")
Back to Journals » The Application of Clinical Genetics » Volume 7
The genetic basis of Lynch syndrome and its implications for clinical practice and risk management
Authors Cohen S, Leininger A
Received 22 April 2014
Accepted for publication 15 May 2014
Published 22 July 2014 Volume 2014:7 Pages 147—158
DOI https://doi.org/10.2147/TACG.S51483
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Video abstract presented by Stephanie A Cohen.
Views: 602
Stephanie A Cohen,1 Anna Leininger2
1Cancer Genetics Risk Assessment Program, St Vincent Health, Indianapolis, IN, USA; 2Minnesota Oncology, Woodbury, MN, USA
Abstract: Lynch syndrome is the most common cause of hereditary colon cancer, and accounts for as much as 3% of all colon and endometrial cancers. The identification and management of individuals with Lynch syndrome have evolved over the past 20 years, yet the syndrome remains vastly underdiagnosed. It is important for clinicians to recognize individuals and families who are at risk in order to be able to manage them appropriately and reduce their morbidity and mortality from this condition. This review will touch on the history of Lynch syndrome, the current knowledge of genotype–phenotype correlations, the cancers associated with Lynch syndrome, and management of individuals who are gene carriers.
Keywords: Lynch syndrome, hereditary cancer, hereditary nonpolyposis colorectal cancer, mismatch repair, mismatch repair genes, immunohistochemistry, microsatellite instability
Overview and genetic basis
Lynch syndrome is a dominantly inherited cancer syndrome in which predisposition to colorectal, endometrial, and other cancers occurs due to an underlying defect in the cellular mismatch repair (MMR) system. MMR proteins form a complex that detects and corrects replication errors. A compromised MMR system leads to accelerated accumulation of somatic mutations, often resulting in carcinogenesis.
MLH1, MSH2, MSH6, and PMS2 are among the genes that produce MMR proteins. Lynch syndrome is caused by a heritable mutation in one copy of an MMR gene. At a phenotypic level, Lynch syndrome is dominant with variable expressivity. Secondary, somatic loss of the corresponding normal allele compromises the function of the entire MMR complex; Lynch syndrome is therefore recessive at the cellular level. An estimated 70%–90% of Lynch syndrome is attributable to deleterious mutations in MLH1 and MSH2, with the remaining 10%–30% distributed approximately equally between MSH6 and PMS2.1–3 Up to 3% of Lynch syndrome is due to mutations in the EPCAM gene, which is involved in epithelial cell adhesion, cell signaling, and proliferation. EPCAM is directly upstream of MSH2, and deletions of the 3′ end of EPCAM result in epigenetic hypermethylation of the MSH2 promoter, causing Lynch syndrome.4
Lynch syndrome exhibits characteristic features of cancer predisposition syndromes, including substantially elevated risks for specific cancers, earlier onset, high rates of multiple primary cancers, and the absence of typical risk factors. Cancers associated with Lynch syndrome include colorectal, endometrial, ovarian, stomach, hepatobiliary, urinary, small bowel, brain/central nervous system, and sebaceous tumors. Cancer risks are strongly influenced by which MMR gene mutation is present but may also vary substantially between and within families, due to broader influences of the genome and gene–environment interaction.
Historical perspective and evolution of descriptive terms
The first colorectal cancer syndrome to be well characterized was called familial adenomatous polyposis (FAP) and was characterized by very early onset, massively prolific development of colorectal polyps. Later, when high rates of colorectal cancer were observed in some families in the absence of florid polyposis, the term “hereditary nonpolyposis colorectal cancer” (HNPCC) was used to describe this new clinical entity, distinguishing it from the previously recognized FAP. Based on clinical observations, the association of colorectal cancers with brain tumors was named Turcot syndrome, and colorectal cancers associated with sebaceous neoplasms and keratoacanthomas were termed Muir–Torre syndrome. Identifying the underlying molecular etiology led to the realization that Turcot syndrome with colorectal cancer and glioblastoma is due to an MMR deficit. Muir–Torre is also caused by underlying MMR defects, and both conditions are now recognized as part of the broader clinical spectrum of Lynch syndrome.
HNPCC became defined by an evolving series of criteria. The first was published in 1991 after an international meeting of researchers and clinicians (the International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer) in Amsterdam, the Netherlands.5 The Amsterdam criteria, which can be remembered using a “3–2–1” mnemonic, were intended to more precisely define a homogeneous population for research purposes. The Amsterdam criteria describe families who do not have FAP and in which three closely related individuals spanning at least two generations have had colorectal cancer, with at least one diagnosis occurring prior to age 50 years. “Closely related” is defined as one of the affected trio being a first degree relative of the other two. With increasing recognition of the extracolonic manifestations of Lynch syndrome over the next decade, criteria were revised in 1999 to include extracolonic cancers.6
The Amsterdam and revised Amsterdam criteria were developed with emphasis on specificity rather than sensitivity, and were intended for use as research criteria. Nevertheless, they became widely used clinically to identify high-risk families, with an estimated sensitivity and specificity of 60% and 70%.7,8 Authors of these criteria were careful to point out that these criteria should not be used to exclude individuals or families with features of Lynch syndrome from mutation analysis. Nevertheless, these criteria continue to be utilized in ways that were not intended, and it is unfortunate that some payers still utilize these criteria to determine eligibility for coverage of genetic testing.
“Lynch syndrome”, named for Dr Henry T Lynch, who was among the first to recognize and describe families with hereditary cancer predisposition, is now the accepted and preferred term to describe a hereditary syndrome caused by germline mutations that disrupt the function of an MMR gene. Although “HNPCC” is still used somewhat interchangeably with “Lynch syndrome”, it fails to recognize the associated extracolonic features and is less specific, as not all family history-defined HNPCC has underlying MMR defects. The eponymous Dr Henry Lynch is internationally recognized for his contributions to the discovery of the syndrome, his descriptions of the natural history, raising awareness by publishing and speaking, and his graciousness and support for organizations that work directly with individuals and families with Lynch syndrome.
Not long after discovery in the mid-1990s, commercial testing became available for MLH1 and MSH2 around the turn of the decade, with genetic testing for four MMR genes plus EPCAM available within 12 years (Figure 1). Families who meet Amsterdam I criteria but do not have an MMR deficit as the underlying etiology (so-called familial colorectal cancer type X) have been described and characterized.9 These families have elevated colorectal cancer risk compared with a general population but not the same magnitude of risk as Lynch syndrome, and do not appear to have elevated risk for extracolonic cancers. The underlying genetic causes remain undefined, although with the recent advent of next-generation sequencing panels, additional genes will likely be implicated in some cases.
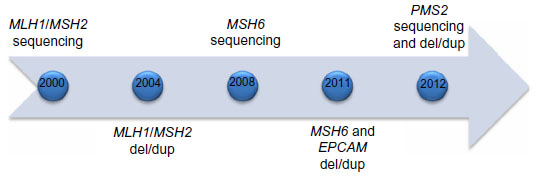 | Figure 1 Approximate time line for availability of mismatch repair gene tests. |
Clinical spectrum of Lynch syndrome
The risks of developing Lynch syndrome-associated cancers are gene and sex influenced. Initial studies tended to overestimate penetrance, due to the purposeful selection bias of the Amsterdam criteria. Penetrance data and cancer risk estimates have continued to evolve as genetic testing becomes more widespread. Generally, cancer risk estimates have trended downward, and the lower penetrance genes PMS2 and MSH6 have been found to account for a higher proportion of Lynch syndrome than previously recognized.10 As gene and age-specific data evolve, it becomes important from a clinical standpoint to review recent literature for the most accurate risk estimates associated with a particular gene. Specific cancer risks associated with Lynch syndrome are reviewed regularly and displayed in tabular format in the National Comprehensive Cancer Network (NCCN) colorectal screening guidelines.11
Individuals with an MLH1 or MSH2 gene mutation have the highest risks and the widest array of cancers attributable to Lynch syndrome. In particular, men with an MSH2 mutation have the highest risk for several types of cancers.12–14 MSH6 carriers have lower colorectal cancer risks but substantial gynecologic cancer risks.15,16 PMS2 carriers have lower colorectal and gynecologic cancer risks.17 Data on extracolonic, nongynecologic cancers specific to MSH6 and PMS2 are sparse. Risks associated with EPCAM deletions are being elucidated. Deletions may occur in the 3′ end of EPCAM, or may span both EPCAM and MSH2. In cases where the deletion is in EPCAM only, the epigenetic silencing of MSH2 occurs only in cells that express EPCAM, and therefore creates a mosaic pattern of MSH2 inactivation. It appears that in people with this cause for Lynch syndrome the risk of colorectal cancer remains high, but endometrial cancer risk is low. Individuals with deletions that span both EPCAM and MSH2 have cancer risks similar to those with MSH2 mutations.4,18
Colorectal cancer
Features of colorectal cancer associated with Lynch syndrome include earlier average age at onset, right-sided predominance, elevated risk of synchronous and metachronous cancers, and rapid adenoma to carcinoma progression compared with sporadic adenomas.19–21 Histologic characteristics of Lynch syndrome-related colon cancers have been observed to be poorly differentiated, with tumor-infiltrating lymphocytes, mucin containing, and with signet ring or cribriform histology.22,23 There appears to be a survival advantage when matched stage for stage with non-Lynch syndrome colorectal cancers.24–26
Colorectal cancer risks are reported to be as high as 75%, with median ages reported from 44 years to 61 years in those with Lynch syndrome.27 These vary according to which gene is involved and are well documented in other publications and summarized in Table 1.12,16,17,28–34 Although 10% of colorectal cancers in the general population occur prior to age 50 years, in Lynch syndrome approximately 50% occur prior to age 50 years, before routine colorectal screening would typically commence.35,36 The rate of synchronous and metachronous colorectal cancers is dramatically elevated in colon and rectal cancer survivors with Lynch syndrome, with approximately 15%–20% developing a second colorectal cancer within 10 years, 40%–50% within 20 years, and >60% within 30 years.37 Finally, the average dwell time from onset of a polyp to onset of carcinoma is much shorter in Lynch syndrome. Polyps may progress to carcinoma within 2–3 years among individuals with Lynch syndrome, compared with from 4 years to >10 years in the general population.38,39
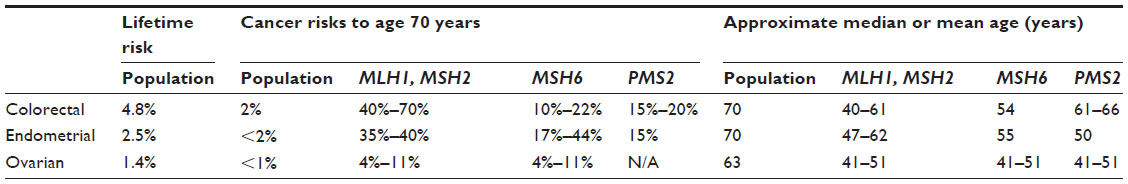 | Table 1 Colorectal and gynecologic cancer risks for people with Lynch syndrome compared with the general US population |
Endometrial cancer
Endometrial cancer is at least as likely as colorectal cancer to be the initial cancer diagnosis in women with Lynch syndrome, and synchronous endometrial/ovarian cancers are more likely.40–42 As many as 26% of female survivors of colorectal cancer due to Lynch syndrome will develop endometrial cancer within 10 years of initial diagnosis.42 Individuals with an MSH2 or MSH6 mutation have the highest risk for endometrial cancer, with a lifetime risk of up to 44% (Table 1).15,33
Lynch syndrome-associated endometrial cancers have primarily endometrioid histology, but other types, including clear cell, are observed.43 MMR-deficient endometrial cancers are more likely to exhibit specific morphological features, including peritumoral lymphocytes, prominent tumor-infiltrating lymphocytes, and heterogeneous tumors displaying two morphologically distinct tumor cell populations.44 The reported average ages of onset vary significantly and, in general, are younger, but there is evidence that the average age of onset may not be as early as previously thought, with the advent of universal screening for Lynch syndrome among individuals with endometrial cancer.16,45 Lynch syndrome is present in 8%–9% of women with early onset endometrial cancers, and 7%–21% of women with synchronous endometrial and ovarian cancers.41,44,46–49 Features of lower uterine segment endometrial cancers, which account for 3.5% of endometrial cancers overall, were observed in 42% of women with Lynch syndrome.50 As many as 30% of women with endometrial cancer of the lower uterine segment may have Lynch syndrome.51,52
Ovarian cancer
Approximately 2% of ovarian cancers are due to Lynch syndrome.53 When selected for early age of onset, specifically those diagnosed under the age of 40 years, the association with Lynch syndrome may be closer to 4%.54
Reported lifetime risks for ovarian cancer in women with Lynch syndrome fall primarily within the range of 6.7%–12% and appear highest for carriers of MSH2 mutations, followed by MSH6 and MLH1 (Table 1).13,16,55,56 Synchronous endometrial cancer is reported in ~22%, and 55% have a synchronous or metachronous Lynch syndrome-related cancer.49 Mean ages for diagnosis of ovarian cancer in Lynch syndrome are primarily in the 40 to 50-year range, with up to 30% of Lynch syndrome-related ovarian cancers diagnosed prior to age 35 years.13,49,53 Although Lynch syndrome-associated ovarian cancers are predominantly epithelial, unlike BRCA-related ovarian cancers, which are characteristically high grade serous, Lynch syndrome-associated ovarian cancers tend to display a higher proportion of endometrioid, clear cell, and mucinous cancers.49,57–60
Gastric cancer
Gastric cancer is reported to occur in approximately 5%–13% of individuals with Lynch syndrome, with considerable variability based on country of origin.13,60 Risks are reported to be higher in MLH1 and MSH2 than other mutation carriers, and higher in males than in females, with a mean age of onset of 55 years (Table 2).13,55,60,61 Lynch syndrome-related gastric cancer is primarily, but not exclusively, the intestinal type, with diffuse-type gastric cancers representing 12.5%–23%.60–62
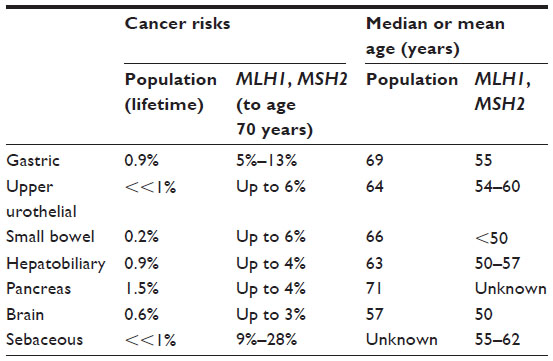 | Table 2 Extracolonic, nongynecologic cancer risks for MLH1/MSH2 carriers compared with the general US population |
Small bowel cancer
Up to 6% of individuals with Lynch syndrome develop small bowel cancer at a median age of <50 years for carriers of MLH1 and MSH2 mutations, and 54 years for MSH6 carriers (Table 2).55,63 As with several Lynch syndrome-associated extracolonic cancers, the risk appears highest in men with MSH2 mutations and lowest in carriers of MSH6 mutations, with scant data available for carriers of PMS2 mutations.55
Urinary tract cancer
Renal pelvis and urothelial (transitional cell) cancers are exceedingly rare in the general population.64 In contrast, people with Lynch syndrome have up to an 8% risk of developing upper urothelial cancers by age 70 years, at a median age of 58–62 years, with the highest risk occurring in men with MSH2 mutations (Table 2).13,65–67 Recent data suggest a two- to four-fold elevated risk of bladder cancer as well, such that for men with MSH2 mutations the risk of developing a urinary tract cancer by age 70 years may approach or exceed 20%.65,68
Sebaceous neoplasms
The presence of sebaceous neoplasms in individuals and families with other internal malignancies was referred to as Muir–Torre syndrome before molecular genetic testing demonstrated a common underlying etiology. Sebaceous neoplasms, particularly carcinomas, are exceedingly rare.69 Sebaceous neoplasms in people with Lynch syndrome are more likely than sporadic neoplasms to occur prior to age 60 years (median age 56 years), be multiple rather than isolated, and occur in the context of a personal or family history of Lynch syndrome-related cancer(s).70 The incidence of sebaceous neoplasms among individuals with Lynch syndrome has been reported to be as high as 9% (Table 2).71,72
Other rare tumors associated with Lynch syndrome
The spectrum of Lynch syndrome-associated tumors is wide, and several very rare cancers in the general population are seen more frequently in Lynch syndrome (Table 2).55 Although the risks for these rare tumors are greatly increased above the general population risks, the absolute risks are low. Individuals with Lynch syndrome have a risk of up to nearly 4% to develop pancreatic cancer by age 70 years. Pancreatic cancers appear most frequently in families with MSH2 mutations, followed by MLH1 and MSH6.73 Up to 4% of people with Lynch syndrome develop hepatobiliary cancer by age 70 years (median 50–57 years), another rare cancer in the general population.13 Finally, individuals with Lynch syndrome have up to a 3% lifetime risk of developing cancers of the brain and central nervous system, particularly glioblastoma.74
Evolving spectrum of Lynch syndrome-associated cancers
Prostate cancer has recently been associated with Lynch syndrome, and data are beginning to emerge regarding the risks and ages of onset. Several studies have found the lifetime risk for prostate cancer in Lynch syndrome to be increased by two- to five-fold.75–77 Additional studies are needed to determine whether Lynch syndrome-associated prostate cancers occur at an earlier average age or are more aggressive.75,76
The relationship between breast cancer and Lynch syndrome remains unresolved. Studies have not consistently demonstrated a higher than expected incidence of breast cancer among individuals with Lynch syndrome.78,79 Several studies have demonstrated evidence of MMR with loss of immunohistochemical staining in breast cancers found among known carriers of a mismatch gene mutation.80–82 As breast cancer is fairly common in the general population, larger studies are needed to determine whether breast cancer is indeed part of the Lynch syndrome cancer spectrum.
Surveillance
A major reason to identify individuals with Lynch syndrome is to optimize surveillance, which ultimately minimizes morbidity and mortality. Surveillance recommendations for individuals with Lynch syndrome differ substantially from those of the general population, due to the accelerated progression from colorectal adenoma to carcinoma and the increased incidence of cancers that can be avoided with prophylactic surgery, such as endometrial and ovarian cancers.83–85 The elevated risk for colorectal cancer to occur at young ages justifies the initiation of surveillance as young as age 20–25 years, depending on the family history and genotype.11 The right-sided predominance of colon cancers with Lynch syndrome necessitates a colonoscopy rather than a sigmoidoscopy.
The NCCN has published guidelines for management of individuals with Lynch syndrome that are regularly reviewed and updated, and there are several other publications that outline recommendations for surveillance.11,86,87 It is notable that the most recent version of the NCCN guidelines reflects evolving evidence that individuals with a PMS2 or MSH6 mutation may have reduced penetrance and thus may not require the same intensity of surveillance.
Surveillance for colon cancer should include annual or biannual colonoscopy and begin around the age of 25 years.86,88,89 Individuals with family members who were diagnosed at very young ages may consider colonoscopy earlier, typically 5–10 years before the earliest age of onset in the family. Recent evidence suggests that individuals with MSH6 or PMS2 mutations may be able to delay initiation of colonoscopy until as late as 30 years, due to the reduced penetrance.11,16,17 Fewer colon cancers are identified when surveillance with polypectomy is performed at 1 to 2-year intervals and cancers are identified at earlier stages with overall improved survival rates.89,90 Studies comparing regular light colonoscopy with use of indigo carmine dye have not noted improved overall survival.91 However, there was documented improvement in detection of very small polyps, and it remains to be determined whether this benefit may ultimately translate into better outcomes.
Endometrial cancer symptoms include abnormal uterine bleeding and pain, which are usually early indicators easily recognized by patients. Patients should be educated to seek medical evaluation if they experience abnormal bleeding. There is no clear management recommendation regarding endometrial biopsy for surveillance, as it is invasive, and there appears to be no evidence to suggest that outcomes are improved.92
Surveillance for other cancers is widely debated due to the lack of evidenced-based improvement of outcomes. Therefore, most groups do not make any specific recommendations for extracolonic cancer surveillance. Some practitioners may consider small bowel X-ray and/or upper endoscopy to screen for cancers of the upper gastrointestinal tract, and urinalysis with cytology to screen for urothelial cancer in individual cases, but there are no guidelines to direct these surveillance methods. Finally, the issue of dermatology screening has been raised, based on a single study that found that almost 10% of individuals with Lynch syndrome had sebaceous adenomas or the Muir–Torre variant of Lynch syndrome.71
Surgical considerations
There continues to be a debate about colectomy versus subtotal colectomy at the time of colon cancer treatment for individuals known to have Lynch syndrome. Surgeons may opt for a subtotal colectomy at the time of a colon cancer diagnosis in an individual with Lynch syndrome, despite lack of evidence demonstrating survival benefit.93,94 Quality of life issues following total abdominal colectomy should be carefully considered, as should access and adherence to surveillance, and management should be tailored on an individual basis.95,96 Few physicians would recommend prophylactic colectomy today, although it was considered early on in the chronicle of Lynch syndrome.
There is little debate with regard to prophylactic removal of the uterus and ovaries once childbearing is complete, due to the lack of effective surveillance of the ovaries and the significant decrease in the risk for both cancers following prophylactic surgery.83 The average age of onset of endometrial cancer in Lynch syndrome is 55 years, and current recommendations suggest total abdominal hysterectomy and bilateral salpingo-oophorectomy by the age of 50 years.97 Thorough pathological examination of surgical specimens is recommended, as gynecologic malignancies may be present already at the time of prophylactic surgery.98,99 Removing ovaries after or near the time of menopause eliminates some of the issues of early surgical menopause, as seen in BRCA1/2 carriers. However, 30% of Lynch syndrome-associated ovarian cancers occur prior to age 35 years, and there is no apparent contraindication for use of hormone replacement in this population. The likelihood of endometrial and ovarian cancer after prophylactic salpingo-oophorectomy is very low.83
Chemoprevention
Trials of aspirin have shown promise in reducing polyp burden among individuals with Lynch syndrome. Use of aspirin for 4 years or longer is associated with a reduction in the risk for colon cancer, although the effect is not evident until at least 5 years after the intervention.100 The optimal age to initiate the recommended dose and necessary duration of aspirin use has not been established, and studies are now ongoing. Birth control pills reduce the risk for both endometrial and ovarian cancer in the general population, and this effect was similar in a small cohort of women with Lynch syndrome.101
Recommendations for treatment and surveillance for Lynch syndrome continue to evolve. NCCN guidelines are reviewed and updated regularly, incorporating new information as it arises. Coordination of care in Lynch syndrome is essential to ensure that patients are getting the most appropriate and up-to-date care. This often requires collaboration between many different specialists, such as gastroenterologists, gynecologic oncologists or gynecologists, primary care providers, and genetic counselors.102
Psychosocial issues
Optimally, genetic testing should be done in a supportive setting with the expertise of a health care practitioner who is familiar with Lynch syndrome and some of the psychosocial issues that may be present. Genetic testing for Lynch syndrome may cause anxiety and distress, although studies have shown that the majority of individuals adapt to their results, and negative effects, if any, appear to be short term.103,104 Knowledge of one’s mutation and risk status may also provide individuals with a sense of control and optimism.105
There may be psychological effects of living with the threat of cancer. This may be influenced by prior experience with illness or death from cancer in the family. Feelings of guilt with regard to passing on the gene mutation or the possibility of passing on the gene mutation may also be present.105
Adherence to surveillance recommendations does not seem to be impacted by anxiety or cancer worry but may be impacted by an individual’s perceived barriers to screening.106 Barriers that have been described include discomfort, embarrassment, and lack of awareness of surveillance recommendations.106–108 Recognizing these factors in individual patients is important so that health care practitioners can maximize compliance with surveillance recommendations.
Identification of Lynch syndrome: universal screening
Lynch syndrome accounts for ~3% of all colorectal and endometrial cancers.2,97,109,110 Identification of Lynch syndrome has traditionally relied on multiple steps, including recognition of typical features and appropriate testing and/or referral to a genetics provider. Although there are some histological features within individual tumors that can indicate a likelihood of MMR deficit, and other clues, such as location within the body system (eg, lower uterine segment endometrial cancer or proximal colon cancer), Lynch syndrome-associated colon and endometrial cancers are not necessarily distinguishable from sporadic colon and endometrial cancers.36,111 Systematic collection, documentation, and assessment of family history are highly variable among health care providers, and rarely is this information readily available to pathologists who may recognize histological features of Lynch syndrome. Given these limitations and the compelling reasons to identify these individuals and their at-risk family members, universal screening has been proposed as a way to adequately identify individuals with Lynch syndrome.
Universal screening for Lynch syndrome is the evaluation of all colon and/or endometrial tumors at the time of diagnosis for evidence of MMR deficit. Microsatellite instability (MSI) and immunohistochemistry (IHC) are two screening methods used to identify affected individuals who may have Lynch syndrome. MSI is a measure of whether the MMR system is functioning. Loss of MMR function, which can be caused by Lynch syndrome or by epigenetic silencing of the MLH1 gene, results in MSI, which can be detected by polymerase chain reaction on a colon tumor specimen. Evidence of MSI suggests further workup to rule out Lynch syndrome. IHC is a demonstration of the presence or absence of MMR proteins in the tumor. Absence of a protein(s) as demonstrated by IHC staining suggests the possibility of a mutation in the corresponding gene.
The Bethesda criteria were developed to define populations for which colon tumor testing with MSI and/or IHC was indicated.112 These evolved over time to include personal and family history of extracolonic cancers.113 However, studies of universal screening of all colorectal and endometrial cancers suggest that as many as 70% of people with Lynch syndrome do not meet Amsterdam or Bethesda guidelines.97,114
Universal screening for Lynch syndrome has been demonstrated to be cost-effective, largely due to the identification of unaffected family members and subsequent prevention of colon and endometrial cancers.1,115–118 The issue of whether or not consent should be obtained prior to screening has been debated. However, in practice, direct informed consent prior to tumor screening is rare.119–121
There are documented challenges to implementation of a universal screening program for Lynch syndrome.122 One challenge has been to establish an effective process for notification and discussion with the patient, with subsequent patient uptake of genetic testing and notification of at-risk relatives.123–125 Individuals identified by universal screening for Lynch syndrome may not return for genetic counseling and testing if it is not apparent to them how it would impact their care and/or if their perception is that further evaluation is not warranted based on their family history.126 There have been several studies attempting new approaches to remove barriers and improve compliance with follow-up genetic counseling and testing, with variable success.127–129 Cascade testing for at-risk relatives is complex and depends upon effective intrafamilial communication.123,130 Factors that correlate with how well information is transmitted among family members include the education of individuals with Lynch syndrome and recommendations by the health care professionals who care for them.123,126,131 Most individuals with Lynch syndrome inform first-degree family members but are less likely to notify more distant relatives, due to lack of closeness and concerns that relatives may not understand the information shared.132 Resources to assist health care providers and families in the process of notifying at-risk relatives include an online tool, informational brochures, and a searchable database to identify genetic counselors (Table 3).
 | Table 3 Lynch syndrome resources |
Molecular testing does not always identify the underlying mutation in screen-positive individuals who are identified by tumor screening. Current technology may not detect all mutations, or there may be additional, yet-to-be-identified genes that cause Lynch syndrome. Biallelic somatic mutations may explain some cases of absent MMR proteins detected by IHC.133 Personal and family history may be helpful in distinguishing sporadic biallelic somatic mutations from true Lynch syndrome with no identifiable mutation.
Finally, the clinical utility of evaluating nonendometrial, extracolonic tumors for evidence of MMR continues to be unclear. The accuracy of screening ovarian tumors for MMR with IHC or MSI is questionable, although abnormal findings certainly warrant further evaluation and consideration of germline testing.134,135 Rare sebaceous tumors (including sebaceous adenomas, epitheliomas, and carcinomas) are strongly associated with the Muir–Torre variant of Lynch syndrome, and 30%–60% of sebaceous tumors are related to an MMR defect.136 However, there are differing opinions on the utility of screening all sebaceous tumors with IHC. Some authors have advocated for IHC screening of all sebaceous tumors.137,138 Others have argued that in the absence of a personal or family history of Lynch syndrome-associated cancers, the positive predictive value of IHC on sebaceous tumors is not high enough to warrant routine screening.70 A recent publication describes a scoring system based on the number of sebaceous neoplasms, age at diagnosis, and personal and family history of Lynch syndrome-associated cancers, and concludes that IHC alone is a poor predictor for identifying people with Lynch syndrome.139
Other Lynch syndrome-associated cancers may display evidence of MMR, but results tend to be less reliable than screening other tumors. There is incomplete concordance between MSI and IHC analysis in gastric tumors, so one group has recommended screening gastric tumors with both MSI and IHC when meeting revised Bethesda criteria.62 Other tumors have much fewer data regarding reliability of MSI and IHC in assessing likelihood for Lynch syndrome. The one exception is small bowel tumors, in which the performance of MSI and IHC appears to be similar to that in colon cancers.140
In conclusion, there are case reports of abnormal IHC among rare and unusual tumors in individuals with confirmed Lynch syndrome, but not enough evidence to routinely screen these tumors for evidence of MMR, nor enough evidence to be able to rule out suspected Lynch syndrome with normal IHC and/or MSI.141
Emerging issues
As the complex puzzle of Lynch syndrome genetics has been pieced together, inherited biallelic mutations in MMR genes have been found to cause constitutional MMR deficiency syndrome, a unique pediatric syndrome.142 Symptoms include extremely early onset cancers, including colon and small bowel cancers. Café-au-lait macules are common, seen among 70% of individuals with inherited biallelic MMR mutations, and the presence of colon polyposis has been reported as well. The average age of onset of colon cancer is around 15–16 years, and the average age of onset of small bowel cancer is 20 years.143,144
When younger family members of childbearing age are identified with Lynch syndrome, the issue of prenatal or preimplantation diagnosis may be raised. A small study of individuals at risk for Lynch syndrome found that childbearing decisions were impacted by whether or not a mutation was identified.145 Most surveyed believed that it was ethical to offer prenatal testing for Lynch syndrome, despite the fact that few intended to do so themselves. Genetic counselors and other specialty reproductive service providers are available to discuss options with families who indicate an interest in exploring reproductive options.
Studies are beginning to focus on gene–environment interactions, to attempt to explain some of the variability between families with mutations in the same gene, or even within families with the same gene mutation. For example, body mass index seems to be a risk factor for colorectal cancer but not endometrial cancer in Lynch syndrome.146,147 This type of information may be helpful in providing a sense of control to individuals at risk, and additional studies are needed in this complex area of gene–environment interactions.
Summary
The last 20 years have seen tremendous gains in knowledge about the underlying cause of Lynch syndrome, as well as increasing understanding of the spectrum of associated disease. Despite clear evidence that the most substantial cancer risks can be readily mitigated, Lynch syndrome remains underdiagnosed. It is critical to adapt to evolving criteria and universal screening to identify individuals with Lynch syndrome. Effective strategies to identify and appropriately manage the health of people with Lynch syndrome will continue to evolve. Astute clinicians can save lives by identifying patients with Lynch syndrome and managing their health accordingly as knowledge continues to evolve.
Disclosure
The authors have no conflicts of interest in this work.
References
Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med. 2009;11(1):42–65. | |
Egoavil C, Alenda C, Castillejo A, et al. Prevalence of Lynch syndrome among patients with newly diagnosed endometrial cancers. PLoS One. 2013;8(11):e79737. | |
Weissman S, Burt R, Church J, et al. Identification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing: National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer Joint Practice Guideline. J Genet Couns. 2012;21(4):484–493. | |
Tutlewska K, Lubinski J, Kurzawski G. Germline deletions in the EPCAM gene as a cause of Lynch syndrome: literature review. Hered Cancer Clin Pract. 2013;11(1):9. | |
Vasen H, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum. 1991;34(5):424–425. | |
Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC. Gastroenterology. 1999;116(6):1453–1456. | |
Liu T, Wahlberg S, Burek E, Lindblom P, Rubio C, Lindblom A. Microsatellite instability as a predictor of a mutation in a DNA mismatch repair gene in familial colorectal cancer. Genes Chromosomes Cancer. 2000;27(1):17–25. | |
Syngal S, Fox EA, Eng C, Kolodner RD, Garber JE. Sensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J Med Genet. 2000;37(9):641–645. | |
Lindor NM. Familial colorectal cancer type X: the other half of hereditary nonpolyposis colon cancer syndrome. Surg Oncol Clin N Am. 2009;18(4):637–645. | |
Hampel H, Stephens JA, Pukkala E, et al. Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology. 2005;129(2):415–421. | |
Provenzale D, Jasperson K, Ahnen DJ, et al. NCCN guidlines v1.2014. Colorectal cancer screening. NCCN Guidelines 2014. Available from: http://www.nccn.org. Accessed June 5, 2014. | |
Ramsoekh D, Wagner A, van Leerdam M, et al. Cancer risk in MLH1, MSH2 and MSH6 mutation carriers; different risk profiles may influence clinical management. Hered Cancer Clin Pract. 2009;7(1):17. | |
Watson P, Vasen HF, Mecklin JP, et al. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer. 2008;123(2):444–449. | |
Lin-Hurtubise KM, Yheulon CG, Gagliano RA, Lynch HT. Excess of extracolonic non-endometrial multiple primary cancers in MSH2 germline mutation carriers over MLH1. J Surg Oncol. 2013;108(7):433–437. | |
Baglietto L, Lindor NM, Dowty JG, et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst. 2010;102(3):193–201. | |
Bonadona V, Bonaiti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011;305(22):2304–2310. | |
Senter L, Clendenning M, Sotamaa K, et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology. 2008;135(2):419–428. | |
Ligtenberg ML, Kuiper R, Geurts van Kessel A, Hoogerbrugge N. EPCAM deletion carriers constitute a unique subgroup of Lynch syndrome patients. Fam Cancer. 2013;12(2):169–174. | |
Järvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 118(5):829–834. | |
de Vos tot Nederveen Cappel WH, Nagengast FM, Griffioen G, et al. Surveillance for Hereditary Nonpolyposis Colorectal Cancer. Dis Colon Rectum. 2002;45(12):1588–1594. | |
Johnson PM, Gallinger S, McLeod RS. Surveillance colonoscopy in individuals at risk for hereditary nonpolyposis colorectal cancer: an evidence-based review. Dis Colon Rectum. 2006;49(1):80–95. | |
Messerini L, Mori S, Zampi G. Pathologic features of hereditary non-polyposis colorectal cancer. Tumori. 1996;82(2):114–116. | |
Shia J, Klimstra DS, Nafa K, et al. Value of immunohistochemical detection of DNA mismatch repair proteins in predicting germline mutation in hereditary colorectal neoplasms. Am J Surg Pathol. 2005; 29(1):96–104. | |
Watson P, Lin KM, Rodriguez-Bigas MA, et al. Colorectal carcinoma survival among hereditary nonpolyposis colorectal carcinoma family members. Cancer. 1998;83(2):259–266. | |
Stigliano V, Assisi D, Cosimelli M, et al. Survival of hereditary non-polyposis colorectal cancer patients compared with sporadic colorectal cancer patients. J Exp Clin Cancer Res. 2008;27:39. | |
Drescher KM, Sharma P, Lynch HT. Current hypotheses on how microsatellite instability leads to enhanced survival of Lynch syndrome patients. Clin Dev Immunol. 2010;2010:170432. | |
Barrow E, Hill J, Evans DG. Cancer risk in Lynch Syndrome. Fam Cancer. 2013;12(2):229–240. | |
Choi YH, Cotterchio M, McKeown-Eyssen G, et al. Penetrance of colorectal cancer among MLH1/MSH2 carriers participating in the colorectal cancer familial registry in Ontario. Hered Cancer Clin Pract. 2009;7(1):14. | |
Kempers MJE, Kuiper RP, Ockeloen CW, et al. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. Lancet Oncol. 2011;12(1):49–55. | |
Baglietto L, Lindor NM, Dowty JG, et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst. 2010;102(3):193–201. | |
Pérez-Cabornero L, Infante M, Velasco E, Lastra E, Miner C, Durán M. Genotype–phenotype correlation in MMR mutation-positive families with Lynch syndrome. Int J Colorectal Dis. 2013;28(9):1195–1201. | |
Goecke T, Schulmann K, Engel C, et al. Genotype-phenotype comparison of German MLH1 and MSH2 mutation carriers clinically affected with Lynch syndrome: a report by the German HNPCC Consortium. J Clin Oncol. 2006;24(26):4285–4292. | |
Stoffel E, Mukherjee B, Raymond VM, et al. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology. 2009;137(5):1621–1627. | |
Plaschke J, Engel C, Krüger S, et al. Lower incidence of colorectal cancer and later age of disease onset in 27 families with pathogenic MSH6 germline mutations compared with families with MLH1 or MSH2 mutations: the German Hereditary Nonpolyposis Colorectal Cancer Consortium. J Clin Oncol. 2004;22(22):4486–4494. | |
American Cancer Society. Colorectal Cancer Facts and Figures 2011–2013. Atlanta, GA: American Cancer Society. 2011. | |
Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26(35):5783–5788. | |
Win A, Parry S, Parry B, et al. Risk of metachronous colon cancer following surgery for rectal cancer in mismatch repair gene mutation carriers. Ann Surg Oncol. 2013;20(6):1829–1836. | |
Edelstein DL, Axilbund J, Baxter M, et al. Rapid development of colorectal neoplasia in patients with Lynch syndrome. Clin Gastroenterol Hepatol. 2011;9(4):340–343. | |
Winawer S, Fletcher R, Rex D, et al. Colorectal cancer screening and surveillance: clinical guidelines and rationale: update based on new evidence. Gastroenterology. 2003;124(2):544–560. | |
Lu KH, Dinh M, Kohlmann W, et al. Gynecologic cancer as a “sentinel cancer” for women with hereditary nonpolyposis colorectal cancer syndrome. Obstet Gynecol. 2005;105(3):569–574. | |
Walsh CS, Blum A, Walts A, et al. Lynch syndrome among gynecologic oncology patients meeting Bethesda guidelines for screening. Gynecol Oncol. 2010;116(3):516–521. | |
Obermair A, Youlden DR, Young JP, et al. Risk of endometrial cancer for women diagnosed with HNPCC-related colorectal carcinoma. Int J Cancer. 2010;127(11):2678–2684. | |
Broaddus RR, Lynch HT, Chen LM, et al. Pathologic features of endometrial carcinoma associated with HNPCC: a comparison with sporadic endometrial carcinoma. Cancer. 2006;106(1):87–94. | |
Garg K, Soslow RA. Lynch syndrome (hereditary non-polyposis colorectal cancer) and endometrial carcinoma. J Clin Pathol. 2009;62(8):679–684. | |
Leenen CH, van Lier MG, van Doorn HC, et al. Prospective evaluation of molecular screening for Lynch syndrome in patients with endometrial cancer ≤70 years. Gynecol Oncol. 2012;125(2):414–420. | |
Soliman PT, Broaddus RR, Schmeler KM, et al. Women with synchronous primary cancers of the endometrium and ovary: do they have Lynch syndrome? J Clin Oncol. 2005;23(36):9344–9350. | |
Walsh MD, Cummings MC, Buchanan DD, et al. Molecular, pathologic, and clinical features of early-onset endometrial cancer: identifying presumptive Lynch syndrome patients. Clin Cancer Res. March 15 2008;14(6):1692–1700. | |
Kim MK, Song SY, Do I-G, et al. Synchronous gynecologic malignancy and preliminary results of Lynch syndrome. J Gynecol Oncol. 2011;22(4):233–238. | |
Watson P, Bützow R, Lynch HT, et al. The clinical features of ovarian cancer in hereditary nonpolyposis colorectal cancer. Gynecol Oncol. 2001;82(2):223–228. | |
Ryan P, Mulligan AM, Aronson M, et al. Comparison of clinical schemas and morphologic features in predicting Lynch syndrome in mutation-positive patients with endometrial cancer encountered in the context of familial gastrointestinal cancer registries. Cancer. 2012;118(3):681–688. | |
Masuda K BK, Yanokura M, et al. Carcinoma of the lower uterine segment (LUS): clinicopathological characteristics and association with Lynch syndrome. Curr Genomics. 2011;12(1):25–29. | |
Westin SN, Lacour RA, Urbauer DL, et al. Carcinoma of the lower uterine segment: a newly described association with Lynch syndrome. J Clin Oncol. 2008;26(36):5965–5971. | |
Malander S, Rambech E, Kristoffersson U, et al. The contribution of the hereditary nonpolyposis colorectal cancer syndrome to the development of ovarian cancer. Gynecol Oncol. 2006;101(2):238–243. | |
Domanska K, Malander S, Masback A, Nilbert M. Ovarian cancer at young age: the contribution of mismatch-repair defects in a population-based series of epithelial ovarian cancer before age 40. Int J Gynecol Cancer. 2007;17(4):789–793. | |
Engel C, Loeffler M, Steinke V, et al. Risks of less common cancers in proven mutation carriers with Lynch syndrome. J Clin Oncol. 2012;30(35):4409–4415. | |
Pal T, Permuth-Wey J, Sellers TA. A review of the clinical relevance of mismatch-repair deficiency in ovarian cancer. Cancer. 2008;113(4):733–742. | |
Chui MH, Gilks CB, Cooper K, Clarke BA. Identifying Lynch syndrome in patients with ovarian carcinoma: the significance of tumor subtype. Adv Anat Pathol. 2013;20(6):378–386. | |
Lynch HT, Casey MJ, Snyder CL, et al. Hereditary ovarian carcinoma: heterogeneity, molecular genetics, pathology, and management. Mol Oncol. 2009;3(2):97–137. | |
Ketabi Z, Bartuma K, Bernstein I, et al. Ovarian cancer linked to lynch syndrome typically presents as early-onset, non-serous epithelial tumors. Gynecol Oncol. 2011;121(3):462–465. | |
Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81(2):214–218. | |
Capelle LG, Van Grieken NC, Lingsma HF, et al. Risk and epidemiological time trends of gastric cancer in Lynch syndrome carriers in The Netherlands. Gastroenterology. 2010;138(2):487–492. | |
Chun N, Ford JM. Genetic testing by cancer site: stomach. Cancer J. 2012;18(4):355–363. | |
Park JG, Kim DW, Hong CW, et al. Germ line mutations of mismatch repair genes in hereditary nonpolyposis colorectal cancer patients with small bowel cancer: International Society for Gastrointestinal Hereditary Tumours Collaborative Study. Clin Cancer Res. 2006;12(11 Pt 1):3389–3393. | |
Gupta R, Paner GP, Amin MB. Neoplasms of the upper urinary tract: a review with focus on urothelial carcinoma of the pelvicalyceal system and aspects related to its diagnosis and reporting. Adv Anat Pathol. 2008;15(3):127–139. | |
van der Post RS, Kiemeney LA, Ligtenberg MJ, et al. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet. 2010;47(7):464–470. | |
Aarnio M, Säily M, Juhola M, et al. Uroepithelial and kidney carcinoma in Lynch syndrome. Fam Cancer. 2012;11(3):395–401. | |
Crockett DG, Wagner DG, Holmäng S, Johansson SL, Lynch HT. Upper urinary tract carcinoma in Lynch syndrome cases. J Urol. 2011;185(5):1627–1630. | |
Skeldon SC, Semotiuk K, Aronson M, et al. Patients with Lynch syndrome mismatch repair gene mutations are at higher risk for not only upper tract urothelial cancer but also bladder cancer. Eur Urol. 2013;63(2):379–385. | |
Dasgupta T, Wilson LD, Yu JB. A retrospective review of 1349 cases of sebaceous carcinoma. Cancer. 2009;115(1):158–165. | |
Roberts M, Riegert-Johnson D, Thomas B, et al. Screening for Muir–Torre syndrome using mismatch repair protein immunohistochemistry of sebaceous neoplasms. J Genet Couns. 2013;22(3):393–405. | |
South CD, Hampel H, Comeras I, Westman JA, Frankel WL, de la Chapelle A. The frequency of Muir–Torre syndrome among Lynch syndrome families. J Natl Cancer Inst. 2008;100(4):277–281. | |
Ponti G, Losi L, Pedroni M, et al. Value of MLH1 and MSH2 mutations in the appearance of Muir–Torre syndrome phenotype in HNPCC patients presenting sebaceous gland tumors or keratoacanthomas. J Invest Dermatol. 2006;126(10):2302–2307. | |
Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302(16):1790–1795. | |
Vasen HFA, Sanders EACM, Taal BG, et al. The risk of brain tumours in hereditary non-polyposis colorectal cancer (HNPCC). Int J Cancer. 1996;65(4):422–425. | |
Haraldsdottir S, Hampel H, Wei L, et al. Prostate cancer incidence in males with Lynch syndrome. Genet Med. Epub January 16, 2014. | |
Ryan S, Jenkins MA, Win AK. Risk of prostate cancer in Lynch syndrome: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2014;23(3):437–449. | |
Raymond VM, Mukherjee B, Wang F, et al. Elevated risk of prostate cancer among men with Lynch syndrome. J Clin Oncol. 2013;31(14):1713–1718. | |
Buerki N, Gautier L, Kovac M, et al. Evidence for breast cancer as an integral part of Lynch syndrome. Genes Chromosomes Cancer. 2012;51(1):83–91. | |
Win A, Lindor N, Jenkins M. Risk of breast cancer in Lynch syndrome: a systematic review. Breast Cancer Res. 2013;15(2):R27. | |
Wen YH, Brogi E, Zeng Z, et al. DNA mismatch repair deficiency in breast carcinoma: a pilot study of triple-negative and non-triple-negative tumors. Am J Surg Pathol. 2012;36(11):1700–1708. | |
D’Arcy C, Wen YH, Stadler ZK, Brogi E, Shia J. Synchronous breast cancers with different morphologic and molecular phenotypes occurring in Lynch syndrome: what does the heterogeneity imply? Am J Surg Pathol. 2011;35(11):1743–1748. | |
Jensen U, Sunde L, Timshel S, et al. Mismatch repair defective breast cancer in the hereditary nonpolyposis colorectal cancer syndrome. Breast Cancer Res Treat. 2010;120(3):777–782. | |
Schmeler KM, Lynch HT, Chen LM, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med. 2006;354(3):261–269. | |
Yurgelun MB, Mercado R, Rosenblatt M, et al. Impact of genetic testing on endometrial cancer risk-reducing practices in women at risk for Lynch syndrome. Gynecol Oncol. 2012;127(3):544–551. | |
Mecklin JP, Aarnio M, Läärä E, et al. Development of colorectal tumors in colonoscopic surveillance in Lynch syndrome. Gastroenterology. 2007;133(4):1093–1098. | |
Vasen HFA, Blanco I, Aktan-Collan K, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013;62(6):812–823. | |
Grover S, Syngal S. Risk assessment, genetic testing, and management of Lynch syndrome. J Natl Compr Canc Netw. 2010;8(1):98–105. | |
Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA. 2006;296(12):1507–1517. | |
Stupart DA, Goldberg PA, Algar U, Ramesar R. Surveillance colonoscopy improves survival in a cohort of subjects with a single mismatch repair gene mutation. Colorectal Dis. 2009;11(2):126–130. | |
Jarvinen HJ, Renkonen-Sinisalo L, Aktan-Collan K, Peltomaki P, Aaltonen LA, Mecklin JP. Ten years after mutation testing for Lynch syndrome: cancer incidence and outcome in mutation-positive and mutation-negative family members. J Clin Oncol. 2009;27(28):4793–4797. | |
Stoffel EM, Turgeon DK, Stockwell DH, et al. Missed adenomas during colonoscopic surveillance in individuals with Lynch syndrome (hereditary nonpolyposis colorectal cancer). Cancer Prev Res (Phila). 2008;1(6):470–475. | |
Lu KH, Daniels M. Endometrial and ovarian cancer in women with Lynch syndrome: update in screening and prevention. Fam Cancer. 2013;12(2):273–277. | |
Baucom RB, Wise PE. Endoscopic and surgical management of hereditary nonpolyposis colorectal cancer. Clin Colon Rectal Surg. 2012;25(2):90–96. | |
Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut. 2011;60(7):950–957. | |
Haanstra JF, de Vos Tot Nederveen Cappel WH, Gopie JP, et al. Quality of life after surgery for colon cancer in patients with Lynch syndrome: partial versus subtotal colectomy. Dis Colon Rectum. 2012;55(6):653–659. | |
Rodriguez-Bigas M, Möeslein G. Surgical treatment of hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome). Fam Cancer. 2013;12(2):295–300. | |
Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66(15):7810–7817. | |
Downes MR, Allo G, McCluggage WG, et al. Review of findings in prophylactic gynaecological specimens in Lynch syndrome with literature review and recommendations for grossing. Histopathology. Epub February 4, 2014. | |
Karamurzin Y, Soslow RA, Garg K. Histologic evaluation of prophylactic hysterectomy and oophorectomy in Lynch syndrome. Am J Surg Pathol. 2013;37(4):579–585. | |
Burn J, Gerdes AM, Macrae F, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. 2011;378(9809):2081–2087. | |
Lu KH, Loose DS, Yates MS, et al. Prospective multicenter randomized intermediate biomarker study of oral contraceptive versus depo-provera for prevention of endometrial cancer in women with Lynch syndrome. Cancer Prev Res (Phila). 2013;6(8):774–781. | |
Rubinstein WS, Weissman SM. Managing hereditary gastrointestinal cancer syndromes: the partnership between genetic counselors and gastroenterologists. Nat Clin Pract Gastroenterol Hepatol. 2008;5(10):569–582. | |
Claes E DL, Evers-Kiebooms, et al. Predictive testing for hereditary non-polyposis colorectal cancer: motivation, illness representations and short-term psychological impact. Patient Educ Couns. 2004;65(2):265–274. | |
Meiser B, Collins V, Warren R, et al. Psychological impact of genetic testing for hereditary non-polyposis colorectal cancer. Clin Genet. 2004;66(6):502–511. | |
Esplen MJ, Stuckless N, Gallinger S, et al. Development and validation of an instrument to measure the impact of genetic testing on self-concept in Lynch syndrome. Clin Genet. 2011;80(5):415–423. | |
Aktan-Collan K, Kääriäinen H, Järvinen H, et al. Psychosocial consequences of predictive genetic testing for lynch syndrome and associations to surveillance behaviour in a 7-year follow-up study. Fam Cancer. 2013;12(4):639–646. | |
Stoffel EM, Mercado RC, Kohlmann W, et al. Prevalence and predictors of appropriate colorectal cancer surveillance in Lynch syndrome. Am J Gastroenterol. 2010;105(8):1851–1860. | |
Ketabi Z, Mosgaard BJ, Gerdes A-M, Ladelund S, Bernstein IT. Awareness of endometrial cancer risk and compliance with screening in hereditary nonpolyposis colorectal cancer. Obstet Gynecol. 2012;120(5):1005–1012. | |
Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med Overseas Ed. 2005;352(18):1851–1860. | |
EGAPP. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. 2009;11(1):35–41. | |
Clarke BA, Cooper K. Identifying Lynch syndrome in patients with endometrial carcinoma: shortcomings of morphologic and clinical schemas. Adv Anat Pathol. 2012;19(4):231–238. | |
Rodriguez-Bigas MA, Boland CR, Hamilton SR, et al. A National Cancer Institute workshop on hereditary nonpolyposis colorectal cancer syndrome: meeting highlights and bethesda guidelines. J Natl Cancer Inst. 1997;89(23):1758–1762. | |
Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–268. | |
Ramsoekh D, Wagner A, van Leerdam ME, et al. A high incidence of MSH6 mutations in Amsterdam criteria II-negative families tested in a diagnostic setting. Gut. 2008;57(11):1539–1544. | |
Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila). 2011;4(1):9–22. | |
Gudgeon J, Williams J, Burt RW, Samowitz W, Snow G, Williams M. Lynch Syndrome screening implementation: business analysis by a healthcare system. Am J Manag Care. 2011;17(8):e288–e300. | |
Mvundura M, Grosse SD, Hampel H, Palomaki GE. The cost-effectiveness of genetic testing strategies for Lynch syndrome among newly diagnosed patients with colorectal cancer. Genet Med. 2010;12(2):93–104. | |
Ladabaum U, Wang G, Terdiman J, et al. Strategies to identify the Lynch syndrome among patients with colorectal cancer. Ann Intern Med. 2011;155(2):69–79. | |
Williams JL, Williams MS. Informed consent and immunohistochemistry screening for Lynch syndrome. Genet Med. 2011;13(9):848–849. | |
Chubak B, Heald B, Sharp RR. Informed consent to microsatellite instability and immunohistochemistry screening for Lynch syndrome. Genet Med. 2011;13(4):356–360. | |
Cohen S. Current Lynch syndrome tumor screening practices: a survey of genetic counselors. J Genet Couns. 2013;23(1):1–10. | |
Bellcross CA, Bedrosian SR, Daniels E, et al. Implementing screening for Lynch syndrome among patients with newly diagnosed colorectal cancer: summary of a public health/clinical collaborative meeting. Genet Med. 2012;14(1):152–162. | |
Jasperson K. Colorectal cancer: cascade genetic testing in Lynch syndrome: room for improvement. Nat Rev Gastroenterol Hepatol. 2013;10(9):506–508. | |
South CD, Yearsley M, Martin E, Arnold M, Frankel W, Hampel H. Immunohistochemistry staining for the mismatch repair proteins in the clinical care of patients with colorectal cancer. Genet Med. 2009;11(11):812–817. | |
Ward RL, Hicks S, Hawkins NJ. Population-based molecular screening for Lynch syndrome: implications for personalized medicine. J Clin Oncol. 2013;31(20):2554–2562. | |
Cragun D, Malo TL, Pal T, Shibata D, Vadaparampil ST. Colorectal cancer survivors’ interest in genetic testing for hereditary cancer: implications for universal tumor screening. Genet Test Mol Biomarkers. 2012;16(6):493–499. | |
Hall MJ, Herda MM, Handorf EA, et al. Direct-to-patient disclosure of results of mismatch repair screening for Lynch syndrome via electronic personal health record: a feasibility study. Genet Med. Epub May 1, 2014. | |
Heald B, Plesec T, Liu X, et al. Implementation of universal microsatellite instability and immunohistochemistry screening for diagnosing Lynch syndrome in a large academic medical center. J Clin Oncol. 2013;31(10):1336–1340. | |
Cragun D, Debate R, Vadaparampil ST, Baldwin J, Hampel H, Pal T. Comparing universal Lynch syndrome tumor-screening programs to evaluate associations between implementation strategies and patient follow-through. Genet Med. Epub March 20, 2014. | |
Sharaf RN, Myer P, Stave CD, Diamond LC, Ladabaum U. Uptake of genetic testing by relatives of lynch syndrome probands: a systematic review. Clin Gastroenterol Hepatol. 2013;11(9):1093–1100. | |
Tomiak E, Samson A, Spector N, et al. Reflex testing for Lynch syndrome: if we build it, will they come? Lessons learned from the uptake of clinical genetics services by individuals with newly diagnosed colorectal cancer (CRC). Fam Cancer. 2014;13(1):75–82. | |
Stoffel EM, Ford B, Mercado RC, et al. Sharing genetic test results in Lynch syndrome: communication with close and distant relatives. Clin Gastroenterol Hepatol. 2008;6(3):333–338. | |
Mensenkamp AR, Vogelaar IP, van Zelst-Stams WA, et al. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology. 2014;146(3):643–646. e648. | |
Coppola D, Nicosia S, Doty A, et al. Uncertainty in the utility of immunohistochemistry in mismatch repair protein expression in epithelial ovarian cancer. Anticancer Res. 2012;32(11):4963–4969. | |
Lee JH, Cragun D, Thompson Z, et al. Association between IHC and MSI testing to identify mismatch repair-deficient patients with ovarian cancer. Genet Test Mol Biomarkers. 2014;18(4):229–235. | |
Abbas O, Mahalingam M. Cutaneous sebaceous neoplasms as markers of Muir–Torre syndrome: a diagnostic algorithm. J Cutan Pathol. 2009;36(6):613–619. | |
Orta L, Klimstra DS, Qin J, et al. Towards identification of hereditary DNA mismatch repair deficiency: sebaceous neoplasm warrants routine immunohistochemical screening regardless of patient’s age or other clinical characteristics. Am J Surg Pathol. 2009;33(6):934–944. | |
Plocharczyk EF, Frankel WL, Hampel H, Peters SB. Mismatch repair protein deficiency is common in sebaceous neoplasms and suggests the importance of screening for Lynch syndrome. Am J Dermatopathol. 2013;35(2):191–195. | |
Roberts ME, Riegert-Johnson DL, Thomas BC, et al. A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir–Torre variant of Lynch syndrome. Genet Med. Epub March 6, 2014. | |
Williams AS, Huang W-Y. The analysis of microsatellite instability in extracolonic gastrointestinal malignancy. Pathology. 2013;45(6):540–552. | |
Karamurzin Y, Zeng Z, Stadler ZK, et al. Unusual DNA mismatch repair–deficient tumors in Lynch syndrome: a report of new cases and review of the literature. Hum Pathol. 2012;43(10):1677–1687. | |
Wimmer K, Kratz CP, Vasen HFA, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium “Care for CMMRD” (C4CMMRD). J Med Genet. 2014;51(6):355–365. | |
Poley JW, Wagner A, Hoogmans MM, et al. Biallelic germline mutations of mismatch-repair genes: a possible cause for multiple pediatric malignancies. Cancer. 2007;109(11):2349–2356. | |
Durno CA, Holter S, Sherman PM, Gallinger S. The gastrointestinal phenotype of germline biallelic mismatch repair gene mutations. Am J Gastroenterol. 2010;105(11):2449–2456. | |
Dewanwala A, Chittenden A, Rosenblatt M, et al. Attitudes toward childbearing and prenatal testing in individuals undergoing genetic testing for Lynch Syndrome. Fam Cancer. 2011;10(3):549–556. | |
Win AK, Dowty JG, English DR, et al. Body mass index in early adulthood and colorectal cancer risk for carriers and non-carriers of germline mutations in DNA mismatch repair genes. Br J Cancer. 2011;105(1):162–169. | |
Win AK, Dowty JG, Antill YC, et al. Body mass index in early adulthood and endometrial cancer risk for mismatch repair gene mutation carriers. Obstet Gynecol. 2011;117(4):899–905. | |
Surveillance, Epidemiology, and End Results Program (SEER). Cancer Stat Fact Sheets. Bethesda, MD: National Institutes of Health. Available from: http://seer.cancer.gov/statfacts/html/breast.html. Accessed June 12, 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.