Moreover, the adhesion-related molecule ICAM-1 was overexpressed in pulmonary tissue, which allowed drained cancer cells to remain in the lung and contribute to the formation of pulmonary metastasis.
Conclusion: Collectively, cigarette smoking may contribute to the pathogenesis and development of pulmonary metastasis in CRC through enhancement of adhesion and inflammation.
Keywords: smoking, colorectal cancer, pulmonary metastasis, adhesion molecules, ICAM-1
Introduction
Colorectal cancer (CRC) is one of the leading causes of cancer-related death and led to 50,681 deaths in 2017 in Japan. Almost every third patient diagnosed with CRC presents with, or develops, metastatic disease [Vital Statistics Japan (Ministry of Health, Labour and Welfare)]. Therefore, it is necessary to clarify the mechanism of metastasis, and in particular pulmonary metastasis, which is especially difficult to diagnose and often difficult to treat.
In general, it is thought that toxic chemicals and stimulants contained in tobacco induce inflammation, change the microcirculation, cause tissue necrosis, and promote alveolar damage.1,2 Smoking is also a risk factor for malignancy of not only the lung but also other organs, such as the pharynx, larynx, esophagus, pancreas, and bladder.3–5 Recently, a meta-analysis clarified that smoking is a risk factor that increas es the incidence and mortality of CRC, and smoking status at the time of CRC diagnosis is correlated with prognosis.6,7 In addition, other reports have shown that smoking may cause pulmonary metastasis of breast cancer.8
Previously, our retrospective study showed that current smokers, compared with former smokers and those who never smoked, might have a significantly increased risk of metachronous pulmonary metastasis following curative resection of CRC.9 Although CRC is a high morbidity cancer that can have a high survival rate when diagnosed and treated early, it is known that prognosis worsens when it metastasizes to other organs. Thus, determining the relationship between smoking and pulmonary metastasis of CRC may lead to not only proof of smoking toxicity and add to the risks of passive smoking, but may also lead to the development of new drug therapies for pulmonary metastasis. Therefore, the aim of this study was to clarify the impact of smoking on pulmonary metastasis in CRC.
Materials and Methods
Cell Culture
The mouse colorectal adenocarcinoma cell line CMT-93 (CCL-223, ATCC, Manassas, VA, USA) was maintained in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, St. Louis, MO, USA) with 10% (v/v) fetal bovine serum and 10,000 units/mL penicillin, 10,000 µg/mL streptomycin, and 25 µg/mL Gibco amphotericin B (Thermo Fisher Scientific, Tokyo, Japan). The cells were cultured at 37°C with 5% CO2.
Animal
We used C57BL/6 male mice, which were purchased from CLEA Japan. The mice were allowed to acclimate for at least 7 days before use and were 6 weeks old at the start of the experimental protocol. The mice were housed in a controlled environment in the laboratory at the Keio University School of Medicine under standard temperature and light and dark cycles.
Ethics approval for the present study was provided by the Ethics Committee at the Laboratory Animal Care and Use Committee at Keio University School of Medicine (approval number: 15006), and the study was performed in accordance with the for the Care and Use of Laboratory Animals (NIH).
Design of Animal Experiment
The schedules for smoke inhalation and tumor cell injection are shown in Figure 1. The smoking device had a limited number of chambers, and it was necessary to carry out the experiment with the minimum number of mice for animal protection, so the experiment was planned with 21 mice each in the control group and smoking group. The smoking group mice were exposed to smoke for 60 min/day on weekdays for 3 weeks. Age-matched control group mice were exposed to air over the same time period. On the 15th day of smoking exposure, the mice were anesthetized with 2.0% isoflurane and 3.5 × 106 CMT-93 cells in a total volume of 100 μL phosphate-buffered saline (PBS) were injected into the tail vein using a 26-gauge needle. Six and eight weeks following injection the extent of pulmonary metastasis was evaluated using in vivo micro computed tomography (CT). We compared the number of mice with pulmonary metastasis, and the number of pulmonary metastases in each mouse between the control and smoking mice. After the second micro CT scan 8 weeks after injection, all of the mice were sacrificed under systemic anesthesia with 2.0% isoflurane by exsanguination, which involved breaking of the inferior vena cava. The lung was extracted, and the right lobe was fixed by filling with 500 μL of 4% paraformaldehyde through the right bronchus and used for pathological examination (Figure 2A). The bronchoalveolar lavage (BAL) was collected from the left lobe by washing three times with 500 μL saline through the left bronchus. Then, the remnant left robe was homogenized and stored at −80°C for subsequent DNA extraction and analysis.
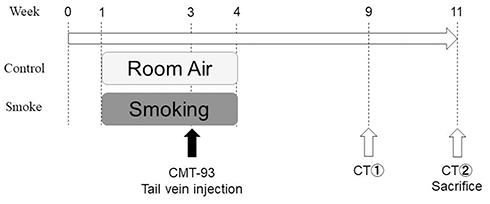 |
Figure 1 Animal experimental protocol, The smoking group mice were exposed to smoke for 60 min/day on weekdays for 3 weeks. The control group mice were exposed to air over the same time period. On the 15th day of smoking exposure, 3.5 × 106 CMT-93 cells were injected into the tail vein. Six and eight weeks following injection, the extent of pulmonary metastasis was evaluated using in vivo micro CT. All mice were sacrificed immediately after the last CT examination and the lungs were extracted. |
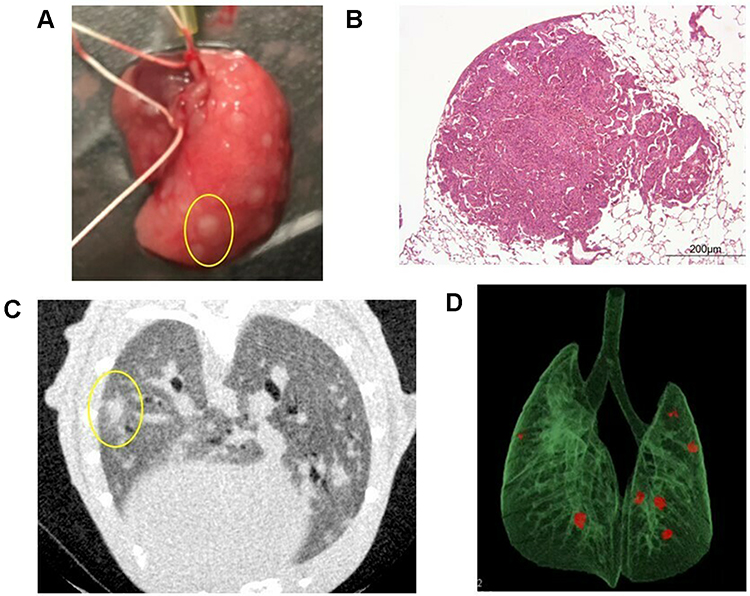 |
Figure 2 Pulmonary metastases. (A) Macroscopic findings of pulmonary metastasis in the right lobe of the lung. In this mouse, many nodules were identified (one is circled in yellow). (B) Hematoxylin-eosin staining of pulmonary metastasis in a mouse. (C) A representative two-dimensional image of a micro CT scan in a smoking mouse. In this mouse, one nodule was identified in the left lung (circled in yellow). (D) CT reconstruction of a pulmonary metastasis from a mouse model. Three-dimensional microstructural image data were reconstructed using Tri/3D-BON software. Red colored nodules indicate pulmonary metastasis. |
Smoke Exposure
Mice inhaled mainstream cigarette smoke (CS) generated from commercially available filtered cigarettes (Marlboro, 12 mg tar/1.0 mg nicotine) through their nose. The SIS-CS system (Shibata Scientific Technology, Tokyo, Japan), which consists of both a CS generator (SG-300) and an inhalation chamber, to which 10 body holders were set at a time, was used. Fresh cigarettes purchased within 1 month of use were used throughout the experiments. The following experimental settings were used to generate CS: stroke volume of 15 mL and 10 puffs/min.
Hematoxylin-Eosin (HE) Staining and Immunostaining
Lung tissues from the mice were embedded in paraffin. The paraffin blocks were cut into 3-μm thick sagittal sections and subjected to hematoxylin-eosin (HE) and immunohistochemical (IHC) staining (Figure 2B). Pathological evaluation using HE staining was conducted to count the number of pulmonary metastases in the largest longitudinal section in the right lobe. In IHC staining, the expression of ICAM-1 was evaluated. Each section was deparaffinized in xylene and soaked for 30 min at room temperature in 0.3% H2O2/methanol to block endogenous peroxidase. The primary antibody anti-ICAM1 (ab171123, Abcam, Cambridgeshire, UK) was diluted 1:100 and applied overnight at 4°C. The primary antibody was visualized using biotinylated goat anti-rabbit immunoglobulin G and the EnVision+ System- HRP Labeled Polymer (Dako North America Inc., Carpinteria, CA, USA), which was applied for 3 min at room temperature. The slide was counterstained with hematoxylin.
ICAM-1 expression was quantified by measuring the area of expression of ICAM-1 as a percentage (0–100%) using an imaging analysis program (ImageJ Version 1.8; NIH, Bethesda, MD, USA). Briefly, five random field images (0.02 × 0.01 inches), from the slide of the maximum longitudinal section from the right lobe, were imaged using a light microscope and colors that were not of interest were removed via replace mode. The adapted images were converted to grey scale and the area of expression was located by adjusting the threshold. The percentage area of expression was denoted by positive pixels on the labeled areas.5,10
The all samples were confirmed by histopathological assessment by an experienced pathologist (M.S).
Enzyme-Linked Immunosorbent Assay (ELISA) for IL-6 and TNF-α
The protein levels of IL-6 and TNF-α in the BAL were measured to evaluate inflammation of the lung. The concentrations of IL-6 and TNF-α in the BAL were determined using a mouse IL-6 Quantikine ELISA kit (R&D systems, Minneapolis, MN, USA) and mouse TNF-α ELISA Development Kit (PromoCell, Heidelberg, Germany).
Micro CT Imaging
Micro CT was used for evaluation of pulmonary metastasis before sacrifice (Figure 2C and D). The X-ray micro-CT system (R_mCT2; Rigaku, Tokyo, Japan) was operated with the following parameters: 90 kV, 160 μA, chest CT; respiratory and cardiac reconstruction mode, 24 × 24 mm field of view (50 × 50 μm pixel size). The scan time of 4.5 min yielded an average exposure of 1653 mGy/scan per whole body. The mice were scanned in the prone position and the inhalation anesthesia, which was administered through a nose cone, consisted of a mix of isoflurane (Pfizer Japan, Tokyo, Japan) and oxygen. Respiratory and cardiac reconstruction mode captured the X-ray view to reconstruct lung images only at the diastolic phase of the heart during the end-expiratory period by simultaneously monitoring the movement of both breathing and the heart under radiographic guidance. The examination was performed twice, at the 6th and 8th weeks, and pulmonary metastases were confirmed by comparing two metachronous images.
Statistical Analysis
All results were expressed as mean value (± standard deviation) without any notation. All statistical analyses were performed using Stata software (Stata Corp., College Station, TX, USA). P values < 0.05 were considered to indicate statistical significance. In terms of pulmonary metastasis of mice, comparisons between two groups were performed with χ2 tests. The others were statistically analyzed using the Mann–Whitney U-test without any notation.
Results
In the aforementioned protocol, 21 mice were used in each group and micro CT and pathological examination were used to evaluate pulmonary metastasis (Table 1). Micro CT imaging revealed that the incidence of pulmonary metastasis was significantly higher in the smoking group (11 vs 3, P = 0.019), and the number of pulmonary metastases per mouse was also higher in the smoking group compared with the control group (3.9 ± 1.4 vs 0.8 ± 0.5, P = 0.019). The maximum tumor diameter of metastatic lesions measured by micro CT was significantly higher in the smoking group (1.39 ± 0.22 vs 0.41 ± 0.2 mm, P = 0.036). In addition, pathological evaluation for pulmonary metastasis was performed using HE staining of the largest longitudinal section in the right lobe. Similarly, the number of mice with pulmonary metastasis and the number of metastases per mouse was higher in the smoking group [12 mice (57%) vs 6 mice (29%), P = 0.061, 3.48 ± 1.5 vs 0.9 ± 0.4, P = 0.063].
 |
Table 1 Comparison of Pulmonary Metastasis |
To evaluate the direct impact of smoking on the lungs, the inflammatory cytokines (ie, IL-6 and TNF-α) in BAL were investigated in mice models that did not undergo tail vein injection with tumor cells. As shown in Figure 3, both were significantly higher in the smoking group than the control group (31.2 ± 6.2 vs 22.1 ± 3.8 pg/mL, P = 0.014, and 145.5 ± 46.7 vs 109.5 ± 17.5, P = 0.047, respectively).
 |
Figure 3 IL-6 and TNF-α in the bronchoalveolar lavage (BAL). The protein levels of IL-6 and TNF-α in the BAL were measured. The BAL was collected from the left lobe by washing three times with 500 μL saline through the left bronchus. The concentrations of IL-6 and TNF-α in the BAL were determined using a mouse IL-6 Quantikine ELISA kit and a mouse TNF-α ELISA Development Kit. (A) IL-6 expression in the BAL was significantly higher in the smoking group compared with the control group (31.2 ± 6.2 vs 22.1 ± 3.8, P = 0.014). (B) TNF-α expression in BAL was significantly higher in the smoking group compared with the control group (145.5 ± 46.7 vs 109.5 ± 17.5, P = 0.047). |
Inflammation is known to induce several adhesion molecules, which can trap circulating tumor cells, leading to metastasis. Thus, the expression of four adhesion molecules (ICAM-1, VCAM-1, E-selectin, and P-selectin) in the lung was measured by RT-PCR in mice models that did not undergo tail vein injection with tumor cells. The results are shown in Supplementary Figure 1. Moreover, IHC staining suggested that ICAM-1 was significantly overexpressed in the smoking group (7.75 ± 3.27 vs 5.48 ± 2.1, P = 0.049, Figure 4). Interestingly, there was some pulmonary metastasis surrounding the area with enhanced ICAM-1 expression (Figure 5).
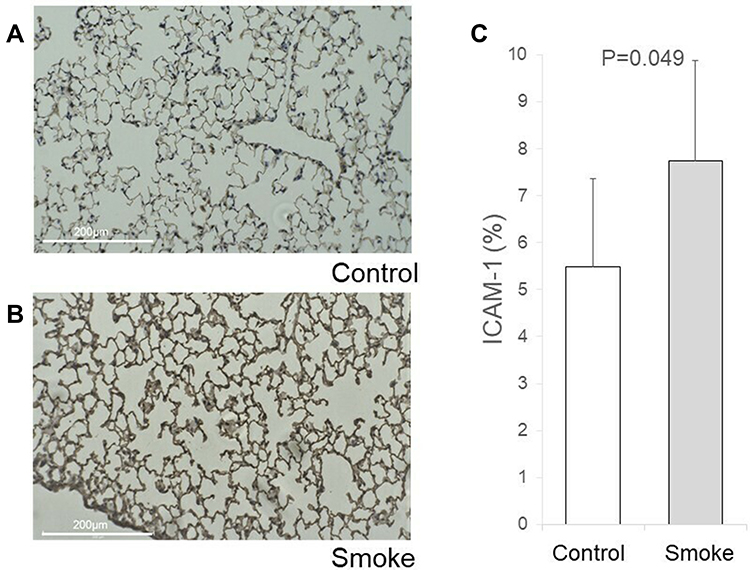 |
Figure 4 Immunohistochemical (IHC) staining of ICAM-1. Lung tissues from the largest longitudinal section in the right lobe of control and smoking mice were stained for ICAM-1. Anti-ICAM1 primary antibody diluted 1:100 was used for IHC staining. ICAM-1 expression was quantified by measuring the percentage area of expression (0–100%) using ImageJ. Briefly, five randomly chosen field images (0.02 × 0.01 inches) from the slide containing the maximum longitudinal section of the right lobe were acquired using a light microscope, and colors that were not of interest were removed via the replace mode. The adapted images were converted to grey scale and the area of expression was located by adjusting the threshold. The percentage area of expression is denoted by positive pixels on the labeled areas. (A) IHC staining of ICAM-1 in control mouse lung. (B) IHC staining of ICAM-1 in smoking mouse lung. (C) The ICAM-1 antibody tended to strongly stain the lungs from the smoking group (7.75 ± 3.27 vs 5.48 ± 2.1, P = 0.049). |
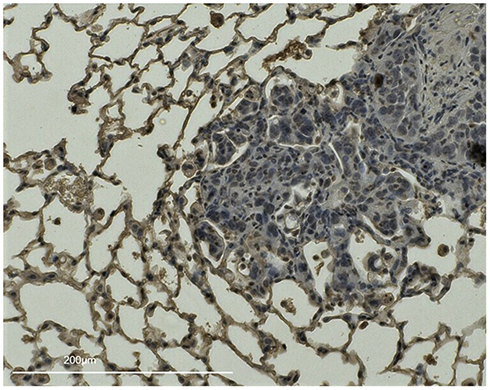 |
Figure 5 IHC staining of ICAM-1 in a smoking mouse lung with pulmonary metastasis, Lung tissues from the largest longitudinal section in the right lobe of a smoking group mouse injected with CMT-93 cells on the 15th day of smoking exposure were stained for ICAM-1. There are some pulmonary metastases surrounded by areas enhanced with ICAM-1. |
Discussion
In this study, there was a significant increase in the number of pulmonary metastases of CRC, as shown radiologically and pathologically, in the smoking group compared to the control group; this is consistent with the results of a previous clinical study.9 The culprit was the trapping of cancer cells by ICAM-1, an adhesion molecule overexpressed due to inflammation caused by smoking. Collectively, smoking is a risk factor for pulmonary metastasis in CRC. To the best of our knowledge, this is the first basic research study to clarify the impact of smoking on pulmonary metastasis in CRC.
In this study, it was determined that IL-6 and TNF-α expression in BAL from mice increased due to smoking. IL-6 and TNF-α are representative inflammatory cytokines; they are the most sensitive mediators of biological reactions and are secreted rapidly in response to inflammation.11,12 Previous studies have discussed the relationship between inflammation and cancer metastasis.10,13 In addition, many reports have shown that smoking-induced inflammation in lung tissue increased permeability between blood vessels and the alveolar epithelium, changed local immune function, and enhanced secretions.1,14 This study also confirmed that smoking induced inflammation in the lungs by elevation of inflammatory cytokines, such as IL-6 and TNF-α, in BAL. Moreover, a comprehensive analysis of various adhesion molecules showed that ICAM-1 was overexpressed. Inflammation is known to induce the expression of cell adhesion molecules such as cadherin, selectin, VCAM-1, and ICAM-1.15–17 In the lung, several researchers have reported that smoking-induced ICAM-1 was expressed on the cell surface. ICAM-1 has also been reported to correlate with inflammatory cytokines such as TNF-α and IL-6, and with pulmonary inflammation and viral infections.18–20 Previous reports have shown that there is an association between adhesion molecules and cancer metastasis15,21. Similarly, in this study, we believe that the expression of ICAM-1 in the lungs promoted the development of pulmonary metastasis of CRC.
Until now, reports have shown that smoking may cause cancer metastasis, but there have been no specific reports on the mechanism. We can assume from these results that smoking-induced inflammation in the lungs enhanced cell adhesion, trapping inflowing cancer cells to engraft and proliferate.
In the present study, a short-duration smoking mouse model was used to clarify the impact of smoking on pulmonary metastasis of CRC. Primary lung cancer is known to occur around emphysematous lesions, which suggests that a “field” that retains avascular and alveolar lesions is necessary as a cancer origin.22 On the other hand, clinical studies have suggested that active smoking, rather than prior smoking likely causes pulmonary metastasis. The reason is that the number of cancer cells drained into the lung is critical and the avascular area due to emphysema does not favor pulmonary metastasis. Thus, we opted to use a short-duration smoking mouse model to ensure that smoking induced acute and active inflammation without any structural changes, which occurs in diseases such as emphysema.23
In the past, some studies showed the etiologic field effect models which is the exposures and genetic factor promote tissue microenvironment of cancer and suggest that exposure may be associated with cancer progression.24,25 Smoking is considered one of the exposures and it influences multiple phases of tumor evolution. Therefore, past report supported that smoking may promote microenvironment of lungs, and causes pulmonary metastases of CRC.
A limitation of this study is that the 3-week smoking duration for the mouse model may not be a reliable reproduction of actual human smoking habits. In addition, this study only used one mouse strain and one colorectal cancer cell line. In the future, other strains, larger animal, other systems, and human tissues experiment is required for validation. Furthermore, in this study we focused on adhesion molecules and the relationship between inflammation and metastasis, but other factors related to migration and the micro-immune environment may also be involved; therefore, further experiments are needed.
Notwithstanding, this is the first study to clarify the impact of smoking on pulmonary metastasis of CRC, and is an important first step to understanding the mechanism of pathogenesis of pulmonary metastasis.
Conclusion
Smoking-induced inflammation of the lung and enhanced expression of cell adhesion molecules might contribute to the pathogenesis of pulmonary metastasis of CRC. Therefore, smoking cessation may improve the prognosis of CRC by preventing pulmonary metastasis; however, further research is needed.
Disclosure
Prof. Dr. Yuko Kitagawa reports grants and/or personal fees from ASAHI KASEI PHARMA CO., LTD., TAIHO PHARMACEUTICAL CO., LTD, CHUGAI PHARMACEUTICAL CO., LTD., DAIICHI SANKYO COMPANY, LIMITED, Merck Serono Co., Ltd., EA Pharma Co., Ltd., Yakult Honsha Co. Ltd., Otsuka Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., Otsuka Pharmaceutical Factory Inc., SHIONOGI & CO., LTD., KAKEN PHARMACEUTICAL CO., LTD., Kowa Pharmaceutical Co., Ltd., Astellas Pharma Inc., MEDICON INC., DAINIPPON SUMITOMO PHARMA Co., Ltd., Taisho Toyama Pharmaceutical Co., Ltd., Kyowa Hakko Kirin Co., Ltd., Pfizer Japan Inc., ONO PHARMACEUTICAL CO., LTD., NIHON PHARMACEUTICAL CO., LTD., Japan Blood Products Organization, Medtronic Japan Co., Ltd., Sanofi K.K., Eisai Co., Ltd., TSUMURA & CO., KCI Licensing, Inc., ABBOTT JAPAN CO., LTD., FUJIFILM Toyama Chemical Co., Ltd., outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Sobus SL, Warren GW. The biologic effects of cigarette smoke on cancer cells. Cancer. 2014;120(23):3617–3626. doi:10.1002/cncr.28904
2. Hackshaw AK, Law MR, Wald NJ. The accumulated evidence on lung cancer and environmental tobacco smoke. BMJ. 1997;315(7114):980–988. doi:10.1136/bmj.315.7114.980
3. Malhotra J, Borron C, Freedman ND, et al. Association between Cigar or Pipe Smoking and Cancer Risk in Men: A Pooled Analysis of Five Cohort Studies. Cancer Prevent Res. 2017;10(12):704–709.
4. Kuang JJ, Jiang ZM, Chen YX, et al. Smoking Exposure and Survival of Patients with Esophagus Cancer: A Systematic Review and Meta-Analysis. Gastroenterol Res Pract. 2016;2016:7682387.
5. Inoue-Choi M, Hartge P, Liao LM, Caporaso N, Freedman ND. Association between long-term low-intensity cigarette smoking and incidence of smoking-related cancer in the national institutes of health-AARP cohort. Int j Cancer. 2018;142(2):271–280.
6. Walter V, Jansen L, Hoffmeister M, Brenner H. Smoking and survival of colorectal cancer patients: systematic review and meta-analysis. Ann Oncol. 2014;25(8):1517–1525.
7. Walter V, Jansen L, Hoffmeister M, Ulrich A, Chang-Claude J, Brenner H. Smoking and survival of colorectal cancer patients: population-based study from Germany. Int j Cancer. 2015;137(6):1433–1445.
8. Murin S, Pinkerton KE, Hubbard NE, Erickson K. The effect of cigarette smoke exposure on pulmonary metastatic disease in a murine model of metastatic breast cancer. Chest. 2004;125(4):1467–1471.
9. Yahagi M, Tsuruta M, Hasegawa H, et al. Smoking is a risk factor for pulmonary metastasis in colorectal cancer. Colorectal Dis. 2017;19(9):O322–o328.
10. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. doi:10.1038/nature01322
11. Tanaka T, Kishimoto T. The biology and medical implications of interleukin-6. Cancer Immunol Res. 2014;2(4):288–294. doi:10.1158/2326-6066.CIR-14-0022
12. Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 2006;25(3):409–416. doi:10.1007/s10555-006-9005-3
13. Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. EMT and inflammation: inseparable actors of cancer progression. Mol Oncol. 2017;11(7):805–823. doi:10.1002/1878-0261.12095
14. Vu T, Jin L, Datta PK. Effect of Cigarette Smoking on Epithelial to Mesenchymal Transition (EMT) in Lung Cancer. J Clin Med. 2016;5(4):4. doi:10.3390/jcm5040044
15. Reymond N, d’Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13(12):858–870. doi:10.1038/nrc3628
16. Jiang M, Xu X, Bi Y, Xu J, Qin C, Han M. Systemic inflammation promotes lung metastasis via E-selectin upregulation in mouse breast cancer model. Cancer Biol Ther. 2014;15(6):789–796. doi:10.4161/cbt.28552
17. Scott DA, Palmer RM. The influence of tobacco smoking on adhesion molecule profiles. Tob Induc Dis. 2002;1(1):7–25. doi:10.1186/1617-9625-1-1-7
18. Blann AD, Kirkpatrick U, Devine C, Naser S, McCollum CN. The influence of acute smoking on leucocytes, platelets and the endothelium. Atherosclerosis. 1998;141(1):133–139. doi:10.1016/S0021-9150(98)00163-4
19. Yu N, Sun YT, Su XM, He M, Dai B, Kang J. Treatment with eucalyptol mitigates cigarette smoke-induced lung injury through suppressing ICAM-1 gene expression. Biosci Rep. 2018;38(4):4. doi:10.1042/BSR20171636
20. Shukla SD, Mahmood MQ, Weston S, et al. The main rhinovirus respiratory tract adhesion site (ICAM-1) is upregulated in smokers and patients with chronic airflow limitation (CAL). Respir Res. 2017;18(1):6. doi:10.1186/s12931-016-0483-8
21. Saiki I. Cell Adhesion Molecules and Cancer Metastasis. Jpn J Pharmacol. 1997;75(3):215–242. doi:10.1254/jjp.75.215
22. Chubachi S, Takahashi S, Tsutsumi A, et al. Radiologic features of precancerous areas of the lungs in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2017;12:1613–1624. doi:10.2147/COPD.S132709
23. Sasaki M, Chubachi S, Kameyama N, et al. Evaluation of cigarette smoke-induced emphysema in mice using quantitative micro-computed tomography. Am J Physiol Lung Cell Mol Physiol. 2015;308(10):L1039–L1045. doi:10.1152/ajplung.00366.2014
24. Lochhead P, Chan AT, Nishihara R, et al. Etiologic field effect: reappraisal of the field effect concept in cancer predisposition and progression. Modern Pathol. 2015;28(1):14–29.
25. Gladstein S, Damania D, Almassalha LM, et al. Correlating colorectal cancer risk with field carcinogenesis progression using partial wave spectroscopic microscopy. Cancer Med. 2018;7(5):2109–2120.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.