")
Back to Journals » Drug Design, Development and Therapy » Volume 16
The Flavagline Compound 1-(2-(dimethylamino)acetyl)-Rocaglaol Induces Apoptosis in K562 Cells by Regulating the PI3K/Akt/mTOR, JAK2/STAT3, and MAPK Pathways
Authors Yang X, Wu X, Wu X, Huang L, Song J, Yuan C, He Z, Li Y
Received 20 January 2022
Accepted for publication 27 June 2022
Published 4 August 2022 Volume 2022:16 Pages 2545—2557
DOI https://doi.org/10.2147/DDDT.S357891
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Jianbo Sun
Xinmei Yang,1– 3 Xijun Wu,4 Xiaosen Wu,5 Lei Huang,1,2 Jingrui Song,1,2 Chunmao Yuan,1,2 Zhixu He,3,6 Yanmei Li1,2
1State Key Laboratory for Functions and Applications of Medicinal Plants, Guizhou Medical University, Guiyang, 550014, People’s Republic of China; 2The Key Laboratory of Chemistry for Natural Products of Guizhou Province and Chinese Academy of Sciences, Guizhou Medical University, Guiyang, 550014, People’s Republic of China; 3Stem Cell and Tissue Engineering Research Center, Guizhou Medical University, Guiyang, 550004, People’s Republic of China; 4Department of Laboratory, The Affiliated Jinyang Hospital of Guizhou Medical University, Guiyang, 550023, People’s Republic of China; 5FuRong Tobacco Research Station, Xiangxi Autonomous Prefecture Tobacco Company Yongshun Branch, Yongshun, 416700, People’s Republic of China; 6Department of Pediatrics, Affiliated Hospital of Guizhou Medical University, Guiyang, 550001, People’s Republic of China
Correspondence: Yanmei Li, State Key Laboratory for Functions and Applications of Medicinal Plants, Guizhou Medical University, Guiyang, 550014, People’s Republic of China, Tel/Fax +86 85183805081, Email [email protected] Zhixu He, Stem Cell and Tissue Engineering Research Center, Guizhou Medical University, Guiyang, 550004, People’s Republic of China, Tel/Fax +86 13595019670, Email [email protected]
Purpose: Chronic myelogenous leukemia (CML) is a hematological malignancy with increased proliferation of cells of the myeloid series. This can disrupt normal hematopoiesis. The 1-(2-(dimethylamino)acetyl)-rocaglaol (MQ-16) is a new synthetic flavagline compound that showed promising activity in chronic myeloid leukemia K562 cells. This study aims to analyze the underlying mechanisms of MQ-16 against CML.
Methods: Growth, cell cycle progression, and apoptosis were assessed in K562 cells following MQ-16 exposure by MTT assay and flow cytometry. The effect of MQ-16 on DNA strands between nucleosomes was examined by 1% agarose gel electrophoresis. PI3K/Akt/mTOR, JAK2/STAT3, and mitogen-activated protein kinase (MAPK) pathway-related proteins were detected in MQ-16–treated K562 cells by Western blot.
Results: MQ-16 significantly inhibited the proliferation of K562 cells and arrested the cell cycle at the G2/M phase in a time- and concentration-dependent manner. MQ-16 induced mitochondria-dependent apoptosis by downregulating the anti-apoptotic proteins Bcl-2 and Bcl-xL and induced time- and concentration-dependent DNA fragmentation. In addition, MQ-16 affected the expression of PI3K/Akt/mTOR, JAK2/STAT3, and MAPK pathway-related proteins.
Conclusion: In summary, MQ-16 appears to be a promising chemotherapeutic drug for treating CML.
Keywords: CML, flavagline, cycle arrest, apoptosis, PI3K/Akt/mTOR, JAK2/STAT3, MAPK
Introduction
Cancer has always been a world health problem of deep concern. A survey reported in the United States in 2021 found that the incidence of lung cancer, colorectal cancer, leukemia and other cancers was relatively high.1 Among them, leukemia can occur at any stage, which was mainly divided into four categories: acute myeloid leukemia, chronic myeloid leukemia (CML), acute lymphocytic leukemia, and chronic lymphocytic leukemia, according to the cumulative cell type of the lesion and the progression of the disease.2 CML is a hematological malignancy that affects blood and bone marrow. In the Western hemisphere, men with CML accounted for 15% of all cases of new-onset leukemia.1 It is characterized by the translocation of chromosomes 9 and 22 via the fusion of Abelson tyrosine-protein kinase 1 (ABL1) and breakpoint cluster region protein (BCR). The product of BCR-ABL1 is a tyrosine kinase with increased activity.3 CML mainly occurs in three stages, chronic, accelerated, and blast phases.4 Before the development of tyrosine kinase inhibitors (TKIs), the median survival time of patients was 5–7 years. With the birth of TKIs, the life expectancy of CML patients has dramatically changed. The CML patients taking long-term TKIs are expected to achieve the survival and “quality of life” of the gender- and age-matched healthy people.5 However, this is insufficient for patients in the accelerated or blast phase because about 5–7% of patients will still progress to the accelerated/blast phase.6 For patients with blast phase CML, the survival period is up to 12 months.7 In addition, For the CML patients, taking TKIs for life and regular physical examinations are expensive, causing heavy financial pressure on families. Therefore, the continued development and search for new therapeutic drugs is our eternal goal.
Meliaceae includes 50 genera with about 1400 species, mainly distributed in tropical and subtropical regions.8 Currently, lignans, flavonoids, terpenoids, bisamides and flavaglines have been isolated from Meliaceae. Nevertheless, flavaglines have only been reported in the Meliaceae family so far.9 Since the discovery of the first compound rocaglamide in 1982, more and more flavaglines compounds have been studied, such as rocaglaol, silvestrol, and aglaiastatin.10 Flavagline compounds have a unique cyclopenta[b]benzofuran skeleton, and they were initially discovered to exert unique effects, including anti-insect, anti-fungal, anti-inflammatory, and neuroprotective activities. Interestingly, it was later proved that they have substantial anti-cancer effects at low nanomolar concentrations without significant toxicity on human umbilical vein endothelial cells.11 Flavaglines have been reported in many studies to induce apoptosis or cell cycle arrest in oral cancer, colorectal cancer, bladder urothelial cancer cells, leukemia, and other malignant cells.12–15 The most studied mechanism of action is related to protein synthesis, which was mainly inhibited by influencing core processes of mRNA translation.16 To further explore its mechanism of action, 1-(2-(dimethylamino)acetyl)-rocaglaol (MQ-16) was synthesized to examine the activity against CML and the underlying molecular mechanism in K562 cells.
Apoptosis is a process of programmed cell death, which can effectively remove damaged cells. To maintain normal physiology and tissue function, damaged, dysfunctional, or unneeded cells can be constantly eliminated by regulating cell death.17 Dysfunction of the normal cell death pathway can cause a variety of diseases, such as cancer. Therefore, inducing apoptosis is an ideal treatment strategy for cancer.18 The PI3K/Akt/mTOR, JAK2/STAT3, and mitogen-activated protein kinase (MAPK) pathways are highly active in cancer cells, and they are related to cell proliferation, apoptosis, differentiation, and inflammation. Inhibition of these pathways confirmed their ability to regulate the growth of a variety of tumor cells.19–21 It is unclear whether these pathways are involved in the anti-proliferative effects of MQ-16 in K562 cells.
In the present study, we explored the underlying anti-cancer mechanisms of MQ-16 in K562 cells by analyzing the cell cycle and apoptosis using flow cytometry. Then, we measured the expression of PI3K/Akt/mTOR, JAK2/STAT3, and MAPK pathway-associated proteins using Western blot. These findings could provide insight for targeting multiple pathways to treat CML and facilitate the development of MQ-16 for the potential treatment of CML.
Materials and Methods
Synthesis for MQ-16
There were two steps required to synthesize MQ-16:1 Rocaglaol was synthesized according to our previous procedure.22 Then To a solution of rocaglaol (30 mg, 0.065 mmol) in 2 mL DCM were added Et3N (28.7 μL, 0.207 mmol), chloroacetyl chloride (11 μL, 0.138 mmol) and a catalytic amount of DMAP at room temperature. The reaction mixture was stirred at room temperature for 10 h. To the solution was added with 30 mL water, and then extracted with ethyl acetate (30 mL). The organic layer was then washed with brine, dried over Na2SO4, and concentrated in vacuum. The crude products were prepared and purified by preparative TLC to obtain pure compound, 1-chloracetylrocaglaol.
2 To a solution of 1-chloracetylrocaglaol (30 mg, 0.056 mmol) in 2 mL DMF were added 2.52 µL dimethylamine in THF (2 M), NaHCO3 (7 mg, 0.084 mmol), and a catalytic amount of CsF at room temperature. The reaction mixture was stirred at room temperature for 10 h. To the solution was added with 30 mL water, and then extracted with ethyl acetate (30 mL). The organic layer was then washed with brine, dried over Na2SO4, and concentrated in vacuum. The crude products were prepared and purified by preparative TLC to obtain pure compound, 1-(2-(dimethylamino)acetyl)-rocaglaol (MQ-16).
MQ-16. m.p. 105–106 ℃;1 H NMR (400 MHz, CDCl3) δ: 7.13–7.06 (7H, m), 6.63 (2H, d, J = 8.9 Hz), 6.23 (1H, d, J = 1.9 Hz), 6.03 (1H, d, J = 1.9 Hz), 5.86 (1H, d, J = 3.7 Hz), 4.10 (1H, dd, J = 13.8, 6.1 Hz), 3.84 (3H, s), 3.76 (3H, s), 3.67 (3H, s), 3.00 (1H, d, J = 16.9 Hz), 2.86 (1H, td, J = 13.9, 4.9 Hz), 2.82 (1H, d, J = 16.9 Hz), 2.33 (1H, dd, J = 13.7, 6.3 Hz), 2.25 (6H, s), 1.77 (1H, s);13 C NMR (100 MHz, CDCl3) δ 169.3, 163.7, 161.0, 158.5, 157.9, 138.5, 128.7, 128.7, 128.0, 128.0, 127.8, 127.8, 127.2, 127.2, 126.2, 112.6, 106.6, 103.0, 93.3, 91.7, 88.2, 79.1, 59.8, 55.6, 55.4, 55.0, 53.8, 44.9, 44.9, 35.7; ESIMS m/z 542.2 [M + Na]+; HRESIMS m/z 520.2338 [M + H]+ (Calcd. for C30H34O7N, 520.2329). Its purity was 98.65% by HPLC analysis.
Reagents and Antibodies
Imatinib was purchased from MedChemExpress (New Jersey, USA). A BCA protein assay kit and DNA extraction phenol reagent were purchased from Solarbio (Beijing, China). An annexin V-FITC/ propidium iodide (PI) kit was purchased from Becton, Dickinson and Company (New Jersey, USA). Hoechst staining kit, Apoptosis-DNA Ladder Extraction Kit, and mitochondrial membrane potential assay kit with JC-1 were purchased from Beyotime (Jiangsu, China). Primary antibodies against Bcl-2 (4223S), Bcl-xL (2762S), cleaved caspase-9 (7237S), caspase-3 (9662S), cleaved caspase-3 (9661S), poly (ADP-ribose) polymerase (PARP; 9542S), p-Chk1 (2348T), Cdc25C (4688S), p-Cdc25C (4901S), p-Cdc2 (4539S), PI3K (4249T), Akt (4685S), p-mTOR (2974T), STAT3 (12640), MEK1/2 (8727T), p-MEK1/2 (9154T), c-Jun NH2 terminal kinase (JNK; 9252T), and p-JNK (4668T) and their respective secondary antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibodies against caspase-9 (ab25758), Bim (ab32158), Chk1 (ab40866), Chk2 (ab109413), p-Chk2 (ab32148), CDK1 (ab133327), cyclin B1 (ab32053), mTOR (ab32028), p-STAT3 (ab76315), Ras (ab52939), extracellular signal-regulated kinase (ERK; ab184699), p-ERK (ab32538), c-Myc (ab32072), p38 (ab47363), and p-p38 (ab178867) were purchased from Abcam (Cambridge, MA, UK). Antibodies against p-PI3K (341468) and p-Akt (11054) were purchased from ZEN BIOSCIENCE (Chengdu, China) and Signalway Antibody (CA, USA), respectively.
Cell Lines and Cell Culture
The K562 human chronic myeloid leukemia cell line and the 7702 human normal liver cells were obtained from ATCC (Manassas, VA, USA) and cultured in RPMI or DMEM medium (Gibco, USA) supplemented with 5% fetal bovine serum (VACCA, USA) at 37℃.
Cell Viability Assays
K562 cells and HL-7702 cells (8 × 103 cells/well) were exposed to MQ-16 (25–400 nM) or Imatinib (0.1875–3 μM) for 72 h. Control cells were treated with an equal volume of dimethyl sulfoxide (DMSO) (final concentration, <0.1%). Ten microliters of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) were added to cells cultured in 96-well plates for 4 h, and the absorbance of formazan products was measured at 490 nm using the Synergy 2 modular Multi-Mode Reader (BioTek, Winooski, VT, USA). Then, we used the FORECAST function to calculate the IC50 at 72 h. K562 cells were seeded in 96-well plates with MQ-16 (50, 100, 200, or 400 nM) for different times, and the cell inhibition rate and the growth curve were then analyzed using GraphPad Prism 8. The inhibition rate was calculated as follows: inhibition rate = (control group OD490 - treatment group OD490)/control group OD490 × 100%.23
Cell Cycle and Apoptosis Analyses
According to the IC50 value of MQ-16, we chose 50–200 nM concentration for subsequent experiments. K562 cells (6 × 105 cells/well) were incubated with MQ-16 or DMSO for 24, 48, or 72 h. Then, cells were collected and washed once or twice with pre-chilled phosphate-buffered solution (PBS). For cell cycle analysis, after fixation in cold 70% ethanol overnight at 4℃, cells were stained with 0.5 mL of PI (50 µg/mL), RNase (5 µg/mL) inhibitor, and Triton X100 (0.5 µg/mL) for 10 min at room temperature in the dark and then analyzed by flow cytometry (ACEA NovoCyte, USA). For apoptosis experiments, cells were incubated in the dark with 50 µL of binding buffer, followed by 2.5 µL of annexin- V and 2.5 µL of PI for 15 min at room temperature, then analyzed by flow cytometry.15
Hoechst 33258 Staining
To further observe apoptosis in K562 cells, cells (1 × 106 cells/well) were incubated with MQ-16 for 48 or 72 h. Then, we carried out the experiment according to the steps of the Hoechst staining kit instructions. The cell samples were collected by centrifugation and fixed with 0.5 mL of fixing solution for 10 min. After discarding the stationary solution, the cells were stained with 0.5 mL of Hoechst 33258 for 5 min. Next, the cells were resuspended in 50 µL of PBS containing a fluorescence quenching agent, and a fluorescence microscope (Leica Microsystems, Germany) was used to observe the nuclear morphology.
Detection of Fragmented DNA by Agarose Gel Electrophoresis
To examine the effect of MQ-16 on DNA strands, K562 cells (1 × 106 cells/well) with or without MQ-16 for 48 or 72 h were collected and washed twice with PBS. According to the apoptosis-DNA ladder extraction Kit instructions, the cells were lysed by incubation with 500 μL sample lysis buffer containing 0.5% proteinase K at 50℃ overnight. Then 500 μL tris-phenol (pH8.0) and chloroform were successively added to the sample for DNA extraction. After centrifugation for 5 min at 12,000 × g at 4℃, about 300 μL supernatant was slowly aspirated, 60 μL 10M CH3COONH4 and 600 μL ethanol absolute were added, and the mixture was inverted several times to mix. At this time, DNA precipitation was observed. Subsequently, the samples were stored at −20℃ for 1 h or overnight to fully precipitate small DNA fragments. After centrifugation for 10 min at 12,000 × g at 4℃, 50–100 μL TE was added to dissolve DNA. Finally, after DNA quantification, DNA fragmentation was detected by 1% agarose gel electrophoresis (80 v, 2 h).
Mitochondrial Membrane Potential Assay
To detect changes in mitochondrial membrane potential (MMP), K562 cells (1 × 106 cells/well) were incubated with or without MQ-16 for 72 h. Following the kit instructions, 1×105-6 × 105 cells were collected and resuspended in 0.5 mL of cell culture medium, and after adding 0.5 mL of JC-1 staining working medium, cells were incubated at 37℃ for 20 min. After washing with JC-1 staining buffer (1×), cells were resuspended with an appropriate amount of JC-1 staining buffer (1×) and observed using a fluorescence microscope.
Western Blotting Analysis
The collected K562 cells (2 × 106 cells/well) were mixed with cell lysis buffer for immunoprecipitation on ice for 1 h. The BCA assay kit was used to determine the concentration of proteins. Proteins (50 μg) were resolved by sodium dodecyl sulfate−polyacrylamide gel electrophoresis and transferred to PVDF membranes via wet transfer. The membranes were blocked with 3% BSA for 1 h, then washed with 1× TBST (20 mM Tris-HCl, 0.1% Tween 20) three times for 5 min each. Next, the membranes were incubated overnight at 4 ℃ with primary antibodies. The next day, the membranes were washed and incubated with the FITC-labeled secondary antibody at room temperature for 2 h. Finally, the membranes were scanned using the Odyssey Platform (LI-COR Biosciences, Lincoln, NE, USA).24
Statistical Analysis
Each experiment was repeated at least three times independently, and statistical analysis was performed using Student’s t-test or one-way ANOVA in GraphPad Prism 8.0 and SPSS 26.0. The data are expressed as the mean ± SD, and P < 0.05 indicated statistical significance.
Results
MQ-16 Suppressed K562 Cell Proliferation
MQ-16 is a new flavagline compound (Figure 1A). Using MTT assay, we found that MQ-16 could significantly inhibit K562 cell proliferation (IC50 161.85 ± 9.44 nM). Cell viability was significantly decreased by 50–400 nM MQ-16 (Figure 1B). Furthermore, MQ-16 inhibited K562 cell growth in a time- and concentration-dependent manner (Figure 1C). The cell morphology analysis also indicated that MQ-16 gradually increased the amount of cell debris in a concentration- and time-dependent manner compared with the control DMSO (Figure 1D) (Figure S1A). MQ-16 has four times less toxicity on HL-7702 (IC50 672.65±24.85 nM) than on K562 cells. Compared with positive control imatinib, MQ-16 has more toxicity on K562 cells and HL-7702 (Table 1).
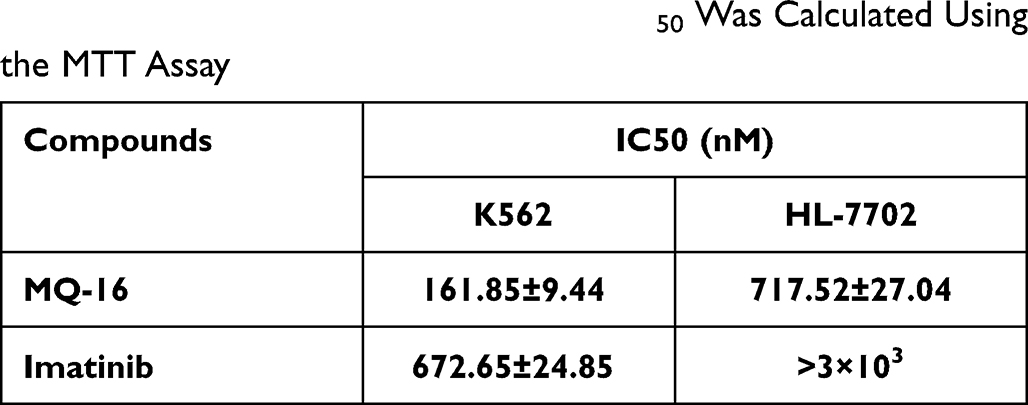 |
Table 1 K562 and HL-7702 Cells Were Exposed to MQ-16 or Imatinib in 96-Well Plates for 72 H. The IC50 Was Calculated Using the MTT Assay |
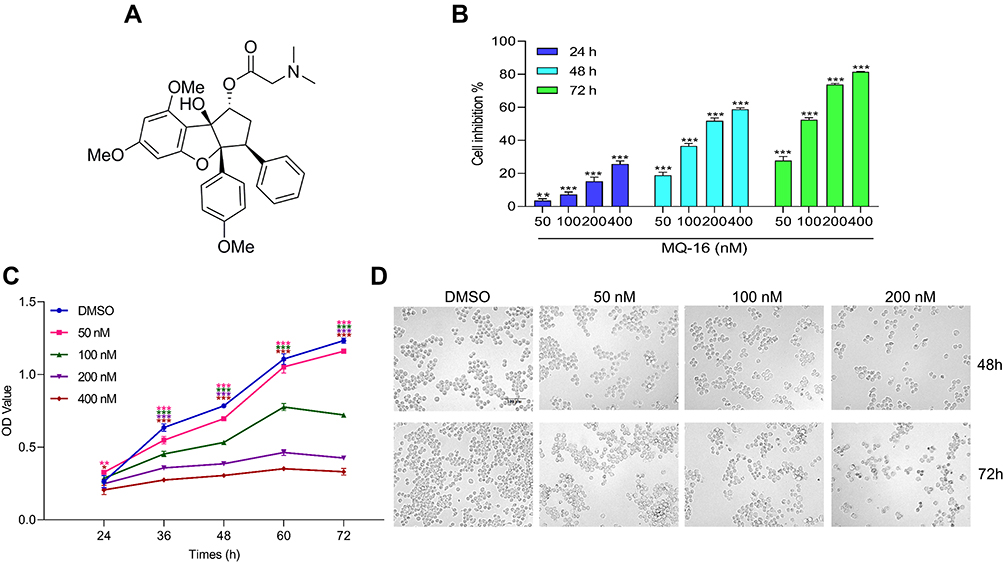 |
Figure 1 MQ-16 inhibited the proliferation of K562 cells. (A) The chemical structure of MQ-16. (B). Cells were treated with MQ-16 (50, 100, 200, or 400 nM) in 96-well plates for 24, 48, or 72 h, and the cell growth inhibition rate was calculated using the MTT assay. **P < 0.01, ***P < 0.001, versus the DMSO group. (C) K562 cells were incubated with MQ-16 for different times, then the growth curve was drawn using GraphPad Prism 8. *P < 0.05, **P < 0.01, ***P < 0.001, versus the DMSO group. (D) The morphology of K562 cells was observed and photographed using an inverted microscope (magnification, ×200; scale bar = 100 μm). |
MQ-16 Induced Cell Cycle Arrest in K562 Cells at the G2/M Phase
To investigate the effect of MQ-16 on cell cycle arrest, we used flow cytometry to detect the cellular DNA content of the cells and found that K562 cells exposed to MQ-16 for 24 h began to accumulate in the G2/M phase. The percentages of K562 cells in the G2/M phase were increased from 14.45% to 27.27%, 54.17%, and 58.04% at 0, 50, 100, and 200 nM for 72 h, respectively (Figure 2A and B). To investigate the molecular mechanisms underlying MQ-16–induced G2/M arrest, the expression of G2/M phase-related proteins was analyzed by Western blot. The downregulation of p-Cdc25C and p-Cdc2, and upregulation of p-Chk1/2 further proved that MQ-16 induced G2/M phase arrest and even caused DNA damage in G2/M phase (Figure 2C and D).
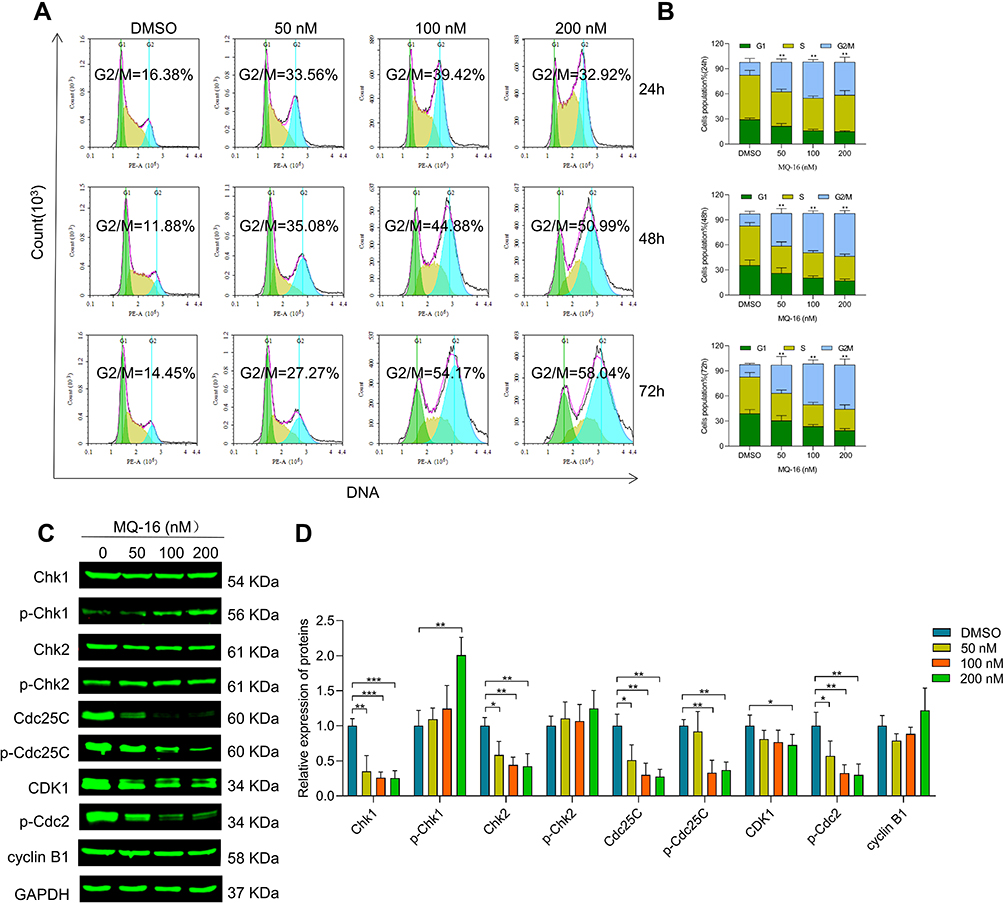 |
Figure 2 MQ-16 induced G2/M arrest in K562 cells. (A) K562 cells were treated with various concentrations of MQ-16 for 24, 48, or 72 h, then the cell cycle distribution was determined using propidium iodide. (B) The percentages of K562 cells in each phase of the cell cycle (G0/G1, S, and G2/M) are presented on the right. Data are presented as the mean ± SD of three independent experiments. **P < 0.01, versus the DMSO group. (C) The expression of Chk1/2, p-Chk1/2, Cdc25C, p-Cdc25C, CDK1, p-Cdc2, and cyclin B1 was detected by Western blot. (D) Quantification of relative protein expression levels of Chk1/2, p-Chk1/2, Cdc25C, p-Cdc25C, CDK1, p-Cdc2, and cyclin B1. GAPDH was used as loading control. *P < 0.05, **P < 0.01, ***P <0.001, versus the DMSO group. |
MQ-16 Induced Apoptosis in K562 Cells
To evaluate the effect of MQ-16 on cell apoptosis, the apoptosis rate, cell nuclear morphology, and DNA fragmentation were examined in K562 cells treated with different concentrations of MQ-16. The results illustrated that MQ-16 induced apoptosis in K562 cells, and the apoptosis rate under the highest concentration of MQ-16 exceeded 30% (Figure 3A and B) (Figure S1B). Hoechst 33258 is a blue fluorescent dye that can penetrate the cell membrane and binds to the minor groove in the DNA double-strand for DNA staining. Compared with the control group, after staining with Hoechst 33258, the nuclei of the MQ-16 group were densely stained, and the color was white (Figure 3C). Also, DNA fragmentation resulted in a time- and concentration-dependent DNA ladder pattern was found by 1% agarose gel electrophoresis (Figure 3D).
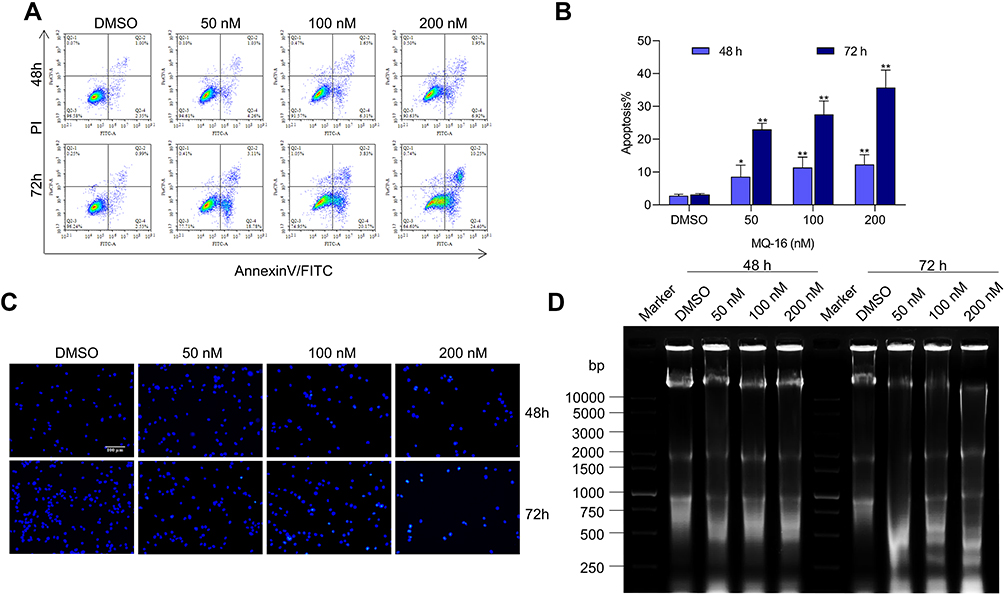 |
Figure 3 MQ-16 induced apoptosis in K562 cells. (A) K562 cells were treated with various concentrations of MQ-16 for 48 or 72 h, then apoptosis was detected by annexin- V/propidium iodide double staining using flow cytometry. (B) The percentage of apoptotic cells is presented below the images. *P < 0.05, **P < 0.01, versus the control. (C) K562 cells were stained with Hoechst 33258 and then observed by fluorescence microscopy (magnification, ×200; scale bar = 100 μm). (D) After treatment with MQ-16 for 48 h, DNA fragmentation in K562 cells was detected by 1% agarose gel electrophoresis. |
MQ-16 Decreased the MMP by Downregulating Bcl-2 Protein
Moreover, the assessment of MMP revealed that the number of cells with green fluorescence gradually increased in a concentration-dependent manner in the MQ-16 groups (Figure 4A). In mammalian cells, the Bcl2 and caspase families are involved in the process of apoptosis.25 To identify the mechanisms underlying MQ-16- induced apoptosis in K562 cells, apoptosis-related proteins were detected using Western blot. The results revealed that Bcl-2 and Bcl-xL were significantly downregulated in K562 cells treated with MQ-16. Whereas cleaved caspase-9, cleaved caspase-3, and cleaved PARP were upregulated by MQ-16 treatment (Figure 4B and C).
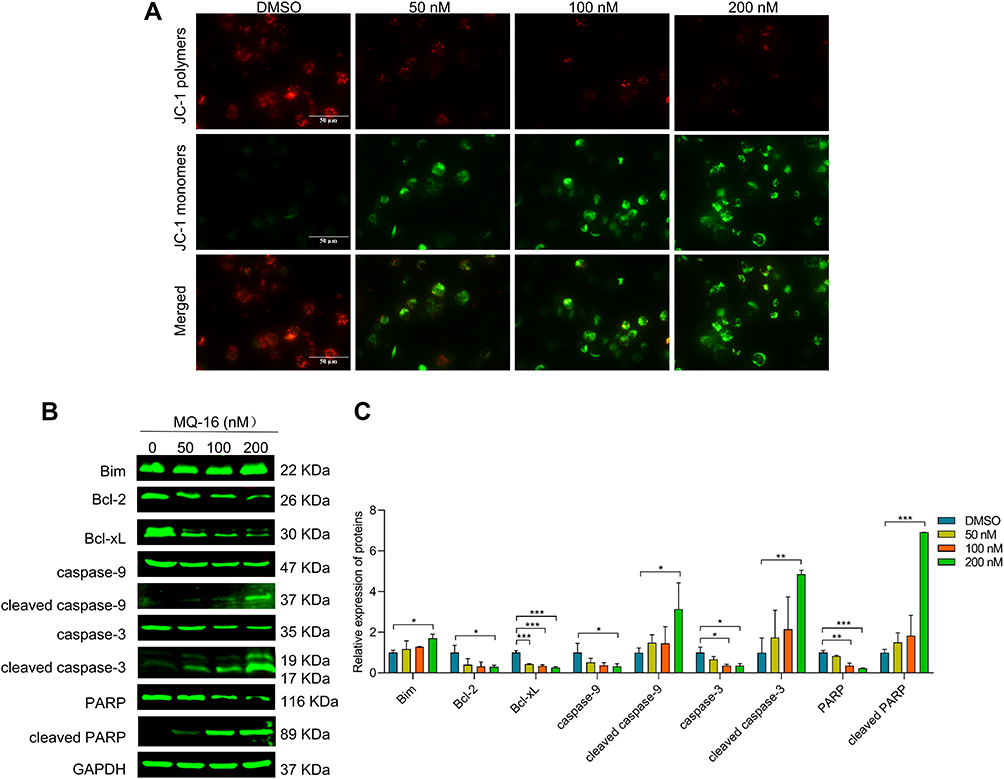 |
Figure 4 MQ-16 decreased the MMP by downregulating the Bcl-2 protein. (A) K562 cells were treated with MQ-16 for 72 h, then incubated with JC-1 staining buffer. Mitochondrial membrane potential was examined by fluorescence microscopy (magnification, ×630; scale bar = 50 μm). (B) The expression of Bim, Bcl-2, Bcl-xL, caspase-3, cleaved caspase-3, caspase-9, cleaved caspase-9, poly (ADP-ribose) polymerase (PARP), and cleaved PARP was measured by Western blot. (C) Quantification of relative protein expression levels of Bim, Bcl-2, Bcl-xL, caspase-3, cleaved caspase-3, caspase-9, cleaved caspase-9, PARP, and cleaved PARP. GAPDH was used as loading control. *P < 0.05, **P < 0.01, ***P <0.001, versus the DMSO group. |
Mechanism of Anticancer Activity of MQ-16
Previous studies revealed the critical roles of the PI3K/Akt/mTOR, JAK2/STAT3, and MAPK signaling pathways in the action of chemotherapeutic drugs.26–28 To further study the mechanism of MQ-16 in K562 cells, we performed Western blot to analyze the expression of related signaling proteins after exposure to MQ-16 for 24 h. The results revealed that the expression of p-PI3K, p-Akt, p-mTOR, p-JAK2, p-STAT3, p-MEK, and p-ERK was significantly reduced. However, p-p38 and p-JNK were activated, and their expression was significantly upregulated (Figure 5A–F).
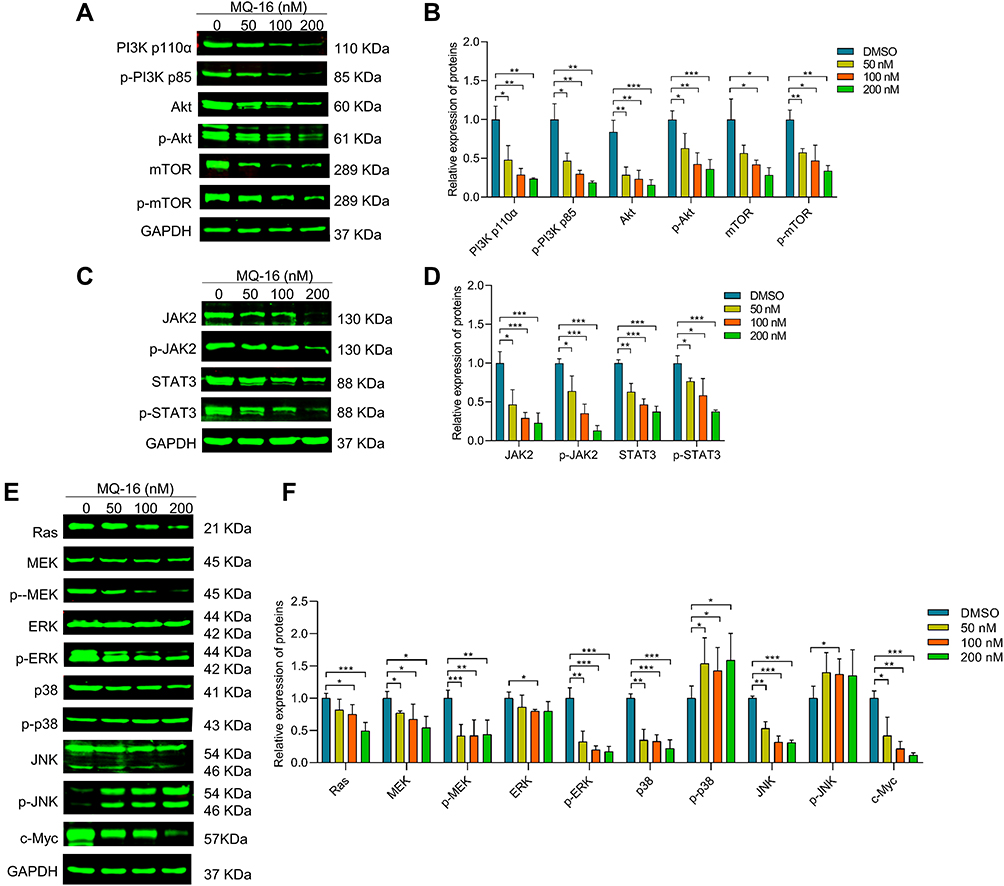 |
Figure 5 The effects of MQ-16 on the PI3K/AKT/mTOR, JAK2/STAT3, and mitogen-activated protein kinase signaling pathways. (A), (C) and (E) K562 cells were incubated with different concentrations of MQ-16 for 24 h. The expression of PI3K, p-PI3K, Akt, p-Akt, mTOR, p-mTOR, JAK2, p-JAK2, STAT3, p-STAT3, Ras, MEK, p-MEK, extracellular signal-regulated kinase (ERK), p-ERK, P38, p-P38, c-Jun NH2 terminal kinase (JNK), p-JNK, and c-Myc was detected by Western blot. (B), (D) and (F) Quantification of relative protein expression levels of PI3K, p-PI3K, Akt, p-Akt, mTOR, p-mTOR, JAK2, p-JAK2, STAT3, p-STAT3, Ras, MEK, p-MEK, ERK, p-ERK, P38, p-P38, JNK, p-JNK, and c-Myc. GAPDH was used as loading control. *P < 0.05, **P < 0.01, ***P <0.001, versus the DMSO group. |
Discussion
Patients with CML are mostly Philadelphia chromosome-positive, and thus, TKIs can slow disease progression and prolong survival. However, extending survival remains challenging in patients transitioning from the chronic phase to the acute phase.29 Flavagline compounds have been proven to inhibit the proliferation of leukemia cells, kill leukemia stem cells, and enhance the efficacy of anti-leukemia chemotherapy drugs.30 In this study, we explored the mechanism of the new synthetic flavagline compound MQ-16, finding that it induced apoptosis in K562 cells, which may have been mediated by the PI3K/Akt/mTOR, JAK2/STAT3, and MAPK signaling pathways.
Endogenous and exogenous stimuli mainly cause DNA damage. There are reports that genotoxic chemotherapeutic drugs can cause DNA damage.31,32 DNA damage activates DNA damage checkpoints and initiates DNA repair procedures. However, in response to severe or irreparable DNA damage, cells undergo permanent cell cycle arrest or apoptosis.33,34 Using flow cytometry, we revealed that MQ-16 markedly induced cell cycle arrest in the G2/M phase. Western blot demonstrated that the expression of phosphorylated cell cycle checkpoint kinase (Chk1/2) was increased, indicating that G2/M DNA damage checkpoints were activated. Chk1/2 further regulates the phosphorylation of Cdc25C and inhibits its activity.35 The regular operation of the cell cycle is inseparable from the strict regulation of cyclins and cyclin-dependent kinases. Different cyclins and cyclin-dependent kinases regulate cells at different stages. G2/M phase is mainly regulated by the cyclin B1 and CDK1 protein complex.36,37 At the protein level, MQ-16 inhibited the expression of CDK1, but its effect on cyclin B1 was not detected.
Apoptosis is an orderly and autonomous process of cell death controlled by various genes. DNA damage that is difficult to repair eventually leads to cell apoptosis.38 Because of the ability of cancer cells to evade apoptosis, the use of chemotherapeutic drugs is restricted in the clinic. Apoptosis has been widely studied as a target for tumor therapy.39 In the present study, apoptosis was induced by MQ-16 in K562 cells in a time- and concentration-dependent manner. Apoptosis is mainly triggered by two classical pathways: the extrinsic pathway (death receptors pathway) and the intrinsic pathway.40 Mitochondria mainly mediate the endogenous apoptosis pathway, and mitochondrial membrane potential is mostly maintained by the Bcl2 family.41 The Bcl2 family is divided into anti-apoptotic proteins such as Bcl2 and Bcl-xL and pro-apoptotic proteins such as Bax and Bim. A balance is maintained between anti-apoptotic and pro-apoptotic proteins to ensure the function of mitochondrial membrane potential.42 MQ-16 significantly reduced the expression of Bcl-2 and Bcl-xL. It promoted the expression of Bim, resulting in dysregulation of the balance of pro-apoptotic and anti-apoptotic proteins and a subsequent reduction of mitochondrial membrane potential. Subsequently, the promoter caspase-9 and the effector caspase-3 were activated. Western blot revealed that cleaved caspase-3 and cleaved PARP were upregulated by MQ-16 in a concentration-dependent manner. Caspase-3 is the most important terminal splicing enzyme involved in apoptosis, and its primary substrate is PARP, which is associated with DNA repair and gene integrity.43,44 Therefore, the upregulation of cleaved PARP and DNA fragmentation further proved that MQ-16 induced apoptosis.
The PI3K/Akt/mTOR, JAK2/STAT3, and MAPK signaling pathways are frequently activated in leukemia and other hematopoietic diseases by upstream mutations of cytokine receptors, abnormal chromosomal translocations, and other genetic mechanisms. Efficient targeting of these signaling pathways may lead to inhibition of cell growth and death of leukemia cells.45 BCR-ABL translocations are present in almost all patients with CML, and most patients are sensitive to imatinib, which inhibits the BCR-ABL oncoprotein.46 Compared with CML treatment with the targeted “upstream” inhibitor BCR-ABL, targeting the downstream signaling pathway to treat CML is an exciting theory due to its importance in survival.47 PI3K is a member of the lipid kinase family, and there are three types, of which Class I PI3K is most closely associated with cancer. Class IA PI3K comprises a regulatory subunit p85 and a catalytic subunit p110.48 When PI3K is activated, and the activated PI3K phosphorylates the substrate PIP2 to generate PIP3, which subsequently recruits Akt to the cell membrane. Akt, a serine/threonine kinase, is closely related to cell survival.49 Akt can regulate a variety of target proteins that control cell proliferation, survival, growth, and other processes, including the anti-apoptotic proteins Bcl-2, Bcl-xL; the pro-apoptotic proteins Bim, Bad and Bax; the DNA damage checkpoint kinase Chk1.50 In addition, Bcl2 family proteins are also regulated by ERK1/2.51 The Ras/Raf/MEK/ERK pathway acts as a complementary pathway to PI3K, and inhibition of the PI3K pathway may lead to feedback activation of MEK/ERK, a phenomenon that may impair its antitumor activity.52 In our study, MQ-16 could inhibit both PI3K and ERK pathways, thereby avoiding feedback activation of MEK/ERK. MAPKs are serine/threonine kinases that mediate intracellular signal transduction. The MAPK family consists of ERK, p38, and JNK. In different cells, ERK and p38, JNK sometimes show different functions in cell survival. Between p38 and ERK, there is a complex interaction. They have been reported to exhibit opposing effects on regulating cell survival and apoptosis.53 The JNK pathway can promote or resist apoptosis, depending on the cell type, the nature of the death stimulus, and the activity of other signaling pathways.54 In this study, MQ-16 activated p38 and JNK, making them involved in the regulation of apoptosis in the opposite role to ERK. Long et al reported that activation of p38 also decreased the phosphorylation level of CDC25C to arrest leukemia cells in the G2/M phase.55 The same trend was observed in this paper, indicating p38 might been also involved in the cycle regulation by MQ-16. A growing number of studies have found that abnormal expression and activation of STAT3 accompany the development of leukemia, suggesting a potential role of STAT3 in the pathogenesis of leukemia.56 STAT3 can be activated by multiple mechanisms, including through JAK/STAT3, Ras/MAPK, and non-receptor tyrosine kinase signaling pathways.57 In leukemia cells, Liu et al reported that inhibiting the phosphorylation levels of ERK and STAT3 inhibited cell growth, and the inhibition of ERK and STAT3 pathways resulted in the downregulation of the anti-apoptotic protein Bcl-2, thereby inducing apoptosis.58 In this study, MQ-16 significantly decreased p-ERK, p-STAT3 and Bcl-2. In summary, the results indicated that MQ-16-induced apoptosis in K562 cells might be regulated by PI3K/Akt/mTOR, JAK2/STAT3, and MAPK pathways (Figure 6).
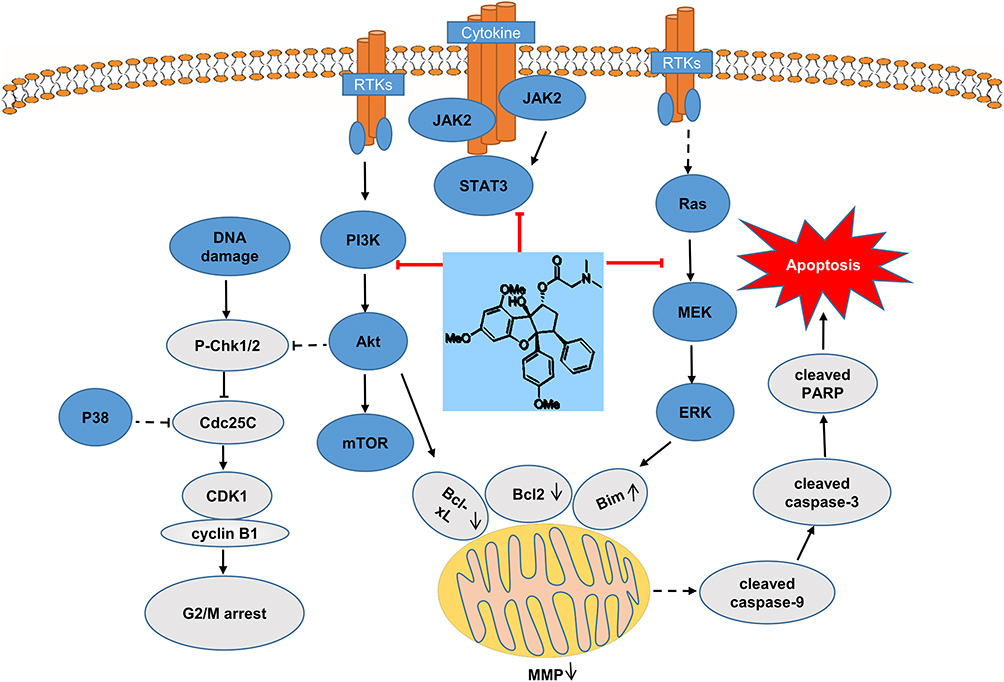 |
Figure 6 Schematic mechanisms of MQ-16 inducing G2/M arrest and apoptosis by regulating PI3K/Akt/mTOR, JAK2/STAT3, and MAPK signaling pathways. |
Conclusion
In conclusion, our present study demonstrated the activity of MQ-16 against K562 cells in vitro. MQ-16 induced G2/M arrest by activating the DNA damage checkpoint. MQ-16 induced mitochondria-dependent apoptosis by regulating the expression of Bcl2 family members and activating the caspase cascade. In addition, apoptosis may be induced by regulating the PI3K/AKT/mTOR, JAK2/STAT3, and MAPK pathways. Moreover, additional experiments showed that MQ-16 also induced apoptosis in human acute T lymphoblastic leukemia Jurkat cells (Figure S2A and B), indicating that MQ-16 might have therapeutic effects on both myeloid leukemia and lymphoid leukemia, and could enter clinical development in the future.
Acknowledgments
This research was supported financially by the grants from the National Natural Science Foundation of China
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. doi:10.3322/caac.21654
2. Juliusson G, Hough R. Leukemia. Prog Tumor Res. 2016;43:87–100.
3. Bartram CR, Klein A, Hagemeijer A, et al. Translocation of c-abl oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1983;306(5940):277–280. doi:10.1038/306277a0
4. Apperley JF. Chronic myeloid leukaemia. Lancet. 2015;385(9976):1447–1459. doi:10.1016/S0140-6736(13)62120-0
5. Hochhaus A, Baccarani M, Silver RT, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34(4):966–984. doi:10.1038/s41375-020-0776-2
6. Hehlmann R. Chronic myeloid Leukemia in 2020. Hemasphere. 2020;4(5):e468. doi:10.1097/HS9.0000000000000468
7. Jain P, Kantarjian HM, Ghorab A, et al. Prognostic factors and survival outcomes in patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era: cohort study of 477 patients. Cancer. 2017;123:4391–4402. doi:10.1002/cncr.30864
8. Paritala V, Chiruvella KK, Thammineni C, et al. Phytochemicals and antimicrobial potentials of mahogany family. Rev Bras Farmacogn. 2015;25:61–83.
9. Ebada SS, Lajkiewicz N, Porco JA Jr, et al. Chemistry and biology of rocaglamides (= flavaglines) and related derivatives from aglaia species (meliaceae). Prog Chem Org Nat Prod. 2011;94:1–58. doi:10.1007/978-3-7091-0748-5_1
10. Kim S, Salim AA, Swanson SM, et al. Potential of cyclopenta[b]benzofurans from Aglaia species in cancer chemotherapy. Anticancer Agents Med Chem. 2006;6(4):319–345. doi:10.2174/187152006777698123
11. Ribeiro N, Thuaud F, Nebigil C, et al. Recent advances in the biology and chemistry of the flavaglines. Bioorg Med Chem. 2012;20(6):1857–1864. doi:10.1016/j.bmc.2011.10.048
12. Uzawa K, Kasamatsu A, Saito T, et al. Growth suppression of human oral cancer cells by candidate agents for cetuximab-side effects. Exp Cell Res. 2019;376(2):210–220. doi:10.1016/j.yexcr.2019.01.016
13. Hausott B, Greger H, Marian B. Flavaglines: a group of efficient growth inhibitors block cell cycle progression and induce apoptosis in colorectal cancer cells. Int J Cancer. 2004;109(6):933–940. doi:10.1002/ijc.20033
14. Yuan G, Chen X, Liu Z, et al. Flavagline analog FL3 induces cell cycle arrest in urothelial carcinoma cell of the bladder by inhibiting the Akt/PHB interaction to activate the GADD45α pathway. J Exp Clin Cancer Res. 2018;37(1):21–32. doi:10.1186/s13046-018-0695-5
15. Song J, Yuan C, Yang J, et al. Novel flavagline-like compounds with potent Fli-1 inhibitory activity suppress diverse types of leukemia. FEBS J. 2018;285(24):4631–4645. doi:10.1111/febs.14690
16. Burgers LD, Fürst R. Natural products as drugs and tools for influencing core processes of eukaryotic mRNA translation. Pharmacol Res. 2021;170:105535. doi:10.1016/j.phrs.2021.105535
17. Rathmell JC, Thompson CB. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell. 2002;109(2):97–107. doi:10.1016/S0092-8674(02)00704-3
18. Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17:395–417. doi:10.1038/s41571-020-0341-y
19. Fresno Vara JÁF, Casado E, de Castro J, et al. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30(2):193–204. doi:10.1016/j.ctrv.2003.07.007
20. Venugopal S, Bar-Natan M, Mascarenhas JO. JAKs to STATs: a tantalizing therapeutic target in acute myeloid leukemia. Blood Rev. 2020;40:100634. doi:10.1016/j.blre.2019.100634
21. Kim EK, Choi E-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802(4):396–405. doi:10.1016/j.bbadis.2009.12.009
22. Yang HJ, Li YN, Yan C, et al. Discovery and synthesis of rocaglaol derivatives inducing apoptosis in HCT116 cells via suppression of MAPK signaling pathway. Fitoterapia. 2021;151:104876. doi:10.1016/j.fitote.2021.104876
23. Yang J, Qiu J, Hu Y, et al. A natural small molecule induces megakaryocytic differentiation and suppresses leukemogenesis through activation of PKCδ/ERK1/2 signaling pathway in erythroleukemia cells. Biomed Pharmacother. 2019;118:109265. doi:10.1016/j.biopha.2019.109265
24. Chen X, Song J, Yuan D, et al. Incaspitolide A extracted from Carpesium cernuum induces apoptosis in vitro via the PI3K/AKT pathway in benign prostatic hyperplasia. Biosci Rep. 2021;41(6):BSR20210477. doi:10.1042/BSR20210477
25. Zaman S, Wang R, Gandhi V. Targeting the apoptosis pathway in hematologic malignancies. Leuk Lymphoma. 2014;55(9):1980–1992. doi:10.3109/10428194.2013.855307
26. Xiao W, Li B, Sun X, et al. DCZ3301, a novel aryl-guanidino inhibitor, induces cell apoptosis and cell cycle arrest via suppressing the PI3K/AKT pathway in T-cell leukemia/lymphoma. Acta Biochim Biophys Sin. 2018;50:643–650. doi:10.1093/abbs/gmy047
27. Zhu M, Shi X, Gong Z, et al. Cantharidin treatment inhibits hepatocellular carcinoma development by regulating the JAK2/STAT3 and PI3K/Akt pathways in an EphB4-dependent manner. Pharmacol Res. 2020;158:104868. doi:10.1016/j.phrs.2020.104868
28. Xiao Y, Deng T, Wang D. Davanone terpenoid inhibits cisplatin-resistant acute myeloid leukemia cancer cell growth by inducing caspase-dependent apoptosis, loss of mitochondrial membrane potential, inhibition of cell migration and invasion and targeting PI3K/AKT/MAPK signalling pathway. J BUON. 2020;25:1607–1613.
29. Braun TP, Eide CA, Druker BJ. Response and resistance to BCR-ABL1-targeted therapies. Cancer Cell. 2020;37(4):530–542. doi:10.1016/j.ccell.2020.03.006
30. Callahan KP, Minhajuddin M, Corbett C, et al. Flavaglines target primitive leukemia cells and enhance anti-leukemia drug activity. Leukemia. 2014;28(10):1960–1968. doi:10.1038/leu.2014.93
31. Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K, et al. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73(1):39–85. doi:10.1146/annurev.biochem.73.011303.073723
32. Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332(2):237–248. doi:10.1016/j.canlet.2012.01.007
33. Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9(4):297–308. doi:10.1038/nrm2351
34. Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016;16(1):20–33. doi:10.1038/nrc.2015.2
35. Liu K, Zheng M, Lu R, et al. The role of CDC25C in cell cycle regulation and clinical cancer therapy: a systematic review. Cancer Cell Int. 2020;20(1):213–228. doi:10.1186/s12935-020-01304-w
36. Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15(6):122–131. doi:10.1186/gb4184
37. Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24(11):1770–1783. doi:10.1200/JCO.2005.03.7689
38. Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12(9):440–450. doi:10.1016/j.molmed.2006.07.007
39. Dixon SC, Soriano BJ, Lush RM, et al. Apoptosis: its role in the development of malignancies and its potential as a novel therapeutic target. Ann Pharmacother. 1997;31(1):76–82. doi:10.1177/106002809703100113
40. Pistritto G, Trisciuoglio D, Ceci C, et al. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging. 2016;8(4):603–619. doi:10.18632/aging.100934
41. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116(2):205–219. doi:10.1016/S0092-8674(04)00046-7
42. Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20(3):175–193. doi:10.1038/s41580-018-0089-8
43. Thomberry NA, Laxebnik Y. Caspases: enemies within. Science. 1998;281(5381):1312–1316. doi:10.1126/science.281.5381.1312
44. Gourley C, Balmaña J, Ledermann JA, et al. Moving from poly (ADP-Ribose) polymerase inhibition to targeting DNA repair and DNA damage response in cancer therapy. J Clin Oncol. 2019;37(25):2257–2269. doi:10.1200/JCO.18.02050
45. McCubrey JA, Steelman LS, Abrams SL, et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008;22(4):708–722. doi:10.1038/leu.2008.27
46. An X, Tiwari AK, Sun Y, et al. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review. Leuk Res. 2010;34(10):1255–1268. doi:10.1016/j.leukres.2010.04.016
47. Steelman LS, Abrams SL, Whelan J, et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008;22(4):686–707. doi:10.1038/leu.2008.26
48. Vogt PK, Hart JR, Gymnopoulos M, et al. Phosphatidylinositol 3-kinase: the oncoprotein. Curr Top Microbiol Immunol. 2010;347:79–104. doi:10.1007/82_2010_80
49. Noorolyai S, Shajari N, Baghbani E, et al. The relation between PI3K/AKT signalling pathway and cancer. Gene. 2019;698:120–128. doi:10.1016/j.gene.2019.02.076
50. Vachhani P, Bose P, Rahmani M, et al. Rational combination of dual PI3K/mTOR blockade and Bcl-2/-xL inhibition in AML. Physiol Genomics. 2014;46(13):448–456. doi:10.1152/physiolgenomics.00173.2013
51. Vitagliano O, Addeo R, D’Angelo V, et al. The Bcl-2/Bax and Ras/Raf/MEK/ERK signaling pathways: implications in pediatric leukemia pathogenesis and new prospects for therapeutic approaches. Expert Rev Hematol. 2013;6(5):587–597.
52. De Luca A, Maiello MR, D’Alessio A, et al. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012;16(Suppl 2):S17–27. doi:10.1517/14728222.2011.639361
53. Xia Z, Dickens M, Raingeaud J, et al. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270(5240):1326–1331. doi:10.1126/science.270.5240.1326
54. Jing L, Anning L. Role of JNK activation in apoptosis: a doubleedged sword. Cell Res. 2005;15:36–42. doi:10.1038/sj.cr.7290262
55. Long Q, Xiao X, Yi P, et al. L20, a Calothrixin B analog, induces intrinsic apoptosis on HEL cells through ROS/γ-H2AX/p38 MAPK pathway. Biomed Pharmacother. 2021;137:111336. doi:10.1016/j.biopha.2021.111336
56. Shi Y, Zhang Z, Qu X, et al. Roles of STAT3 in leukemia (review). Int J Oncol. 2018;53(1):7–20. doi:10.3892/ijo.2018.4386
57. Pilati C, Zucman-Rossi J. Mutations leading to constitutive active gp130/JAK1/STAT3 pathway. Cytokine Growth Factor Rev. 2015;26:499–506. doi:10.1016/j.cytogfr.2015.07.010
58. Liu C, Zeng Y, Dai LH, et al. Mogrol represents a novel leukemia therapeutic, via ERK and STAT3 inhibition. Am J Cancer Res. 2015;5(4):1308–1318.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.