")
Back to Journals » The Application of Clinical Genetics » Volume 8
The genetics of Ménière’s disease
Authors Chiarella G , Petrolo C, Cassandro E
Received 5 October 2014
Accepted for publication 25 November 2014
Published 8 January 2015 Volume 2015:8 Pages 9—17
DOI https://doi.org/10.2147/TACG.S59024
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Giuseppe Chiarella,1 C Petrolo,1 E Cassandro2
1Department of Experimental and Clinical Medicine, Audiology and Phoniatrics Unit, Magna Graecia University of Catanzaro, Catanzaro, Italy; 2Department of Medicine and Surgery, University of Salerno, Salerno, Italy
Abstract: Our understanding of the genetic basis of Ménière’s disease (MD) is still limited. Although the familial clustering and the geographical and racial differences in incidence strongly suggest a certain role for genetic factors in the development of MD, no convincing evidence for an association with any gene exists, at present. In this review, starting from rational bases for a genetic approach to MD, we explored the numerous reports published in literature and summarize the recent advances in understanding of the genetic fundaments of the disease.
Keywords: Mènière’s disease, gene, vertigo, etiology, pathogenesis
Introduction
Ménière’s disease (MD) is a disorder of the inner ear characterized by both cochlear and vestibular dysfunction, defined by the symptom complex of fluctuating sensorineural hearing loss, vertigo, tinnitus, and aural fullness. It is a chronic illness, with recurrence of symptoms without prevision, as they fluctuate within periods of activity and may be in remission for much time.
The prevalence of MD has been reported in literature by numerous investigators to be between 17 and 513 of 100,000 individuals, depending on the geographical localization of study.1–4 Bilateral disease is reported to develop in 8% to 20% of cases, rarely simultaneously.5 MD in children and younger people is reported only in a few rare cases, the majority of reported children are 10 years and older.6–8 Most cases of MD are sporadic (SMD), but there are numerous affected persons who report other family members with similar symptoms suggesting the possibility of familial MD (FMD).9,10 No clinical differences have been reported between sporadic and familial form but patients affected with FMD seem to suffer from earlier onset and more severe manifestation of the disease.11–13
In this review, starting from rational bases for a genetic approach to MD, we will explore the numerous reports published in literature and summarize the recent advances in understanding of the genetic fundaments of this disease.
Limits to a successful genetic approach to MD
In an effort to develop uniform reporting criteria for MD, the American Academy of Otolaryngology-Head and Neck Surgery Committee on Hearing and Equilibrium published guidelines for diagnosing this entity in 1995.14 To be diagnosed with definite MD, a patient must show two or more episodes of characteristic vertigo (each episode longer than 20 minutes), documented hearing loss (usually fluctuating low frequency sensorineural loss seen on serial audiograms), and presence of aural fullness and/or tinnitus in the affected ear. Other causes of vertigo must be excluded.
A general problem in most of the investigations is the definition of MD. Although the American Academy of Otolaryngology-Head and Neck Surgery criteria are almost always cited, often there are patients included only showing “partial MD”, with not all of the symptoms. Because of these poorly defined samples, the resulting numbers concerning frequency of familial and/or isolated MD cannot always be considered as accurate.
MD has a clinical heterogeneity and the time course of the episodes of vertigo and hearing loss is variable. Another obstacle for an accurate selection of familial cases is that the onset of the disease is almost always in adult age.15
It is simple to understand how the diagnosis based on clinical criteria exposes to high risk of bias in study of epidemiology and generally in the selection of patients affected by MD.
Beyond the clinical diagnosis with no truly objective data we have some other problems approaching MD.
First of all, the histological feature of MD patients has been historically addressed in endolymphatic hydrops (EH)16 with the consequent speculation that this condition is the base of the MD clinical picture. However, recent studies on temporal bone17 described similar conditions of EH in specimens from people with and without the characteristic symptoms of MD. On the other side, Wangemann18 described patients with MD without hydrops on histologic examination hypothesizing that the presence of hydrops is neither essential nor specific to MD. This unreliability is confirmed in a recent, very interesting, review from Foster and Breeze (2013). Autopsy data from numerous studies do not support the interpretations that the association of MD and EH is an epiphenomenon or that MD causes EH; this opens the door to the probability that EH causes MD: however, if it is causative, hydrops alone is insufficient to cause MD, indicating that there must be one or more additional cofactors that cause asymptomatic hydrops to become symptomatic MD.19
As a consequence, MD pathophysiology is poorly understood. There are many proposed theories, several intrinsic (genetic, anatomic, metabolic, endocrine, autoimmune, or vascular), other extrinsic (allergic, viral, or traumatic). None of these hypotheses has really been accepted: each theory is in need of confirmation.
Intriguing possibilities (also for genetic implications that we will see later) are MD as migraine variant, or as a consequence of recrudescent herpes virus infection or of an autoimmune mechanism.
Up to 56% of all patients with MD has concomitant migraine.20 This suggests a common pathway for symptoms in MD and migraine.21 Cha et al suggested that the frequent association of episodic vertigo, migraine, and MD in closely related individuals, including identical twins, supports the heritability of a “migraine-Ménière’s syndrome”, with variable expression of the individual features of hearing loss, episodic vertigo, and migraine headaches.22 Moreover, it is reported that agents for migraine prophylaxis have shown benefit in some patients with MD.23 The genetic implications of migraine is quite well documented with numerous loci identified that determine some mechanisms of ion channel dysfunction, potentially superposable on MD symptoms.9
The viral infection theory has been taken into account for the ease of transmission and the diffusion of the potential infectious agents. The best similarity with the clinical course of MD with intermittent symptoms within periods of quiescence, recurrence provoked by stress, variable severity, and symptoms that tend to decrease with time are from neurotrophic DNA viruses, capable of establishing a latent infection in sensory nerve ganglia: herpes simplex virus (HSV) and varicella zoster virus (VZV).24 However, investigations about the role of HSV and VZV in MD have produced inconclusive results. Welling et al25 in 1997 found no viral DNA in the vestibular ganglia of 22 patients. Vrabec, in 2003, with a more sensitive polymerase chain reaction–based approach, reported 100% positive DNA in 25 MD patients undergoing vestibular neurectomy and 80% in controls group.26 More recently Gartner et al27 were unable to find genomic DNA of these viruses in patients with definite MD, stating that reactivation of HSV and VZV in vestibular ganglion does not seem to play a role in the pathogenesis of MD.
Autoimmune mechanisms appear to be associated with the pathophysiology of MD.28,29 The evidences that support this hypothesis include the finding of elevated levels of autoantibodies or circulating immune complexes in the serum of some patients,29,30 and the association with a functional variant of a lymphoid protein phosphatase, LYP, which inhibits T cell receptors response in patients with bilateral ear involvement.31 Moreover, different autoantibodies such as antinuclear antibodies, antiphospholipid antibodies, and antibodies against a 68kD protein have been studied in small series of patients with MD, showing conflicting results.28,29,32–34 Recently, a case-control study tested the hypothesis that impaired clearance of immune complexes by innate immune cell receptors may be important in MD. No association between polymorphisms in these receptors or levels of circulating immune complexes and MD was found. Furthermore, MD displays an elevated prevalence of systemic autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, and ankylosing spondylitis.35 Finally, recent findings from Chiarella et al36,37 seem to confirm the role of inflammatory response in MD pathogenesis, suggesting an intriguing potential role of a set of plasma proteins that could be used as an effective tool for a biomarker-oriented diagnosis and staging of MD.
Rationale for genetic suspicion for MD
There are several characteristics of MD that support a genetic background.
An ethnic susceptibility is well documented in literature. Various reports indicate that women (56%) are more susceptible than men, with nearly identical age of onset for the disease.38 There is an ethnic bias in susceptibility to MD, with the disease being extremely rare in individuals of African ancestry, slightly more prevalent in those individuals of Asian ancestry. A significant disease burden is observed within native Latino American individuals but MD remains primarily a disease of Caucasians (83%),38 with an estimated prevalence of 218 cases per 100,000 persons.39
Inheritance of MD has been described since 1941.40 Since then, several further publications supporting a probable autosomal dominant inheritance have followed, with incomplete penetrance estimated at about 60%.4,39,41–43 Despite that, recently, in a very large cohort some families with an autosomal recessive inheritance were reported.12
The familial form of MD is reported with a frequency between 7% and 15%. Siblings of MD patients display a 10-fold increased risk of developing the disease.9,11,43,44 Several MD pedigrees have been reported, mostly in Caucasians. Some Brazilian families with concurrent MD and migraine are also described.45
Sometimes, individuals in successive generations manifest the MD phenotype at an earlier age and/or with more severe manifestations, strongly suggesting the presence of anticipation, a phenomenon associated with expanded trinucleotide diseases or dynamic mutations. This has been described in numerous cases of FMD.4,41,42,44,46 Although, some authors suggest that this observation could be biased by the difference in health care availability among different generations.47
Genetic studies on MD
In Table 1 the reports on genetics studies on MD are summarized.
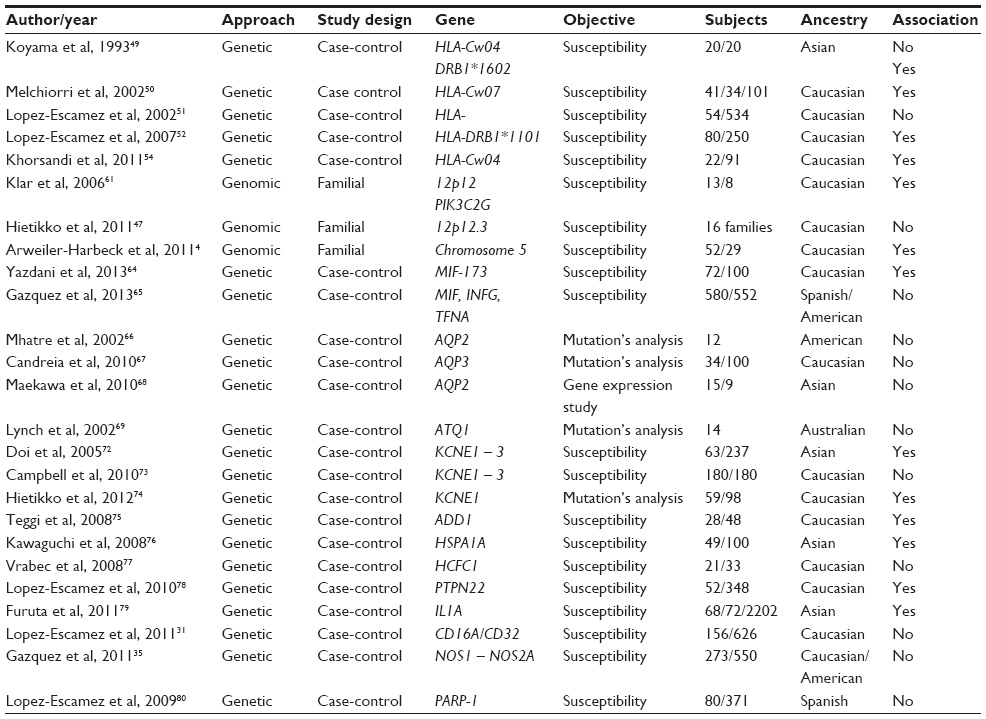 | Table 1 Reports on genetics studies on Ménière’s disease |
Linkage studies
Linkage analysis relies on evaluation of genomic markers to identify the region of the genome that segregates with the disease. Genetic markers of MD have not been identified precisely.
HLA
Based on the hypothesis of an autoimmune cause or of a dysfunction in immune function for the production of EH in MD, early investigations focused on analyzing human leukocyte antigens’ (HLAs) associations in susceptibility of MD.
Multiple studies have been performed, with different HLA associations or no association documented. Among the earliest studies, Xenellis et al48 detected an association between the Cw07 antigen and MD in 41 British patients and Koyama et al49 with the allele DRB1*1602 in a Japanese population. Only one study provides replication of the Cw07 association, with a significant increase in the distribution of the HLA-Cw07 allele in MD patients in comparison to the patients affected by other inner ear diseases or healthy controls, suggesting a predisposing role for the HLA-Cw07 specificities in MD.50 These results were not consistent and not confirmed by successive studies: Lopez-Escamez et al,51 with a larger sample, failed to demonstrate differences in phenotypic frequencies in Cw07 and no specific association with allele DRB1 1602 in a Spanish population, suggesting that the immunological alterations reported are probably epiphenomenon of the inner ear damage. Later, Lopez-Escamez et al52 observed an association between HLA-DRB1 *1101 with bilateral MD.
Because of the observed association between both sporadic and familial cases of MD and HLA class I haplotypes, Morrison et al53 suggested the possibility of an MD locus lying between the HLA-C and HLA-A loci on the short arm of chromosome 6.
More recently Khorsandi et al54 reported the association of definite MD with HLA-Cw04 allele.
DFNA9
DFNA9 is a type of autosomal dominant nonsyndromic (DFNA) hearing loss. It is localized to chromosome 14q12-13 and the responsible gene is cochlin (COCH). Among the DFNA types of deafness this has unique features, it is the only DFNA type of hearing loss with concomitant vestibular impairment that shares some features with MD, including vertigo, tinnitus, and aural fullness.55 Based on these similarities, COCH gene has been screened in a large number of MD patients of different ethnicities but no associations have been found.
Considering the studies of SMD form, Sanchez et al56 reported that the known mutations in exons 4 and 5 of the COCH gene have a low prevalence in MD, not confirming association of COCH with MD. Usami et al57 concluded that COCH mutations may not be a major cause for typical MD. In other studies on FMD, Fransen et al58 reported the presence of auditory and vestibular symptoms in a large Belgian family with hereditary hearing loss, designated DFNA9. They described close linkage between the disease and markers surrounding DFNA9 locus. Mutation analysis revealed a missense mutation changing Pro to Ser (P51S). In this study and in other reports it is not clear if DFNA9 patients who suffer from recurrent vertigo represent true cases of MD.41,59
Another experience proposed a unique FMD locus on chromosome 14q distinct from COCH (in a region overlapping COCH). Mutation analysis did not detect any of the reported COCH mutations in DFNA9, or any novel mutations. The authors concluded that their familial locus is not in COCH, without proposing an alternative candidate gene.39
DFNA9 and MD are finally considered separate entities: DFNA9 is characterized by early-onset high-frequency hearing loss and vertigo, both of which progress to severe impairment, whereas the hearing loss and vertigo of MD are late onset and low frequency at first. Moreover, although some temporal bones from individuals with DFNA9 have shown the presence of EH, the characteristic finding in DFNA9 is microfibrillar deposits in the stria vascularis, a feature not detected in MD.60
Chromosome 12
Klar et al61 studied a large Swedish family segregating for MD for linkage to loci in known familial forms of cochleovestibular dysfunction using a genome wide set of microsatellite markers. This family did show linkage for several markers on chromosome 12. When combined with two additional families, the locus was narrowed to 12p12.3. Within this region only a single known gene is identified, encoding for phosphatidylinositol 3-kinase class 2 gamma (PIK3C2G). Activation of phosphatidylinositol 3-kinase contributes to the differentiation of cells in vestibular epithelia from rats, with a proposed role of these kinases in regeneration of hair cells in mammalian ears.62 This finding obtained additional support from a successive work by the same group of researchers63 that refined the region to 1,48 Mb and screened two candidate genes on chromosome 12 (RERGL and PIK3C2G). Mutation analysis involving the coding regions of the gene did not detect any mutations. Moreover, no linkage with this chromosomal region was observed in a series of 16 Finnish MD families,47 suggesting genetic heterogeneity among families affected with MD. Arweiler-Harbeck et al4 in their genome-wide linked analysis of 17 German families with 2–4 generations positive for definite MD, documented a weakly positive score for locus on chromosome 12, that in addition appears clearly different from the region identified by Klar et al,61 and found a probable candidate region for MD on chromosome 5.
Macrophage migration inhibitory factor (MIF)
More recently, again based on the involvement of immune system in MD, Yazdani et al64 explored the association between MIF-173 G/C polymorphism and MD in an Iranian population. MIF plays a key role in immune-mediated reactions. This study’s result indicates the potential role of MIF in definite MD. In the same year, Gázquez et al65 found a significant association with the allele containing five repeats of CATT within the MIF gene in patients with MD in the Spanish cohort but not in the American set of the study.
Candidate genes association studies
In this approach, genes hypothesized to be involved in MD are screened in affected individuals. The selection accuracy of the gene is fundamental for the result. All genes for MD have been selected based on a theory of pathogenesis, but also if some studies found significance for a marker in the candidate gene, none have been replicated in a second population sample.
Genes involved in ionic composition or water transport
Aquaporin
Some authors focused on mutations in aquaporin (AQP), a transmembrane protein expressed in the endolymphatic sac that transports water and other solutes through the cell membrane. AQP gene family has a role in fluid transport and AQP1, 2, 3, 4, and 5 have been identified within the inner ear. Mhatre et al,66 in unrelated patients, tested the hypothesis that mutations in AQP2 gene are linked to MD in humans. No sequence alterations in AQP2 was identified in any of the 12 individuals affected with MD. Two other studies67,68 searched for mutations in several AQP genes, AQP1–AQP4, in affected individuals; however, no causative mutations were identified.
Antiquitin
Without preliminary linkage study, Lynch et al69 selected as candidate gene antiquin (ATQ) for mutation analysis from a series of FMD cases. The choice was based on a presumed effect in maintenance of fluid balance from the similarity with the pea protein 26 g70 that is utilized to counteract water stress in some plant species. No differences were found between affected and unaffected individuals.
KCNE genes
Investigations into the genes KCNE1 and KCNE3, two voltage-gated potassium channel genes expressed in the inner ear,71 have been similarly inconclusive: on the hypothesis that a dysfunction of these ion transporters may have a role in the development of MD, Doi et al72 found that single nucleotide polymorphisms (SNPs) in each gene KCNE1 (112G/A – rs1805127) and KCNE3 (198T/C – rs11702354) showed significant allele frequency differences in a Japanese population affected with MD suggesting that these are susceptibility genes for sporadic forms of MD. Campbell et al73 found no association in the Caucasian population and could not duplicate the Doi results. In a more recent study of Hietikko et al74 the genotype with the A-allele seemed to protect against SMD. However, they found four novel sequence variations in the KCNE1 gene in three SMD and one FMD patient, the mutation seems to be disease causing but needs a larger sample to be confirmed.
Adducin
Adducin (ADD) is a heterodimeric cytoskeleton protein consisting of three subunits (alpha, beta, and gamma) coded by three different genes (ADD1, ADD2, and ADD3). ADD1 Gly460Trp polymorphism is associated with salt-sensitive hypertension and increased Na-K pump activity in transfected cells. Teggi et al75 have not found any significant difference in the distribution of ADD2 and ADD3 polymorphism genotypes. The frequency of ADD1 Trp allele (rs4961) is significantly increased in patients with MD compared with controls. These data support the possibility that increased Na, K-ATPase activity may be one of the pathologic mechanisms inducing hyperosmolarity in endolymph which may cause hydrops.
Other candidate genes
HSP70
On the observation that MD might be caused or triggered by psychological stress, Kawaguchi et al76 examined two SNPs in heat shock 70 kD protein 1A (HSPA1A) which is thought to be involved in the cellular stress response. The SNP 190 G/C was found to be a factor associated with MD: there was significance for one SNP (rs1043618) but not for the other (rs1008438). Both SNPs have been associated with other diseases like stroke, heart disease, open angle glaucoma, Parkinson’s disease, and noise-induced hearing loss. Interestingly no clinical correlation exists between the other diseases associated with these SNPs and MD. Consequently we are in need of future follow-up to establish if the altered function acts as a cause of MD or, more likely, in concert with other still unknown factors to produce it.
Host cell factor C1 (HCFC1)
Vrabec et al77 reported a study of a series of SNPs in multiple candidate genes based on a suspected role in MD pathogenesis among patients with MD and selected control individuals. The most significant finding was that the minor allele at each SNP site was significantly more common in controls, suggesting a protective effect of the minor allele in multiple SNPs in the same haplotype block on the X chromosome that includes HCFC1. The functional consequences of the SNPs in HCFC1 are unknown but the documented interaction between HCFC1 and the HSV viral protein VP16 suggests a model of MD based on HSV reactivation.26 In neurons, HCFC1 provokes viral reactivation under stress conditions. So, in this model of MD pathophysiology, the reactivation of latent virus infection could be the mechanism underlying the appearance of MD symptoms and herpes virus infection is a necessary cofactor.
PTPN22
MD has also been associated with a polymorphism in the gene PTPN22 (rs2476601,1858C/T), encoding a lymphoid protein phosphatase suggesting that the genotype may confer differential susceptibility to bilateral MD in the Spanish population and supporting an autoimmune etiology for bilateral forms of MD.78
Interleukin-1
Furuta et al79 documented the association of interleukin-1 gene (IL1) polymorphism to both MD and sudden sensorineural hearing loss.
All of the candidate genes examined, could be plausibly linked to MD and each report outlines a verisimilar mechanism of the disease. The accuracy of these reported associations relies on replication in following studies. The real problem is that most of these studies analyzed familial and sporadic patients together, and principally, that no positive replications of previously described candidate genes have been reported. Actually, a recent study of Hietikko et al74 reports on the screening of previously MD associated genes AQP2, KCNE1, KCNE3, COCH, HCFC1, and ADD1 on 59 individuals (38 sporadic and 21 independent FMD samples) with a control population of 98 persons. Only rs1805127 (KCNE1) remained significant after correction for multiple testing. No association with MD was observed for any of the other genes and no significant haplo/diplotypes were observed.
Conclusion
Based on the published reports analyzed in this review, we can conclude that no convincing evidence for an association with a specific gene has been found, at present.
In fact, all the findings in literature indicate a genetic heterogeneity.
The phenotypic diversity of MD, without available objective tools or measures for certain diagnosis, makes the selection of patients hard, with the adult onset of the disease possibly complicating the assembly of appropriate control groups. Moreover, the predisposing or influencing factors of MD may differ between ethnic groups and even between families affected with FMD.
All these considerations, taken together, illustrate the challenge involved in the candidate gene approach to the study of MD.
A very good consideration9 is that MD is a complex disease and, as a consequence, the genetic contribution to MD is also complex. The single gene, or the genes, potentially implicated may not be sufficient to determine the disease by themselves, so, in future research, we have to consider with great attention the combined effect of environmental factors on a susceptible genetic background and the possibility of gene interactions.
On the other hand, the adult onset of MD undermines the regroup of individuals with a common phenotype in association studies. A method to approach adult-onset complex diseases may be carefully selected case-control association studies.
Today, genome-wide studies have gained an elevated level of definition from recent technological advances in sequencing and in sequence analysis. The true problem of genome-wide association is mainly the costs and the evolving methods of statistical analysis.
Undoubtedly, association studies have important limits in defining new disease genes. Inaccurate phenotype definition, as well as difficulties in the interpretation of DNA sequence alterations can lead to premature claims of association. Confirming the DNA sequence alteration by traditional molecular techniques, linkage analysis in selected pedigrees, and replication of the association in at least a second population is essential to confirm any preliminary finding.
In summary, reports based on a candidate gene’s approach provided very little information on the etiology of MD, whereas genetic studies based on linkage with polymorphic traits may provide more information about the development of the disease. These studies will need the collection of larger, well-defined case-control groups affected with FMD for use in genome-wide analyses and/or exome sequencing.
Acknowledgment
The authors want to thank Professor Nicola Perrotti for his precious contribution.
Disclosure
The authors have no conflicts of interest to disclose.
References
Havia M, Kentala E, Pyykko I. Prevalence of Ménière’s disease in general population of southern Finland. Otolaryngol Head Neck Surg. 2005;133(5):762–768. | |
Stahle J, Stahle C, Arenberg K. Incidence of Ménière’s disease. Arch Otolaryngol. 1978;104(2):99–102. | |
Watanabe Y, Mizukoshi K, Shojaku H, et al. Epidemiological and clinical characteristics of Ménière’s disease in Japan. Acta Otolaryngol. 1995;519:206–210. | |
Arweiler-Harbeck D, Horsthemke B, Jahnke K, Hennies HC. Genetic aspects of familial Meniere’s disease. Otol Neurotol. 2011;32(4):695–700. | |
Clemmens C, Ruckenstein M. Characteristics of patients with unilateral and bilateral Meniere’s disease. Otol Neurotol. 2012;33(7):1266–1269. | |
Choung YH, Park K, Kim CH, Kim K. Rare cases of Ménière’s disease in children. J Laryngol Otol. 2006;120(4):343–352. | |
Hausler R, Toupet M, Guidetti G, Basseres F, Montandon P. Ménière’s disease in children. Am J Otolaryngol. 1987;8(4):187–193. | |
Brantberg K, Duan M, Falahat B. Ménière’s disease in children aged 4–7 years. Acta Otolaryngol. 2012;132(5):505–509. | |
Vrabec JT. Genetic investigations of Ménière’s disease. Otolaryngol Clin North Am. 2010;43(5):1121–1132. | |
Eppsteiner RW, Smith RJ. Genetic disorders of the vestibular system. Curr Opin Otolaryngol Head Neck Surg. 2011;19(5):397–402. | |
Morrison AW, Bailey ME, Morrison GA. Familial Meniere’s disease: clinical and genetic aspects. J Laryngol Otol. 2009;123(1):29–37. | |
Requena T, Espinosa-Sanchez JM, Cabrera S, et al. Familial clustering and genetic heterogeneity in Meniere’s disease. Clin Genet. 2014;85(3):245–252. | |
Hietikko E, Sorri M, Männikkö M, Kotimäki J. Higher prevalence of autoimmune diseases and longer spells of vertigo in patients affected with familial Ménière’s disease: A clinical comparison of familial and sporadic Ménière’s disease. Am J Audiol. 2014;23(2):232–237. | |
No authors listed. Committee on Hearing and Equilibrium guidelines for the diagnosis and evaluation of therapy in Meniere’s disease. Otolaryngol Head Neck Surg. 1995;113(3):181–185. | |
Belinchon A, Perez-Garrigues H, Tenias JM. Evolution of symptoms in Meniere’s disease. Audiol Neurootol. 2012;17(2):126–132. | |
Hallpike C, Cairns H. Observations on the pathology of Ménière’s syndrome (Section of Otology). Proc R Soc Med. 1938;31(11):1317–1336. | |
Merchant SN, Adams JC, Nadol JB Jr. Pathophysiology of Meniere’s syndrome: are symptoms caused by endolymphatic hydrops? Otol Neurotol. 2005;26(1):74–81. | |
Wangemann P. K1 cycling and the endocochlear potential. Hear Res. 2002;165(1–2):1–9. | |
Foster CA, Breeze RE. Endolymphatic hydrops in Ménière’s disease: cause, consequence, or epiphenomenon? Otol Neurotol. 2013;34(7):1210–1214. | |
Radtke A, Lempert T, Gresty MA, et al. Migraine and Ménière’s disease: is there a link? Neurology. 2002;59(11):1700–1704. | |
Ibekwe TS, Fasunla JA, Ibekwe PU, Obasikene GC, Onakoya PA, Nwaorgu OG. Migraine and Meniere’s disease: two different phenomena with frequently observed concomitant occurrences. J Natl Med Assoc. 2008;100(3):334–338. | |
Cha YH, Kane MJ, Baloh RW. Familial clustering of migraine, episodic vertigo, and Ménière’s disease. Otol Neurotol. 2008;29(1):93–96. | |
Bikhazi P, Jackson C, Ruckenstein MJ. Efficacy of antimigrainous therapy in the treatment of migraine-associated dizziness. Am J Otol. 1997;18(3):350–354. | |
Spruance SL. The natural history of recurrent oral-facial herpes simplex virus infection. Semin Dermatol. 1992;11(3):200–206. | |
Welling DB, Daniels RL, Brainard J, Western LM, Prior TW. Detection of viral DNA in endolymphatic sac tissue from Ménière’s disease patients. Am J Otol. 1994;15(5):639–643. | |
Vrabec JT. Herpes simplex virus and Meniere’s disease. Laryngoscope. 2003;113(9):1431–1438. | |
Gartner M, Bossart W, Linder T. Herpes virus and Ménière’s disease. ORL J Otorhinolaryngol Relat Spec. 2008;70(1):28–31. | |
Riente L, Bongiorni F, Nacci A, et al. Antibodies to inner ear antigens in Meniere’s disease. Clin Exp Immunol. 2004;135(1):159–163. | |
Nacci A, Dallan I, Monzani F, et al. Elevated antithyroid peroxidase and antinuclear autoantibody titers in Meniere’s disease patients: more than a chance association? Audiol Neurotol. 2010;15(1):1–6. | |
Yoo TJ, Shea J Jr, Ge X, et al. Presence of autoantibodies in the sera of Ménière’s disease. Ann Otol Rhinol Laryngol. 2001;110(5 Pt 1):425–429. | |
Lopez-Escamez JA, Saenz-Lopez P, Gazquez I, et al. Polymorphisms of CD16A and CD32 Fcgamma receptors and circulating immune complexes in Ménière’s disease: a case-control study. BMC Med Genet. 2011;12:2. | |
Suslu N, Yilmaz T, Gursel B. Utility of immunologic parameters in the evaluation of Ménière’s disease. Acta Otolaryngol. 2009;129(11):1160–1165. | |
Gazquez I, Requena T, Espinosa JM, Batuecas A, Lopez-Escamez JA. Genetic and clinical heterogeneity in Meniere’s disease. Autoimmun Rev. 2012;11(12):925–926. | |
Greco A, Gallo A, Fusconi M, Marinelli C, Macri GF, de Vincentiis M. Meniere’s disease might be an autoimmune condition? Autoimmun Rev. 2012;11(10):731–738. | |
Gazquez I, Soto-Varela A, Aran I, et al. High prevalence of systemic autoimmune diseases in patients with Ménière’s disease. PLoS One. 2011;6(10):e26759. | |
Chiarella G, Saccomanno M, Scumaci D, et al. Proteomics in Ménière disease. J Cell Physiol. 2012;227(1):308–312. | |
Chiarella G, Di Domenico M, Petrolo C, et al. A proteomics-driven assay defines specific plasma protein signatures in different stages of Ménière’s disease. J Cell Biochem. 2014;115(6):1097–1100. | |
Ohmen JD, White CH, Li X, et al. Genetic evidence for an ethnic diversity in the susceptibility to Ménière’s disease. Otol Neurotol. 2013;34(7):1336–1341. | |
Morrison AW, Johnson KJ. Genetics (molecular biology) and Meniere’s disease. Otolaryngol Clin North Am. 2002;35(3):497–516. | |
Brown MR. Ménière’s syndrome. Arch Neurol Psychiatry. 1941;46: 561–565. | |
Frykholm C, Larsen HC, Dahl N, Klar J, Rask-Andersen H, Friberg U. Familial Ménière’s disease in five generations. Otol Neurotol. 2006;27(5):681–686. | |
Arweiler DJ, Jahnke K, Grosse-Wilde H. Ménière disease as an autosome dominant hereditary disease. Laryngorhinootologie. 1995;74(8):512–515. | |
Klockars T, Kentala E. Inheritance of Meniere’s disease in the Finnish population. Arch Otolaryngol Head Neck Surg. 2007;133(1):73–77. | |
Morrison AW. Anticipation in Meniere’s disease. J Laryngol Otol. 1995;109(6):499–502. | |
Oliveira CA, Ferrari I, Messias CI. Occurrence of familial Ménière’s syndrome and migraine in Brasilia. Ann Otol Rhinol Laryngol. 2002; 111(3 Pt 1):229–236. | |
Fung K, Xie Y, Hall SF, Lillicrap DP, Taylor SA. Genetic basis of Familial Ménière’s disease. J Otolaryngol. 2002;31(1):1–4. | |
Hietikko E, Kotimäki J, Kentala E, et al. Finnish familial Ménière disease is not linked to chromosome 12p12.3, and anticipation and cosegregation with migraine are not common findings. Genet Med. 2011;13(5):415–420. | |
Xenellis J, Morrison AW, McClowskey D, Festenstein H. HLA antigens in the pathogenesis of Ménière’s disease. J Laryngol Otol. 1986;100(1):21–24. | |
Koyama S, Mitsuishi Y, Bibee K, Watanabe I, Terasaki PI. HLA associations with Ménière’s disease. Acta Otolaryngol. 1993;113(5):575–578. | |
Melchiorri L, Martini A, Rizzo R, et al. Human leukocyte antigen-A, -B, -C and -DR alleles and soluble human leukocyte antigen class I serum level in Ménière’s disease. Acta Otolaryngol Suppl. 2002;(548):26–29. | |
Lopez-Escamez JA, Lopez-Nevot A, Cortes R, et al. Expression of A, B, C and DR antigens in definite Meniere’s disease in a Spanish population. Eur Arch Otorhinolaryngol. 2002;259(7):347–350. | |
Lopez-Escamez JA, Vilchez JR, Soto-Varela A, et al. HLA-DRB1*1101 allele may be associated with bilateral Ménière’s disease in southern European population. Otol Neurotol. 2007;28(7):891–895. | |
Morrison AW, Mowbray JF, Williamson R, Sheeka S, Sodha N, Koskinen N. On genetic and environmental factors in Menière’s disease. Am J Otol. 1994;15(1):35–39. | |
Khorsandi MT, Amoli MM, Borghei H, et al. Associations between HLA-C alleles and definite Meniere’s disease. Iran J Allergy Asthma Immunol. 2011;10(2):119–122. | |
Manolis EN, Yandavi N, Nadol JB Jr, et al. A gene for nonsyndromic autosomal dominant progressive postlingual sensorineural hearing loss maps to chromosome 14q12–13. Hum Mol Genet. 1996;5(7):1047–1050. | |
Sanchez E, Lopez-Escamez JA, Lopez-Nevot MA, et al. Absence of COCH mutations in patients with Ménière disease. Eur J Hum Genet. 2004;12(1):75–78. | |
Usami S, Takahashi K, Yuge I, et al. Mutations in the COCH gene are a frequent cause of autosomal dominant progressive cochleo-vestibular dysfunction, but not of Meniere’s disease. Eur J Hum Genet. 2003;11(10):744–748. | |
Fransen E, Verstreken M, Verhagen WI, et al. High prevalence of symptoms of Meniere’s disease in three families with a mutation in the COCH gene. Hum Mol Genet. 1999;8(8):1425–1429. | |
Verstreken M, Declau F, Wuyts FL, et al. Hereditary otovestibular dysfunction and Ménière’s disease in a large Belgian family is caused by a missense mutation in the COCH gene. Otol Neurotol. 2001;22(6):874–881. | |
Khetarpal U. DFNA9 is a progressive audiovestibular dysfunction with a microfibrillar deposit in the inner ear. Laryngoscope. 2000;110(8):1379–1384. | |
Klar J, Frykholm C, Friberg U, Dahl N. A Meniere’s disease gene linked to chromosome 12p12.3. Am J Med Genet B Neuropsychiatr Genet. 2006;141B(5):463–467. | |
Montcouquiol M, Corwin JT. Intracellular signals that control cell proliferation in mammalian balance epithelia: Key roles for phosphatidylinositol-3 kinase, mammalian target of rapamycin, and S6 kinases in preference to calcium, protein kinase C, and mitogen-activated protein kinase. J Neurosci. 2001;21(2):570–580. | |
Gabrikova D, Frykholm C, Friberg U, et al. Familiar Ménière’s disease restricted to 1.48 Mb on chromosome 12p12.3 by allelic and haplotype association. J Hum Genet. 2010;55(12):834–837. | |
Yazdani N, Khorsandi Ashtiani MT, Zarandy MM, et al. Association between MIF gene variation and Meniere’s disease. Int J Immunogenet. 2013;40(6):488–491. | |
Gázquez I, Moreno A, Requena T, et al. Functional variants of MIF, INFG and TFNA genes are not associated with disease susceptibility or hearing loss progression in patients with Ménière’s disease. Eur Arch Otorhinolaryngol. 2013;270(4):1521–1529. | |
Mhatre AN, Jero J, Chiappini I, et al. Aquaporin-2 expression in the mammalian cochlea and investigation of its role in Meniere’s disease. Hear Res. 2002;170(1–2):59–69. | |
Candreia C, Schmuziger N, Gurtler N. Molecular analysis of aquaporin genes 1 to 4 in patients with Meniere’s disease. Cell Physiol Biochem. 2010;26(4–5):787–792. | |
Maekawa C, Kitahara T, Kizawa K, et al. Expression and translocation of aquaporin-2 in the endolymphatic sac in patients with Meniere’s disease. J Neuroendocrinol. 2010;22(11):1157–1164. | |
Lynch M, Cameron TL, Knight M, et al. Structural and mutational analysis of antiquitin as a candidate gene for Meniere disease. Am J Med Genet. 2002;110(4):397–399. | |
Lee P, Kuhl W, Gelbart T, Kamimura T, West C, Beutler E. Homology between a human protein and a protein of the green garden pea. Genomics. 199415;21(2):371–378. | |
Wang W, Kim HJ, Lee JH, et al. Functional significance of K+ channel β-subunit KCNE3 in auditory neurons. J Biol Chem. 2014;289(24):16802–16813. | |
Doi K, Sato T, Kuramasu T, et al. Ménière’s disease is associated with single nucleotide polymorphisms in the human potassium channel genes, KCNE1 and KCNE3. ORL J Otorhinolaryngol Relat Spec. 2005;67(5):289–293. | |
Campbell CA, Della Santina CC, Meyer NC, et al. Polymorphisms in KCNE1 or KCNE3 are not associated with Ménière disease in the Caucasian population. Am J Med Genet A. 2010;152A(1):67–74. | |
Hietikko E, Kotimäki J, Okuloff A, Sorri M, Männikkö M. A replication study on proposed candidate genes in Ménière’s disease, and a review of the current status of genetic studies. Int J Audiol. 2012;51(11):841–845. | |
Teggi R, Lanzani C, Zagato L, et al. Gly460Trp alpha-adducin mutation as a possible mechanism leading to endolymphatic hydrops in Ménière’s syndrome. Otol Neurotol. 2008;29(6):824–828. | |
Kawaguchi S, Hagiwara A, Suzuki M. Polymorphic analysis of the heat-shock protein 70 gene (HSPA1A) in Ménière’s disease. Acta Otolaryngol. 2008;128(11):1173–1177. | |
Vrabec JT, Liu L, Li B, Leal SM. Sequence variants in host cell factor C1 are associated with Ménière’s disease. Otol Neurotol. 2008;29(4):561–566. | |
Lopez-Escamez JA, Saenz-Lopez P, Acosta L, et al. Association of a functional polymorphism of PTPN22 encoding a lymphoid protein phosphatase in bilateral Ménière’s disease. Laryngoscope. 2010;120(1):103–107. | |
Furuta T, Teranishi M, Uchida Y, et al. Association of interleukin-1 gene polymorphisms with sudden sensorineural hearing loss and Ménière’s disease. Int J Immunogenet. 2011;38(3):249–254. | |
Lopez-Escamez JA, Moreno A, Bernal M, et al. Poly(ADP-ribose) polymerase-1 (PARP-1) longer alleles spanning the promoter region may confer protection to bilateral Meniere’s disease. Acta Otolaryngol. 2009;129(11):1222–1225. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.