")
Back to Journals » The Application of Clinical Genetics » Volume 7
The genetic basis of ankylosing spondylitis: new insights into disease pathogenesis
Authors Tsui F, Tsui HW, Akram A, Haroon N, Inman R
Received 30 November 2013
Accepted for publication 9 March 2014
Published 22 May 2014 Volume 2014:7 Pages 105—115
DOI https://doi.org/10.2147/TACG.S37325
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Florence WL Tsui,1,2 Hing Wo Tsui,1 Ali Akram,1,3 Nigil Haroon,1–3 Robert D Inman1–3
1Genetics and Development Division, Toronto Western Research Institute, University Health Network, 2Department of Immunology, 3Institute of Medical Science, University of Toronto, Toronto, ON, Canada
Abstract: Ankylosing spondylitis (AS) is a complex disease involving multiple risk factors, both genetic and environmental. AS patients are predominantly young men, and the disease is characterized by inflammation and ankylosis, mainly at the cartilage–bone interface and enthesis. HLA-B27 has been known to be the major AS-susceptibility gene for more than 40 years. Despite advances made in the past few years, progress in the search for non-human leukocyte antigen susceptibility genes has been hampered by the heterogeneity of the disease. Compared to other complex diseases, such as inflammatory bowel disease (IBD), fewer susceptibility loci have been identified in AS. Furthermore, non-major histocompatibility-complex susceptibility loci discovered, such as ERAP1 and IL23R, are likely contributors to joint inflammation. Identification and confirmation of functional variants remains a significant challenge of investigations involving genome-wide association studies (GWAS). It remains unclear why none of the AS-susceptibility genes identified in GWAS appear to be directly involved in the ankylosing process. Numerous reviews have recently been published on the genetics of AS. Therefore, aside from a brief summary of what AS GWAS has successfully achieved thus far, this review will focus on directions that could address unanswered questions raised by GWAS.
Keywords: ankylosing spondylitis, genome-wide association studies, risk loci, ankylosis, joint and gut inflammation, clinical subsets
Introduction
Ankylosing spondylitis
Ankylosing spondylitis (AS) is a subset of spondyloarthritis (SpA), which is characterized by inflammation of the sacroiliac joints, peripheral inflammatory arthropathy, and the absence of rheumatoid factor. Other subsets of SpA include reactive arthritis, psoriatic arthritis, colitic arthropathies (inflammatory bowel disease [IBD]-related SpA), and undifferentiated SpA. With a prevalence of 0.1%–1.4%, AS is an underrecognized form of chronic arthritis.1 It can lead to significant spinal disease and peripheral arthritis, which can manifest as chronic back pain and a progressive spinal ankylosis. The disease strikes predominantly men between the ages of 20 and 40 years, in their peak productive years, leading to significant loss of work productivity and decreased quality of life.
Diagnosis, progression, and current management of AS
AS is usually diagnosed according to the modified New York criteria,2 which include a combination of such clinical features as limited motion of the lumbar spine, persistent lower-back pain, limited chest expansion, and radiographic evidence of sacroiliitis. The hallmark of AS is neo-ossification at the site of joint inflammation.3 Although joint inflammation can be detected early in the disease process, eg, in the first year of symptoms, using magnetic resonance-imaging technology, this is not a definitive diagnosis test for AS. The subsequent spinal structural changes, as visualized on radiographs, appear relatively late. This explains in part why it can take 5–10 years to confirm a diagnosis of AS after the initial onset of symptoms.4,5
Nonsteroidal anti-inflammatory drugs (NSAIDs) are the first-line drug treatment for AS patients with symptomatic disease.6 Continuous use of NSAIDs appears to slow radiographic progression.7,8 If NSAID treatment fails, biologics, particularly tumor necrosis factor (TNF)-α inhibitors, are used in patients with active disease. The TNF blockers adalimumab, etanercept, infliximab, golimumab, and certolizumab have all been proven to be highly effective in controlling inflammation and in improving the quality of life of most AS patients. Whether these TNF blockers could halt progression of structural changes remains a controversial issue. Some studies showed no structural impact,9 but a recent study indicated that TNF inhibitors impact structural damage, such as new syndesmophyte appearance, growth of existing syndesmophytes, and development of ankylosis in AS.10 However, a substantial proportion of AS patients (~25%) do not respond to TNF-inhibitor therapy, and some initial responders developed secondary unresponsiveness.
Joint ankylosis
Current concepts regard new bone formation at the enthesis as a pathological response to injury, and that joint inflammation precedes ossification. Early stages of ankylosis involve squaring of the vertebral bodies and formation of syndesmophytes. Total spinal ankylosis and kyphosis are found in the most severe cases. Radiographic changes of the cervical and lumbar spine in AS patients are scored using the modified Stokes AS Spine Score (mSASSS).11 Scoring of sequential radiographs from the same patient over time determines the change in mSASSS per year. Systematic evaluation of ankylosis progression in AS patients is subjective and time-consuming.12,13
There remains some uncertainty whether joint inflammation and ankylosis in AS are linked events or independent processes.14–16 For clinicians, this critical issue affects the management strategy for SpA: early intervention with anti-TNF therapy would prevent ankylosis development only if inflammation and ankylosis are sequential and linked processes.
Genetics and AS
Based on family and twin studies, it has been established for a long time that AS has a strong genetic component. The sibling recurrence risk of AS is 9.2% (λs is 82) compared to 0.1% in the general population.17 The heritability of AS is estimated to be >95%. The strong association with human leukocyte antigen (HLA)-B27 was discovered in the early 1970s. Population studies have indicated that 2% of HLA-B27-positive individuals develop AS,18 implying that other factors – genetic, environmental, or stochastic – contribute importantly to disease development. The most direct evidence that genetic polymorphisms other than HLA-B27 contribute to AS is the difference in concordance rates reported for HLA-B27-positive monozygotic twins (17 of 27 [63%]) and HLA-B27-positive dizygotic twins (four of 15 [27%]).19 These same statistics indicate that the predisposition to AS is not entirely genetically determined. Nongenetic factors might correspond to an environmental effect, such as a specific microbial infection, as implicated in reactive arthritis, or it might correspond to a stochastic event in development, such as the emergence of specific immune cells.
Recent genome-wide association study findings
A number of updated reviews on AS genetics,20–24 including genome-wide association study (GWAS) results, have recently been published, and thus we only briefly summarize the key findings here. Based on the most recent report on AS susceptibility loci detected by ImmunoChip (Illumina, San Diego, CA, USA) genotyping of the largest cohort (more than 10,000 individuals for both cases and controls examined),25 there are at least 25 AS non-major histocompatibility complex (MHC) immune-related risk loci (summarized in Table 1), eleven of which have been identified previously.26 A major achievement of these studies relates to the identification of important biological pathways likely responsible for AS pathogenesis.
Role of the interleukin-23-related pathway
 | Table 1 Summary of ankylosing spondylitis-susceptibility genes identified by genome-wide association studies |
AS-associated genetic variants relating to this pathway include interleukin (IL)-23R, IL-12β, Tyk2, IL27, and IL-6R. Susceptibility to loci involved in this pathway is shared among many inflammatory diseases, including IBD and psoriasis. It remains to be elucidated how influence on one common pathway leads to the development of different inflammatory diseases. A recent elegant study in Cell illustrates the power of using a combination of genetic, clinical, and functional analyses in both mice and humans to unravel how a noncoding single-nucleotide polymorphism (SNP) can influence the disease process.27 Intriguingly, this FOXO3 variant (rs12212067), which regulates cytokine production in monocytes, was not associated with susceptibility to the rheumatoid arthritis or Crohn’s disease.
It is established that IL-23 drives the differentiation of CD4-positive Th17 cells, which produce IL-17. IL-17 in turn can facilitate the production of other factors (such as IL-6, IL-8, TNF, chemokines, matrix metalloproteinases, and receptor activator of nuclear factor κB ligand) from a wide range of cell types.28 A French SpA study illustrated that variants at loci in the IL-23/Th17 pathway influence expression levels of genes involved in the differentiation of Th17/Th1 cells, and it is likely that the pathological outcome is dictated by combinatorial assortments of multiple variants.29 A comprehensive discussion on how IL-23/IL-17 pathways impact on AS pathogenesis is beyond the scope of this review. An excellent review on this aspect has just been published.28
Role of aminopeptidases
In addition to ERAP1 and ERAP2, two other aminopeptidases (LNPEP and NPEPPS) are associated with AS,23 reiterating the importance of antigen presentation in AS pathogenesis. Both protective and susceptible ERAP1 variants associated with AS have been identified. The relative attributable risk of ERAP1 to AS is about 25%, whereas that of HLA-B27 is about 50%. These two genes combined provide the two most powerful disease risk factors to AS. Intriguingly, the association of ERAP1 is restricted to HLA-B27-positive AS patients.25 One recent functional study showed that ERAP1 variants affect HLA-B27 antigen presentation and stability in vivo.30 Protective variants lead to less ERAP1 activity, and less efficient trimming of HLA-B27 ligands. Another study supported the notion that AS-associated ERAP1 variants alter the composition and length of HLA-B27 ligands.31 A more in-depth review on the role of ERAP1 in AS pathogenesis will be discussed in later sections.
ERAP2 is unique to humans, and does not exist in mice. However, a high-frequency variant, when present in homozygosity (about 25% of the population), results in the absence of ERAP2 protein in these individuals. In ERAP2-deficient human B cells, surface MHC-I expression is reduced.32 It remains unclear whether the absence of ERAP2 might alter/modulate antigen presentation in these individuals, especially patients with such diseases as AS and Crohn’s disease in which disease-associated ERAP2 variants exist. Intriguingly, one ERAP2 variant (rs2549782) confers natural resistance to human immunodeficiency virus-1 infection.33 Results from the most recent GWAS indicated that ERAP2 variants are associated with AS in HLA-B27-negative cases.25
Despite substantial sequence homology, similar overall domain organization and structures between ERAP1 and ERAP2, the N-terminal peptide specificities between these two aminopeptidases are quite different, as explained by their crystal structures.34 We showed that an ERAP1 ERAP2 haplotype (rs27044[G] rs30187[T] rs2549782[T]) is associated with familial AS.35 A recent study using sequencing haplotypes in 20 individuals showed that this haplotype occurs naturally.36 Amino acid variants coded by ERAP2 rs2549782 (N392K) alter both the specificity and activity of ERAP2. Amino acid variants coded by ERAP1 rs27044 (Q730E) and rs30187 (K528R) affect peptide-trimming activity. To date, there has been only one study that assessed the effects of naturally occurring ERAP haplotypes.36 Importantly, results from this study showed that ERAP SNPs, when assessed in combination (as a haplotype), showed different effects compared to those assessed singly. Though there are human cells deficient in ERAP2, currently no ERAP1-deficient human cells are available for accurate assessments of the effects of natural ERAP haplotypes, and this poses a limitation on these types of studies. It is expected that different ERAP haplotypes would impact on natural killer cell and cytotoxic T-lymphocyte functions.
Additional AS risk loci contribute to variations in T-cell lineages (EOMES, IL7R, RUNX3, and ZM1Z1 for CD8+ T cells, and BACH2 and SH2B3 for CD4+ T cells). Variants in G-protein-coupled receptors (GPR35, GPR37, GPR65, and GPR25) were also identified, but their involvement with AS pathogenesis is less clear. It has been estimated that all these non-MHC risk loci only account for 4.3% of heritability in AS, while HLA-B27 contribute 20.1%, implying that a majority of risk loci (about 75%) remain undefined.
Another insight emerged from GWAS relates to common risk loci shared among some inflammatory diseases. Most notably is the largest number of loci shared between AS and IBD37,38 (12 and 11 AS loci shared with Crohn’s disease and ulcerative colitis [UC] respectively). At least 163 risk loci have been identified in IBD and about 28 of them were shared between Crohn’s disease and ulcerative colitis. The significant number of risk loci shared between AS and IBD supports the recent concept that gut involvement contributes to disease pathogenesis in a large subset (up to 60%) of AS patients. This issue is further addressed in a later section of this review.
Limitations of GWAS
In general, despite the wealth of new information obtained from GWAS, some unexpected challenges also emerged. Following are examples: 1) The number of risk loci uncovered was not only high (in terms of thousands in some complex traits) but also increased proportionally with the cohort size analyzed.39 There are some indications that variants from different steps of the same biological pathway could contribute similarly to the eventual clinical outcome; 2) The effect size of individual risk loci are usually very modest (odds ratio ~1.05–1.4), and GWAS can only detect common variants with a minor allele frequency of >0.05. For minor causal variants, deep sequencing of the regions of interest is required;40,41 3) Identification of causal variants with direct or indirect functional relevance to disease risk has proved to be difficult. It is challenging to prove that functional consequences of variants (usually assessed singly) contribute to disease pathogenesis. For complex traits, different combinations of risk factors can lead to similar clinical outcomes, leading to disease heterogeneity. In IBD, of 163 risk loci identified, only one (nucleotide-binding oligomerization domain-containing protein 2) has been shown to correlate with clinical outcomes.42 More importantly, the lack of large cohorts with detailed clinical parameters and well-characterized disease outcome renders meaningful analyses of genotypes obtained in GWAS a major challenge; 4) Known AS susceptibility locus (such as the IL1 gene cluster) can be missed. In the earlier AS genetic studies, positive and negative results were obtained with respect to the association of variants within the IL1 gene cluster in AS.43–47 This locus was not detected in recent AS GWAS. However, a recent French study showed that IL1A is associated with AS susceptibility or sacroiliitis in AS.48 Likely reasons contributing to the discrepancies among studies include disease heterogeneity, study design, and power limitations. The precise role of IL-1 in AS pathogenesis remains unclear, though it could contribute singly or in combination with other pathways with susceptibility variants (such as IL-23). Despite these inconsistent results from different studies, the latest International Genetics of Ankylosing Spondylitis Consortium GWAS25 concluded that AS is associated with the IL1R1–IL1R2 locus located on chromosome 2q11. There were two signals, one in each of the IL1Rgenes.
Are there risk loci relating to neo-ossification/ankylosis in AS patients?
In the AS GWAS result published in 2010,26 ANTXR2 (CMG2) was identified as one of the risk loci. Unfortunately, no SNP in this locus was included in the most recent AS GWAS, and thus it is unclear whether this association is replicable. ANTXR2 is not associated with AS in the Han Chinese,49 and the minor allele frequency was too low to analyze this in Koreans. Anthrax toxin-receptor 2 could potentially affect new bone formation, as it is a membrane-bound molecule that can interact with low-density lipoprotein receptor-related protein (LRP)-6.50 LRP6 is an important surface receptor in the Wnt/β–catenin pathway, and thus can affect osteoblastic activity. More work in discerning whether ANTXR2 plays a role in AS pathogenesis is warranted.
A recent GWAS performed in Han Chinese with AS49 detected two risk loci likely with relevance to bone formation (HAPLN1–EDIL3 at 5q14.3 and ANO6 at 12q12.1). HAPLN1 has been shown to be involved with osteophyte formation51 in Japanese women with spinal osteoarthritis. EDIL3 has an inhibitory effect on Wnt/β-catenin signaling.52 ANO6 plays a role in osteoclastogenesis.53 The most recent GWAS on East Asians using the ImmunoChip failed to replicate association of these two loci.25 One explanation is the low frequencies of the variants. This scenario is supported by the absence of association in IL23R variants for Han Chinese AS, but recent sequencing of this locus revealed a few rare variants with potential functional relevance.54 However, it is also possible that the absence of ANTRX2, HAPLN1–EDIL3, and ANO6 association in Han Chinese AS GWAS might be due to other ethnic differences.
There are a few studies implicating the role of IL-23 in excess bone ossification in AS. In a French population, an IL-23R variant was associated with radiographic sacroiliitis in AS.55 A more indirect finding relates to elevated levels of IL-23 detected in bone marrow cells from AS spinal facet joints obtained in corrective surgery.56 A mouse study also illustrated that IL-23 could drive enthesitis via entheseal resident T cells (positive for IL-23R, retinoic acid receptor-related orphan receptor-γt, and CD3) in an IL-22-dependent manner.57
Considering the strength of association of ERAP1 with AS, we questioned whether genes involved in the antigen-processing pathway could also be a marker of severity in addition to susceptibility. For this study,58 a total of 241 Caucasian patients with AS from spondylitis clinics in Toronto and Edmonton were included. Those patients who had at least two full sets of radiographs for mSASSS scoring at a minimum interval of 1.5 years were included in the analysis for genetic predictors of progression. Genotyping was done for a panel of 13 coding region SNPs in the ERAP1, LMP2, LMP7, TAP1, and TAP2 genes. In the univariate analysis for predictors of baseline radiographic severity, the duration of disease was the strongest, followed by male sex and LMP2 and ERAP1 variants. In multivariate analysis, the only genetic predictor that remained strongly associated with severity was LMP2. This is very interesting, as LMP2 has previously been reported to be associated with AS and uveitis in AS patients. Due to the association with uveitis, LMP2 may in fact be a marker of a more aggressive form of AS that could lead to more structural damage. Moreover, the proteasome helps to break down β-catenin, and abnormalities in LMP2 could lead to excess Wnt/β-catenin signaling and osteoblastic activity.
A candidate-gene approach to unravel ankylosis-related risk loci in AS; the role of ANKH and TNAP
Inorganic pyrophosphate (PPi) plays an important role in regulating mineralization and bone formation.59 The sources of PPi for the cartilage matrix include intracellular-to-extracellular transport of PPi through the cell membrane in association with a membrane transport system that involves the ANKH (human homolog of progressive ankylosis) protein60 and generation of PPi at the cell surface by tissue-nonspecific alkaline phosphatases (TNAPs) or ectonucleotidases.61,62
Using a candidate-gene approach, our linkage- and family-based association analyses63 demonstrated that North American Caucasian AS patients have excess sharing of the ANKH gene region, and that AS is significantly associated with a specific ANKH haplotype.63 Significantly, we further showed that there are sex differences in the ANKH variants associated with AS.64 Intriguingly, there is heterogeneity even in multiplex AS families. There were three types of families in our cohort of multiplex AS families: 1) families with affected individuals of both sexes, 2) families with only men affected, and 3) families with only women affected. In the first type of families with affected individuals of both sexes, two ANKH SNPs (rs28006[C] rs25957[C]) were associated with AS only in affected women. This haplotype was transmitted to affected women 79% of the time (15 of 19), but to affected men only 27% of the time (three of eleven). This effect is substantial, as the odds ratio for increased risk approaches 3.0 (0.79/0.27=2.92). In another haplotype (rs26307[C] rs27356[C]), the frequency of transmission was 70% to affected men (21 of 30) and 43% to affected women (13 of 30). In the subset of families with only men affected, 94% of the time (16 of 17), this haplotype was transmitted to affected men. There were too few informative families with only women affected with this haplotype, and thus we do not have a reliable assessment of the frequency at which this haplotype was transmitted to affected women in this subset for comparison.
Using Family-Based Association Test (Harvard School of Public Health, Boston, MA, USA) analysis in the subset of AS families with affected individuals of both sexes, we also found that a TNAP variant (Tyr263His) is significantly associated with AS.65 Furthermore, this TNAP variant is significantly associated with AS in affected men (dominant model, P=0.005). More impressively, the frequency of transmission of this allele was 92.3% (12 of 13) to affected men and only 64.7% (eleven of 17) to affected women. It appears that in this subset of AS families with affected individuals of both sexes, an ANKH polymorphism in the intron 1, close to exon 2 region, predisposes the affected women to AS, while a TNAP variant (possibly Tyr263His) predisposes the affected men to AS. The TNAP Tyr263His is a functional variant,66 and is associated with bone mineral density in elderly women.67 Osteoporosis is a common occurrence in AS. Investigation on whether this functional TNAP variant (Tyr263His) is associated with osteoporosis in AS patients is warranted.
In our cohort of North American Caucasian multiplex families, both ANKH and TNAP were associated with AS (summarized in Figure 1). Subsequently, there have been reports of ANKH but not TNAP association in Japanese68 and Han Chinese69 cohorts. In an earlier UK study, no ANKH association was detected.70 A recent presentation at the annual meeting of the American College of Rheumatology reported that ANKH variants were associated with disease severity in Korean AS.71
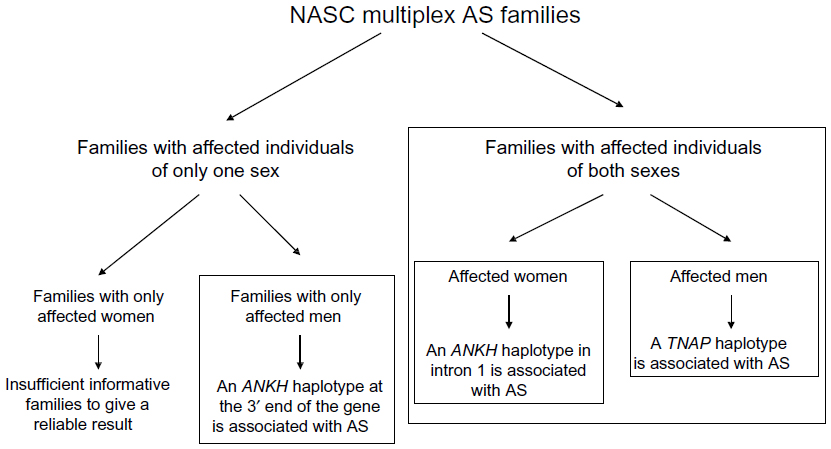 | Figure 1 Summary of family-based association analyses using multiplex ankylosing spondylitis (AS) families from the North American Spondylitis Consortium (NASC). |
Gene–gene interactions: the role of ERAP1 and HLA-B27 in AS
The discovery of the association of ERAP with AS opened new opportunities for investigating AS. Gain or loss of ERAP1 function could be used in experimental settings to further decipher the mechanisms by which ERAP1 contributes to the overall pathogenesis of AS. The mechanisms by which ERAP1 and HLA-B27 contribute to AS are under study. The main function of ERAP1 is to trim peptides within the ER into the right length before binding to MHC-I molecules. Abnormal function of ERAP1 can lead to generation of anomalous peptides of incorrect length and sequence, which subsequent to MHC-I binding (eg, HLA-B27) can lead to peptide–MHC-I complex misfolding. These misfolded proteins could either accumulate within the ER, thus contributing to the ER stress, or be transported to the cell surface as a free heavy chain. Both of these mechanisms have been implicated as contributing to AS pathogenesis, but the degree of their contribution to the overall pathogenesis of AS remains speculative. Loss of ERAP1 function leads to decreased surface expression of MHC-I molecules. Reduced surface expression has been suggested to contribute to the overall pathogenesis of AS. Recent data from our laboratory analyzing immune response to influenza virus have shown a change in the overall flu-peptide repertoire in mice deficient in ERAP function (ERAP1-/-).72 Our results confirm the findings of other investigators: ERAP1-/- mice had changes in the presentation of viral epitopes of different types, indicating the importance of ERAP in viral peptide generation and presentation. While these studies implicate the presence of an arthritogenic peptide in AS, the identity of this epitope is unknown. Since loss of ERAP1 function seems to be protective in AS, ERAP1 may be involved in the generation of arthritogenic peptides. However, to date, there has been no direct evidence either confirming or refuting this hypothesis.
Gene–gene interactions: the role of ERAP1 in AS patients following influenza infection
ERAP1 plays an integral role in the immune response following an infection. It can either enhance or diminish the immune response by changing the peptide repertoire presented by antigen-presenting cells.73–75 ERAP1, for instance, has been shown to affect the repertoire of peptides generated following influenza infection, one of the most commonly encountered human viruses. However, there are no reports investigating the host response to flu infection in AS patients. Although there are suggestions that an arthritogenic peptide or peptides plays a central role in AS,76,77 so far no such peptides have been discovered. Prior flu infection may influence the presentation of such arthritogenic peptides. In addition, AS is a multigenic disease, and as such, studies of AS patients with prior flu infection would be complex to analyze.77 Given these limitations, there is a high need for an AS animal model to investigate the role of these aforementioned factors in vivo. An animal model could also be useful to address gene–gene interaction in AS, which is highlighted by the fact that ERAP1 is associated only with B27-positive AS and not with B27-negative AS.
To overcome this limitation, we have used human HLA transgenic (HLA Tg) mice, which lack both ERAP1 and endogenous MHC-I molecule expression and which express HLA-B27 (ie, Tg HLA-B27/ERAP-/-) to investigate the interaction of ERAP1 and HLA-B27 in AS. These two genes are the strongest genetic associations with AS to date,78 yet B27/ERAP-/- mice manifest no articular abnormalities (unpublished results). It should be noted in this regard that loss-of-function variants of ERAP1 are protective in AS. On the other hand, since the flu peptides presented by B27 are well known, this animal model can provide important insights into the role of ERAP in generating B27-specific antigenic peptides. Initial studies of flu infection of Tg HLA-B27/ERAP-/- mice suggest that ERAP1 may play a central role in the phenomenon of immunodominance after flu infection.79 This animal model could prove valuable in deciphering the interaction of ERAP1 and HLA-B27 in an in vivo context.
Animal models with axial ankylosis
Though some AS patients have peripheral arthritis, AS is mainly an axial disease. An informative animal model could unravel the underlying mechanisms that lead to the development of axial inflammation and eventual ankylosis. Because AS is a multifactorial complex disease, it is unlikely that a perfect animal model for AS exists. Since HLA-B27 showed the strongest association with AS, transgenic rats highly expressing HLA-B27 and human β2-microglobulin could be a reasonable model. These transgenic rats have spontaneous peripheral and axial inflammation, as well as gut disease.80 With elevated β2-microglobulin expression, ankylosis developed with concurrent suppression of gut disease and an unfolded protein response.81
Two mouse models overexpressing TNFα (hTNFtg82 and TNF AU-rich elements [ΔARE]83) develop systemic inflammation, gut disease, and sacroiliitis, but no ankylosis. A recent study84 reported that mechanotransduction can lead to enthesitis and neo-ossification at entheseal sites in tail-suspended TNFΔARE mice. Enthesitis induction involves a number of pathways, including signaling via IL-23R-positive entheseal resident cells.
The proteoglycan-induced spondylitis (PGISp) model mimics many features of human AS, including axial inflammation and ankylosis.85,86 The spines of the PGISp mice showed decreased levels of Wnt-signaling antagonists (such as Dickkopf-related protein 1 and sclerostin [SOST]).87 There is evidence suggesting that enhanced Wnt/β-catenin signaling contributes to ankylosis in AS patients. Dickkopf-related protein 1, an antagonist of Wnt/β-catenin signaling, has been reported to be either dysfunctional88 or present at lower-than-normal levels in AS patients.89 Serum levels of SOST are also lower in AS patients than healthy individuals.90 Our recent work on ank/ank (progressive ankylosis) mice that have peripheral and spinal ankylosis showed enhanced Wnt/β-catenin signaling in the joints of these mutant mice.91
Old male DBA/1 mice develop peripheral arthritis and it has been shown that noggin (NOG) can rescue the enthesopathy.92 In contrast, we showed that NOG treatment of ank/ank mice led to more severe ankylosis, with concurrent generation of high levels of immunoglobulin (Ig)-G immune complexes (ICs) in which the autoantigens are either NOG (a bone morphogenetic protein-signaling antagonist) or SOST (a Wnt/β-catenin signaling antagonist).93 These results from the mutant ank/ank mice led to our novel finding of similar NOG/SOST IgG ICs in humans, and AS patients have significantly elevated serum levels of these ICs.93 An intriguing possibility is that ICs involving autoantibodies against NOG and SOST at their interacting sites may mimic the inhibitory interaction that naturally occurs between these two proteins. By this mechanism, these autoantibodies would be predicted to play a physiological role in bone homeostasis in normal individuals. However, overabundance of these autoantibodies would lead to reduced levels of functional NOG and SOST, resulting in enhanced bone morphogenetic protein and β-catenin signaling, neo-ossification and eventual spinal ankylosis.
Lessons from GWAS: how might they help in disease management?
Biologics involving inhibition of TNFα have proven to have dramatic efficacy in about 70% of AS patients, but they do not cure the disease. In many patients, challenges remain regarding how best to achieve and maintain remission. As mentioned earlier, treatment targeting IL-1 using anakinra was not very promising.94 IL6R is one of the AS-susceptibility genes detected in GWAS, yet tocilizumab (anti-human IL-6-receptor monoclonal antibody)95 and sarilumab (a fully human monoclonal antibody against IL-6Rα)96 appear to have no efficacy in AS patients. Genetic studies implicated the importance of the IL-23 signaling pathway. A recent prospective clinical trial showed promising efficacy and safety in the use of ustekinumab97 (anti-IL-12/23p40) to treat AS patients. Promising results were shown in IL-17 blockade.98 More studies are warranted to explore novel therapies for AS patients especially for TNF-inhibitor nonresponders.
Future directions
As mentioned earlier, AS is a very heterogeneous complex disease. Different combinations of risk loci, together with other nongenetic factors, would lead to a similar clinical outcome, ie, AS disease. Studies using patient cohorts with bias toward certain subsets would result in controversial and inconsistent results. Genetic analysis of AS subsets would lead to the identification of subset-specific risk loci. The association of ERAP1 being restricted to HLA-B27-positive AS patients represents a good example.
It has been known for decades that AS patients have extra-articular manifestations. A recent systematic meta-analysis99 confirmed that about 25.8% of AS patients have acute anterior uveitis (AAU); 9.3% and 6.8% have concomitant psoriasis and IBD, respectively. Intriguingly, the large overlap of AS patients with AAU is associated with disease duration, implying that the disease process in AS renders the patient susceptible to AAU. In contrast, it was reported that occurrence of IBD or psoriasis in AS was not associated with disease duration. In some such AS patients, IBD and psoriasis might be present prior to the diagnosis of AS. The implication of this is that certain AS subsets might have different etiologic pathways underlying the disease. For example, histopathological findings demonstrated that 50%–60% of AS patients have evidence of gut inflammation,100 although this is clinically evident in only 5%–10% of AS patients.101 It may be that AS patients with subclinical versus clinical gut inflammation represent distinct subsets of the disease. It is possible that AS patients whose gut and joint inflammation might be triggered by specific microbial exposure and thus leading to distinctive, pathogenic immune responses. In support of this, we recently found that higher-than-normal levels of NOG and SOST IgG immune complexes were detected in sera of AS patients, as well as in patients with both AS and IBD.93 A recent GWAS has already revealed common risk loci between AS and IBD (such as ERAP1 and IL23R).25 It is anticipated that performing GWAS using samples from AS patients with subclinical versus clinical gut inflammation would unravel novel risk loci specific for this AS subset.
In summary, recent GWAS findings provide invaluable clues on pathways key to the development of AS. The logical lines of investigation to follow should focus on the biology of the risk factors, especially in terms of how these risk factors would influence disease pathogenesis and clinical outcome.
Acknowledgment
This work was funded by the Arthritis Society of Canada, the Canadian Institutes of Health Research (CIHR), and the CIHR Institute of Musculoskeletal Health and Arthritis (IMHA).
Disclosure
The authors report no conflicts of interest in this work.
References
Pal B. Ankylosing spondylitis, a seronegative spondyloarthritis. Practitioner. 1987;231:785–793. | |
Van der Linden S, Valkenburg H, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteria. Arthritis Rheum. 1984;27:361–368. | |
Dakwar E, Reddy J, Vale FL, Uribe JS. A review of the pathogenesis of ankylosing spondylitis. Neurosurg Focus. 2008;24:E2. | |
Feldtkeller E, Khan MA, van der Heijde D, van der Linden S, Braun J. Age at disease onset and diagnosis delay in HLA-B27 negative vs positive patients with ankylosing spondylitis. Rheumatol Int. 2003;23: 61–66. | |
Rudwaleit M, van der Heijde D, Khan MA, Braun J, Sieper J. How to diagnose axial spondyloarthritis early. Ann Rheum Dis. 2004;63: 535–543. | |
Song IH, Poddubnyy DA, Rudwaleit M, Sieper J. Benefits and risks of ankylosing spondylitis treatment with nonsteroidal anti-inflammatory drugs. Arthritis Rheum. 2008;58:929–938. | |
Wanders A, van der Heijde D, Landewé R, et al. Nonsteroidal antiinflammatory drugs reduce radiographic progression in patients with ankylosing spondylitis: a randomized clinical trial. Arthritis Rheum. 2005;52:1756–1765. | |
Poddubnyy D, Rudwaleit M, Haibel H, et al. Effect of nonsteroidal anti-inflammatory drugs on radiographic spinal progression in patients with axial spondyloarthritis: results from the German Spondyloarthritis Inception Cohort. Ann Rheum Dis. 2012;71:1616–1622. | |
Van der Heijde D, Landewé R, Einstein S, et al. Radiographic progression of ankylosing spondylitis after up to two years of treatment with etanercept. Arthritis Rheum. 2008;58:1324–1331. | |
Haroon N, Inman RD, Learch TJ, et al. The impact of tumor necrosis factor α inhibitors on radiographic progression in ankylosing spondylitis. Arthritis Rheum. 2013;65:2645–2654. | |
Averns HL, Oxtoby J, Taylor HG, Jones PW, Dziedzic K, Dawes PT. Radiological outcome in ankylosing spondylitis: use of the Stoke Ankylosing Spondylitis Spine Score (SASSS). Br J Rheumatol. 1996;35:373–376. | |
Wanders AJ, Landewé RB, Spoorenberg A, et al. What is the most appropriate radiologic scoring method for ankylosing spondylitis? A comparison of the available methods based on the outcome measures in rheumatology clinical trials filter. Arthritis Rheum. 2004;50: 2622–2632. | |
Creemers MC, Franssen MJ, van’t Hof MA, Gribnau FW, van de Putte LB, van Riel PL. Assessment of outcome in ankylosing spondylitis: an extended radiographic scoring system. Ann Rheum Dis. 2005;64:127–129. | |
Baraliakos X, Listing J, Rudwaleit M, et al. The relationship between inflammation and new bone formation in patients with ankylosing spondylitis. Arthritis Res Ther. 2008;10:R104. | |
Lories RJ, Dougados M. Inflammation and ankylosis: still an enigmatic relationship in spondyloarthritis. Ann Rheum Dis. 2012;71:317–318. | |
Maksymowych WP, Elewaut D, Schett G. Motion for debate: the development of ankylosis in ankylosing spondylitis is largely dependent on inflammation. Arthritis Rheum. 2012;64:1713–1719. | |
Brown MA, Laval SH, Brophy S, Calin A. Recurrence risk modeling of the genetic susceptibility to ankylosing spondylitis. Ann Rheum Dis. 2000;59:883–886. | |
Braun J, Bollow M, Remlinger G, et al. Prevalence of spondylarthropathies in HLA-B27 positive and negative blood donors. Arthritis Rheum. 1998;41:58–67. | |
Brown MA, Kennedy LG, MacGregor AJ, et al. Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA and the environment. Arthritis Rheum. 1997;40:1823–1828. | |
Diaz-Peña R, López-Vázquez A, López-Larrea C. Old and new HLA associations with ankylosing spondylitis. Tissue Antigens. 2012;80: 205–213. | |
Reveille JD. Genetics of spondyloarthritis – beyond the MHC. Nat Rev Rheumatol. 2012;8:296–304. | |
Robinson PC, Brown MA. The genetics of ankylosing spondylitis and axial spondyloarthritis. Rheum Dis Clin North Am. 2012;38:539–553. | |
Robinson PC, Brown MA. Genetics of ankylosing spondylitis. Mol Immunol. 2014;57:2–11. | |
Jin GX, Duan JZ, Guo WL, Li L, Cui SQ, Wang H. Association between IL-1RN gene polymorphisms and susceptibility to ankylosing spondylitis: a large Human Genome Epidemiology review and meta-analysis. Genet Mol Res. 2013;12:1720–1730. | |
International Genetics of Ankylosing Spondylitis Consortium (IGAS), Cortes A, Hadler J, et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet. 2013;45:730–738. | |
Australo-Anglo-American Spondyloarthritis Consortium (TASC), Reveille JD, Sims AM, et al. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat Genet. 2010;42:123–127. | |
Lee JC, Espéli M, Anderson CA, et al. Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathway. Cell. 2013;155:57–69. | |
Smith JA, Colbert RA. The interleukin-23/interleukin-17 axis in spondyloarthritis pathogenesis: Th17 and beyond. Arthritis Rheum. 2014;66: 231–241. | |
Coffre M, Rournier M, Rybczynska M, et al. Combinatorial control of Th17 and Th1 cell functions by genetic variations in genes associated with the interleukin-23 signaling pathway in spondyloarthritis. Arthritis Rheum. 2013;65:1510–1521. | |
García-Medel N, Sanz-Bravo A, Van Nguyen D, et al. Functional interaction of the ankylosing spondylitis-associated endoplasmic reticulum aminopeptidase I polymorphism and HLA-B27 in vivo. Mol Cell Proteomics. 2012;11:1416–1429. | |
Chen L, Fischer R, Peng Y, et al. Critical role of endoplasmic reticulum aminopeptidase I in determining the length and sequence of peptides bound and presented by HLA-B27. Arthritis Rheum. 2014;66: 284–294. | |
Andrés AM, Dennis MY, Kretzschmar WW, et al. Balancing selection maintains a form of ERAP2 that undergoes nonsense-mediated decay and affects antigen presentation. PLoS Genet. 2010;6:e1001157. | |
Cagliani R, Riva S, Biasin M, et al. Genetic diversity at endoplasmic reticulum aminopeptidases is maintained by balancing selection and is associated with natural resistance to HIV-1 infection. Hum Mol Genet. 2010;19:4705–4714. | |
Birtley JR, Saridakis E, Stratikos E, Mavridis IM. The crystal structure of human endoplasmic reticulum aminopeptidase 2 reveals the atomic basis for distinct roles in antigen processing. Biochemistry. 2012;51: 286–295. | |
Tsui FW, Haroon N, Reveille J, et al. Association of an ERAP1 ERAP2 haplotype with familial ankylosing spondylitis. Ann Rheum Dis. 2010;69:733–736. | |
Reeves E, Edwards CJ, Elliott T, James E. Naturally occurring ERAP1 haplotypes encode functionally distinct alleles with fine substrate specificity. J Immunol. 2013;191:35–43. | |
Lees CW1, Barrett JC, Parkes M, Satsangi J. New IBD genetics: common pathways with other diseases. Gut. 2011;60:1739–1753. | |
Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. | |
Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. | |
Lin DY, Tang ZZ. A general framework for detecting disease associations with rare variants in sequencing studies. Am J Hum Genet. 2011;89:354–367. | |
Beaudoin M, Goyette P, Boucher G, et al. Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet. 2013;9:e1003723. | |
Wolters FL, Russel MG, Sijbrandij J, et al. Disease outcome of inflammatory bowel disease patients: general outline of a Europe-wide population-based 10-year clinical follow-up study. Scand J Gastroenterol Suppl. 2006;243:46–54. | |
Djouadi K, Nedeiec B, Tamouza R, et al. Interleukin 1 gene cluster polymorphisms in multiplex families with spondylarthropathies. Cytokine. 2001;13:98–103. | |
Maksymowych WP, Rahman P, Reeve JP, Gladman DD, Peddle L, Inman RD. Association of the IL1 gene cluster with susceptibility to ankylosing spondylitis: an analysis of three Canadian populations. Arthritis Rheum. 2006;54:974–985. | |
Timms AE, Crane AM, Sims AM, et al. The interleukin 1 gene cluster contains a major susceptibility locus for ankylosing spondylitis. Am J Hum Genet. 2004;75:587–595. | |
Van der Paardt M, Crusius JB, García-González MA, et al. Interleukin-1 beta and interleukin-1 receptor antagonist gene polymorphisms in ankylosing spondylitis. Rheumatology (Oxford). 2001;40:1359–1364. | |
Sims AM, Timms AE, Bruges-Armas J, et al. Prospective meta-analysis of interleukin 1 gene complex polymorphisms confirms associations with ankylosing spondylitis. Ann Rheum Dis. 2008;67:1305–1309. | |
Monnet D, Kadi A, Izac B, et al. Association between the IL-1 family gene cluster and spondyloarthritis. Ann Rheum Dis. 2012;71:885–890. | |
Lin Z, Bei JX, Shen M, et al. A genome-wide association study in Han Chinese identifies new susceptibility loci for ankylosing spondylitis. Nat Genet. 2011;44:73–77. | |
Deuquet J, Lausch E, Superti-Furga A, van der Goot FG. The dark sides of capillary morphogenesis gene 2. EMBO J. 2012;31:3–13. | |
Urano T, Narusawa K, Shiraki M, et al. Single-nucleotide polymorphism in the hyaluronan and proteoglycan link protein 1 (HAPLN1) gene is associated with spinal osteophyte formation and disc degeneration in Japanese women. Eur Spine J. 2011;20:572–577. | |
Takai A, Inomata H, Arakawa A, Yakura R, Matsuo-Takasaki M, Sasai Y. Anterior neural development requires Del1, a matrix-associated protein that attenuates canonical Wnt signaling via the Ror2 pathway. Development. 2010;137:3293–3302. | |
Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010;468:834–838. | |
Davidson SI, Jiang L, Cortes A, et al. Brief report: High-throughput sequencing of IL23R reveals a low-frequency, nonsynonymous single-nucleotide polymorphism that is associated with ankylosing spondylitis in a Han Chinese population. Arthritis Rheum. 2013;65:1747–1752. | |
Kadi A, Cosstantino F, Izac B, et al. Brief report: The IL23R nonsynonymous polymorphism rs11209026 is associated with radiographic sacroiliitis in spondyloarthritis. Arthritis Rheum. 2013;65: 2655–2660. | |
Appel H, Maier R, Bleil J, et al. In situ analysis of interleukin-23- and interleukin-12-positive cells in the spine of patients with ankylosing spondylitis. Arthritis Rheum. 2013;65:1522–1529. | |
Sherlock JP, Joyce-Shaikh B, Turner SP, et al. IL-23 induces spondyloarthropathy by acting on ROR-γ+CD3+CD4–CD8– entheseal resident T cells. Nat Med. 2012;18:1069–1076. | |
Haroon N, Maksymowych WP, Rahman P, Tsui FW, O’Shea FD, Inman RD. Radiographic severity of ankylosing spondylitis is associated with polymorphism of the large multifunctional peptidase 2 gene in the Spondyloarthritis Research Consortium of Canada cohort. Arthritis Rheum.2012;64:1119–1126. | |
Meyer JL. Can biological calcification occur in the presence of pyrophosphate? Arch Biochem Biophys. 1984;231:1–8. | |
Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289: 265–270. | |
Goding JW, Grobben B, Slegers H. Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim Biophys Acta. 2003;1638:1–19. | |
Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol. 2001;281:C1–C11. | |
Tsui FW, Tsui HW, Cheng EY, et al. Novel genetic markers in the 5′-flanking region of ANKH are associated with ankylosing spondylitis. Arthritis Rheum. 2003;48:791–797. | |
Tsui HW, Inman RD, Paterson AD, Reveille JD, Tsui FW. ANKH variants associated with ankylosing spondylitis: gender differences. Arthritis Res Ther. 2005;7:R513–R525. | |
Tsui HW, Inman RD, Reveille JD, Tsui FW. Association of a TNAP haplotype with ankylosing spondylitis. Arthritis Rheum. 2007;56: 234–243. | |
Goseki-Sone M, Sogabe N, Fukushi-Irie M, et al. Functional analysis of the single nucleotide polymorphism (787T>C) in the tissue-nonspecific alkaline phosphatase gene associated with BMD. J Bone Miner Res. 2005;20:773–782. | |
Sogabe N, Oda K, Nakamura H, et al. Molecular effects of the tissue-nonspecific alkaline phosphatase gene polymorphism (787T>C) associated with bone mineral density. Biomed Res. 2008;29:213–219. | |
Furuichi T, Maeda K, Chou CR, et al. Association of the MSX2 gene polymorphisms with ankylosing spondylitis in Japanese. J Hum Genet. 2008;53:419–424. | |
Liu Z, Cui Y, Zhou X, Zhang X, Han J. Association of mineralization-related genes TNAP and ANKH polymorphisms with ankylosing spondylitis in the Chinese Han population. Biosci Trends. 2013;7:89–92. | |
Timms AE, Zhang Y, Bradbury L, Wordsworth BP, Brown MA. Investigation of the role of ANKH in ankylosing spondylitis. Arthritis Rheum. 2003;48:2898–2902. | |
Kim TH, Young BJ, Bang SY, et al. Genetic variants in OPG and ANKH are associated with severe ankylosis in patients with AS in Korean cohort. Arthritis Rheum. 2013;65 Suppl S10. Abstract 1901. | |
Akram A, Inman RD. Influenza infection of MHC-1 transgenic mice reveals that ERAP is necessary and sufficient for generation of the B27-specific immunodominant epitopes. Arthritis Rheum. 2013;65 Suppl:S531. | |
Akram A, Inman RD. Co-expression of HLA-B7 and HLA-B27 alleles is associated with B7-restricted immunodominant responses following influenza infection. Eur J Immunol. 2013;43:3254–3267. | |
Blanchard N, Shastri N. Coping with loss of perfection in the MHC class I peptide repertoire. Curr Opin Immunol. 2008;20:82–88. | |
Kanaseki T, Shastri N. Endoplasmic reticulum aminopeptidases associated with antigen processing regulates quality of processed peptides presented by MHC class I molecules. J Immunol. 2008;181:6275–6282. | |
Haroon N. Endoplasmic reticulum aminopeptidase 1 and interleukin-23 receptor in ankylosing spondylitis. Curr Rheumatol Rep. 2010;14: 383–389. | |
Haroon M, Inman RD. Endoplasmic reticulum aminopeptidases: biology and pathogenic potential. Nat Rev Rheumatol. 2010;6:461–467. | |
Evans DM, Spencer CC, Pointon JJ, et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet. 2011;43:761–767. | |
Akram A, Inman RD. Immunodominance: a pivotal principle in host response to viral infections. Clin Immunol. 2012;143:99–115. | |
Hammer RE, Maika SD, Richardson JA, Tang JP, Taurog JD. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human β2m: an animal model of HLA-B27-associated human disorders. Cell. 1990;63:1099–1112. | |
Tran TM, Dorris MI, Saturntira N, et al. Additional human beta-microglobulin curbs HLA-B27 misfolding and promotes arthritis and spondylitis without colitis in male HLA-B27-transgenic rats. Arthritis Rheum. 2006;54:1317–1327. | |
Redlich K, Görtz B, Hayer S, et al. Overexpression of tumor necrosis factor causes bilateral sacroiliitis. Arthritis Rheum. 2004;50:1001–1005. | |
Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. | |
Jacques P, Lambrecht S, Verheugen E, et al. Proof of concept: enthesitis and new bone formation in spondyloarthritis are driven by mechanical strain and stromal cells. Ann Rheum Dis. 2014;73:437–445. | |
Glant TT, Mikecz K, Arzoumanian A, Poole AR. Proteoglycan-induced arthritis in BALB/c mice. Clinical features and histopathology. Arthritis Rheum. 1987;30:201–212. | |
Bárdos T, Szabó Z, Czipri M, et al. A longitudinal study on an autoimmune murine model of ankylosing spondylitis. Ann Rheum Dis. 2005;64:981–987. | |
Haynes KR, Pettit AR, Duan R, et al. Excessive bone formation in a mouse model of ankylosing spondylitis is associated with decreases in Wnt pathway inhibitors. Arthritis Res Ther. 2012;14:R253. | |
Daoussis D, Liossiss SC, Solomou EE, et al. Evidence that Dkk-1 is dysfunctional in ankylosing spondylitis. Arthritis Rheum. 2010;62: 150–158. | |
Kwon SR, Lim MJ, Suh CH, et al. Dickkopf-1 level is lower in patients with ankylosing spondylitis than in healthy people and is not influenced by anti-tumor necrosis factor therapy. Rheumatol Int. 2012;32: 2523–2527. | |
Appel H, Ruiz-Heiland G, Listing J, et al. Altered skeletal expression of sclerostin and its link to radiographic progression in ankylosing spondylitis. Arthritis Rheum. 2009;60:3257–3262. | |
Las Heras F, Pritzker KPH, So A, et al. Aberrant chondrocyte hypertrophy and activation of β-catenin signaling precede joint ankylosis in ank/ank mice. J Rheumatol. 2012;39:583–593. | |
Lories RJ, Derese I, Luyten FP. Modulation of bone morphogenetic protein signaling inhibits the onset and progression of ankylosing spondylitis. J Clin Invest. 2005;115:1571–1579. | |
Tsui FW, Tsui HW, Las Heras F, Pritzker KP, Inman RD. Serum levels of novel noggin and sclerostin-immune complexes are elevated in ankylosing spondylitis. Ann Rheum Dis. Epub July 26, 2013. | |
Haibel H, Rudwaleit M, Listing J, Sieper J. Open label trial of anakinra in active ankylosing spondylitis over 24 weeks. Ann Rheum Dis. 2005;64:296–298. | |
Dudler J, Aubry-Rozier V. Tocilizumab in axial spondyloarthropathies: about 18 cases. Ann Rheum Dis. 2011;70 Suppl 3:128. | |
Sieper J, Braun J, Kay J, et al. Sarilumab for the treatment of ankylosing spondylitis: results of a Phase II, randomized, double-blind, placebo-controlled study (ALIGN). Ann Rheum Dis. Epub February 18, 2014. | |
Poddubnyy D, Hermann KG, Callhoff J, Listing J, Sieper J. Ustekinumab for the treatment of patients with active ankylosing spondylitis: results of a 28-week, prospective, open-label, proof-of-concept study (TOPAS). Ann Rheum Dis. Epub January 3, 2014. | |
Baeten D, Baraliakos X, Braun J, et al. Anti-interleukin-17A monoclonal antibody seculinumab in treatment of ankylosing spondylitis: a randomized, double-blind, placebo-controlled trial. Lancet. 2013;382: 1705–1713. | |
Stolwijk C, van Tubergen A, Castillo-Ortiz JD, Boonen A. Prevalence of extra-articular manifestations in patients with ankylosing spondylitis: a systematic review and meta-analysis. Ann Rheum Dis. Epub September 2, 2013. | |
Van Praet L, van den Bosch FE, Jacques P, et al. Microscopic gut inflammation in axial spondyloarthritis: a multiparametric predictive model. Ann Rheum Dis. 2013;72:414–417. | |
Bremander A, Petersson IF, Bergman S, Englund M. Population-based estimates of common comorbidities and cardiovascular disease in ankylosing spondylitis. Arthritis Care Res (Hoboken). 2011;63:550–556. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.