")
Back to Journals » International Journal of Women's Health » Volume 7
The genetics of premature ovarian failure: current perspectives
Authors Chapman C, Cree L, Shelling A
Received 7 July 2015
Accepted for publication 3 September 2015
Published 23 September 2015 Volume 2015:7 Pages 799—810
DOI https://doi.org/10.2147/IJWH.S64024
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Elie Al-Chaer
Chevy Chapman, Lynsey Cree, Andrew N Shelling
Department of Obstetrics and Gynecology, Faculty of Medical and Health Sciences, University of Auckland, Auckland, New Zealand
Abstract: Premature ovarian failure (POF) is a common cause of infertility in women, characterized by amenorrhea, hypoestrogenism, and elevated gonadotropin levels in women under the age of 40. Many genes have been identified over the past few years that contribute to the development of POF. However, few genes have been identified that can explain a substantial proportion of cases of POF. The unbiased approaches of genome-wide association studies and next-generation sequencing technologies have identified several novel genes implicated in POF. As only a small proportion of genes influencing idiopathic POF have been identified thus far, it remains to be determined how many genes and molecular pathways may influence idiopathic POF development. However, owing to POF’s diverse etiology and genetic heterogeneity, we expect to see the contribution of several new and novel molecular pathways that will greatly enhance our understanding of the regulation of ovarian function. Future genetic studies in large cohorts of well-defined, unrelated, idiopathic POF patients will provide a great opportunity to identify the missing heritability of idiopathic POF. The identification of several causative genes may allow for early detection and would provide better opportunity for early intervention, and furthermore, the identification of specific gene defects will help direct potential targets for future treatment.
Keywords: premature ovarian failure, POF, next-generation sequencing, genome-wide association studies, genetics, ovarian aging
Introduction
The end of a woman’s reproductive lifespan is marked by the occurrence of menopause, defined as being the last menstruation that occurs for a woman, but is caused by the exhaustion of the ovarian reserve.1 In the general female population, across many ethnicities and over recent human history, the average age of natural menopause has remained at 50–52 years. However, aberrations in ovulatory processes, as outlined in Figure 1, may cause a pathogenic early depletion of ovarian follicles, which may result in premature menopause. Menopause before the age of 40 years is defined as premature ovarian failure (POF), also known as primary ovarian insufficiency.2 POF is described as the premature cessation of ovarian function and is characterized by 4–6 months of amenorrhea, a rise in serum follicle-stimulating hormone (FSH) levels to greater than 40 mIU/L, and hypoestrogenism.2,3 POF is a common disease, occurring in 1% of all women and 0.1% of women below the age of 30 years.2 As a consequence of hypoestrogenism, POF is associated with a greater risk of osteoporosis, osteoarthritis, and cardiovascular disease.4,5 Women suffering from POF experience similar symptoms to natural menopause; however, these symptoms are also accompanied by an earlier loss of fertility. Therefore, women at risk of POF who delay childbearing until after their 30s may experience problems conceiving and carrying a pregnancy to full term.1 This loss of fertility can be due to an accelerated loss of follicles, an inability of the remaining follicles to respond to ovulatory signals, an initially reduced ovarian reserve at the time of birth, or a combination of all.2 As the development of POF occurs without any early discernible signs or symptoms, the diagnosis of POF currently occurs at the end stage of the disease, with a large proportion of these patients having not previously conceived before the time of diagnosis.2
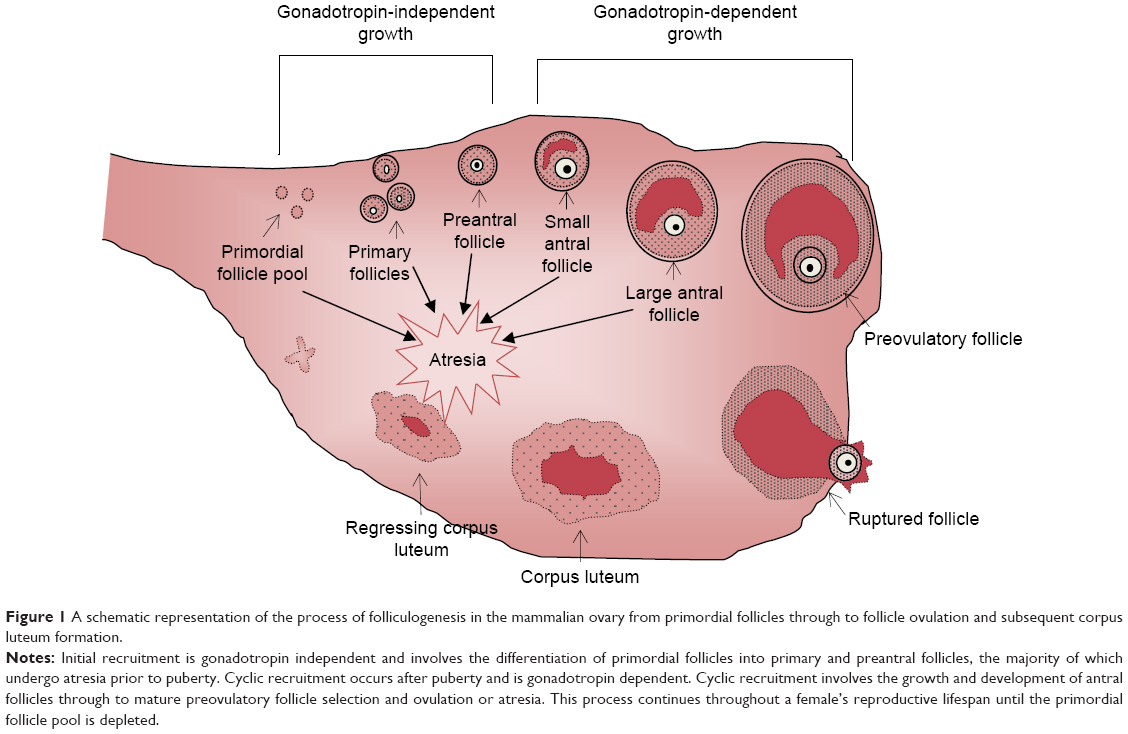 | Figure 1 A schematic representation of the process of folliculogenesis in the mammalian ovary from primordial follicles through to follicle ovulation and subsequent corpus luteum formation. |
POF is a heterogeneous disease that can develop secondary to many factors such as infection, metabolic disease, autoimmune disorders, or iatrogenic causes such as radiation, chemotherapy, or physical damage to the ovary.6 Additionally, some POF cases are syndromic, where the POF phenotype accompanies a specific disease, such as Turner’s syndrome or blepharophimosis–ptosis–epicanthus inversus syndrome (BPES) type 1. However, 50%–90% of cases are idiopathic and likely involve a substantial genetic contribution.7 A genetic basis of menopause is supported by heritability estimates of menopausal age between mother–daughter pairs ranging from 44% to 65% and recent reports of several genetic loci with significant associations with age at natural menopause identified through genome-wide association studies (GWAS).1,8–10 The importance of genetics in the etiology of POF is supported by the observation that in approximately 10%–30% of idiopathic cases, a first-degree relative is also affected, as well as the observation that a woman with an affected mother is six times more likely to develop POF.4
Genetic analyses of POF patients have identified several chromosomal abnormalities, single gene mutations, and genetic polymorphisms, from multiple different biological pathways, associated with POF development.11 However, the genetic defects investigated thus far have been shown to contribute only a small percentage toward the development of POF.4 The diverse etiology of POF is in line with these findings and suggests that the pathogenesis of nonsyndromic POF is unlikely to be caused by a single gene or genetic defect, but rather supports the view of POF being a heterogeneous genetic disease involving the interaction of multiple genetic defects and environmental factors.1,12 Therefore, current research is focused on increasing the understanding of the genetic basis of idiopathic POF. This information will provide a greater insight into POF pathogenesis and allow for the identification of biomarkers, which may have the ability to predict early onset, providing women at risk with the opportunity to plan their families earlier.
In this review, we aim to provide an overview of the current knowledge of the genetic basis of POF development. To review current literature, we did a descriptive review of the literature based on our existing knowledge in the field and by searching publication databases with the terms “premature ovarian failure”, “primary ovarian insufficiency”, “genetics”, and “mutation”. In addition, we will provide an update on the recent advances in identifying novel genetic factors associated with POF through means of GWAS and next-generation sequencing (NGS).
X-chromosome defects
X-chromosome abnormalities have long been recognized as the most common genetic cause of POF, estimated to account for as much as 12% of cases.13–15 These abnormalities usually involve either whole or partial deletions, duplications, or translocations of the X-chromosome. X monosomy (45X), known as Turner’s syndrome, has an incidence of 1:2,500.16 In phenotypic women, Turner’s syndrome is associated with short stature, gonadal dysgenesis, and primary amenorrhea.16 In women with Tuner’s syndrome, oocyte loss usually begins early in childhood as a result of accelerated follicle atresia.16 Trisomy X (47XXX) is the most common form of aneuploidy, occurring in 1:1,000 females and may manifest in many mosaic forms, ie, 45X/47XXX, 46XX/47XXX, or 47XXX/48XXXX.17 Although most cases do not present with major medical problems, the 47XXX genotype has been associated with hypergonadotropic POF in some cases.17 Cytogenetic analysis in POF patients have identified a “critical region” on the long arm of the X-chromosome, necessary for ovarian development and function.7 Deletions are commonly localized at Xq21.3-q27 (POF1) in POF patients, whereas balanced X: autosomal translocations occur frequently at Xq13.3-q21.1 (POF2).2,18 Although no single gene has been shown to be consistently associated with POF within this critical region, the strong association of POF with X-chromosome abnormalities suggests that X-chromosome genes are critical for normal ovarian function, and any structural abnormality in this region may contribute toward POF development.2,6
Aside from Turner’s syndrome, a premutation in the fragile-X mental retardation (FMR1) gene, located at Xq27.3, is the most commonly known congenital cause of POF.15 The 5′UTR region of the gene contains an expandable CGG repeat region, whereby <45 repeats is considered normal, while >200 repeats is considered a full mutation and is associated with fragile-X syndrome in male carriers.12 Repeats within the range of 55–199 are known as a premutation and are associated with an increased risk of POF in women.19,20 FMR1 premutations have been identified in 6% of sporadic POF cases and up to 13% in familial forms.20 The FMR1 protein is highly expressed in fetal ovary germ cells, as well as in the granulosa cells of maturing follicles.21 There are two prevailing explanations for the association of the FMR1 premutation and POF. The FMR1 protein is a RNA-binding protein, which has a suppressive effect on the translation of a subset of mRNAs, the overexpression of which may impair the expression of oocyte development genes, resulting in a decrease in the size of the initial follicular pool.7,12 Alternatively, it has been proposed that an accumulation of mutated FMR1 mRNA may have a long-term toxic effect on the ovary and lead to excessive follicle atresia.22 Some recent studies have suggested that there is no association between CGG repeats of intermediate size and POF compared with controls; therefore, some of this data may need to be re-evaluated in light of recent findings.23
Candidate genes
Given that the main phenotype of POF is infertility, identifying the causal gene in nonsyndromic POF families has proven difficult, as the majority of these cases have small or no family histories.6 Therefore, researchers have relied on candidate gene-driven studies in order to identify potentially causative genes involved in POF development.2 By screening genes known to play roles in folliculogenesis and ovarian function, as well as genes identified in the use of genetically modified mouse models, several POF candidate genes have been recognized. These genes are summarized in Table 1. However, the field has been limited by many underpowered studies, especially when only a single gene or a few gene variants have been studied at once, without providing adequate statistical analysis for the likelihood of the increased chance of identifying a false positive by not correcting for multiple testing.24 In addition, the identification of relevant candidate genes is restricted by our current understanding of disease, which in many cases is limited.
 | Table 1 POF candidate genes and their functions in relation to POF pathogenesis |
Oocyte-specific transcription factors
The initiation of primordial follicle formation and their subsequent differentiation into primary follicles is dependent on the expression of oocyte-specific genes, which are regulated by specific transcription factors.14 The screening of transcription factors regulating folliculogenesis in multiple POF populations has led to the identification of several causative mutations in the NR5A1, NOBOX, FIGLA, and FOXL2 genes.25–27 The nuclear receptor subfamily five group A member 1 (NR5A1), also known as steroidogenic factor 1, is a nuclear receptor located at 9q33.3 and is implicated in early gonadal differentiation.27,28 NR5A1 modulates steroidogenesis via the regulation of genes involved in the hypothalamic–pituitary–steroidogenic axis, such as STAR, CYP11A1, CYP17A1, CYP19A1, LH, and INHA.28 The conditional knockout of NR5A1 in mouse granulosa cells causes infertility, as a result of hypoplastic ovaries and a reduced number of oocytes.29 To date, several NR5A1 mutations have been identified in four families with a history of 46XX POF, as well as in 2/25 idiopathic POF patients, which were absent in the 700 controls.27 All of these mutations resulted in impaired transactivational activity of gonadal promoters regulating follicle growth, thus leading to altered folliculogenesis and subsequent POF.30
Both newborn ovary homeobox (NOBOX) and factor in germline alpha (FIGLA) encode oocyte-specific transcription factors, which regulate oocyte-specific genes necessary for the early stages of folliculogenesis.31 Through the use of knockout mouse models, NOBOX is thought to regulate the expression of a variety of oocyte-specific genes including BMP-15 and GDF-9.31 The deletion of NOBOX expression in these mice results in an accelerated loss of oocytes postnatally, representing a phenotype similar to nonsyndromic POF in humans.28 Two separate studies in Caucasian POF patients have identified several NOBOX mutations, which were absent in the control populations.26 Functional analysis of these mutations revealed a significant decrease in NOBOX DNA binding activity, resulting in the impaired regulation of genes critical for folliculogenesis.26 However, no NOBOX mutations were identified in studies of Chinese and Japanese POF patients, suggesting that NOBOX mutations may be rare in these populations.32,33 FIGLA regulates the expression of zona pellucida genes, the knockout of which in mice also mimics the phenotype of nonsyndromic POF, with an accelerated postnatal loss of primordial follicles.34 A study by Zhao et al35 in 2008 identified two novel FIGLA coding variants in a cohort of 100 Chinese POF women. These variants were absent in the 304 Chinese controls.35 The suggested mechanisms through which these variants led to POF are through haploinsufficiency (p.G6sfsX66), and a dominant negative effect, through altered heterodimerization (p.140delN).35
The forkhead box L2 (FOXL2) gene, located at 3q23, is constitutively expressed primarily in granulosa cells in the ovaries from embryogenesis through to adult life.25 Mutations in the FOXL2 gene are responsible for BPES type 1, of which female carriers also present with POF.6,36 FOXL2 is believed to play a role in granulosa cell differentiation, which is supported by in vitro studies localizing FOXL2 expression to undifferentiated granulosa cells, and through knockout mouse models that show the failure of granulosa cell differentiation, leading to the premature activation and depletion of primordial follicles.37 In addition, FOXL2 mutations have been identified in ~5% of nonsyndromic POF patients (without BPES), suggesting that FOXL2 mutations may also be associated with idiopathic POF.25
More recent studies have highlighted the involvement of the spermatogenesis and oogenesis-specific basic helix–loop–helix (SOHLH) 1 and 2 transcription factors in POF development. SOHLH 1 and 2 are expressed exclusively in primordial follicles up until the primary follicle stage, and are master regulators of oocyte-specific genes critical for early follicle growth and differentiation, including NOBOX, FIGLA, BMP-15, and GDF-9.14 Both SOHLH 1 and 2 knockout female mouse models present with infertility and accelerated follicle loss, due to defects in primordial cell differentiation, with both models exhibiting atrophied ovaries devoid of oocytes by 6 weeks of age.38 A recent study by Qin et al14 identified four novel potentially deleterious heterogeneous SOHLH2 variants in a large cohort of Chinese and Serbian patients with POF. The study suggested that as two of these variants, −210G > T and 530+6T > G, resulted in reduced mature protein production, the primordial to primary follicle transition will be altered, leading to accelerated follicle loss and contributing to the POF phenotype in these patients.14 Additionally, a study conducted by Zhao et al39 identified three novel SOHLH1 variants potentially causative to POF in 364 Chinese that were absent in 400 controls. However, replication studies in larger populations, especially those in different ethnicities, are required to validate a causative association of SOHLH1 and 2 variants with POF.
Folliculogenesis growth factors
Bone morphogenetic protein 15 (BMP-15), located at Xq11.2, is a member of the transforming growth factor β (TGF-β) superfamily and encodes an oocyte-specific growth and differentiation factor.40 BMP-15 is involved in stimulating folliculogenesis and promoting follicle maturation by regulating granulosa cell differentiation and proliferation.41 The essential role of BMP-15 in folliculogenesis is illustrated by knockout mice models that present with subfertility due to defective ovulation processes.42 Many heterozygous variants in the BMP-15 gene proregion, essential for proprotein dimerization, have been identified in several populations of POF patients with variable frequencies.43–46 These variants result in either a reduced production of mature BMP-15 protein or the production of variants with impaired activity.47 Although the mechanism through which BMP-15 variants influence POF development is presently unknown, it is proposed that BMP-15 haploinsufficiency may lead to defective granulosa cell signaling and impaired antiapoptotic mechanisms resulting in increased follicle atresia.47 However, an association of BMP-15 variants and POF has not been found in all studies.48 Additionally, the variants have also been identified in a small number of control participants, which may diminish their significance. Conversely, it may suggest that these are relatively common variants that contribute to an individual’s predisposition toward POF in combination with other predisposing factors.42
The growth differentiation factor 9 (GDF-9) gene, a homologue of BMP-15, is also a member of the TGF-β superfamily and is located at 5q23.2. Like BMP-15, GDF-9 is oocyte specific and regulates primordial follicle development and stimulates granulosa cell proliferation and follicle maturation.49 GDF-9 is also thought to play a role in steroidogenesis and the modulation of FSH sensitivity in granulosa cells.43 GDF-9 null mice are completely infertile, suggesting that GDF-9 stimulation is essential for primordial follicles to progress.50 To date, several GDF-9 variants have been identified in POF cohorts of varying ethnicities, all of which are heterozygous and located in the proregion.43,46 However, several other studies have found no association between GDF-9 variants and POF, suggesting GDF-9 variants may be a rare contributor to POF.43,48,51
Inhibin alpha (INHA) encodes the alpha subunit of inhibin A and inhibin B, which together with their corresponding beta units (INHBA and INHBB), a class of dimeric glycoproteins, also belonging to the TGF-β superfamily.52 Inhibins are produced primarily by granulosa cells and act on the pituitary by inhibiting FSH production.53 Decreased inhibin production is associated with increased FSH production, leading to increased follicle recruitment and an increased depletion of the follicular pool.13 Indeed, women with idiopathic POF have lower serum levels of inhibins and higher serum FSH than that of age-matched fertile controls, suggesting that inhibins play an important role in normal ovarian function.52 Given the important biological role of inhibins in regulating folliculogenesis, INHA has long been a candidate gene for POF.13 Genetic studies on INHA variants have identified three polymorphisms associated with POF.52 One such polymorphism, a missense 769G > A variation, has shown a significant association in multiple POF populations across several ethnicities.13,54,55 Functional studies show this polymorphism results in a decrease in inhibin activity, suggesting that women carrying this variant have reduced FSH inhibition and an increased rate of follicular depletion, predisposing these women to POF.56 However, the association of INHA variants with POF is controversial due to a lack of association found in a larger Italian and German cohort consisting of 611 POF patients and 1,084 controls, and in recent meta-analysis.56,57 As the 769G > A variant is rare in some ethnicities, it is possible this variant may increase the risk of POF in some ethnicities more than others, thus contributing toward the discrepancies in the literature.49,52
Ovarian steroidogenesis
The FSH and luteinizing hormone receptors (FSHR and LHR) and their corresponding ligands, FSH and LH, play a critical role during the gonadotropic-dependent stages of follicle growth and development. FSH and LH are secreted by the pituitary gland in response to the pulsatile stimulation of gonadotropin-releasing hormone from the hypothalamus.58 The binding of FSH to its receptor, located on the granulosa cells of small antral follicles, causes the upregulation of granulosa cell estrogen production, increased follicle growth, and the recruitment of follicles into a pool of growing preovulatory follicles.59 LHR activation is involved in oocyte maturation, the promotion of ovulation, and ovarian steroidogenesis.60 The hypothalamic–pituitary–gonadal loop is closed by the negative feedback of estradiol and inhibins on the pituitary.61 Because of its central role in follicle recruitment, FSHR has long been considered a candidate gene for POF.6 The first FSHR mutation, a homozygous C566T variant, was reported by Aittomäki et al62 which was found to be causative for primary amenorrhea in six Finnish POF families.62 However, the FSHR C566T variant is rare outside of the Finnish population.63,64 Mutations in the FSHR gene are associated with FSH resistance resulting in a raised basal serum FSH and estrogen deficiency, indicative of a reduced ovarian reserve. Women with complete FSH resistance have hypoplastic ovaries and primary amenorrhea, similar to FSHR knockout mice, which are sterile due to a lack of puberty and incomplete follicular development.65 Several other FSHR variants have been identified in POF women with secondary amenorrhea, including a novel heterozygous substitution found recently in a New Zealand woman with mild ovarian dysfunction and a family history of POF.66 However, FSHR variants are rare and have not been shown to have a direct role in POF development.6,11 LH resistance is characterized by oligomenorrhea or secondary amenorrhea due to the failure of ovulation.27,67 However, the occurrence of inactivating LHR variants in POF women is rare, probably because the degree of LH resistance must be severe in order to produce the POF phenotype.28
Estrogen receptor 1 (ESR1), located at 6q25.1, encodes ER-α and is one of the two estrogen receptor subtypes; the other of which is ER-β, encoded by ESR2.68 The estrogen receptors are ligand-dependent transcription factors located on thecal and granulosa cells, as well as a variety of other tissues, which regulate the expression of a variety of genes involved in cellular growth and proliferation.69 Estrogenic signaling plays a critical role in follicular growth and maturation by maintaining FSH-induced follicular growth, regulating FSH activity, and inhibiting follicle apoptosis.70 Indeed, ESR1 knockout mice models are born fertile, but gradually lose their fertility due to the inability of follicles to progress to the antral stage, resulting in the presence of multiple large atretic follicles around 20–22 days of age.71 Several studies have investigated the relationship between the two most common ESR1 variants, PvuII (397T > C) and XbaI (351A > G), and POF development; however, the results are conflicting. Some studies reported an association between these variants and an increased risk of POF,70,72,73 while others have found either no association74 or that the XbaI variant in particular is associated with a reduced risk of POF.69,75 Additionally, the data on PvuII/XbaI haplotypes are also inconsistent, for instance, Cordts et al73 found the C/A haplotype to be associated with an increased risk of POF, whereas a study by Yang et al5 identified an association with a decrease in risk. In an unpublished New Zealand study conducted in a predominantly Caucasian cohort of 245 women, our team identified an association between the C/A haplotype and a reduced risk of low ovarian reserve, supporting a protective effect against POF development.
Genome-wide association studies
The genes mentioned previously were chosen as candidate genes for POF due to their known roles in folliculogenesis or ovarian function. Although the candidate gene approach has yielded some significant findings, it fails to identify novel genes from other pathways, which may be associated with POF development.76 A more recent strategy for identifying novel causative genes is to perform GWAS. In contrast to the candidate gene approach, which assesses one or a few genetic regions, GWAS studies employ an unbiased approach by investigating the entire genome, allowing for the identification of genetic variants without any assumptions as to their underlying mechanisms or biological pathways.77 GWAS identify potential causal genes by analyzing the association of genetic markers or single nucleotide polymorphisms (SNPs), between large case-control groups of unrelated individuals.78 GWAS have identified novel gene–disease associations, opening up new areas of research, often where previously there had been limited understanding of the disease etiology.24 A significant amount of interest has developed around identifying the genes governing menopausal age, with several loci significantly associated with age at natural menopause identified through multiple large-scale GWAS studies.8–10 These studies identified 17 novel susceptibility loci, over several chromosomes, involved in hormonal regulation, immune function, and DNA repair, which account for 2.5%–4.1% of the variation in the age at natural menopause.8–10,76 It has always been assumed that genes that regulate the age at natural menopause would also be involved in the more pronounced POF phenotype. To date, only a few GWAS studies have been conducted in POF patients, and these have often been in only small populations. The results of these studies will need to be replicated in larger studies, and across different ethnicities, to confirm their importance and validity.
Kang et al79 reported the first two-stage POF GWAS study in 101 cases and 87 controls of Korean descent. This study identified a strong association between the parathyroid hormone-responsive B1 (PTHB1) gene and POF. The physiological function of PTHB1 and its role in ovarian function remain unknown.80 However, PTHB1 variants have been identified in patients with Bardet–Biedl syndrome, a rare multisystemic genetic condition that can sometimes be associated with POF, suggesting that PTHB1 may be a novel candidate gene for POF.80 Another GWAS study by Knauff et al81 in 99 unrelated idiopathic POF patients and 235 unrelated controls of Caucasian descent identified a significant association between the ADAMTS19 gene and POF.81 ADAMTS19 encodes a zinc-dependent metalloprotease, which has been shown to be upregulated in female mice gonads during sexual differentiation, suggesting that ADAMTS19 is a biologically plausible POF candidate gene.81 However, this finding was not confirmed in an independent replication study of 60 POF patients and 90 controls,77 and so its role is currently unclear.
Following these initial POF GWAS studies, researchers have investigated the risk of early menopause (<45 years of age) and POF, for the 17 genetic variants known to be associated with the natural age of menopause.8–10 Genetic variants in four loci, 20q12.3 (MCM8), 19q13.42 (BRSK1/TMEM150B), 5q35.2 (UIMC1/HK3), and 6q24.2 (SYCP2L), previously shown to be associated with variations in the age at natural menopause, were shown to increase the risk of both early menopause and POF.1,82,83 These findings suggest that normal menopause and POF are interrelated traits, which are influenced by common underlying genetic variants, with POF representing the extreme end of the normal distribution of the age at natural menopause.1 In support of this hypothesis, our team identified an association between a TMEM150B variant and a BRSK1/TMEM150B haplotype, with an increased risk of low ovarian reserve in an unpublished study of 245 New Zealand women, hence predisposing carriers toward POF.
Little is known about the physiological functions of these newly identified variants, which is a common problem associated with GWAS, as genes are identified based on their statistical significance, and not their biological relevance. However, it is believed MCM8 is involved in the initiation of DNA replication and cellular proliferation, BRSK1 in oocyte polarity and growth, and TMEM150B in damage-induced apoptosis.8,9 The overlap of the association of natural menopause variants with POF risk highlights a significant influence of DNA damage and repair pathways in the natural process of folliculogenesis. As an accumulation of DNA damage is a major contributor to the aging process, the association of these ubiquitously expressed DNA repair genes with earlier onset menopause aligns with the hypothesis that early menopause and POF are symptoms of an individual who suffers from an overall premature aging phenotype.10,22 However, the lack of functional characterization of these novel variants limits the ability to interpret GWAS findings, as they provide no biological insight into disease pathogenesis. Therefore, further studies, including replication in larger independent populations and the development of animal models, are required to provide functional support to the GWAS candidate gene associations and further improve our knowledge of the mechanisms regulating ovarian aging and possibly aging in general.82 Currently, the ReproGen Consortium is undertaking the largest GWAS study on early menopause and POF to date. The results of this project will help illuminate whether common genetic variants regulating normal menopausal age variation also influence the phenotypic extremes of the menopausal age distribution, as well as provide novel insights into the mechanisms involved in regulating ovarian aging.76
While GWAS studies have identified some important and relevant genes, future studies will require larger datasets and meta-analysis of combined GWAS to provide even greater statistical power to identify smaller genetic associations. To date, for other diseases, GWAS studies have only been able to explain a small amount of the genetic variation related to that disease. This suggests that there are either more genes to be identified, other genetic mechanisms involved, or the GWAS technology is unable to identify all the relevant variants.24 It is unlikely that any really significant common SNPs will be present in POF; therefore, it is more likely that there will be more importance placed on individual rare SNPs that will require new sequencing technology to identify.
Copy number variants
In addition to candidate gene and SNP-based GWAS studies, genome-wide structural variation technologies, such as array comparative genomic hybridization, have been applied to complex diseases such as POF for the detection of genetic imbalances, such as deletions and duplications, also known as copy number variations (CNVs).84 CNVs are DNA regions >1 kb that vary in number between individuals and contribute toward phenotypic variation and disease susceptibility by altering transcriptional and translational levels, disrupting regulatory elements, or influencing gene dosage levels of adjacent or nearby genes.85 Aboura et al86 conducted the first study to assess the presence and prevalence of CNV in both sporadic and familial POF cases. The researchers identified five statistically significant potential candidate CNVs in genes involved in reproductive disease (DNAH5 and NAIP), reproductive endocrinology (DUSP22 and NUPR1), and folliculogenesis (AKT1).86 Another CNV study, conducted with higher resolution, identified a further 44 potentially causative microdeletions and microduplications in 74 POF patients.87 Interestingly, the rearrangements affected genes involved in meiosis (PLCB1, RB1CC1, and MAP4K4), DNA repair (RBBP8), and folliculogenesis (IMMP2L, FER1L6, and MEIG1),87 which are biological processes already implicated in POF pathogenesis, as identified in other POF genetic studies. McGuire et al88 conducted a SNP array-based study in 88 POF women and discovered 25 novel microdeletions and microduplications. The large majority of these CNVs were located on autosomes, suggesting that autosomal gene defects may have a larger contribution toward POF development than X-chromosome abnormalities, which coincides with the genetic heterogeneity and complex etiology of POF.77,88 Two of the genes associated with the autosomal microdeletions are involved in DNA double-strand break repair during meiosis (SYCE1) and mitotic cell progression (CPEB1). SYCE1 disruption leads to germ cell apoptosis in knockout mice, whereas CPEB1-deficient mice lack follicles as a result of oocyte loss.88 Therefore, these two genes are possible candidate POF genes, whereby reduced copy number/haploinsufficiency may lead to accelerated follicle loss and POF in humans.88 The consistent association of genetic defects in DNA repair and replication genes with POF suggests that this pathway significantly influences POF development and highlights a potential area to focus on for possible targeted treatments. Further studies in much larger POF cohorts are warranted to validate these findings and to identify novel CNVs. Animal studies should also be utilized to elucidate the role of these novel variants in ovarian development and aging; however, creating animal models of gene duplications is more technically challenging to perform than simple gene deletions.
MicroRNAs
The analysis of microRNAs (miRNAs) is another promising strategy recently applied to POF gene research. miRNAs are small noncoding single-stranded RNA molecules, approximately 22–24 nucleotides in size, that are involved in posttranslational gene regulation through repression or cleavage of specific RNA targets.89 miRNAs regulate cell differentiation, cell cycle progression, and apoptosis and, through the use of mouse models, have been shown to play a role in oocyte maturation and folliculogenesis.90 The first POF miRNA microarray analysis, by Zhaou et al demonstrated a differential miRNA expression profile in a very small cohort of three POF patients, compared to three normal women. One of the upregulated miRNAs, mir-23a, was demonstrated to promote granulosa cell apoptosis via the repression of X-linked inhibitor of apoptosis (XIAP) expression, suggesting the differential expression of mir-23a may be a potential contributor to POF development.91 However, because of the extremely small sample size in this study, the role of miRNAs in the etiology of POF is unclear. More recently, Dang et al92 investigated the differential expression of miRNAs in a large cohort of Chinese women, and identified 22 significantly upregulated and 29 significantly downregulated miRNAs in 140 POF patients, compared to 140 controls. Among them, mir-22-3p was significantly downregulated in POF patients, and a negative association between serum mir-22-3p and FSH levels was identified.92 The researchers suggest mir-22-3p may regulate pituitary FSH secretion, as its expression has been identified in the pig pituitary, whereby the decreased expression may contribute toward POF pathogenesis.92 These findings highlight how defects in the miRNA regulatory networks can impact on folliculogenesis. Further studies with larger sample sizes and different ethnicities are warranted to investigate the role of miRNAs in POF pathogenesis.
NGS and future directions
Over the last two decades, considerable efforts have been made to identify genetic causes and risk factors of idiopathic POF; however, only a limited number of genes have been validated as causative. The failure thus far to identify the genetic basis of idiopathic POF is due, in part, to the lack of familial cases large enough to allow causal gene detection through traditional genetic linkage analysis approaches. Additionally, ovarian development and function is influenced by several different molecular pathways, each regulated by many genes, making candidate gene selection difficult. The selection of relevant candidate genes is limited by our current understanding of POF, and it is likely many biologically relevant molecular pathways are underrepresented in our current genetic studies. Although the application of GWAS in POF research has accelerated the identification of novel candidate genes and genetic risk factors through unbiased genome analysis, it has not identified any genes that have a significant role in the development of POF. Sequencing-based approaches to gene identification have previously been limited due to its expense, limited throughput, and also due to technical limitations, as traditional Sanger sequencing can only analyze ~700 base pairs per reaction.93 Recently, significant advances in NGS technologies have provided a more efficient means of large-scale analysis of the entire human genome.61 NGS, such as whole-exome sequencing (WES), targeted NGS and whole-genome sequencing are high throughput DNA sequencing technologies that allow for the analysis of millions of base pairs in parallel across the entire exome or genome in a single reaction.61 Since the initial introduction of NGS in 2005, the cost of sequencing entire genomes has greatly reduced, resulting in NGS being used for both the discovery of novel disease-related genes and improving diagnostic genetic screening for both monogenic and polygenic diseases.94
To date, four studies using WES have mainly been conducted in consanguineous families with inherited POF, of which pathogenic variants in the stromal antigen 3 (STAG3), HFM1, MCM8, and MCM9 genes were identified.95–98 Interestingly, these genes are implicated in DNA replication and repair, meiosis, and chromosome stability, further supporting the importance of these pathways in idiopathic POF pathogenesis. Although the exact mechanism of how defects in DNA repair pathways and genomic instability contribute toward POF development is not yet known, it is likely that an accumulation of DNA damage and chromosomal instability in the ovary would lead to accelerated follicle atresia, therefore predisposing women to POF.96 Indeed, these findings coincide with several knockout mouse models of meiotic and DNA repair genes, such as DMC1, MSH5, STAG3, and SYCE1, which present with accelerated follicle loss, due in part to increased double-stranded DNA breaks.96 Additionally, a more recent targeted NGS study of 70 candidate POF genes by Fonseca et al93 reported an association between mutations in the ADAMTS19 and BMP receptor 2 (BMPR2) genes with POF pathogenesis, in 12 unrelated idiopathic POF women. Both of these genes are plausible candidate genes for POF. ADAMTS19 has previously been reported as associated with POF,81 and defects in BMPR2 may disrupt the downstream signaling of its ligands, BMP-15 and GDF-9, therefore disrupting folliculogenesis and contributing to POF pathogenesis.93
Most NGS studies conducted thus far have only been performed in small cohorts and are usually consanguineous POF families. This focus on consanguineous families has been partially to ensure a high likelihood for success, as the identified genes would be recessive, and therefore would be homozygously mutant in the affected individuals. As NGS technology gets cheaper, it is expected that there will be more studies in larger cohorts of unrelated idiopathic POF patients, which will identify novel rare gene variants and will identify further genetic regions contributing to POF development. Any new identified gene variants will produce a larger suite of candidate genes for more thorough analysis. Additionally, as the role of these newly identified variants play in POF pathogenesis is not known, future functional studies, both in in vitro human cell lines and in vivo mouse models, are necessary to elucidate the mechanisms through which these variants influence POF risk. Nevertheless, these preliminary findings suggest NGS technologies are powerful new tools, useful for identifying novel genetic regions and pathways involved in POF pathogenesis. The continued discovery and validation of POF-related genetic variants will allow for the development of new diagnostic and predictive tests, as well as the potential for novel targeted treatments.
Conclusion
As a result of recent technological advances, knowledge of the molecular basis and pathophysiology of idiopathic POF is rapidly growing. The unbiased approaches of GWAS and NGS technologies have highlighted defects in the folliculogenesis and DNA damage and repair pathways as significant contributors toward POF pathogenesis, providing a tangible focus for potential targeted treatments. However, it is unclear at present how we might design targeted therapies to defects affecting molecular mechanisms such as DNA damage and repair. Because of POF’s diverse etiology and genetic heterogeneity, we expect to see the contribution of many new and novel molecular pathways that will greatly enhance our understanding of the regulation of ovarian function. As only a small proportion of genes influencing idiopathic POF have been identified thus far, it remains to be determined how many genes and molecular pathways may influence idiopathic POF development. Future NGS studies in large cohorts of well-defined, unrelated, idiopathic POF patients will provide a great opportunity to identify the missing heritability of idiopathic POF.
Disclosure
The authors report no conflicts of interest in this work.
References
Perry JRB, Corre T, Esko T, et al. A genome-wide association study of early menopause and the combined impact of identified variants. Hum Mol Genet. 2013;22(7):1465–1472. | ||
Shelling AN. Premature ovarian failure. Reproduction. 2010:140(5):633–641. | ||
Bachelot A, Rouxel A, Massin N, et al. Phenotyping and genetic studies of 357 consecutive patients presenting with premature ovarian failure. Eur J Endocrinol. 2009;161:179–187. | ||
Qin Y, Sun M, You L, et al. ESR1, HK3 and BRSK1 gene variants are associated with both age at natural menopause and premature ovarian failure. Orphanet J Rare Dis. 2012;7:5. | ||
Yang J, Cho L, Lim Y, et al. Estrogen receptor-1 genetic polymorphisms for the risk of premature ovarian failure and early menopause. J Women’s Health. 2010;19(2):297–304. | ||
Woad KJ, Watkins WJ, Prendergast D, Shelling AN. The genetic basis of premature ovarian failure. Aust N Z J Obstet Gynecol. 2006;46(3):242–244. | ||
Cordts E, Christofolini D, Amaro dos Santos A, Bianco B, Barbosa C. Genetic aspects of premature ovarian failure: a literature review. Arch Gynecol Obstet. 2011;283:635–643. | ||
He C, Kraft P, Chen C, et al. Genome-wide association studies identify loci associated with age at menarche and age at menopause. Nat Genet. 2009;41(6):724–728. | ||
Stolk L, Guangju Z, van Meurs J, et al. Loci at chromosomes 13, 19 and 20 influence age at natural menopause. Nature. 2009;(41):645–647. | ||
Stolk L, Perry J, Chasman D, et al. Meta-analyses identify 13 loci associated with age at menopause and highlight DNA repair and immune pathways. Nat Genet. 2012;44(3):260–268. | ||
Pu D, Xing Y, Gao Y, Gu L, Wu J. Gene variation and premature ovarian failure: a meta-analysis. Eur J Obstet Gynecol Reprod Biol. 2014;182:226–237. | ||
Dixit H, Rao L, Padmalatha V, et al. Genes governing premature ovarian failure. Reprod Biomed Online. 2010;20:724–740. | ||
Shelling AN, Burton KA, Chand AL, et al. Inhibin: a candidate gene for premature ovarian failure. Hum Reprod. 2000;15(12):2644–2649. | ||
Qin Y, Jiao X, Dalgleish R, et al. Novel variants in the SOHLH2 gene are implicated in human premature ovarian failure. Fertil Steril. 2014;101(4):1104–1109.e6. | ||
Goswami D, Conway GS. Premature ovarian failure. Hum Reprod. 2005;11:391–410. | ||
Bianco B, Nunes Lipay MV, Guedes AD, Verreschi IT. Clinical implications of the detection of the Y-chromosome mosaicism in Turner’s syndrome: report of 3 cases. Fertil Steril. 2008;90(4):1197.e17–1197.e20. | ||
Villanueva AL, Rebar RW. Triple-X syndrome and premature ovarian failure. Obstet Gynecol. 1983;62(3 Suppl):70s–73s. | ||
Therman E, Laxova R, Susman B. The critical region on the human Xq. Hum Genet. 1990;85:455–461. | ||
Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, et al. Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study–preliminary data. Am J Med Genet. 1999;83:322–325. | ||
Allen EG, Sullivan AK, Marcus M, et al. Examination of reproductive aging milestones among women who carry the FMR1 premuation. Hum Reprod. 2007;22:2142–2152. | ||
Hergersberg M, Matsuo K, Gassmann M, et al. Tissue-specific expression of a FMR1/beta-galactosidase fusion gene in transgenic mice. Hum Mol Genet. 1995;4:359–366. | ||
Wood MA, Rajkovic A. Genomic markers of ovarian reserve. Semin Reprod Med. 2013;31:399–415. | ||
Voorhuis M, Onland-Moret NC, Janse F, et al. The significance of fragile X mental retardation gene 1 CGG repeat sizes in the normal and intermediate range in women with primary ovarian insufficiency. Hum Reprod. 2014;29(7):1585–1593. | ||
Shelling AN. Progress in the study of genetic disease: bringing new light to complex problems. Postgrad Med J. 2009;85(1008):505–507. | ||
Harris S, Chand A, Winship I, Gersak K, Aittomaki K, Shelling A. Identification of novel mutations in FOXL2 associated with premature ovarian failure. Mol Hum Reprod. 2002;8:729–733. | ||
Qin Y, Choi Y, Zhao H, Simpson JL, Chen ZJ, Rajkovic A. NOBOX homeobox mutation causes premature ovarian failure. Am J Hum Genet. 2007;81:576–581. | ||
Lourenco D, Brauner R, Lin L, et al. Mutations in NR5A1 associated with ovarian insufficiency. N Engl J Med. 2009;360:1200–1210. | ||
Persani L, Rossetti R, Cacciatore C. Genes involved in human premature ovarian failure. J Mol Endocrinol. 2010;45:257–279. | ||
Jeyasuria P, Ikeda Y, Jamin SP, et al. Cell-specific knockout of steroidogenic factor 1 reveals its essential roles in gonadal function. Mol Endocrinol. 2004;18:1610–1619. | ||
Bashamboo A, McElreavey K. NR5A1/SF-1 and development and function of the ovary. Ann Endocrinol (Paris). 2010;71:177–182. | ||
Rajkovic A, Pangas SA, Ballow D, Suzumori N, Matzuk MM. NOBOX deficiency disrupts early folliculogenesis and oocyte-specific gene expression. Science. 2004;305:1157–1159. | ||
Zhao XX, Suzumori N, Yamaguchi M, Suzumori K. Mutational analysis of the homeobox region of the human NOBOX gene in Japanese women who exhibit premature ovarian failure. Fertil Steril. 2005;83:1843–1844. | ||
Qin Y, Shi Y, Zhao Y, Carson SA, Simpson JL, Chen ZJ. Mutation analysis of NOBOX homeodomain in Chinese women with premature ovarian failure. Fertil Steril. 2009;91:1507–1509. | ||
Liang L, Soyal SM, Dean J. FIGalpha, a germ cell specific transcription factor involved in the coordinate expression of the zona pellucida genes. Development. 1997;124:4939–4947. | ||
Zhao H, Chen ZJ, Qin Y, et al. Transcription factor FIGLA is mutated in patients with premature ovarian failure. Am J Hum Genet. 2008;82:1342–1348. | ||
Crisponi L, Deiana M, Loi A, et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet. 2004;27:159–166. | ||
Schmidt D, Ovitt CE, Anlag K, et al. The murine winged-helix transcription factor FOXL2 is required for granulosa cell differentiation and ovary maintenance. Development. 2004;131:933–942. | ||
Choi Y, Yuan D, Rajkovic A. Germ cell-specific transcriptional regulator Sohlh2 is essential for early mouse folliculogenesis and oocyte-specific gene expression. Biol Reprod. 2008;79(6):1176–1182. | ||
Zhao H, Li G, Dalgleish R, et al. Transcription factor SOHLH1 potentially associated with primary ovarian insufficiency. Fertil Steril. 2015;103(2):548–553.e5. | ||
Hashimoto O, Moore RK, Shimasaki S. Posttranslational processing of mouse and human BMP-15: potential implication in the determination of ovulation quota. PNAS. 2005;102:5426–5431. | ||
Dube JL, Wang P, Elvin J, Lyons KM, Celeste AJ, Matzuk MM. The bone morphogenetic protein 15 gene is X-linked and expressed in oocytes. Mol Endocrinol. 1998;12:1809–1817. | ||
Tiotiu D, Alvaro Mercadal B, Imbert R, et al. Variants of the BMP15 gene in a cohort of patients with premature ovarian failure. Hum Reprod. 2010;25(6):1581–1587. | ||
Chand AL, Ponnampolam A, Harris SE, Winship IM, Shelling AN. Mutational analysis of GDF9 and BMP15 as candidate genes in premature ovarian failure. Fertil Steril. 2006;86(4):1009–1012. | ||
Di Pasquale E, Rossetti R, Marozzi A, et al. Identification of new variants of human BMP15 gene in a large cohort of women with premature ovarian failure. J Clin Endocrinol Metab. 2006;91(5):1976–1979. | ||
Dixit H, Rao L, Padmalatha V, et al. Missence mutations in the BMP15 gene are associated with ovarian failure. Hum Genet. 2006;119(4):408–415. | ||
Laissue P, Christin-Maitre S, Touraine P, et al. Mutations and sequence variants in GDF9 and BMP15 in patients with premature ovarian failure. Eur J Endocrinol. 2006;154(5):739–744. | ||
Rossetti R, Di Pasquale E, Marozzi A, et al. BMP15 mutations associated with primary ovarian insufficiency cause a defective production of bioactive protein. Hum Mutat. 2009;30(5):804–810. | ||
Takebayashi K, Takakura K, Wang H, Kimura F, Kasahara K, Noda Y. Mutation analysis of the growth differentiation factor-9 and -9B genes in patients with premature ovarian failure and polycystic ovary syndrome. Fertil Steril. 2000;74:976–979. | ||
Persani L, Rossetti R, Cacciatore C, Fabre S. Genetic defects of ovarian TGF-B-like factors and premature ovarian failure. J Endocrinol Invest. 2011;34:244–251. | ||
Dong J, Albertini DF, Nishimori K, Rajendra Kumar T, Lu N, Matzuk MM. Growth differentiation factor is required during early ovarian folliculogenesis. Nature. 1996;383:531–535. | ||
Wang B, Wen Q, Ni F, Zhou S, Wang J, Cao Y, Ma X. Analyses of growth differentiation factor 9 (GDF9) and bone morphogenetic protein 15 (BMP15) mutation in Chinese women with premature ovarian failure. Clin Endocrinol. 2010;72:135–136. | ||
Rah H, Jeon YJ, Ko JJ, et al. Association of inhibin α gene promoter polymorphisms with risk of idiopathic primary ovarian insufficiency in Korean women. Maturitas. 2014;77(2):163–167. | ||
Robertson DM, Cahir N, Findlay JK, Burger HG, Groome N. Biological and immunological characterization of inhibin forms in human follicular fluid and plasma. J Clin Endocrinol. 1997;82:889–896. | ||
Dixit H, Deendayal M, Singh L. Mutational analysis of the mature peptide region of inhibin genes in Indian women with ovarian failure. Hum Reprod. 2004;19(8):1760–1764. | ||
Marozzi A, Porta C, Vegetti W, et al. Mutation analysis of the inhibin alpha gene in a cohort of Italian women affected by ovarian failure. Hum Reprod. 2002;17(7):1741–1745. | ||
Chand AL, Harrison CA, Shelling AN. Inhibin and premature ovarian failure. Hum Reprod Update. 2010;16(1):39–50. | ||
Corre T, Schuettler J, Bione S, et al. A large-scale association study to assess the impact of known variants of the human INHA gene on premature ovarian failure. Hum Reprod. 2009;24(8):2023–2028. | ||
Mohiyiddeen L, Newman WG, McBurney H, Mulugeta B, Roberts SA, Nardo LG. Follicle-stimulating hormone receptor gene polymorphisms are not associated with ovarian reserve markers. Fertil Steril. 2012;97(3):677–681. | ||
Dias JA, Cohen BD, Lindau-Shepard B, Nechamen CA, Peterson AJ, Schmidt A. Molecular, structural, and cellular biology of follitropin and follitropin receptor. Vitam Horm. 2002;64:249–322. | ||
Ascoli M, Fanelli F, Segaloff DL. The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr Rev. 2002;23:141–174. | ||
Laissue P. Etiological coding sequence variants in non-syndromic premature ovarian failure: from genetic linkage analysis to next generation sequencing. Mol Cell Endocrinol. 2015;411:243–257. | ||
Aittomäki K, Dieguez Lucena J, Pakarinen P, et al. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell. 1995;82(6):959–968. | ||
da Fonte Kohek MB, Batista MC, Russell AJ, et al. No evidence of the inactivating mutation (C566T) in the follicle-stimulating hormone receptor gene in Brazilian women with premature ovarian failure. Fertil Steri. 1998;70:565–567. | ||
Layman LC, Amde S, Cohen DP, Jin M, Xie J. The Finnish follicle-stimulating hormone receptor gene mutation is rare in North American women with 46,XX ovarian failure. Fertil Steril. 1998;69:300–302. | ||
Abel MH, Wootton AN, Wilkins V, Huhtaniemi I, Knight PG, Charlton HM. The effect of a null mutation in the follicle-stimulating hormone receptor gene on mouse reproduction. Endocrinology. 2000;141:1795–1803. | ||
Woad KJ, Prendergast D, Winship IM, Shelling AN. FSH receptor gene variants are rarely associated with premature ovarian failure. Reprod Biomed Online. 2013;26(4):396–399. | ||
Latronico AC, Chai Y, Arnhold IJ, Liu X, Mendonca BB, Segaloff DL. A homozygous microdeletion in helix 7 of the luteinizing hormone receptor associated with familial testicular and ovarian resistance is due to both decreased cell surface expression and impaired effector activation by the cell surface receptor. Mol Endocrinol. 1998;12:442–450. | ||
Lee S, Kang D, Hudgins-Spivey S, et al. Theca-specific estrogen receptor-α knockout mice lose fertility prematurely. Endocrinology. 2009;150(8). | ||
de Mattos CS, Trevisan CM, Peluso C, et al. ESR1 and ESR2 gene polymorphisms are associated with human reproduction outcomes in brazilian women. J Ovarian Res. 2014;7:114. | ||
M’Rabet N, Moffat R, Helbing S, Kaech A, Zhang H, de Geyter C. The CC-allee of the pvull polymorphic variant in intron 1 of the a-estrogen receptor gene is significanlty more prevalent amoung infertile woman at risk of premature aging. Fertil Steri. 2012;98(4):965–972. | ||
Drummond AE, Findlay JK. The role of estrogen in folliculogenesis. Mol Cell Endocrinol. 1999;151(1–2):57–64. | ||
Bretherick KL, Hanna CW, Currie LM, Fluker MR, Hammond GL, Robinson WP. Estrogen receptor α gene polymorphisms are associated with idiopathic premature ovarian failure. Fertil Steril. 2008;89(2):318–324. | ||
Cordts E, Santos A, Peluso C, Bianco B, Barbosa C, Christofolini D. Risk of premature ovarian failure is associated with the PvuII polymorphism at estrogen receptor gene ESR1. J Assist Reprod Genet. 2012;29:1421–1425. | ||
Silva IV, Dias Rezende LC, Lanes SP, et al. Evaluation of PvuII and XbaI polymorphisms in the estrogen receptor alpha gene (ESR1) in relation to menstrual cycle timing and reproductive parameters in post-menopausal women. Maturitas. 2010;67:363–367. | ||
Corbo RM, Ulizzi L, Piombo L, Martinez-Labarga C, De Stefano GF, Scacchi R. Estrogen receptor alpha polymorphisms and fertility in populations with different reproductive patterns. Mol Hum Reprod. 2007;13(8):537–540. | ||
He C, Murabito J. Genome-wide association studies of age at menarche and age at natural menopause. Mol Cell Endocrinol. 2014;382:767–779. | ||
Stigliani S, Anserini P, Nicoletti AJ, et al. Insight into the genomics of premature ovarian failure. Mol Genet Med. 2013;7(3). | ||
Christin-Maitre S, Tachdijan G. Genome-wide association study and premature ovarian failure. Ann Endocrinol. 2010;71:218–221. | ||
Kang H, Lee SK, Kim MH, et al. Parathyroid hormone-responsive B1 gene is associated with premature ovarian failure. Hum Reprod. 2008;23:1457–1465. | ||
Mukerjee G, Dorfman R. High genetic heterogeneity of premature ovarian insufficiency. J IHP. 2014;58–63. | ||
Knauff EAH, Franke L, van Es MA, et al. Genome-wide association study in premature ovarian failure patients suggests ADAMTS19 as a possible candidate gene. Hum Reprod. 2009;24(9):2372–2378. | ||
Chen CTL, Fernández-Rhodes L, Brzyski RG, et al. Replication of loci influencing ages at menarche and menopause in hispanic women: the women’s health initiative SHARe study. Hum Mol Genet. 2011;21(6):1419–1432. | ||
Murray A, Bennett CE, Perry JRB, et al. Common genetic variants are significant risk factors for early menopause: results from the breakthrough generations study. Hum Mol Genet. 2011;20(1):186–192. | ||
Shelling AN, Ferguson LR. Genetic variation in human disease and a new role for copy number variants. Mutat Res. 2007;622(1–2):33–41. | ||
Knauff EAH, Blauw HM, Pearson PL, et al. Copy number variants on the X chromosome in women with primary ovarian insufficiency. Fertil Steril. 2011;95(5):1584–1588.e1. | ||
Aboura A, Dupas C, Tachdijan G, et al. Array comparative genomic hybridization profiling analysis reveals deoxyribonucleic acid copy number variations associated with premature ovarian failure. J Clin Endocrinl Metab. 2009;94(11):4540–4546. | ||
Ledig S, Ropke A, Wieacker P. Copy number variants in premature ovarian failure and ovarian dysgenesis. Sex Dev. 2010;4(4–5):225–232. | ||
McGuire MM, Bowden W, Engel NJ, Ahn HW, Kovanci E, Rajkovic A. Genomic analysis using high-resolution single-nucleotide polymorphism arrays reveals novel microdeletions associated with premature ovarian failure. Fertil Steril. 2011;95(5):1595–1600. | ||
Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. | ||
Murchison EP, Stein P, Xuan Z, et al. Critical roles for Dicer in the female germline. Genes Dev. 2007;21:682–693. | ||
Yang X, Zhou Y, Peng S, et al. Differentially expressed plasma microRNAs in premature ovarian failure patients and the potential regulatory function of mir-23a in granulosa cell apoptosis. Soc Reprod Fertil. 2012;144:235–244. | ||
Dang Y, Zhao S, Qin Y, Han T, Li W, Chen Z. MicroRNA-22-3p is down-regulated in the plasma of Han Chinese patients with premature ovarian failure. Fertil Steril. 2015;103(3):802–807.e1. | ||
Fonseca DJ, Patino LC, Suarez YC, et al. Next generation sequencing in women affected by nonsyndromic premature ovarian failure displays new potential causative genes and mutations. Fertil Steril. 2015;104(1):154–162. | ||
Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med. 2014;370:2418–2425. | ||
Caburet S, Arboleda VA, Llano E, et al. Mutant cohesin in premature ovarian failure. N Engl J Med. 2014;370:943–949. | ||
Wood-Trageser MA, Gurbuz F, Yatsenko SA, et al. MCM9 mutations are associated with ovarian failure, short stature, and chromosomal instability. Am J Hum Genet. 2014;95:754–762. | ||
AlAsiri S, Basit S, Wood-Trageser MA, et al. Exome sequencing reveals MCM8 mutation underlies ovarian failure and chromosomal instability. J Clin Invest. 2015;125(1):258–262. | ||
Wang J, Zhang W, Jiang H, Wu BL. Mutations in HFM1 in recessive primary ovarian insufficiency. N Engl J Med. 2014;370:972–974. | ||
Dixit H, Rao LK, Padmalatha V, et al. Mutational screening of the coding region of growth differentiation factor 9 gene in Indian women with ovarian failure. Menopause. 2005;12:749–754. | ||
Zhao H, Qin Y, Kovanci E, et al. Analyses of GDF9 mutation in 100 Chinese women with premature ovarian failure. Fertility and Sterility. 2007;88:1474–1476. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.