")
Back to Journals » Research and Reviews in Parkinsonism » Volume 4
The genetics of Parkinson’s disease: review of current and emerging candidates
Received 22 February 2014
Accepted for publication 3 April 2014
Published 17 June 2014 Volume 2014:4 Pages 63—75
DOI https://doi.org/10.2147/JPRLS.S38954
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Caroline Ran, Andrea Carmine Belin
Department of Neuroscience, Karolinska Institutet, Stockholm, Sweden
Abstract: Parkinson’s disease (PD) is the second most common neurodegenerative disorder, affecting more than 1% of the population over the age of 65 worldwide. Certain rare forms of the disease are monogenetic, and there is increasing evidence that multiple genetic risk factors are also important for common forms of PD. We have summarized the results from candidate gene and genome-wide association findings in sporadic PD as well as linkage and next-generation sequencing studies of familial PD. To date, 19 genetic loci, PARK1–19, have been reported for rare forms of PD, including autosomal-dominant and autosomal-recessive PD. At 14 of these loci, genes have been identified carrying mutations that are linked to affected family members. These genes have also been shown to constitute candidate genes for idiopathic forms of PD, since they may also carry other mutations that merely increase risk. Multiple genetic factors combine in different ways to increase or decrease risk, and several of these risk factors need to be identified in order to begin unraveling the causative pathways leading to the different forms of PD. In this review, we present current and emerging PD candidate genes to help explain the pathways leading to neurodegeneration.
Keywords: polymorphism, linkage, synuclein, mitochondria, toxins, GWAS
Introduction
Parkinson’s disease (PD) is a common neurodegenerative disorder in individuals over the age of 65, affecting approximately 1%–2% with no ethnic preference. It is characterized primarily by the degeneration of dopamine (DA) neurons in substantia nigra (SN), but there is also evidence of degeneration in other neurons as well.1 Another typical PD characteristic is the presence of proteinaceous intracellular inclusions, called Lewy bodies (LB), in the brain stem and elsewhere.2 Although there is a relatively large body of knowledge on the pathology and pathophysiology of PD, there is little understanding of its etiology. The main principles for treating PD today – replacing DA, improving the effect of DA, or inhibiting the breakdown of DA – do not cure the disease; they only reduce the symptoms caused by the loss of DA neurons. In order to create a curative treatment for PD, the underlying causes must be understood. While less common forms of PD are monogenic, representing 5%–10% of all PD cases, there is increasing evidence that multiple genetic factors are also important for sporadic PD. Several genetic approaches have been used to screen for genetic risk factors, linkage studies, candidate gene studies, genome-wide association studies (GWASs), and next-generation sequencing. To date, the PARK1–15 and PARK17–19 loci have been identified as risk factors for rare familial forms of PD, while the PARK16 locus was identified as a risk factor in sporadic PD. At 14 of these loci, genes referred to as PARK genes have been identified which carry mutations that are linked to affected family members in several PD families. These genes also constitute candidate genes for sporadic PD, since those same genes may carry other mutations that increase risk or confer protection. Positive as well as negative genetic association studies in PD have been reported for genetic variants in a plethora of candidate genes, but results from replication studies have sometimes been ambiguous.3GWASs have resulted in identifying new candidate gene loci as well as the replication of findings from linkage studies. In this review, we summarize the latest results from candidate gene and genome-wide association findings in sporadic PD, as well as linkage and next-generation sequencing studies of familial PD to help explain the pathways leading to neurodegeneration in PD.
Linkage studies
SNCA/PARK1/PARK4
The first linkage study on PD was conducted by Polymeropoulos et al, who mapped a PD locus to chromosome 4q21, known as PARK1.4 A nonsynonymous mutation (A53T) was later identified in the α-synuclein gene (SNCA) in Greek PD families with an autosomal dominant pattern of inheritance.5 Although this mutation only constitutes a rare cause of PD in three unrelated families of Greek origin, it highlighted the genetic variability in PD. The discovery of the point mutation A53T in SNCA was subsequently followed by the identification of A30P and E46K in unrelated German and Spanish PD families, with the latter also showing LB dementia.6 More recently, H50Q in exon 4 of SNCA was identified in one individual of British origin affected by PD.7 SNCA encodes the α-synuclein protein, for which the normal physiological function has not yet been determined, although α-synuclein has a potential role in synaptic plasticity and vesicle dynamics.8 It has also been shown in a number of studies that α-synuclein is abundantly expressed in the brain and that it constitutes a major component of LBs and Lewy neurites.9 The role of LBs as pathogenic, passive bystanders or possibly protective entities in PD has not been elucidated. The gene dose of SNCA appears to be critical, since SNCA duplication and triplication have also been found to cause PD.6 A triplication of the SNCA locus (incorrectly designated PARK4) results in a doubling in α-synuclein protein levels.10 Moreover, a repetitive sequence in the promoter region and single nucleotide polymorphisms (SNPs) in intronic sequences have also been found to be associated with PD,11,12 suggesting that regulation of gene transcription activity and messenger (m)RNA stability might play a role in α-synuclein-associated PD. SNCA is one of the few genes reported to be associated with PD that has been repeatedly replicated in GWASs.13–22
LRRK2/PARK8
Leucine-rich repeat kinase 2 (LRRK2), localized at the PARK8 locus, was suggested as a candidate gene for PD in 2004.23,24 Many LRRK2 mutations have now been linked to PD. The G2019S mutation in LRRK2 is the largest single known cause of PD today.25 It contributes to both sporadic and familial disease in populations of different ethnic origins. We identified G2019S in Swedish sporadic PD patients and analyzed LRRK2 gene expression in the brain.26 We found that this gene has a relationship to the DA system, evidenced by its high expression in striatum (but not in DA neurons) in rodents and human postmortem tissue.27 Regarding other reported mutations in LRRK2, numerous replication studies have been performed on sporadic cohorts, showing variable results.4 However, the LRRK2 locus has been confirmed to be associated with PD in several GWASs13,16,19,20 Also a recent meta-analysis based on 61 independent studies in Asian populations further confirmed LRRK2 mutations as risk factors for PD.28 Despite extensive research, the exact function of LRRK2 remains unknown. LRRK2 has been shown to bind to a large number of proteins via two protein-protein interaction domains, a leucine-rich repeat domain and a carboxy terminal WD40 domain. LRRK2 also harbors two enzymatic domains, one kinase and one GTPase domain, and is probably involved in quite diverse cellular functions such as mitochondrial function, apoptosis, membrane trafficking, and microtubule polymerization that may contribute to PD pathogenicity.29 Interestingly, LRRK2 is potentially interacting with many other PD candidate genes described in this review; ie, Ras-related protein Rab7-L1 (RAB7L1), vacuolar protein sorting 35 homologue (VPS35),30 SNCA,31,32 V-akt murine thymoma viral oncogene homologue 1 (AKT1),33 and microtubule-associated protein Tau (MAPT).34
Glucocerebrosidase/GBA
Mutations in the glucocerebrosidase gene (GBA), originally identified as the cause of Gaucher’s disease, have been associated with both familial and sporadic PD.35,36 GBA is located on chromosome 1q21 and encodes a lysosomal protein, glucocerebrosidase (GCase), which cleaves the beta-glycosidic linkage of glucosylceramide, an intermediate formed during glycolipid metabolism. Genetic variations in GBA are known to alter GCase activity.37 GBA mutations might lead to lysosomal dysfunction or interfere with the binding of α-synuclein to its putative receptor at the lysosomal membrane.38 This could result in reduced α-synuclein degradation, leading to cell toxicity and potentially PD when affecting DA neurons, for example.
The most common GBA mutations found in PD are L444P and N370S. Parkinson patients carrying heterozygous mutations in GBA are characterized by relatively early disease onset (≤50 years of age), but are otherwise clinically similar to patients with sporadic PD and respond well to L-DOPA (L-3,4-dihydroxyphenylalanine).39 Known GBA mutations affect α-synuclein protein levels both in cells and in transgenic mice.40 This is confirmed by the finding that induced pluripotent stem cell-derived DA neurons from Gaucher’s disease patients carrying N370S show increased levels of α-synuclein compared to DA neurons derived from healthy individuals.37 They also show lower GCase protein levels and enzymatic activity. Mazzulli et al demonstrated that elevated α-synuclein levels caused by low GCase leads to further reduction of lysosomal GCase activity and increase of the GCase retained in the endoplasmic reticulum in murine cortical neurons,37 thus suggesting that improper GCase handling results in increased α-synuclein accumulation.
VPS35/PARK17
Recently, VPS35 was identified through exome sequencing, a next-generation sequencing technique, as a risk gene for PD in families with several affected members originating from Austria and Switzerland.41,42 VPS35 is a component of the retromer complex involved in the transport of specific membrane proteins, in particular acid hydrolase receptors, from endosomes to the trans-Golgi network to avoid their degradation in lysosomes; dysfunction of this pathway could potentially lead to PD pathogenesis.43 The first description of this form of autosomal dominant PD came from a Swiss pedigree.44 Patients were described to have an initially asymmetric Parkinsonism with tremor similar to classic sporadic PD. Sequencing of the Swiss pedigree, as well as an independent Austrian PD pedigree, revealed that all affected family members carried the D620N mutation in the VPS35 gene.41,42 It is noteworthy that seven mutation carriers were detected in middle-aged neurologically healthy individuals, which indicates that the penetrance is high but might not be complete. The D620N mutation has also been reported in PD in Germany.45
In silico analysis of the substitution of aspartic acid at position 620 to the basic amino acid asparagine predicted the mutation to be deleterious.41,42 The substitution is likely to affect protein conformation at the C-terminus, since the asparagine residue forms fewer chemical bonds, thus making the protein more flexible.42 Taken together, the D620N substitution has been reported in 0.1% of the investigated PD patients and thereby accounts for very few PD cases. Additional rare variants in VPS35 have also been identified and could potentially influence the risk of developing PD; several of these variants were identified only in one individual, while others were found in patient populations and in neurologically healthy controls.41,42 These data were confirmed in a large multicenter replication study performed on almost 9,000 PD patients, in which D620N was found in seven patients while the other mutations were found in both patients and control subjects.46 Because of the rarity of these genetic variants, further studies in large cohorts are required to determine the contribution of VPS35 mutations to develop PD.
EIF4G1/PARK18
Analysis of a French pedigree with PD revealed significant linkage to chromosome 3q26-q28.47 One mutation, R1205H, in the eukaryotic translation initiation factor 4-gamma 1 (EIF4G1) segregated with disease. EIF4G1 is the scaffold protein of the RNA translation initiation complex. The R1205H mutation is located close to the putative binding site of EIF3E, another component of the complex; when analyzed in silico, the mutation was predicted to have a deleterious effect on protein function. Indeed, the R1205H substitution decreased the binding between EIF4G1 and EIF3E in transfected cell lines. In addition, cells with mutated EIF4G1 had a more pronounced loss of mitochondrial membrane potential when exposed to stress.47 Mitochondrial dysfunction has also been implicated as an important risk factor in the pathogenesis of PD, since it can lead to apoptosis of midbrain DA neurons.48
Replication studies have so far failed to detect additional EIF4G1 R1205H mutation carriers among PD cases49–51 or detected the mutation exclusively in control subjects;52 however, it is possible that the replication studies were underpowered to detect this rare mutation (0.2% of the PD patients in the initial study). It should also be noted that one of the replication studies was performed on a cohort with a different ethnic background,50 while haplotype analysis in the discovery study indicated that mutation carriers were likely to share an ancestral founder.47 It is also worth noting that the healthy control individuals carrying R1205H came from a general population cohort and might be susceptible to the development of PD later in life.52 Nevertheless, it cannot be excluded that R1205H is not itself disease causing and that the real disease-causing variant at the large multigene 3q26-q28 locus remains to be discovered.
Numerous mutations in EIF4G1 have been discovered in the extensive genetic analyses described above,47,49–52 but the evidence of pathogenicity is ambiguous. Many mutations were found in too few patients, segregation analysis was not practicable, or variants were also reported in control subjects; however, missense mutation A502V is a promising candidate. In conformity with R1205H, A502V was predicted to be deleterious to protein function and is expected to alter the binding of the EIF4E component of the translation initiation complex.47
DNAJC6/PARK19
Recently, pathogenic DnaJ (Hsp40) homologue subfamily C, member 6 (DNAJC6) mutations have been reported in two independent consanguineous families with juvenile Parkinsonism using next-generation sequencing.53,54 Also, member 13 of the DnaJ (Hsp40) homologue subfamily C (DNAJC13) has been discovered to harbor mutations potentially causing PD.55 DNAJC6 on chromosome 1p31.3 encodes auxilin, which is a neuron-specific co-chaperone protein. Together with the molecular chaperone, Hsp70c, auxilin is responsible for the removal and recycling of clathrin from coated endocytosis vesicles.56 DNAJC6’s malfunction leads to impaired synaptic vesicle recycling and disturbed clathrin-mediated endocytosis.53 Dopamine receptors are internalized by clathrin-mediated endocytosis to be degraded by the endosomal system; thus, mutations in DNAJC6 can lead to an abnormal DA receptor metabolism, which can be an important factor for developing PD.57
The first pathogenic mutation in DNAJC6 was found in two Palestinian brothers homozygous for the potentially deleterious mutation c.801-2 A>G,53 while unaffected family members were heterozygous for the genetic variation. In a second study, another DNAJC6 mutation, c.182A>T, was found to segregate with Parkinsonism in a Turkish family.54 Patients in both families have rare forms of juvenile-onset Parkinsonism. Replication studies have so far been negative;58,59 however, this does not exclude DNAJC6 as a candidate gene for PD. More research is needed to understand the possible implication of this gene in PD, and should tentatively be focused on PD patients with juvenile or early onset disease.
Summary of linkage studies
Linkage studies first revealed the existence of a genetic component in PD etiology. After decades of genetic research, the disease is now viewed as a complex genetic disorder. Mutations in genes first identified as disease causing for familial PD have also been discovered in sporadic disease, and SNPs in those genes have also been found to associate with sporadic disease. Herein, we have summarized findings on a few important genes identified through linkage studies (Table 1): SNCA, which was the first gene discovered to harbor PD-associated mutations, as well as LRRK2 and GBA, which together constitute the most common known causes for sporadic PD today. Not all genes found in familial studies are relevant in sporadic PD, nor have they been replicated. VPS35, EIF4G1, and DNAJC6 are the most recent candidate genes, none of which have yet been established as risk factors for common PD. Even so, these genes are of great interest, since they offer indications of molecular pathways that may be disrupted in PD pathology. VPS35, for instance, is a member of the retromer complex, which is involved in the transport of proteins between the Golgi network and endosomes. Alterations in this pathway could potentially interfere with normal lysosome function leading to PD pathogenesis, thus shedding more light on the hypothesis suggesting a role for defective lysosomes in PD as indicated by an involvement of GBA, for example. Concerning VPS35 in particular, the PD-associated mutation has been suggested to cause an endosome phenotype where endosomes are relocalized to a perinuclear position.60 Somehow this impairs transport of the lysosomal protease cathepsin D to the lysosomal compartment, which might affect lysosome function. Furthermore, this ties VPS35 to another risk gene for PD, SNCA, since α-synuclein is a known cathepsin D substrate.61
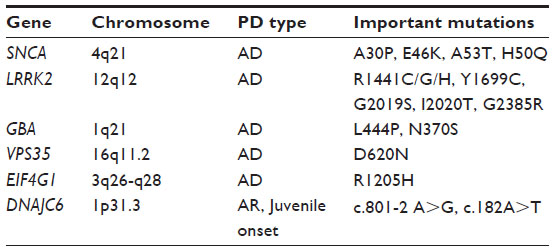 | Table 1 Current candidate genes of interest identified with linkage studies in families with hereditary forms of Parkinson’s disease |
Candidate gene studies
Candidate genes with several different functional properties have been proposed to be involved in the pathogenesis of PD. In this section, we describe candidate genes involved in four areas that have all been suggested to be important in the development of PD: abnormal protein aggregation, detoxifying enzymes, mitochondrial function, and oxidative stress. We also describe a recent candidate gene, integrin alpha 8 (ITGA8), with no known link to PD pathophysiology.
Abnormal protein aggregation
In many neurological disorders, there is evidence of abnormal protein aggregation in postmortem brain tissue of patients. In PD, these aggregations are observed as LB, which are large proteinaceous inclusions in the cytoplasm of neurons, usually seen in the DA neurons of SN. The main component of these inclusions is α-synuclein, but other proteins are also present. Attempts to deduce the composition of these protein aggregations have been difficult, since they display unspecific binding to antibodies.
MAPT/Tau is also a potential component of LBs. Although not regarded as part of the pathological hallmarks of PD, neurofibrillary tangles, which are large intracellular aggregations of hyperphosphorylated tau, have also been noted in postmortem brains of PD patients. Interestingly, tau pathology is more pronounced in PD patients suffering from cognitive decline.62,63
The interest in MAPT increased in the late 1990s after the discovery that MAPT was associated with two disorders related to PD, frontotemporal dementia with Parkinsonism and progressive supranuclear palsy.64–66 The first reported association between MAPT and late onset PD was made by Pastor et al in 2000.67 The H1 haplotype described in supranuclear palsy by Baker et al was found to be overrepresented in PD patients compared to control subjects.65,67 Since these publications, MAPT association with PD has been confirmed both in association studies and in GWASs.13–18,20–22 Exactly how tau influences the pathological changes in the Parkinsonian brain is unclear. Tau binds to microtubules and promotes their stabilization. When phosphorylated, tau is detached from microtubules, with subsequent disruption of microtubule organization. Most pathological genetic variations seen in tau affect the capacity of tau to bind microtubules and to aggregate.68 Interestingly, expression of the tau 4R isoform, which has four microtubule binding repeats and is a product of alternative splicing of exon 10, is increased in PD patients. This is likely to affect the capacity of binding to microtubules, which is critical for normal neuronal function.
Detoxifying enzymes
A few genes associated with PD encode detoxifying enzymes. SNPs and mutations in genes that affect enzymatic activity and or expression levels are prone to affect the risk of developing PD in concert with exposure to environmental risk factors. This combination of effects might mask genetic correlation, making these genes more difficult to study.
Pesticide exposure is one environmental factor that has been suggested to increase the risk to develop PD. The arylesterase paraoxonase 1 (PON1) hydrolyzes organophosphates (such as pesticides) and harbors the L54M mutation. L54M impairs enzyme activity, leads to decreased PON1 expression levels, and has been reported to be associated with PD.69 The minor allele of a PON1 promoter polymorphism, rs854571, has been reported to be more common among controls than PD cases, thus suggesting a protective effect.70 rs854571 is in strong linkage disequilibrium (LD) with another PON1 promoter polymorphism, rs854572, reported to increase PON1 gene expression.71
Mitochondria
Several genes involved in mitochondrial function have been suggested as candidate genes for PD. One such gene is DNA polymerase gamma (POLG1), which codes for the catalytic subunit of the heterotrimeric mitochondrial DNA polymerase and is involved in replication and repair of mitochondrial DNA.72 In addition to its 5′ to 3′ polymerase activity, POLG1 also has a 3′ to 5′ exonuclease activity that is important in the repair process. POLG1 has an N-terminal polyglutamine tract that is encoded by a CAG sequence in exon 2. Variations in repeat length, possibly affecting POLG1 function, have been reported to be associated with PD by several research groups.73
Oxidative stress
Oxidative stress has also been proposed as a mechanism underlying the development of PD. There are currently two hypotheses; the first postulates that the DA neurons are more susceptible to oxidative stress, while the second postulates that increased oxidative stress has global effects in the PD brain. The latter explanation is in line with the notion that none of the many genes discovered to date which have been postulated to cause or increase risk for PD are specific for the DA system and that PD pathology often includes other parts of the nervous system. Dysfunction of mitochondria and impaired protein degradation, for example, are two cellular events that occur during PD which could increase cellular stress and oxidative stress in particular.74
AKT1 on chromosome 14q32.32 encodes a serine/threonine protein kinase that is a potent promoter of cell survival which could possibly protect the PD brain from oxidative stress or, when impaired, increase levels of stress. In 2008, a protective AKT1 haplotype was found to associate with PD in Greece,75 but this genetic association could not be replicated by us in Swedish PD patients.76 While GWASs have not confirmed AKT1 as a protective gene in PD, AKT proteins are less phosphorylated in the brains of PD patients compared to healthy control subjects; therefore, it seems likely that the AKT/PKB (Protein Kinase B) family is somehow implicated in PD.77,78 Whether AKT is a cause or a consequence of PD remains to be established.
ITGA8
The ITGA8 locus on chromosome 10p13 reached genome-wide significance level in a large meta-analysis performed on the studies available in the PDGene database.3,79 The database included 828 individual association studies from more than 50 countries and was combined with GWAS data of all currently published GWASs, which resulted in a total of 16,452 PD patients and 48,810 control subjects. Among several markers in ITGA8, one intronic SNP, rs7077361, was found to associate with PD.
ITGA8 encodes the α8 subunit of the integrin family of receptors. The gene contains many SNPs, some of which are potentially deleterious nonsynonymous variations.80 The integrin α8 subunit forms a complex exclusively with the β1 subunit and is expressed in smooth muscle, myofibroblasts of the lung, and in the mesangial cells of the kidney glomeruli. ITGA8 knockout mice have severe kidney malformation or no kidneys at all.81,82 Moreover, ITGA8 seems to be involved in memory function: mice lacking the iα8 subunit in forebrain excitatory neurons display a reduction in long-term plasticity, as measured in acute hippocampal slices.83 Despite the results of the meta-analysis, there is no obvious link between ITGA8 and PD pathophysiology.
GWAS
Genome-wide technology enables large-scale, hypothesis-free searches of associated SNPs in the whole genome. Although there were great expectations that these studies would rapidly identify new susceptibility loci for PD, the results of GWASs in PD have been somewhat disappointing. The first GWASs done in Parkinson cohorts were underpowered due to small sample sizes and did not reach genome-wide significance.84–86 Also, there were no replications of genetic loci between the different studies. In 2009, the first significant GWASs were published.19,20 SNCA at the PARK4 loci was confirmed to associate with PD in two independent cohorts. In the European population, MAPT was also confirmed as a risk locus for PD.20 In the replication phase of this study, Satake et al were the first to identify two new PD-associated loci by means of GWAS,19 PARK16 at 1q32 and the bone marrow stromal cell antigen 1 (BST1) at 4p15 (Table 2). Furthermore, LRRK2 at 12q12 was confirmed as a risk gene for PD. Since 2009, SNCA, MAPT, and LRRK2 have all been replicated in GWASs.13–15,18,21,22 Three other candidate loci have also been discovered through GWASs, HLA-DRA, SREBF/RAI1, and FAM47E/SCARB2 (Table 2): 1) human leukocyte antigen (HLA) locus harboring HLA-DRA expressed by antigen-presenting cells;15 2) a locus was found on chromosome 17 tagged by rs11868035, which potentially could map to SCAP SREBF chaperone (SREBF) and/or retinoic acid induced 1 (RAI1);13 and 3) a locus marked by rs6812193 on chromosome 4 which lies within the family with sequence similarity 47 member E (FAM47E) gene. This SNP is also close to a more interesting candidate gene, scavenger receptor class B member 2 (SCARB2). SCARB2 encodes the lysosomal integral membrane protein type 2, a protein which directs GCase to the lysosomes.13 This finding strengthens the involvement of GBA in PD, and highlights the importance of correctly functioning lysosomes in the brain.
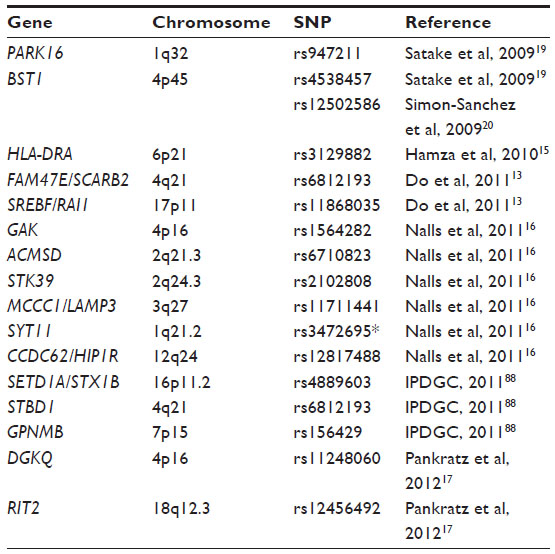 | Table 2 Candidate genes for Parkinson’s disease identified through genome-wide association studies and meta-analysis |
The lack of significance in GWASs described herein has several explanations. Insufficient power in terms of too few participants has been a major problem that could be overcome with GWAS meta-analysis. It is also possible that both genetic risk factors and protection factors are different depending on ethnic background, which could potentially have influenced results from GWASs where large sample sizes are used from cohorts with different ethnic backgrounds. Differences in PD phenotypes such as disease onset, cognitive impairments, symptom severity, and tremor should also be considered. Moreover, there is also the possibility that risk and protective factors are heterogeneous and mirror the variability of Parkinsonian phenotypes, thus indicating that the different forms of PD be considered and analyzed as different disorders. In recognition of this, many studies now exclude PD patients who carry known pathogenic mutations such as G2019S in LRRK2 and L444P in GBA. It is important not to consider results from GWASs as identification of pathogenic variations leading to PD, but rather as indicators of which loci might harbor genes and genetic variations that could lead to PD pathology.
Three meta-analysis studies performed on pre-existing GWAS datasets have identified a few new PD loci (listed in Table 2), which include 1) aminocarboxymuconate semialdehyde decarboxylase (ACMSD), a candidate gene which falls in the group of detoxifying enzymes that prevents the accumulation of the neurotoxic metabolite quinolinate, which is a product of tryptophan metabolism; 2) serine threonine kinase 39 (STK39), a kinase protein activated by cellular stress; 3) chromosomal loci 3q27, which tags either methylcrotonoyl-CoA carboxylase 1 (MCCC1) or lysosomal-associated membrane protein 3 (LAMP3); 4) synaptotagmin 11 (SYT11), which encodes a calcium-sensing protein involved in membrane trafficking in synaptic transmission and also constitutes a known substrate of Parkin; 5) chromosomal loci 12q24, which tags either coiled-coil domain containing 62 (CCDC62) or huntingtin-interacting protein 1 related (HIP1R);16 6) chromosomal loci 16p11.2, which tags either SET domain containing 1A (SETD1A), a component of a histone methyltransferase, or syntaxin 1B (STX1B), a receptor for vesicle transport; 7) starch-binding domain 1 (STBD1); 8) glycoprotein nmb (GPNMB);87 and 9) Ras-like without CAAX (RIT2).17
DGKQ/GAK
Cyclin G associated kinase (GAK) and diacylglycerol kinase theta (DGKQ) were suggested as susceptibility genes in an early GWAS where a genetic locus encompassing both genes at chromosome 4p16 reached P-values close to significance in the order of magnitude 10–6.86 Further support for the involvement of this locus in PD susceptibility was provided by two additional GWASs.21,22 The association was finally confirmed for rs1564282 in GAK in a meta-analysis which included the discovery study material16 and in a replication study performed on an American case-control cohort.88 Since then, many replications of GAK have been reported in PD populations of different ethnicities. Recently, there have also been reports of association with rs11248060 in DGKQ.17,79,89
Further studies on the PD associated locus at chromosome 4p16 should include both genes in order to be able to draw conclusions about their possible involvement in PD. GAK encodes a ubiquitously expressed protein kinase, which is active during the cell cycle.90 Of interest to PD pathophysiology is the involvement of GAK in uncoating of clathrin-coated vesicles.91 Many genes involved in vesicle transport and the endosomal-lysosomal pathway have been suggested as risk-genes for PD; ie, GBA, VPS35, and SCARB2. Functional data support the pathogenicity of GAK gene dysregulation and GAK mutations. Cathepsin D binds to GAK and expression levels of the cathepsin D gene correlate with those of GAK,92 indicating that GAK could have an impact on lysosomal function. Moreover, GAK mRNA levels are known to be decreased in SN pars compacta of PD patients compared to aged control individuals.93 In addition, the disease-associated SNP, rs1564282, is associated with increased SNCA mRNA expression in the frontal cortex. The relationship between α-synuclein and GAK has been confirmed in SNCA transfected human embryonic kidney 293 (HEK293) cells, where α-synuclein levels were increased by inhibition of GAK expression.92 This effect on α-synuclein is probably due to the decrease in lysosomal cathepsin D.
Less is known about the possible implication of DGKQ in PD. DGKQ is a member of the diaglycerol kinase (DGK) gene family. These proteins are ubiquitously expressed and responsible for the conversion of diaglycerol (DAG) to phosphatidic acid (PA) by phosphorylation. All DGK members share a well-conserved kinase domain responsible for the enzymatic activity, and different regulatory domains and motifs confer their specificity. The phosphorylation of DAG is an important step in phosphatidylinositol (PI) turnover, and it is also involved in lipid signaling. Both DAG and PA are biologically active signaling molecules. DGK proteins have been shown to take part in multiple cellular processes. In the central nervous system, DGK proteins are known to affect Ca2+ signaling, as well as the trafficking and fusing of synaptic vesicles at nerve terminals.94
PARK16
The PARK16 locus was first associated with PD in a Japanese GWAS.19 PARK16 is located in a gene-rich area on chromosome 1q32, and several SNPs within the locus strongly associate with disease. Using LD analysis, five candidate genes have been suggested for PARK16: 1) SLC45A3 (Solute carrier family 45 member 3); 2) NUCKS1 (nuclear casein kinase cyclin-dependent kinase substrate 1); 3) RAB7L1 (or RAB29); 4) SLC41A1 (Solute carrier family 41 member 1); and 5) PM20D1 (Peptidase M20 containing domain 1). Since then, PARK16 has been replicated at a genome-wide significance level in European and American PD populations.20 A close-up study of three genes at the PARK16 locus (NUCKS1, RAB7L1, and SLC41A1) in a British population revealed the presence of rare mutations in both RAB7L1 and SLC41A1.95 One common SNP in RAB7L1 was also found to be associated with an increased risk of PD, thus strengthening the hypothesis of involvement of this locus, and in particular for RAB7L1, in PD. Three rare mutations were also found in SLC41A1 in Chinese PD patients.96
It is of great importance that there are large global variations in the genotype frequencies at this locus95 and that this locus might, therefore, exert different degrees of risk of PD in different populations. The results from replication studies are indeed ambiguous as a replication study performed on two Asian cohorts, genotyping the most significant SNPs from the initial GWAS reports19,20 suggested SNPs in the PARK16 loci to have a protective effect on PD which was also observed in a Chilean and in a Taiwanese PD population.97–99 Of note, the Chilean population displays minor allele frequencies similar to those of the Asian populations. Yet other studies show a lack of association.99–101 This variability might be accounted for by differences in genetic background. Considering the replication studies, it seems likely that PARK16 is a susceptibility locus for Asian populations.
As previously described, GWASs highlight genetic loci harboring SNPs and mutations that might confer increased or decreased risk of disease; additional investigations of other SNPs within these loci are needed to confirm the involvement of PARK16 in PD patients with European descent. The differences across populations might also reflect the possibility that the true disease-causing SNP is merely in LD with the SNPs described here and that the LD blocks are different in Asian and European populations. Alternatively, the PARK16 locus might of course contain several independent risk and protective genes for PD.
SLC41A1 encodes a magnesium (Mg2+)/sodium (Na+) exchanger which is believed to be important to maintain Mg2+ homeostasis. Phosphorylation of SLC41A1 by protein kinase A affects the Mg2+ efflux, probably by modulating Mg2+ affinity.102,103 SLC41A1 has been reported to be associated with three very different human disorders: pre-eclampsia, nephronophthisis-like phenotype, and now PD. How this gene might be involved in the pathophysiology of PD remains to be elucidated.104
Little is known about RAB7L1. RAB genes encode small GTP-binding proteins involved in membrane trafficking. The RAB7L1 protein has the conserved domains important for GTP-binding and membrane association, and it is probably involved in exo- and endocytosis.105 In support of this, RAB7L1 has been found to be localized to the Golgi apparatus and to membranous transport intermediates.106 RAB7L1 is ubiquitously expressed, but at very low levels in the brain.105 Interestingly, RAB7L1 seems to interact with LRRK2. The risk effect of the LRRK2 locus was shown to be highly dependent on the absence of the protective PARK16 allele in a reanalysis of preexisting GWAS data.30 In addition, an SNP in LD with PD risk SNP, rs947211, was found to be associated with alternative splicing of RAB7L1. Healthy carriers of this allele displayed lower RAB7L1 protein levels, comparable to the decrease of RAB7L1 found in PD patients that is similar between carriers and noncarriers.30 Malfunctioning of RAB7L1 is hypothesized to affect the intracellular sorting processes between Golgi and lysosomes, for which further support has been presented by knocking down RAB7L1. Attenuation of RAB7L1 produces swollen lysosomes with reduced MPR (cation-independent mannose 6-phosphate receptor), which is necessary to maintain lysosomal function.30 These results do not necessarily mirror the situation in the human brain in vivo, but render RAB7L1 a promising candidate gene for PD as both an LRRK2 interaction partner and as a mediator in the protein sorting and lysosomal defect pathway.
NUCKS1 encodes a nuclear protein with unknown function and does not share homology with any known gene product. The gene displays characteristics of a housekeeping gene: it is highly expressed in brain and all peripheral tissues,107,108 the promoter lacks TATA and CAAT boxes, and the 5′ region is enriched in CpG pairs.107 The NUCKS1 DNA sequence contains numerous putative phosphorylation sites, and the protein is readily phosphorylated by Cdk-1 (Cyclin-dependent kinase 1).108 Although NUCKS1 association with PD has not yet been validated in replication studies, rs947211, an SNP in PARK16 that associates with PD, influences the expression levels of NUCKS1.19
BST1/CD157
The PD locus on chromosome 4p15 was first discovered in the same Japanese GWAS as PARK16.19 The BST1 finding has been replicated in populations of European and Australian origin.18,21 BST1 also turned out to be associated with PD in a larger meta-analysis performed on five cohorts from Europe and USA in which the two previously mentioned studies were also included.16 Replication studies on BST1 show conflicting results, which is most likely due to low power or population heterogeneity.101,109,110 Larger replication studies seem to support BST1 as a candidate gene for PD that has protective genetic variations. The effect is more prevalent in Asian than Caucasian PD populations.79,111,112 BST1 is a cell surface protein bound to the membrane by glycosylphosphatidylinositol linkage and possesses both (Adenosine diphosphate) ADP ribosyl cyclase and cyclic ADP ribose hydrolase enzymatic activities.113,114 BST1 has also been ascribed several additional modes of action which include receptor activity. How this membrane-bound protein with extracellular enzymatic activity can affect intracellular events and signaling has not been elucidated.115 Interestingly, BST1 seems to be an important factor in the immune system, since it is highly expressed in bone marrow cells from patients with rheumatoid arthritis and facilitates immature B-cell proliferation and growth.114,115
The HLA-locus
Another group of genes that have been studied in PD that are related to the immune system are located at the HLA region on chromosome 6p21. This locus was initially discovered in a GWAS performed on an American PD case-control material.15 The HLA region is one of the most polymorphic regions in the human genome, containing several genes coding for class I, II, and III HLA molecules. Evidence of the HLA locus being associated with PD is quite substantial and concerns different genes within the locus. The first report of HLA involvement in PD was published before the GWAS era in 2003, when an HLA-DQB1 allele was suggested to be associated with PD.116 This was followed by a GWAS finding supporting HLA-DRA,15 and several publications on DRA-DRB.16,117,118
LD analysis has revealed that several of the HLA-DR loci might represent true associations with PD;119 however, published data on this locus are more in favor of HLA-DRB, which is the more polymorphic gene. Since HLA genes are closely located on chromosome 6, it is also possible that they act in synergy.
Here, we have reviewed evidence of the association of two loci involved in the immune system: BST1 and HLA. There is clear evidence for the involvement of inflammatory reactions in PD. HLA genes as well as other immunological genes involved in antigen processing and presentation have been found to be differentially expressed in idiopathic PD.120 Whether inflammatory and/or immunological mechanisms are causative in PD or are merely a consequence of pathology remains to be elucidated. In any case, the evidence indicates that common variations in genes implicated in inflammation and other immunological pathways should be considered potential risk factors for PD.
Chromosome 18q12.3/RIT2
Although the RIT2 gene on chromosome 18q12.3 was suggested as a candidate gene for PD by Do et al,13 it did not reach genome-wide significance. There is, however, conflicting evidence as to whether RIT2 is a candidate gene for PD. In a recent study, the RIT2 gene was implicated as a candidate gene in a replication meta-analysis that did not include the discovery dataset.17 In contrast, an independent replication study in a Taiwanese population did not show association. It is plausible that the variation is of no importance in the Asian populations, since allele frequencies are very different between Asian and Caucasian populations at this locus.121 RIT2 encodes RIT2, a small GTPase of the Ras family, which is neuron specific and capable of binding calmodulin.122 Singularly, RIT2 has a direct connection to DA since RIT2 colocalizes with, and is capable of interacting with, the DA transporter (DAT). This interaction is specific and does not extend to other types of transporters. DAT is found at the plasma membrane, but can be internalized by protein kinase C-mediated endocytosis. It seems that this mechanism also depends on RIT2 GTPase activity.123
The 18q12.3 genetic region also contains the gene synaptotagmin 4 (SYT4), a potential PD risk gene through its involvement in vesicle trafficking and endocytosis, as well as its relation to SYT11, proposed to associate with PD by Nalls et al.16 Synaptotagmins are a family of proteins that are involved in synaptic transmission. They have Ca2+ binding domains that regulate the binding of phospholipids, so they are important in the regulation of vesicle fusion and endocytosis at synaptic terminals. In particular, SYT4 and SYT11 are different from other types of synaptotagmins in that they carry a conserved point mutation within a Ca2+ binding domain which renders them incapable of Ca2+ and phospholipid binding.124 All functions of synaptotagmins are not Ca2+ dependent, however, and SYT4 and SYT11 are capable of forming dimers with other isoforms as well as interacting with other proteins. In the cortex of mice, SYT4 is localized to intracellular membranous organelles primarily in the Golgi apparatus, but also in vesicles.125 SYT4 can be found in both dendrites and axons, but not in presynaptic vesicles, thus making it more likely to be involved in membrane transport and exocytosis than participating in synaptic transmission as does its binding partner SYT1. SYT4 knockout mice have impaired motor coordination and impaired hippocampal dependent memory function.126
Conclusion
In order to develop better ways of combating PD – or better yet, prevent clinical symptoms altogether – one must elucidate the causes of disease and take into consideration that there are different forms of the disorder, as well as that any given phenotype may be the effect of different genotypes plus environmental exposures. The identification of new genetic risk and protective factors for PD gives insight into the pathophysiological mechanisms, and thus may potentially aid in the development of novel classes of drugs based on causative factors, and eventually prevent and/or counteract disease. PD patients suffer from major physical and mental discomfort, so the development of more efficient treatments with less or no side effects is of the utmost importance. At the time of a clinical diagnosis of PD, the majority of the DA neurons in SN pars compacta have already been lost; therefore, a challenge for the future is to identify new markers which can be used for presymptomatic diagnosis of PD and for future neuroprotective treatments to become effective. The identification of genetic markers for PD is important in that these markers may allow diagnosis before disease onset and could result in lifelong preventive medications. Today, a diagnosis of PD by a neurologist can only be definitely confirmed by postmortem brain analysis. Findings from genetic research may lead to the development of diagnostic/prognostic methods based on blood samples, or the even less invasive buccal test. Results from several research groups involving genetic studies suggest that PD is not one, but several diseases. Genetic research can give rise to new drugs for treatment based on causative factors with different drug targets. In the future, we will most likely see more individual treatments for PD, based on genetic information, which should lead to a reduction in side effects and more efficient treatments.
In particular, we would like to stress the significance of the retromer associated sorting and the lysosomal pathways. The body of evidence in support of the involvement of these pathways is steadily increasing; and many of the genes discussed in the present review are implicated as risk genes by sorting and lysosomal degradation events: GBA, VPS35, RAB7L1, DGKQ, and GAK, as well as the less-studied genes SYT4, SCARB2, and LAMP3; impairment of these pathways is potentially detrimental in many ways (for example, they could cause impaired protein degradation and accumulation). Indeed, it has been suggested that improper GCase handling results in increase of α-synuclein accumulation, which also provides a link between α-synuclein pathology and the more recently identified candidate genes. α-synuclein has been demonstrated to have a role in PD by several methods, through genetic findings in linkage studies and GWASs, as well as by the fact that it constitutes a major component of LBs and Lewy neurites. Also, LRRK2 might be involved in this retromer-lysosome pathway, since it has been demonstrated that RAB7L1 as well as VPS35 knockout mimics the LRRK2 knockout mice phenotype of lysosomal swelling and neurite collapse.
In this review, we have summarized the most recent and important results from candidate gene and genome-wide association findings in sporadic PD, as well as in linkage and next-generation sequencing studies of familial PD in order to help explain the different possible pathways leading to neurodegeneration in PD.
Acknowledgments
We would like to thank Dr Mathew Birdsall Abrams for critically reading through this review. This work was supported by the Swedish Research Council, the Swedish Brain Foundation, the Swedish Parkinson Foundation, the Swedish Brain Power Initiative, Åke Wibergs Stiftelse, and Karolinska Institutet Funds.
Disclosure
The authors report no conflicts of interest in this work.
References
Parkinson J. An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci. 2002;14(2):223–236; discussion 222. | |
Tretiakoff C. Contributions a l’etude de l’anatomie pathologique du locus niger de soemmering avec quelques deductions relatives a la pathogenie des troubles de tonus musculaire et de la maladie de Parkinson [A study of the pathological anatomy of the locus niger of Soemerring and its relevance to the pathogenesis of changes in muscular tone in Parkinson’s disease]. Paris: 1919. French. | |
PD Gene (database on Internet). PDGENE - Field synopsis of genetic assocation studies in PD. Available from: http://www.pdgene.org/. Accessed June 3, 2014. | |
Polymeropoulos MH, Higgins JJ, Golbe LI, et al. Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science. 1996;274(5290):1197–1199. | |
Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047. | |
Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7(4):306–318. | |
Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord. 2013;28(6):811–813. | |
Bellani S, Sousa VL, Ronzitti G, Valtorta F, Meldolesi J, Chieregatti E. The regulation of synaptic function by alpha-synuclein. Commun Integr Biol. 2010;3(2):106–109. | |
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. | |
Miller DW, Hague SM, Clarimon J, et al. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology. 2004;62(10):1835–1838. | |
Mueller JC, Fuchs J, Hofer A, et al. Multiple regions of alpha-synuclein are associated with Parkinson’s disease. Ann Neurol. 2005;57(4):535–541. | |
Pals P, Lincoln S, Manning J, et al. Alpha-Synuclein promoter confers susceptibility to Parkinson’s disease. Ann Neurol. 2004;56(4):591–595. | |
Do CB, Tung JY, Dorfman E, et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011;7(6):e1002141. | |
Edwards TL, Scott WK, Almonte C, et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. 2010;74(2):97–109. | |
Hamza TH, Zabetian CP, Tenesa A, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010;42(9):781–785. | |
Nalls MA, Plagnol V, Hernandez DG, et al; International Parkinson Disease Genomics Consortium. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377(9766):641–649. | |
Pankratz N, Beecham GW, DeStefano AL, et al; PD GWAS Consortium. Meta-analysis of Parkinson’s disease: identification of a novel locus, RIT2. Ann Neurol. 2012;71(3):370–384. | |
Saad M, Lesage S, Saint-Pierre A, et al; French Parkinson’s Disease Genetics Study Group. Genome-wide association study confirms BST1 and suggests a locus on 12q24 as the risk loci for Parkinson’s disease in the European population. Hum Mol Genet. 2011;20(3):615–627. | |
Satake W, Nakabayashi Y, Mizuta I, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41(12):1303–1307. | |
Simón-Sánchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41(12):1308–1312. | |
Simón-Sánchez J, van Hilten JJ, van de Warrenburg B, et al. Genome-wide association study confirms extant PD risk loci among the Dutch. Eur J Hum Genet. 2011;19(6):655–661. | |
Spencer CC, Plagnol V, Strange A, et al; UK Parkinson’s Disease Consortium; Wellcome Trust Case Control Consortium 2. Dissection of the genetics of Parkinson’s disease identifies an additional association 5′ of SNCA and multiple associated haplotypes at 17q21. Hum Mol Genet. 2011;20(2):345–353. | |
Paisán-Ruíz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44(4):595–600. | |
Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–607. | |
Healy DG, Falchi M, O’Sullivan SS, et al. International LRRK2 Consortium. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7(7):583–590. | |
Carmine Belin A, Westerlund M, Sydow O, et al. Leucine-rich repeat kinase 2 (LRRK2) mutations in a Swedish Parkinson cohort and a healthy nonagenarian. Mov Disord. 2006;21(10):1731–1734. | |
Galter D, Westerlund M, Carmine A, Lindqvist E, Sydow O, Olson L. LRRK2 expression linked to dopamine-innervated areas. Ann Neurol. 2006;59(4):714–719. | |
Wu X, Tang KF, Li Y, et al. Quantitative assessment of the effect of LRRK2 exonic variants on the risk of Parkinson’s disease: a meta-analysis. Parkinsonism Relat Disord. 2012;18(6):722–730. | |
Kett LR, Dauer WT. Leucine-rich repeat kinase 2 for beginners: six key questions. Cold Spring Harb Perspect Med. 2012;2(3):a009407. | |
MacLeod DA, Rhinn H, Kuwahara T, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77(3):425–439. | |
Qing H, Zhang Y, Deng Y, McGeer EG, McGeer PL. Lrrk2 interaction with alpha-synuclein in diffuse Lewy body disease. Biochem Biophys Res Commun. 2009;390(4):1229–1234. | |
Qing H, Wong W, McGeer EG, McGeer PL. Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem Biophys Res Commun. 2009;387(1):149–152. | |
Ohta E, Kawakami F, Kubo M, Obata F. LRRK2 directly phosphorylates Akt1 as a possible physiological substrate: impairment of the kinase activity by Parkinson’s disease-associated mutations. FEBS Lett. 2011;585(14):2165–2170. | |
Kawakami F, Yabata T, Ohta E, et al. LRRK2 phosphorylates tubulin-associated tau but not the free molecule: LRRK2-mediated regulation of the tau-tubulin association and neurite outgrowth. PLoS One. 2012;7(1):e30834. | |
Balicki D, Beutler E. Gaucher disease. Medicine (Baltimore). 1995; 74(6):305–323. | |
Lwin A, Orvisky E, Goker-Alpan O, Lamarca ME, Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab. 2004;81(1):70–73. | |
Mazzulli JR, Xu YH, Sun Y, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146(1):37–52. | |
Wong K, Sidransky E, Verma A, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. 2004;82(3):192–207. | |
Clark LN, Ross BM, Wang Y, et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology. 2007;69(12):1270–1277. | |
Cullen V, Sardi SP, Ng J, et al. Acid β-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter α-synuclein processing. Ann Neurol. 2011;69(6):940–953. | |
Vilarino-Guell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89(1):162–167. | |
Zimprich A, Benet-Pagès A, Struhal W, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89(1):168–175. | |
Bonifacino JS, Hurley JH. Retromer. Curr Opin Cell Biol. 2008;20(4):427–436. | |
Wider C, Skipper L, Solida A, et al. Autosomal dominant dopa-responsive parkinsonism in a multigenerational Swiss family. Parkinsonism Relat Disord. 2008;14(6):465–470. | |
Kumar KR, Weissbach A, Heldmann M, et al. Frequency of the D620N mutation in VPS35 in Parkinson disease. Arch Neurol. 2012;69(10):1360–1364. | |
Sharma M, Ioannidis JP, Aasly JO, et al. GEOPD consortium. A multi-centre clinico-genetic analysis of the VPS35 gene in Parkinson disease indicates reduced penetrance for disease-associated variants. J Med Genet. 2012;49(11):721–726. | |
Chartier-Harlin MC, Dachsel JC, Vilariño-Güell C, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet. 2011;89(3):398–406. | |
Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J. 2012;31(14):3038–3062. | |
Lesage S, Condroyer C, Klebe S, et al; French Parkinson’s Disease Genetics Study Group. EIF4G1 in familial Parkinson’s disease: pathogenic mutations or rare benign variants? Neurobiol Aging. 2012;33(9):2233.e1–2233.e5. | |
Li K, Tang BS, Guo JF, et al. Analysis of EIF4G1 in ethnic Chinese. BMC Neurol. 2013;13:38. | |
Tucci A, Charlesworth G, Sheerin UM, Plagnol V, Wood NW, Hardy J. Study of the genetic variability in a Parkinson’s Disease gene: EIF4G1. Neurosci Lett. 2012;518(1):19–22. | |
Schulte EC, Mollenhauer B, Zimprich A, et al. Variants in eukaryotic translation initiation factor 4G1 in sporadic Parkinson’s disease. Neurogenetics. 2012;13(3):281–285. | |
Edvardson S, Cinnamon Y, Ta-Shma A, et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One. 2012;7(5):e36458. | |
Köroğlu Ç, Baysal L, Cetinkaya M, Karasoy H, Tolun A. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat Disord. 2013;19(3):320–324. | |
Vilariño-Güell C, Rajput A, Milnerwood AJ, et al. DNAJC13 mutations in Parkinson disease. Hum Mol Genet. 2014;23(7):1794–1801. | |
Ungewickell E, Ungewickell H, Holstein SE, et al. Role of auxilin in uncoating clathrin-coated vesicles. Nature. 1995;378(6557):632–635. | |
Paspalas CD, Rakic P, Goldman-Rakic PS. Internalization of D2 dopamine receptors is clathrin-dependent and select to dendro-axonic appositions in primate prefrontal cortex. Eur J Neurosci. 2006;24(5):1395–1403. | |
Foo JN, Liany H, Tan LC, et al. DNAJ mutations are rare in Chinese Parkinson’s disease patients and controls. Neurobiol Aging. 2014;35(4):935.e1–935.e2. | |
Jesús S, Gómez-Garre P, Carrillo F et al. Analysis of c.801-2A>G mutation in the DNAJC6 gene in Parkinson’s disease in southern Spain. Parkinsonism Relat Disord. 2014;20(2):248–249. | |
Follett J, Norwood SJ, Hamilton NA, et al. The Vps35 D620N mutation linked to Parkinson’s disease disrupts the cargo sorting function of retromer. Traffic. 2014;15(2):230–244. | |
Cullen V, Lindfors M, Ng J, et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain. 2009;2:5. | |
Compta Y, Parkkinen L, O’Sullivan SS, et al. Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: which is more important? Brain. 2011;134(Pt 5):1493–1505. | |
Horvath J, Herrmann FR, Burkhard PR, Bouras C, Kövari E. Neuropathology of dementia in a large cohort of patients with Parkinson’s disease. Parkinsonism Relat Disord. 2013;19(10):864–868; discussion 864. | |
Goedert M, Crowther RA, Spillantini MG. Tau mutations cause frontotemporal dementias. Neuron. 1998;21(5):955–958. | |
Baker M, Litvan I, Houlden H, et al. Association of an extended haplotype in the tau gene. Hum Mol Genet. 1999;8(4):711–715. | |
Conrad C, Andreadis A, Trojanowski JQ, et al. Genetic evidence for the involvement of tau. Ann Neurol. 1997;41(2):277–281. | |
Pastor P, Ezquerra M, Muñoz E, et al. Significant association between the tau gene A0/A0. Ann Neurol. 2000;47(2):242–245. | |
Wray S, Lewis PA. A tangled web – tau and sporadic Parkinson’s disease. Front Psychiatry. 2010;1:150. | |
Carmine A, Buervenich S, Sydow O, Anvret M, Olson L. Further evidence for an association of the paraoxonase 1 (PON1) Met-54 allele with Parkinson’s disease. Mov Disord. 2002;17(4):764–766. | |
Belin AC, Ran C, Anvret A, et al. Association of a protective paraoxonase 1 (PON1) polymorphism in Parkinson’s disease. Neurosci Lett. 2012;522(1):30–35. | |
Brophy VH, Hastings MD, Clendenning JB, Richter RJ, Jarvik GP, Furlong CE. Polymorphisms in the human paraoxonase (PON1) promoter. Pharmacogenetics. 2001;11(1):77–84. | |
Bolden A, Noy GP, Weissbach A. DNA polymerase of mitochondria is a gamma-polymerase. J Biol Chem. 1977;252(10):3351–3356. | |
Anvret A, Westerlund M, Sydow O, et al. Variations of the CAG trinucleotide repeat in DNA polymerase γ (POLG1) is associated with Parkinson’s disease in Sweden. Neurosci Lett. 2010;485(2):117–120. | |
Zhou C, Huang Y, Przedborski S. Oxidative stress in Parkinson’s disease: a mechanism of pathogenic and therapeutic significance. Ann N Y Acad Sci. 2008;1147:93–104. | |
Xiromerisiou G, Hadjigeorgiou GM, Papadimitriou A, Katsarogiannis E, Gourbali V, Singleton AB. Association between AKT1 gene and Parkinson’s disease: a protective haplotype. Neurosci Lett. 2008;436(2):232–234. | |
Ran C, Westerlund M, Anvret A, et al. Genetic studies of the protein kinase AKT1 in Parkinson’s disease. Neurosci Lett. 2011;501(1):41–44. | |
Malagelada C, Jin ZH, Greene LA. RTP801 is induced in Parkinson’s disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J Neurosci. 2008;28(53):14363–14371. | |
Timmons S, Coakley MF, Moloney AM, O’ Neill C. Akt signal transduction dysfunction in Parkinson’s disease. Neurosci Lett. 2009;467(1):30–35. | |
Lill CM, Roehr JT, McQueen MB, et al; 23andMe Genetic Epidemiology of Parkinson’s Disease Consortium; International Parkinson’s Disease Genomics Consortium; Parkinson’s Disease GWAS Consortium; Wellcome Trust Case Control Consortium 2). Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS Genet. 2012;8(3):e1002548. | |
Ekwa-Ekoka C, Diaz GA, Carlson C, et al. Genomic organization and sequence variation of the human integrin subunit alpha8 gene (ITGA8). Matrix Biol. 2004;23(7):487–496. | |
Müller U, Wang D, Denda S, Meneses JJ, Pedersen RA, Reichardt LF. Integrin alpha8beta1 is critically important for epithelial-mesenchymal interactions during kidney morphogenesis. Cell. 1997;88(5):603–613. | |
Schnapp LM, Breuss JM, Ramos DM, Sheppard D, Pytela R. Sequence and tissue distribution of the human integrin alpha 8 subunit: a beta 1-associated alpha subunit expressed in smooth muscle cells. J Cell Sci. 1995;108 (Pt 2):537–544. | |
Chan CS, Chen H, Bradley A, Dragatsis I, Rosenmund C, Davis RL. α8-integrins are required for hippocampal long-term potentiation but not for hippocampal-dependent learning. Genes Brain Behav. 2010;9(4):402–410. | |
Fung HC, Scholz S, Matarin M, et al. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2006;5(11):911–916. | |
Maraganore DM, de Andrade M, Lesnick TG, et al. High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet. 2005;77(5):685–693. | |
Pankratz N, Wilk JB, Latourelle JC, et al; PSG-PROGENI and GenePD Investigators, Coordinators and Molecular Genetic Laboratories. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009;124(6):593–605. | |
International Parkinson’s Disease Genomics Consortium (IPDGC); Wellcome Trust Case Control Consortium 2 (WTCCC2). A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet. 2011;7(6):e1002142. | |
Rhodes SL, Sinsheimer JS, Bordelon Y, Bronstein JM, Ritz B. Replication of GWAS associations for GAK and MAPT in Parkinson’s disease. Ann Hum Genet. 2011;75(2):195–200. | |
Chen YP, Song W, Huang R, et al. GAK rs1564282 and DGKQ rs11248060 increase the risk for Parkinson’s disease in a Chinese population. J Clin Neurosci. 2013;20(6):880–883. | |
Kimura SH, Tsuruga H, Yabuta N, Endo Y, Nojima H. Structure, expression, and chromosomal localization of human GAK. Genomics. 1997;44(2):179–187. | |
Eisenberg E, Greene LE. Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic. 2007;8(6):640–646. | |
Dumitriu A, Pacheco CD, Wilk JB, et al. Cyclin-G-associated kinase modifies α-synuclein expression levels and toxicity in Parkinson’s disease: results from the GenePD Study. Hum Mol Genet. 2011;20(8):1478–1487. | |
Grünblatt E, Mandel S, Jacob-Hirsch J, et al. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J Neural Transm. 2004;111(12):1543–1573. | |
Mérida I, Avila-Flores A, Merino E. Diacylglycerol kinases: at the hub of cell signalling. Biochem J. 2008;409(1):1–18. | |
Tucci A, Nalls MA, Houlden H, et al. Genetic variability at the PARK16 locus. Eur J Hum Genet. 2010;18(12):1356–1359. | |
Yan Y, Tian J, Mo X, et al. Genetic variants in the RAB7L1 and SLC41A1 genes of the PARK16 locus in Chinese Parkinson’s disease patients. Int J Neurosci. 2011;121(11):632–636. | |
Tan EK, Kwok HH, Kwok HK, et al. Analysis of GWAS-linked loci in Parkinson disease reaffirms PARK16 as a susceptibility locus. Neurology. 2010;75(6):508–512. | |
Ramirez A, Ziegler A, Winkler S, et al. Association of Parkinson disease to PARK16 in a Chilean sample. Parkinsonism Relat Disord. 2011;17(1):70–71. | |
Vilariño-Güell C, Ross OA, Aasly JO, et al. An independent replication of PARK16 in Asian samples. Neurology. 2010;75(24):2248–2249. | |
Mata IF, Yearout D, Alvarez V, et al. Replication of MAPT and SNCA, but not PARK16-18, as susceptibility genes for Parkinson’s disease. Mov Disord. 2011;26(5):819–823. | |
Pihlstrøm L, Axelsson G, Bjørnarå KA, et al. Supportive evidence for 11 loci from genome-wide association studies in Parkinson’s disease. Neurobiol Aging. 2013;34(6):1708.e7–1708.e13. | |
Goytain A, Quamme GA. Functional characterization of human SLC41A1, a Mg2+ transporter with similarity to prokaryotic MgtE Mg2+ transporters. Physiol Genomics. 2005;21(3):337–342. | |
Kolisek M, Nestler A, Vormann J, Schweigel-Röntgen M. Human gene SLC41A1 encodes for the Na+/Mg²+ exchanger. Am J Physiol Cell Physiol. 2012;302(1):C318–C326. | |
Fleig A, Schweigel-Röntgen M, Kolisek M. Solute Carrier Family SLC41, what do we really know about it? Wiley Interdiscip Rev Membr Transp Signal. 2013;2(6):227–239. | |
Shimizu F, Katagiri T, Suzuki M, et al. Cloning and chromosome assignment to 1q32 of a human cDNA (RAB7L1) encoding a small GTP-binding protein, a member of the RAS superfamily. Cytogenet Cell Genet. 1997;77(3–4):261–263. | |
Spanò S, Liu X, Galán JE. Proteolytic targeting of Rab29 by an effector protein distinguishes the intracellular compartments of human-adapted and broad-host Salmonella. Proc Natl Acad Sci U S A. 2011;108(45):18418–18423. | |
Grundt K, Haga IV, Aleporou-Marinou V, Drosos Y, Wanvik B, Østvold AC. Characterisation of the NUCKS gene on human chromosome 1q32.1 and the presence of a homologous gene in different species. Biochem Biophys Res Commun. 2004;323(3):796–801. | |
Ostvold AC, Norum JH, Mathiesen S, Wanvik B, Sefland I, Grundt K. Molecular cloning of a mammalian nuclear phosphoprotein NUCKS, which serves as a substrate for Cdk1 in vivo. Eur J Biochem. 2001;268(8):2430–2440. | |
Miyake Y, Tanaka K, Fukushima W, et al; Fukuoka Kinki Parkinson’s Disease Study Group. Lack of association between BST1 polymorphisms and sporadic Parkinson’s disease in a Japanese population. J Neurol Sci. 2012;323(1–2):162–166. | |
Zhu LH, Luo XG, Zhou YS, et al. Lack of association between three single nucleotide polymorphisms in the PARK9, PARK15, and BST1 genes and Parkinson’s disease in the northern Han Chinese population. Chin Med J (Engl). 2012;125(4):588–599. | |
Liu J, Xiao Q, Wang Y, et al. Analysis of genome-wide association study-linked loci in Parkinson’s disease of Mainland China. Mov Disord. 2013;28(13):1892–1895. | |
Sharma M, Ioannidis JP, Aasly JO, et al; GEO-PD Consortium. Large-scale replication and heterogeneity in Parkinson disease genetic loci. Neurology. 2012;79(7):659–667. | |
Hirata Y, Kimura N, Sato K, et al. ADP ribosyl cyclase activity of a novel bone marrow stromal cell surface molecule, BST-1. FEBS Lett. 1994;356(2–3):244–248. | |
Kaisho T, Ishikawa J, Oritani K, et al. BST-1, a surface molecule of bone marrow stromal cell lines that facilitates pre-B-cell growth. Proc Natl Acad Sci U S A. 1994;91(12):5325–5329. | |
Ortolan E, Vacca P, Capobianco A, et al. CD157, the Janus of CD38 but with a unique personality. Cell Biochem Funct. 2002;20(4):309–322. | |
Lampe JB, Gossrau G, Herting B, et al. HLA typing and Parkinson’s disease. Eur Neurol. 2003;50(2):64–68. | |
Ahmed I, Tamouza R, Delord M, et al. Association between Parkinson’s disease and the HLA-DRB1 locus. Mov Disord. 2012;27(9):1104–1110. | |
Saiki M, Baker A, Williams-Gray CH, et al. Association of the human leucocyte antigen region with susceptibility to Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2010;81(8):890–891. | |
Hill-Burns EM, Factor SA, Zabetian CP, Thomson G, Payami H. Evidence for more than one Parkinson’s disease-associated variant within the HLA region. PLoS One. 2011;6(11):e27109. | |
Botta-Orfila T, Sànchez-Pla A, Fernández M, Carmona F, Ezquerra M, Tolosa E. Brain transcriptomic profiling in idiopathic and LRRK2-associated Parkinson’s disease. Brain Res. 2012;1466:152–157. | |
Lin CH, Chen ML, Yu CY, Wu RM. RIT2 variant is not associated with Parkinson’s disease in a Taiwanese population. Neurobiol Aging. 2013;34(9):2236.e1–2236.e3. | |
Lee CH, Della NG, Chew CE, Zack DJ. Rin, a neuron-specific and calmodulin-binding small G-protein, and Rit define a novel subfamily of ras proteins. J Neurosci. 1996;16(21):6784–6794. | |
Navaroli DM, Stevens ZH, Uzelac Z, et al. The plasma membrane-associated GTPase Rin interacts with the dopamine transporter and is required for protein kinase C-regulated dopamine transporter trafficking. J Neurosci. 2011;31(39):13758–13770. | |
von Poser C, Ichtchenko K, Shao X, Rizo J, Südhof TC. The evolutionary pressure to inactivate. A subclass of synaptotagmins with an amino acid substitution that abolishes Ca2+ binding. J Biol Chem. 1997;272(22):14314–14319. | |
Ibata K, Hashikawa T, Tsuboi T, et al. Non-polarized distribution of synaptotagmin IV in neurons: evidence that synaptotagmin IV is not a synaptic vesicle protein. Neurosci Res. 2002;43(4):401–406. | |
Ferguson GD, Anagnostaras SG, Silva AJ, Herschman HR. Deficits in memory and motor performance in synaptotagmin IV mutant mice. Proc Natl Acad Sci U S A. 2000;97(10):5598–5603. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.