")
Back to Journals » The Application of Clinical Genetics » Volume 8
The genetics of familial hypercholesterolemia and emerging therapies
Authors Vogt A
Received 7 July 2014
Accepted for publication 28 August 2014
Published 28 January 2015 Volume 2015:8 Pages 27—36
DOI https://doi.org/10.2147/TACG.S44315
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Anja Vogt
Medizinische Klinik und Poliklinik IV, Klinikum der Unversität München, Munich, Germany
Abstract: Familial hypercholesterolemia (FH) results in very high levels of atherogenic low-density lipoprotein (LDL) cholesterol from the time of birth. Mutations of the genes encoding for the LDL receptor, apolipoprotein B and proprotein convertase subtilisin/kexin type 9, are causes for this autosomal dominant inherited condition. Heterozygous FH is very common, while homozygous FH is rare. Affected individuals can experience premature cardiovascular disease; most homozygous patients experience this before the age of 20 years. Since effective LDL cholesterol lowering therapies are available, morbidity and mortality are decreased. The use of statins is the first choice in therapy; combining other lipid-lowering medications is recommended to lower LDL cholesterol sufficiently. In some cases, lipoprotein apheresis is necessary. In heterozygous FH, these measures are effective to lower LDL cholesterol, but in severe cases and in homozygous FH there remains an unmet need. Emerging therapies, such as the recently approved microsomal triglyceride transfer protein inhibitor and the apolipoprotein B antisense oligonucleotide, might offer further options for these patients with very high cardiovascular risk. Early diagnosis and early treatment are important to reduce cardiovascular events and premature death.
Keywords: familial hypercholesterolemia, LDL cholesterol, atherosclerosis, genetics, new therapies, mipomersen
Introduction
Familial hypercholesterolemia (FH) is associated with very high levels of low-density lipoprotein (LDL) cholesterol (LDL-C) from the time of birth. This can lead to myocardial infarctions or death early in life, both of which can be prevented or at least delayed.1–4 FH has been known for a long time; the therapeutical measures to treat it are improving, and there are many recommendations and guidelines for detecting and managing affected persons.3,5–7 Despite these factors, FH is underdiagnosed and undertreated worldwide.1,3 It is challenging to convey the importance of this condition to the general population, because high levels of LDL-C do not cause any symptoms and because affected persons generally feel healthy. The severity of this condition has to be explained repeatedly. Because of the exposure to high levels of LDL-C from birth, the lifetime exposure is very different than for persons acquiring hypercholesterolemia later in life.
FH is inherited in an autosomal dominant way and can be present in a homozygous or a heterozygous form. FH is one of the most frequent monogenetic disorders. Heterozygous FH (heFH) is very common; in a study from 2001, one out of every 500 people was affected.8 In a more recent study, heFH was suspected to be even more frequent: one out of 250 people was affected.4 Homozygous FH (hoFH) is thought to be very rare (one out of 1,000,000 people are thought to be affected), but increased and improved diagnostics using a molecular approach indicate that hoFH might be much more common; they indicate that one out of 300,000 people are affected.9 In founder regions, monogenetic hypercholesterolemia is even more frequent. The European Society of Cardiology/European Atherosclerosis Society guideline for the management of dyslipidemia states very clearly that FH per se is a high risk constellation and has to be treated immediately without any cardiovascular risk estimation.6 The American Heart Association/American College of Cardiology (AHA/ACC) guidelines are not specific about FH, but patients with FH who are in the category of individuals with LDL-C levels >4.9 mmol/L (>190 mg/dL) qualify for intensive therapy.10
Background
Genetics
FH is inherited in an autosomal dominant way; FH is a type of autosomal dominant hypercholesterolemia (ADH). The long known classical FH (ADH1) is defined by mutations of the gene LDLR, which encodes for the LDL receptor (LDL-R). Because of the defective receptors that result, the uptake of LDL-C into the cells is either decreased or inhibited.11 This was first described by Brown and Goldstein.12 Now, more than 1,700 different variants of mutations in the LDLR, which include exonic substitutions, exonic small rearrangements, large rearrangements, promoter variants, and intronic variants, are known worldwide; mutations in the LDLR are the most common causes for FH.13 LDL-C uptake is decreased also if there is a mutation in the gene APOB, which encodes for apolipoprotein B (ApoB), the protein that recognizes LDL-C and enables the binding of LDL-C to the LDL-R. This condition is known as ADH2, or familial defective ApoB100 (FDB).14 One common mutation (R3500Q) and three rare mutations are known.15 The effects of mutations in proprotein convertase subtilisin/kexin type 9 (PCSK9) were deciphered comparatively recently. PCSK9 binds to the LDL-R/LDL-C complex and promotes the degradation of the LDL-R in lysosomes, resulting in lower numbers of LDL-R. Gain of function mutations of PCSK9 (ADH3 and for which >20 mutations are known) result in lower expression of LDL-R on the cell surface and higher levels of LDL-C; conversely, loss of function mutations result in higher expressions of LDL-R on the cell surface and lower levels of LDL-C.1,16 In individuals with loss of function mutations and lower levels of LDL-C, the incidence of cardiovascular disease (CVD) is significantly reduced. This supports the suggestion that low levels of LDL-C from the time of birth are associated with lower cardiovascular risk.16
Autosomal recessive hypercholesterolemia is very rare and can resemble autosomal dominant hoFH clinically. It is caused by mutations in the LDL-R adaptor protein 1 gene (LDLRAP1), which encodes for the putative adaptor protein that binds to the LDL-R and thereby promotes the internalization of the LDL-R/LDL-C complex into the liver cells.17
Homozygous FH can be due to simple homozygosity (same mutation in the same gene on both alleles) or due to compound heterozygosity (two different mutations in the same gene). Double heterozygosity, a rare form of hoFH, is a condition in which there are mutations on two different alleles encoding for FH; one mutation is usually on LDLR, and the second is on one of the other three loci. The levels of LDL-C in patients with hoFH depend on the receptor activity independent of the causative defect. Patients who are receptor negative (those who have <2% activity) are more severely affected than patients who are receptor defective (those who have 2%–25% activity).18 Patients with ADH1 seem to be affected by CVD more frequently than patients with ADH2.19
LDLR mutations are the most common reasons for FH; they account for over >95% of the cases. Mutations in other genes are rare; APOB mutations make up 2%–5% of cases, and mutations in PCSK9 and LDLRAP1 each make up <1% of all FH cases.18
In sitosterolemia, the intestinal uptake of plant sterols is increased due to mutations in two ATP binding cassette transporter genes, ABCG5 and ABCG8, both of which are autosomal recessive. Homozygosity is rare, but the clinical appearance is similar to hoFH with very high levels of LDL-C, xanthomata of the tendons, and early CVD. Unlike in the plasma of patients with hoFH, plant sterols levels are very high in plasma of patients with sitosterolemia.18,20
Clinical manifestations
LDL-C and cardiovascular disease
In FH, LDL-C is markedly elevated from the time of birth. In heFH, levels range from 8–15 mmol/L (310–580 mg/dL) and in hoFH, from 12–30 mmol/L (460–1,160 mg/dL). These high levels increase the risk for premature CVD drastically in men and women of both groups. In heFH, CVD typically develops before the age of 55 years in men and 60 years in women. In hoFH, CVD occurs very early in life. If untreated, CVD causes death usually before the age of 20 years, but sometimes as early as in the first year of life.1 The Simon–Broom Registry showed an increased cardiovascular mortality (relative risk) in women (125-fold) and in men (50-fold) aged 20–39 years.2 Stenoses of the aortic valve and of the aortic root are typical in hoFH.21
External signs of hypercholesterolemia
If a child is presenting with xanthomata, these obvious signs should lead to a prompt and complete workup. Xanthomata occur in hoFH and are typically planar (eg, buttocks) or at the tendons (eg, Achilles tendons). If a child is treated for FH, xanthomata might not develop; this has to be kept in mind when diagnosing FH on the basis of clinical features. An arcus lipoides before the age of 45 years, and especially in childhood, is also a sign of FH.
Diagnostics
The diagnosis of FH can be made clinically or genetically. However, in most countries, there are no screening projects or prevention programs, and it is still a coincidence if a person is diagnosed properly.
Clinical diagnosis
The measuring of LDL-C is pathbreaking in most cases. To further elucidate the risk with regard to lipid disorders, high-density lipoprotein (HDL) cholesterol (HDL-C), triglycerides, and lipoprotein a (Lp[a]) should be measured because in FH, HDL-C as a protective factor is often reduced, and Lp(a) as an additional and independent risk factor is often elevated.22 To define the global cardiovascular risk, all cardiovascular risk factors that aggravate the risk of FH should be recorded. Secondary causes for hypercholesterolemia, such as hypothyroidism, liver dysfunction, renal dysfunction, and diabetes mellitus (DM), have to be excluded.23 If a person is on a lipid-lowering medication, high levels of LDL-C might be overlooked.
Drawing a detailed pedigree is the cornerstone for diagnosing FH. Data about levels of cholesterol, cardiovascular events in first-degree relatives, and the age at the first event are recorded. It has to be noticed that parents of affected children or young adults are still young themselves. A negative history of cardiovascular events, therefore, does not exclude the diagnosis of FH; but, the family history has to be taken repeatedly over the course of time, and second-degree relatives have to be included in the family history.
There are different criteria to define FH clinically. The Dutch Lipid Clinic Criteria (Table 1) and the Simon–Broome Criteria include lipid values, family history, cardiovascular events of the person to be diagnosed and of that person’s relatives, and the existence of tendon xanthomata. Depending on the results, the diagnosis of FH is “definite”, “probable”, or “possible”. The Make Early Diagnosis to Prevent Early Death criteria are based on either total cholesterol or LDL-C in the patient and on histories of the first-, second-, and third-degree relatives.24
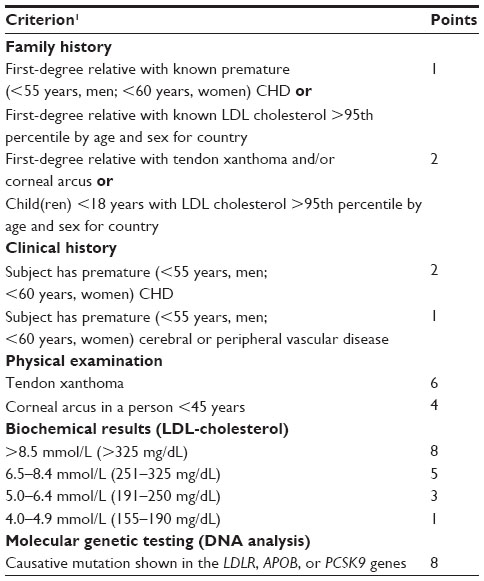 | Table 1 Dutch Lipid Clinic Network criteria for diagnosis of heterozygous FH in adults |
Genetic diagnosis
Genetic testing allows a definite diagnosis in most cases and is recommended by most of the guidelines regarding FH.1,5–7 Especially if LDL-C is not markedly elevated, the detection of a causative mutation is important for treatment decisions. An effective and cost-effective way is cascade screening. The first diagnosed patient is called the index patient. His or her first-degree relatives are screened only for the detected mutation. This is repeated for the first-degree relatives (older and younger) of every newly detected person who is affected. Because of autosomal dominant inheritance, the chance of finding the mutation in a first-degree relative is 50%. It is noteworthy that although a mutation is detected, LDL-C might not be elevated; conversely, although LDL-C is elevated markedly and FH is diagnosed on the basis of the clinical criteria, a mutation might not be detected. Since an unknown mutation does not rule out FH, the clinical diagnosis determines the approach. In the first case, it was recommended that the patient should not be started on lipid-lowering therapy, but that he or she be monitored and has follow-up measurements taken. In the second case, the initiation of lipid-lowering therapy was recommended.1
The knowledge of the underlying genetic mutation might increase the willingness to start treatment early and the compliance with regular and effective therapy. This has to be evaluated individually, and local laws regarding genetic testing have to be followed. Involving a specialized lipid center might be helpful.
Technical assessments
Findings of technical examinations may help affected persons to understand their risks which, otherwise, are difficult to realize. The guidelines mention different methods. Echocardiography is important because in FH, supravalvular stenosis of the aorta and stenosis of the aortic valve are typical.6,21,25,26 Exercise electrocardiograms and other stress tests are valuable for early detection of CVD. More elaborate and expensive methods, such as cardiac computed tomography or cardiac magnetic resonance imaging, are not recommended and should be considered on an individual basis. Duplex sonography of the carotid arteries is a well-documented, validated, and noninvasive method. A trial on children showed that the intima-media thickness increases significantly from age 12 years for those with heHF, unlike for children with normal LDL-levels.27 Ultrasounds of the abdomen would detect calcifications of the abdominal aorta, and measurement of the ankle–brachial index would indicate peripheral arterial disease.
Clinical experiences show that any result of a technical measure facilitates the understanding of the disease. Negative results may motivate patients to preserve their health, and presentation of early alterations may motivate patients to reduce the levels of the alterations or to keep them stable. Regular follow ups to detect changes over time are reasonable, and intervals should be chosen on the basis of the first findings. In cases of positive findings, a cardiologist or angiologist should be involved for further diagnostics or treatment.
Treatment targets
According to the AHA/ACC guidelines, individuals with LDL-C >4.9 mmol/L (>190 mg/dL) qualify for intensive therapy.10 Most patients with FH would be in this category. The Consensus Statement of the European Atherosclerosis Society recommends an LDL-C level of <3.5 mmol/L (<135 mg/dL) for children, of <2.5 mmol/L (<100 mg/dL) for adults, and of <1.8 mmol/L (<70 mg/dL) for adults with established CVD or DM in heFH and hoFH.1 These targets are based solely on the diagnosis of FH as a high risk constellation without any risk calculation.6 If it is not possible to reach the target value, a LDL-C reduction of at least 50% should be obtained. This is also recommended in the guidelines of the United States National Lipid Association and the National Institute for Health and Care and Excellence in the United Kingdom.3,5,6 Treatment should start early in life and should be rigorous enough to reduce and postpone cardiovascular sequelae. Reducing cardiovascular morbidity and mortality are the ultimate targets. It should be mentioned again that other potential cardiovascular risk factors have to be addressed, too.
Treatment
Treatment options comprise lifestyle factors, statin-based drug therapy, and lipoprotein apheresis (Table 2).
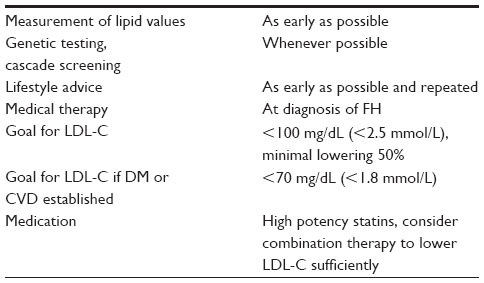 | Table 2 Clinical approach of the diagnosis of and therapy for FH |
Lifestyle
Establishing a healthy lifestyle is crucial, even if LDL-C cannot be lowered drastically.5–7 Lifestyle comprises diet and drinking, physical behavior, and not smoking. The last is the most important; passive smoking also has to be avoided by affected persons. Counseling should be offered repeatedly as patients get older, and smoking parents of affected children have to be included in the treatment. Physical exercise should become a routine early in life. Regarding LDL-C, in the diet, saturated fat should be reduced and unsaturated fatty acids and fiber, especially soluble fiber, should be increased. Additionally, nuts can lower LDL-C. Functional food (eg, margarine, milk) enriched with phytosterols is mentioned by some guidelines and is recommended by some authors, but this is still debated, since endpoint trials are lacking.5,6,28 Rare cases of sitosterolemia that can present phenotypically like FH would have to be ruled out before phytosterols are used.
Other healthy diet patterns, such as reducing soft drinks, alcoholic drinks, and short chain carbohydrates, are important to maintain weight or to reduce a high body weight, and to address metabolic syndrome and DM. The global benefits of establishing and maintaining a healthy lifestyle are beyond lipid values and, therefore, should be the focus of counseling.
Lipid-lowering medications
Statins are widely accepted and are recommended as the first-line therapy for lowering LDL-C.1,5,6 Although the data of prospective and randomized trials in FH are not available, retrospective analyses show substantial benefit. An immediate start with the highest dose of one of the most potent statins was recommended recently.1 LDL-C should be lowered as fast and as far as possible. After initiation with a statin, ezetimibe, nicotinic acid, or bile acid sequestrants should be added to reduce LDL-C as effectively as possible.5
Functional LDL-Rs are crucial for deploying the effect of statins. Their LDL-C lowering capacity in heFH is significant, but in hoFH is limited; this capacity differs widely and depends on the person’s underlying genetic makeup. Since the time that statins were first available, the incidence of cardiovascular events has decreased in primary and secondary prevention in non-FH patients.29 The recommendations for FH patients are based on these data and on studies showing that statins lower LDL-C and CVD events in FH patients as well. The Simon–Broome Register Group showed a declining cardiovascular mortality in heFH after the launch of statins.2 These observations were made repeatedly. Treatment with statins (mostly simvastatin and atorvastatin) was effective at reducing the risk of myocardial infarction to that of nonaffected individuals.30
Ezetimibe reduces intestinal cholesterol absorption, and its LDL-C lowering effect depends on upregulated LDL-R. The drug is proven to be safe and effective in heFH, and the combination of a statin with ezetimibe lowers LDL-C significantly more than statin monotherapy.31 Long-term treatment either with a statin alone or with a statin and ezetimibe was administered to 89 FH children and adolescents whose data were evaluated retrospectively. Among these patients, cardiovascular risk decreased significantly and no cases of CVD developed.32
Bile acid sequestrants lower LDL-C when given as a monotherapy and when given in combination with statin and ezetimibe therapy.33,34 Nicotinic acid lowers not only LDL-C, but Lp(a) and triglycerides as well. In addition, nicotinic acid increases HDL-C. Thus, if locally available, nicotinic acid can be used to further reduce LDL-C and to improve the levels of other unfavorable lipoproteins.35
Although FH and the therapies for it have been known for many years now, and although multiple treatment modalities are available, Pijlman et al36 showed in a cross-sectional study in 2010 that the majority of FH patients were not receiving optimal treatment. In this study, LDL-C was not reduced <2.5 mmol/L (<100 mg/dL) in 79% of patients, and only 21% of patients were on combined therapy.36 Firstly, this shows that the available therapies are not used sufficiently, perhaps because of the lack of awareness by patients or doctors and perhaps because of adverse effects, such as statin-induced myopathy. Secondly, this suggests that new measures might be necessary in cases of statin intolerance or in cases in which statins are unable to sufficiently lower LDL-C. Repeated information and support is necessary, and affected persons should be cared for in specialized lipid clinics. Currently, there are some new LDL-C lowering agents under investigation; these will potentially increase the armamentarium of LDL-C lowering drugs. Two of these agents were launched recently (see the following sections).
Apheresis
In hoFH and severe cases of heFH, LDL-C levels cannot be lowered sufficiently by lipid-lowering medications, even if they are used in the highest doses and maximum combinations.36 In these cases, LDL-C can be reduced significantly by lipoprotein apheresis. This extracorporeal technic decreases ApoB100-containing particles (LDL-C and Lp[a]); apheresis is applied either in addition to the maximally tolerated lipid-lowering medication or alone if lipid-lowering medications are not tolerated. Several technics are available, all reducing LDL-C acutely by about 60%–70%. LDL-C increases again rapidly in the first days following a kinetic of first order.37 Therefore, apheresis has to be repeated regularly, and the time-averaged lipoprotein levels have to be calculated.38 Usually, apheresis has to be repeated every week; in some patients who respond very well to the concomitant lipid-lowering medications, a biweekly treatment might be sufficient.37,39 The recommendations for starting lipoprotein apheresis in FH differ worldwide. In the United States, apheresis is recommended for heFH in patients without CVD whose LDL-C levels are at least 7.76 mmol/L (300 mg/dL) and in patients with established CVD whose LDL-C levels are at least 5.17 mmol/L (200 mg/dL).5 In Germany, apheresis is indicated in hoFH without a threshold or further requirements. Apheresis is also indicated in patients with severe hypercholesterolemia whose LDL-C levels cannot be decreased to goal despite the maximal lipid-lowering medication. These, usually, are patients with severe heFH or with established CVD.40 Lipoprotein apheresis has been in use for over 30 years and is generally well tolerated and safe.41,42 The use of angiotensin-converting enzyme inhibitors is the only contraindication, because of a bradykinin reaction. Patients who use these inhibitors must change their antihypertensive therapy before they start apheresis. Examples of mild and usually easy-to-handle side effects are venous access problems or hypotension.39 FH patients undergoing lipoprotein apheresis have significantly reduced LDL-C levels and profit from this treatment option.37,43 Cardiovascular events can be reduced in hoFH patients.44 Cardiovascular morbidity and mortality can be significantly reduced in heFH, but they cannot be eliminated.45 This leads to the postulation that in hoFH, apheresis should be initiated as early in childhood as technically possible (as early as venous access is possible). Since lipoprotein apheresis is time consuming, expensive, and not available ubiquitously, new treatment options are awaited to reduce the necessity for lipoprotein apheresis.
Emerging therapies
Several new lipid-modifying drugs are currently in development. Agents to lower LDL-C target cholesteryl ester transfer protein (primarily developed to increase levels of HDL-C), PCSK-9, microsomal triglyceride transfer protein (MTP), or ApoB.46,47 All of these are tested in addition to baseline statin therapy and, to varying degrees, they are shown to lower LDL-C more than statin therapy alone. The effect of PCSK-9-inhibitors in FH and especially in hoFH is dependent on functional LDL-R; ongoing trials are addressing these high-risk patients. Agents acting independently of LDL-R might be more effective in FH. Recently, two agents were approved as orphan drugs for hoFH: the MTP-inhibitor lomitapide and the ApoB synthesis inhibitor mipomersen. The US Food and Drug Administration approved both agents; the European Medicines Agency approved lomitapide, but not mipomersen.
ApoB antisense oligonucleotide
Mipomersen is an ApoB antisense oligonucleotide that targets the encoding messenger (m)RNA. This leads to the inhibition of protein translation and the reduced production of ApoB100, the backbone protein of very LDL (VLDL). Less VLDL is assembled in the liver and released into the circulation; this results in lower levels of LDL-C. Another effect is that triglycerides, normally transported via VLDL into the circulation, accumulate in the cells. This can lead to fat accumulation in the liver and to elevated transaminases.48,49
The LDL-C- and Lp(a)-lowering effects were seen in all of the Phase II and Phase III trials, in which different populations were examined. In Phase III trials, 200 mg of mipomersen was injected subcutaneously once per week.50–53
A multicenter, randomized (2:1), double-blind study in hoFH compared the effects of 200 mg mipomersen, delivered subcutaneously, with those of the placebo; both treatments were given for 26 weeks and in addition to lipid-lowering therapy.54 All of the patients were older than 12 years of age and had been diagnosed either clinically or genetically. In the 51 patients, LDL-C was lowered significantly more in the mipomersen group (24.7%, baseline 11.4 mmol/L) than in the placebo group (3.3%, baseline 10.4 mmol/L). As expected, the most common side effects were injection side reactions (76% with mipomersen, 24% with placebo). Alanine aminotransferase rose only in the mipomersen group (12%).
In a 26-week, randomized (2:1) trial, patients with heFH and coronary heart disease were given 200 mg of mipomersen, injected subcutaneously, in addition to ongoing lipid-lowering therapy.55 LDL-C decreased significantly by 28.0% in the mipomersen group, but increased by 5.2% in the placebo group. As side effects, injection site reactions, flu-like symptoms, and alanine aminotransferase elevations were reported.
The FOCUS FH trial,56 ongoing as of this writing, compares two different regiments of mipomersen in patients with FH. Mipomersen might reduce the necessity for lipoprotein apheresis,57 and this question is addressed in trial NCT01598948, which is ongoing as of this writing.58
Mipomersen is now indicated in addition to lipid-lowering medications to reduce LDL-C, ApoB100, and non-HDL-C in patients with hoFH. It is contraindicated in moderate or severe hepatic impairment (Child–Pugh B or C), active liver disease, and cases of unexplained persistent elevations of serum transaminases. The most common adverse reactions in trials were injection site reactions (84%), flu-like symptoms (30%), nausea (14%), headaches (12%), and elevations in serum transaminases, specifically alanine transaminase (10%). To monitor these side effects, mipomersen is available only through a restricted prescribing and distribution program called the KYNAMRO Risk Evaluation and Mitigation Strategy.59 These side effects might reduce patients’ compliance, and since endpoint data are not available yet, the clinical benefit has to be evaluated individually.
MTP-inhibitor
The enzyme MTP is essential for the assembly of VLDL in the liver and chylomicrons in the intestine. By inhibiting MTP, the lower levels of VLDL result in lower levels of LDL-C. Triglycerides are held back in the liver; this leads to increased liver fat content. The dietary fat content has to be reduced to avoid gastrointestinal side effects. In FH, this approach to lower LDL-C is effective because it is independent of LDL-R.
In a single-arm, open-label, Phase III study, 29 patients with hoFH received the MTP-inhibitor lomitapide in addition to lipid-lowering therapy (apheresis was allowed) to assess efficacy and safety; this trial lasted 78 weeks. The dose was escalated up to 60 mg per day, and the median dose was 40 mg per day. Six patients discontinued the treatment. LDL-C was reduced by 50% from baseline to week 26, by 44% to week 56, and by 38% to week 78. Gastrointestinal symptoms were the most common side effects. In addition, the liver fat content increased, and four patients had elevated amino transaminase levels.60 This trial was extended to show persistent efficacy, and this long-term study is ongoing. Lomitapide is only available through the JUXTAPID Risk Evaluation and Mitigation Strategy Program.61
Special considerations
Women
Female patients have to be informed that all medications have to be stopped before a pregnancy is planned, and that medications can be started only after family planning is completed. Because it may not be possible for women to receive effective therapy for many years, it is of utmost importance that the therapy is very strict before and after that time period. In very severe cases, lipid apheresis can be offered during the time that no medical therapy is possible.
Children
LDL-C levels are very high from the time of birth, and children are in danger of experiencing cardiovascular events very early in life. If FH is detected very early, these sequelae can be avoided or delayed by the early onset of therapy (medication, apheresis, or both). It is important to care for FH families. It is important for them to understand that although the knowledge of the genetic condition can be frightening, that knowledge also puts them in a position to act actively for the health of their children. Elevated levels of LDL-C do not hurt and the deleterious effects are very difficult to imagine. Although genetic diagnosis is helpful, potential local laws regarding genetic testing of children have to be followed. If genetic testing is done, it is mandatory that very careful genetic education and explanations about the meaning of the results and about potential implications (eg, insurance policies) are provided before and after testing.
Guidelines of different countries give different advice as when to start statin therapy. Mostly, the ages between 8 years and 14 years are suggested, but the decision has to be made individually in every case and has to be based on lipid values, family history, and clinical appearance. One trial showed that a 2-year treatment with pravastatin in 8–18-year-old children with FH induced a significant regression of carotid intima-media thickness without any adverse events; this finding suggests “the younger, the better” when treating children.62 However, a Cochrane Review stated that only short-term trials in FH children were done.63 These trials raised no safety concerns, but long-term safety data are needed. While routine monitoring in adults is not always recommended, in children this monitoring of development and potential side effects is imperative until long-term data are available.6 Registries could improve our knowledge.
The psychosocial function of children with FH (number [n]=152) and healthy peers (n=62) was determined by a semistructured interview. Of the FH group, 25% had experienced the loss of or a CVD event in a parent, and this resulted in lower scores. Otherwise, FH children had no greater psychological dysfunction than their peers.64 The positive view that knowledge enables early treatment and consequently allows children to grow up healthy should be explained repeatedly. A transition clinic of pediatricians and internists could improve the treatment of those on the border between childhood and adulthood.
Quality of life
Some evidence is available about the quality of life of patients with FH. One study evaluated the psychosocial concerns of families with FH. For this study, 145 parents completed a questionnaire. None thought that not having their child diagnosed with FH would be better, but 11% believed that their quality of life would be better without the diagnosis, and 20% attributed familial conflicts to the FH diagnosis. Of the children, 22% (more boys than girls) worried about CVD.65 In a cohort of elderly FH patients, of which all were ≥65 years of age (n=37, 73% coronary heart disease) all showed the same quality of life as controls.66 The quality of life of seven hoFH patients on LDL apheresis was not significantly different from that of healthy subjects, except for that of two patients with severe coronary heart disease.43
Patients’ organizations
Worldwide patients’ organizations are established by FH patients and relatives who have the experience of not being addressed and treated optimally. Their aims are to raise the awareness for and to increase the knowledge of FH, to offer a platform for affected individuals and their families for sharing experiences, and to help establish and improve the care of persons with FH (http://www.cholco.org, http://www.fhjourneys.com, http://www.fh-foundation.org, http://www.learnyourlipids.com).
Conclusion
FH is a prime example of a highly effective preventive medicine. The genetic background and the pathophysiology are known, and diagnostic tools are available. The approach is defined in various national and international guidelines and recommendations. Therapy is safe and cost effective, and it reduces morbidity and mortality significantly. Nevertheless, FH is still underdiagnosed and undertreated. Nationally organized screening programs to detect FH early in life could improve the medical care and the morbidity-free life span. Wherever such a program is not established, everyone who cares for individuals with elevated LDL-C or for patients with early cardiovascular events is responsible to initiate and follow up the diagnostic and therapeutical steps. Based on a healthy lifestyle, which should be connoted positively, medical therapy should be started early and continued effectively. The use of statins is the first choice, and combination therapy should be considered to lower LDL-C significantly. Long-term strategies for the medical care are warranted to keep affected individuals on therapy.
The emerging medications might allow more effective treatment. They also offer the possibility to increase the awareness for FH.
In general, it should be emphasized repeatedly that although FH is a serious condition, it is very well known, and the therapy is available and normally very effective in reducing FH-related diseases. Thus, affected individuals and their families can handle the situation positively and actively.
Disclosure
Anja Vogt has received speaker honoraria for presentations and advisory board activities by Merck Sharp and Dohme; Genzyme, a Sanofi company; Kaneka; Fresenius; B Braun; and Amgen. Anja Vogt has received research support by Merck Sharp and Dohme. The author reports no other conflicts of interest in this work.
References
Nordestgaard BG, Chapman MJ, Humphries SE, et al; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478–3490a. | |
Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management. Scientific Steering Committee on behalf of the Simon Broome Register Group. Atherosclerosis. 1999;142(1):105–112. | |
National Institute for Health and Care Excellence [webpage on the Internet]. Identification and management of familial hypercholesterolaemia. London, UK: National Institute for Health Care and Excellence; 2008. Available from: http://www.nice.org.uk/CG071. Accessed | |
Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Familial hypercholesterolemia in the Danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab. 2012;97(11):3956–3964. | |
Goldberg AC, Hopkins PN, Toth PP, et al; National Lipid Association Expert Panel on Familial Hypercholesterolemia. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5(Suppl 3):S1–S8. | |
Catapano AL, Chapman J, Wiklund O, Taskinen MR. The new joint EAS/ESC guidelines for the management of dyslipidaemias. Atherosclerosis. 2011;217(1):1. | |
Watts GF, Gidding S, Wierzbicki AS, et al. Integrated guidance on the care of familial hypercholesterolemia from the International FH Foundation. J Clin Lipidol. 2014;8(2):148–172. | |
Goldstein JL, Brown MS. Molecular medicine. The cholesterol quartet. Science. 2001;292(5520):1310–1312. | |
Sjouke B, Kusters DM, Kindt I, et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome. Eur Heart J. Epub Feburary 28, 2014. | |
Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 Suppl 2):S1–S45. | |
Goldstein JL, Brown MS. Familial hypercholesterolemia: identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol. Proc Natl Acad Sci U S A. 1973;70(10):2804–2808. | |
Brown MS, Goldstein JL. Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Proc Natl Acad Sci U S A. 1974;71(3):788–792. | |
Usifo E, Leigh SE, Whittall RA, et al. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Ann Hum Genet. 2012;76(5):387–401. | |
Innerarity TL, Mahley RW, Weisgraber KH, et al. Familial defective apolipoprotein B-100: a mutation of apolipoprotein B that causes hypercholesterolemia. J Lipid Res. 1990;31(8):1337–1349. | |
Soufi M, Sattler AM, Maerz W, et al. A new but frequent mutation of apoB-100-apoB His3543Tyr. Atherosclerosis. 2004;174(1):11–16. | |
Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264–1272. | |
Garuti R, Jones C, Li WP, et al. The modular adaptor protein autosomal recessive hypercholesterolemia (ARH) promotes low density lipoprotein receptor clustering into clathrin-coated pits. J Biol Chem. 2005;280(49):40996–41004. | |
Cuchel M, Bruckert E, Ginsberg HN, et al; for the European Atherosclerosis Society Consensus Panel on Familial Hypercholesterolaemia. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35(32):2146–2157. | |
Vogt A, Keller C, Heigl C, Weiss N, Zöllner N. [Two forms of familial hypercholesterolemia: differences in cardiovascular risk factors, cardiac and extracardiac atherosclerosis.]. Dtsch Med Wochenschr. Epub August 15, 2014. German. | |
Wang J, Joy T, Mymin D, Frohlich J, Hegele RA. Phenotypic heterogeneity of sitosterolemia. J Lipid Res. 2004;45(12):2361–2367. | |
Allen JM, Thompson GR, Myant NB, Steiner R, Oakley CM. Cadiovascular complications of homozygous familial hypercholesterolaemia. Br Heart J. 1980;44(4):361–368. | |
Nenseter MS, Lindvig HW, Ueland T, et al. Lipoprotein(a) levels in coronary heart disease-susceptible and -resistant patients with familial hypercholesterolemia. Atherosclerosis. 2011;216(2):426–432. | |
Jansen AC, van Aalst-Cohen ES, Tanck MW, et al. The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: data in 2400 patients. J Intern Med. 2004;256(6):482–490. | |
Williams RR, Hunt SC, Schumacher MC, et al. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol. 1993;72(2):171–176. | |
Sprecher DL, Schaefer EJ, Kent KM, et al. Cardiovascular features of homozygous familial hypercholesterolemia: analysis of 16 patients. Am J Cardiol. 1984;54(1):20–30. | |
Rafeiyian S, Mojtahedzadeh S, Hekmat M, et al. Supravalvular and valvular aortic stenosis in heterozygous familial hypercholesterolemia. Med Princ Pract. 2007;16(4):315–317. | |
Wiegman A, de Groot E, Hutten BA, et al. Arterial intima-media thickness in children heterozygous for familial hypercholesterolaemia. Lancet. 2004;363(9406):369–370. | |
Gylling H, Plat J, Turley S, et al; European Atherosclerosis Society Consensus Panel on Phytosterols. Plant sterols and plant stanols in the management of dyslipidaemia and prevention of cardiovascular disease. Atherosclerosis. 2014;232(2):346–360. | |
Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366(9493):1267–1278. | |
Versmissen J, Oosterveer DM, Yazdanpanah M, et al. Efficacy of statins in familial hypercholesterolemia: a long term cohort study. BMJ. 2008;337:a2423. | |
van der Graaf A, Cuffie-Jackson C, Vissers MN, et al. Efficacy and safety of coadministration of ezetimibe and simvastatin in adolescents with heterozygous familial hypercholesterolemia. J Am Coll Cardiol. 2008;52(17):1421–1429. | |
Elis A, Zhou R, Stein EA. Treatment of familial hypercholesterolaemia in children and adolescents in the last three decades. Cardiol Young. 2014;24(3):437–441. | |
Tonstad S, Sivertsen M, Aksnes L, Ose L. Low dose colestipol in adolescents with familial hypercholesterolaemia. Arch Dis Child. 1996;74(2):157–160. | |
Huijgen R, Abbink EJ, Bruckert E, et al; Triple Study Group. Colesevelam added to combination therapy with a statin and ezetimibe in patients with familial hypercholesterolemia: a 12-week, multicenter, randomized, double-blind, controlled trial. Clin Ther. 2010;32(4):615–625. | |
Nordestgaard BG, Chapman MJ, Ray K, et al; European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844–2853. | |
Pijlman AH, Huijgen R, Verhagen SN, et al. Evaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: a large cross-sectional study in The Netherlands. Atherosclerosis. 2010;209(1):189–194. | |
Thompson GR. Lipoprotein apheresis. Curr Opin Lipidol. 2010;21(6):487–491. | |
Kroon AA, Aengevaeren WR, van der Werf T, et al. LDL-Apheresis Atherosclerosis Regression Study (LAARS). Effect of aggressive versus conventional lipid lowering treatment on coronary atherosclerosis. Circulation. 1996;93(10):1826–1835. | |
Winters JL. Lipid apheresis, indications, and principles. J Clin Apher. 2011;26(5):269–275. | |
Gemeinsamer Bundesausschuss. Richtlinie des Gemeinsamen Bundesausschusses zu Untersuchungs- und Behandlungsmethoden der vertragsärztlichen Versorgung. Berlin, Germany: Bundesanzeiger; 2014. German. | |
Thompson GR, Lowenthal R, Myant NB. Plasma exchange in the management of homozygous familial hypercholesterolaemia. Lancet. 1975;1(7918):1208–1211. | |
Thompson GR; HEART-UK LDL Apheresis Working Group. Recommendations for the use of LDL apheresis. Atherosclerosis. 2008;198(2):247–255. | |
Græsdal A, Bogsrud MP, Holven KB, et al. Apheresis in homozygous familial hypercholesterolemia: the results of a follow-up of all Norwegian patients with homozygous familial hypercholesterolemia. J Clin Lipidol. 2012;6(4):331–339. | |
Keller C. LDL-apheresis in homozygous LDL-receptor-defective familial hypercholesterolemia: the Munich experience. Atheroscler Suppl. 2009;10(5):21–26. | |
Mabuchi H, Koizumi J, Shimizu M, et al. Long-term efficacy of low-density lipoprotein apheresis on coronary heart disease in familial hypercholesterolemia. Hokuriku-FH-LDL-Apheresis Study Group. Am J Cardiol. 1998;82(12):1489–1495. | |
Norata GD, Ballantyne CM, Catapano AL. New therapeutic principles in dyslipidaemia: focus on LDL and Lp(a) lowering drugs. Eur Heart J. 2013;34(24):1783–1789. | |
Kramer W. Novel drug approaches in development for the treatment of lipid disorders. Exp Clin Endocrinol Diabetes. 2013;121(10):567–580. | |
Rader DJ, Kastelein JJ. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 2014;129(9):1022–1032. | |
Parhofer KG. Mipomersen: evidence-based review of its potential in the treatment of homozygous and severe heterozygous familial hypercholesterolemia. Core Evid. 2012;7:29–38. | |
Yu RZ, Geary RS, Flaim JD, et al. Lack of pharmacokinetic interaction of mipomersen sodium (ISIS 301012), a 2′-O-methoxyethyl modified antisense oligonucleotide targeting apolipoprotein B-100 messenger RNA, with simvastatin and ezetimibe. Clin Pharmacokinet. 2009;48(1):39–50. | |
Akdim F, Visser ME, Tribble DL, et al. Effect of mipomersen, an apolipoprotein B synthesis inhibitor, on low-density lipoprotein cholesterol in patients with familial hypercholesterolemia. Am J Cardiol. 2010;105(10):1413–1419. | |
Akdim F, Tribble DL, Flaim JD, et al. Efficacy of apolipoprotein B synthesis inhibition in subjects with mild-to-moderate hyperlipidaemia. Eur Heart J. 2011;32(21):2650–2659. | |
Akdim F, Stroes ES, Sijbrands EJ, et al. Efficacy and safety of mipomersen, an antisense inhibitor of apolipoprotein B, in hypercholesterolemic subjects receiving stable statin therapy. J Am Coll Cardiol. 2010;55(15):1611–1618. | |
Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375(9719):998–1006. | |
Stein EA, Dufour R, Gagne C, et al. Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation. 2012;126(19):2283–2292. | |
Sanofi (Genzyme, a Sanofi Company). A study of the safety and efficacy of two different regimens of mipomersen in patients with familial hypercholesterolemia and inadequately controlled low-density lipoprotein cholesterol (FOCUS FH). Available from: http://clinicaltrials.gov/show/NCT01475825. NLM identifier: NCT01475825. Accessed October 20, 2014. | |
Vogt A, Parhofer KG. The potential of mipomersen, an ApoB synthesis inhibitor, to reduce necessity for LDL-apheresis in patients with heterozygous familial hypercholesterolemia and coronary artery disease. Expert Opin Pharmacother. 2013;14(6):691–697. | |
Ludwig-Maximilians – University of Munich. Effect of mipomersen on LDL-cholesterol in patients treated by regular apheresis (MICA). Available from: http://clinicaltrials.gov/ct2/show/NCT01598948. NLM identifier: NCT01598948. Accessed October 20, 2014. | |
Genzyme, A Sanofi Company [webpage on the Internet]. KYNAMRO risk evaluation and mitigation strategy (REMS). Cambridge, MA: Genzyme Corporation; 2013. Available from: http://www.kynamrorems.com. Accessed October 20, 2014. | |
Cuchel M, Meagher EA, du Toit Theron H, et al; Phase 3 HoFH Lomitapide Study investigators. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381(9860):40–46. | |
Aegerion Pharmaceuticals, Inc. [webpage on the Internet]. JUXTAPID REMS Program. Cambridge, MA: Aegerion Pharmaceuticals; 2012. Available from: http://www.juxtapidremsprogram.com. Accessed | |
Rodenburg J, Vissers MN, Wiegman A, et al. Statin treatment in children with familial hypercholesterolemia: the younger, the better. Circulation. 2007;116(6):664–668. | |
Vuorio A, Kuoppala J, Kovanen PT, et al. Statins for children with familial hypercholesterolemia. Cochrane Database Syst Rev. 2014;7:CD006401. | |
Tonstad S, Nøvik TS, Vandvik IH. Psychosocial function during treatment for familial hypercholesterolemia. Pediatrics. 1996;98(2 Pt 1):249–255. | |
Tonstad S. Familial hypercholesterolaemia: a pilot study of parents’ and children’s concerns. Acta Paediatr. 1996;85(11):1307–1313. | |
Hyttinen L, Kekäläinen P, Vuorio AF, Sintonen H, Strandberg TE. Health-related quality of life in elderly patients with familial hypercholesterolemia. Int J Technol Assess Health Care. 2008;24(2):228–234. | |
Vogt A. Diagnostik und Therapie. Schwere, asymptotische Hypercholesterinämie im jungen Lebensalter [Diagnosis and therapy. Severity, asymptotic hypercholesterolemia at a young age]. CardioVasc. 2013;13(6):39–43. German. | |
Vogt A. Schwere, asymptotische Hypercholesterinämie im jungen Lebensalter. Diagnostik und Therapie. Cardiovasc. 2013;13(6):39–44. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.