")
Back to Journals » Open Access Journal of Clinical Trials » Volume 11
The European Medicines Agency Clinical Data Website Enables Insights Into Clinical Development Timelines And Strategy
Authors Lehmann S , Allard R , Boehler YB
Received 18 April 2019
Accepted for publication 4 September 2019
Published 15 October 2019 Volume 2019:11 Pages 37—56
DOI https://doi.org/10.2147/OAJCT.S205842
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Sarah Lehmann,1,2 René Allard,2 Yvonne-Beatrice Boehler1
1Faculty of Applied Natural Sciences, TH Koeln - University of Applied Sciences, Leverkusen, Germany; 2Grünenthal Innovation, Drug Development, Data Sciences’ Grünenthal GmbH, Aachen, Germany
Correspondence: René Allard Herrengrabenweg 34, Basel 4054, Switzerland
Tel +41 6176 824 0864
Email [email protected]
Purpose: The clinical study report (CSR) documents of a full clinical development pathway (CDP) have been publicly available on the European Medicines Agency (EMA; Amsterdam, Netherlands) clinical data website (ECDW) since October 2016. Our analysis aimed to determine the extent to which the available clinical development program could be assessed.
Methods: The documents available on the ECDW up to April 1, 2018 and the corresponding European Public Assessment Report (EPAR) were reviewed. Information extracted from the available CSRs focused on dates, phase of development, module leaf structure, and number of protocol amendments. Data analyses included generalized activity normalization time table (GANTT) charts and network analyses.
Results: Of the 86 available CDPs, 55 were initial marketing authorizations covering a diverse range of clinical developments from generics to advanced therapy in the electronic common technical documents (eCTDs). Non-redacted dates were available in 444 CSRs from 15 CDPs to perform retrospective project clinical development management analyses. In these 15 marketing authorizations, the median timespan to submission was 9.3 years (range: 6.2–22.2). The timespan within these 15 clinical developments ranged from 5.9 to 21.4 years (median 8.3). The median time to first-subject-in in the first controlled clinical study pertinent to the claimed indication (CCSPCI) was 4.4 years (range: 0–12.1); the duration of the CCSPCI ranged from 2.4 to 16.9 years (median: 4.4; interquartile range: 4.2–7.0). Four CDPs had concurrent subject enrolment, while seven CDPs had seamless study designs. Subject participation ranged from 52% to 97% of a clinical development timeline.
Conclusion: The publication of CSR documents by the EMA has enabled insights into timelines and project management aspects of the clinical development of medications.
Keywords: drug approval, clinical studies as topic, health information management, EMA Policy 0070, information dissemination
Introduction
The International Council for Harmonisation (ICH) E8 provides guidance for sponsors regarding the process of clinical development of pharmaceuticals for human use.1 The target product profile (TPP),2 a Food and Drug Administration guidance (FDA), provides guidance for a drug development program in terms of target product labelling concepts. It is frequently used within pharmaceutical companies to set individual milestones that need to be reached in the critical path of therapeutic development. In addition, the TPP permits an efficient dialogue with the competent authorities during the drug development process. A clinical development plan and TPP can increase the probability that safety and efficacy data are available in a timely manner and decrease the total time (ie, the critical path) of drug development.2 On March 16, 2004, a report entitled “Innovation/stagnation: challenge and opportunity on the critical path to new medical products”3 was released in the United States of America (USA). This report suggested that there was substantial opportunity to increase the pace of discovery and development of new medical products.3 There are few publications focusing on the actual clinical development timelines. A communication regarding clinical development timelines in the USA was published by the FDA in 2017.4 However, similar information from Europe is currently not available, possibly due to the different authorization procedures (ie, centralized vs national procedures).5
Project management is an integral part of clinical development. Regulatory Intelligence attempts to integrate relevant external knowledge into the internal processes of the company.6 The competitive advantage and profitability of an organization are determined by identifying drivers in the value chain.7,8 Integration of knowledge has been made possible by the European Medicines Agency (EMA; Amsterdam, Netherlands) clinical transparency initiatives, which have made an unprecedented amount of data available to the public. Article 12 of Regulation (EEC) No. 2309/93 required the EMA to make the assessment report of a medicinal product, including the reasons for granting marketing authorization, available on “request from any interested person.”9 Since 2004, the publication of European Public Assessment Reports (EPARs) is required for withdrawn or refused medicinal products, and must be publicly available.5,9 The EPAR provides an overview of the clinical studies submitted to the EMA for marketing approval and summarizes the evaluation performed by the agency.10
In 2012, the EMA and EU regulatory bodies published “Open Clinical Trial Data for All? A view from the Regulators,”11 and subsequently held a “Workshop on-clinical-trial data and transparency.”12 After workshops and a consultation process with stakeholders, the “European Medicines Agency policy on publication of clinical data for medicinal products for human use” (Policy 0070) was issued based on Article 80 of Regulation (EC) No. 726/2014.13 EMA Policy 0070 was adopted on October 2, 2014 with entry into force on January 1, 2015.14,15 The first dossiers were uploaded on the EMA clinical data website (ECDW) on October 20, 2016.16 Detailed guidance on the implementation of Policy 0070 can be found in three external guidance documents published in 2016 and 2017.17,19 EMA Policy 0070 requires that “As a general rule all clinical reports submitted as part of a regulatory application will be subject to publication” on an EMA website.19 The fourth guidance published in October 2018 was not relevant to this analysis. Documents from Modules 2 and 5 of the electronic Common Technical Document (eCTD) are accessible on the ECDW. For the first time, a regulatory authority has made clinical study reports of a full clinical development pathway (CDP) publicly available. The EMA reported that, by December 31, 2017, a total of 54 procedures were published, including 36 initial marketing authorizations (IMA) and 18 extensions of indication. These 54 procedures comprised a total of 3279 documents and >1.3 million pages.20
The Yale University Open Data Access (YODA) Project21 and ClinicalStudyDataRequest.com (CSDR)22 are two of the earliest platforms used by commercial sponsors to make documents and data available to researchers prior to the EMA initiative. The publication of the clinical study reports (CSRs) in a CDP by the EMA has created the unique opportunity to analyze the clinical development project timelines. The information available publicly on the EMA websites (EPAR and ECDW) is illustrated in Figure 1. Initially, the diversity of available CDPs was analyzed to enable the structuring of data by different sponsors into a database. Thereafter, an analysis of CDP timelines and the individual clinical studies with visualization techniques was performed to better understand the interrelationships between clinical development, strategies employed, and the impact on clinical development timelines. The rationale for embarking on this analysis was to determine the clinical development strategies followed by sponsors to achieve EMA marketing authorizations, and assess the timelines as the initial step for resource estimations.
 |
Figure 1 Simplified flow processes in the development of medicinal products and the corresponding process of publicly available information on the EMA website. Abbreviations: eCTD, electronic Common Technical Document; EMA, European Medicines Agency; EPAR, European Public Assessment Report; HTA, Health Technology Assessment. |
Materials And Methods
The initial marketing applications available on the ECDW were downloaded and analyzed by one reviewer; the cut-off date was April 1, 2018. The methodology used to access and navigate through the ECDW has been described in detail by Billiones and Schneider.23 Data extraction of a predetermined list of variables (four broad categories: dates, phase of development, module leaf structure, and number of clinical protocol amendments) was performed from March 2 to May 31, 2018. A total of 3974 documents on the ECDW16 were reviewed. A second analyst independently extracted the timeframe and clinical study report details. Reconciliation of the extracted data was performed after each eCTD.
eCTD Similarity Or Diversity
The similarity or diversity of CDPs (ie, whether the eCTD was an IMA or extension of an existing marketing authorization), EMA ATC category, and the number of studies (by phase and eCTD module category) were analyzed. As the dossiers were different in leaf structure and content, the data extraction process and analyses were iteratively performed for each eCTD using the Deming Cycle Approach.24,25
The eCTD is organized in a structured manner into five modules, as described in ICH M4.26 Module 1 is region specific, while Modules 2, 3, 4, and 5 are intended to be common for all regions. The granularity is detailed in accordance with Appendix 4 of the eCTD specification and structured in a “tree and leaf” structure. The documents are labeled (named) based on their organization in the eCTD. Thus, a document named “m5351-rdea594-203-app1619-sap” indicates that the document is the Statistical Analysis Plan of the RDEA594-203 clinical study report which is part of Module 5 of the eCTD in the subsection 3.5.1. (ie, a clinical study that has been submitted in support of the label, a confirmatory study).
The variability of the available IMA CDPs was assessed by extracting data on the following variables for each CDP: total number of clinical studies, the number of participants in the clinical development, and the breakdown of the clinical development documentation. The “phase of clinical development” was extracted from the study protocols. A further analysis, based on the ICH M426 document name (eg, m5351-XXX in the EMA clinical data website16) was performed in parallel.
Clinical Study Reports Available For CDP Timeline Evaluation
This analysis evaluated the clinical development timelines and focused on the IMAs. The documents within an IMA were evaluated for the availability of dates. The variable “dates” included the dates of available documents (ie, clinical study protocols, clinical study reports, statistical analysis plans), as well as the dates of first-subjects-in/entered and dates of last-subject-out or study recruitment periods. These data were extracted from the CSRs and used to calculate timelines/timeframes for the construction of GANTT charts and project evaluation. The dates of marketing submissions were obtained from the corresponding EPAR for the marketing authorization, available on the EMA EPAR website. The IMA was considered a core CDP if a timeline reconstruction was possible from the available documents. Furthermore, core CDPs had Phases I, II, and III of development. If an overall timeline reconstruction was not possible at a detailed level, then the IMA was clustered into a non-core CDP set. For example, specific dates were redacted in the CSRs for the “autologous CD34+ enriched cell fraction human ADA treatment” CDP. However, the clinical overview stated that the development took place from 2000 to 2014. There were two categories with CDPs in which timeline reconstruction was not possible (ie, “CDPs with redacted dates” and “CDPs with no CSRs”). Subsequently, the core CDPs were clustered by the EMA published Anatomical Therapeutic Chemical Classification System (ATC) code of the IMAs to permit future comparisons for a specific therapeutic indication.
The number of CDPs, the timespan of each CDP (start year to end year, inclusive), and the breakdown of each CDP by the number of CSRs was analyzed for the core and non-core CDP sets. Examples of active substances withdrawn from the market (eg, drisapersen) with published CDPs, were included. The breakdown by phase of development in the core and non-core CDP sets was compared for homogeneity. Generic CDPs were not included in the analyses; a different regulatory strategy was followed and only had Module 5.3.1.2 (Comparative BA and Bioequivalence CSRs) components. Details are shown Supplemental Table S1.
Core CDP Analyses
The core CDP set allowed a more precise timespan (based on the number of days) as all dates could be extracted. Analysis of project management aspects (eg, GANTT), network analysis, and program evaluation and review technique (PERT) was performed as described in standard project management reference works.25 The number of subjects that participated in a CDP was extracted from the corresponding EPAR. Details regarding the timeline assessments are shown in Supplemental Table S2.
Visualization Of Clinical Development Pathways
Reconstruction of the clinical study activities in a CDP was performed by retrospectively fitting the actual dates from the eCTD. The GANTT approaches included plotting dates of documents as well as dates of subject participation (study periods) in a clinical study. A GANTT chart for the eluxadoline CDP was generated to demonstrate the ability to illustrate concurrent and sequential subject participation elements.
A network analysis based on FSI dates for the lesinurad CDP was generated following the 1984 BS 6046: Part 1 schematic.25 Events/milestones were numbered sequentially according to the FSI dates. The RDEA594-101 study was the first event/milestone, as it was the first EMA study of the 42 clinical studies in the CDP to enroll subjects. One open-label and two double-blind extension studies (accounting for the 45 CSRs) were excluded in the representation. Subsequently, the studies were arranged by event/milestone (based on date of FSI) and category (ie, the first category was studies in which the medication was used as “monotherapy in patients”). As the analysis was performed from a retrospective point of view, each activity label had three numbers. The event/milestone was entered in the left half of the circle, the planned duration of a subject in the clinical study (as planned by the protocol) was entered in the upper right quadrant, and the duration of the study and FSI to LSO were entered in the lower right quadrant. The phase of development was color coded. The final event milestone (ie, 43 in the lesinurad CDP) was eCTD submission to the EMA. The planned date in the upper right quadrant was unknown and therefore labeled N/A (Not Available).
Analyses Of Phase I Clinical Studies
In addition to the number of Phase I clinical studies, there was a special interest in determining which CDPs included mass balance, renal and hepatic impairment, as well as thorough QT/QTc prolongation/QTc-like studies (QT-prolongation).
Clinical Protocol Study Amendment Analysis
Information regarding study protocol amendments was obtained from the protocol in the CSR appendix 16.1. The number of protocol amendments was available in the final protocol uploaded on the ECDW. If the interim report was available, the last version of the CSR for the clinical study was analyzed.
All calculations, including median and interquartile ranges, were performed using Excel® in the Microsoft Office 365 Enterprise version (Redmond, WA, USA).
Results
In the first 18 months (October 20, 2016 to April 1, 2018) after the clinical data website became operational, the EMA published clinical study documents for 86 marketing authorizations.
The diversity of the 55 IMA CDPs sorted by ATC is shown in Supplemental Table S1. Of the 3974 documents available on the ECDW up to April 1, 2018, 610 (15%) were CSRs. The therapeutic areas (by ATC clusters) with the most submissions were “other antineoplastic agents” (L01X, 12 CDPs), “direct-acting antivirals” (J05A, seven CDPs), and “immunosuppressants” (L04, five CDPs). In J05 (ie, direct-acting antiviral), CDPs had the oldest CSRs. The earliest Module 5.3 document was dated 1993. The development lifecycle was ongoing in 22 CDPs (ie, there were ongoing studies for which reports were expected after marketing authorization). Pediatric populations were included in the CDPs of 10 IMA submissions (Supplemental Table S1).
CSRs Available For CDP Timeline Evaluation
In total, 610 CSRs were available for the 55 IMAs, including 349 Phase I, 80 Phase II, 124 Phase III, 15 Phase IV, 33 multiple-phase, and 9 clinical studies without a designated phase. Thirteen IMAs did not have CSRs. These IMA submissions were based on CHMP/EWP/QWP/401/98 Rev 1, and included the 11 CDPs (palonosetron [2 IMAs], ertapenem, caspofungin acetate, bortezomib [two IMAs], pemetrexed [two IMAs], docetaxel, methotrexate, and lutetium chloride. In addition, migalastat had no CSR posted due to a legal dispute, and chlorhexidine was an Article 58 submission based on literature. Seven additional CDPs (amlodipine + valsartan, emtricitabine + tenofovir disoproxil [two IMAs], alendronic acid + cholecalciferol, rasagiline, aripiprazole, and sildenafil) consisted of Phase I studies and were excluded from further analysis. Ten IMAs were excluded because of missing dates or redacted dates in documents. Five IMAs had tenofovir (disoproxil or alafenamide) as an active ingredient. Thus, tenofovir was present in multiple categories. Eight IMAs (chenodeoxycholic acid, enoxaparin [two IMAs], tenofovir disoproxil, emtricitabine + tenofovir disoproxil, autologous CD34+ enriched cell fraction that contains CD34+ cells transduced with retroviral vector that encodes for the human ADA cDNA sequence, begelomab, and zonisamide) had all study periods dates in the documents redacted. Although specific dates were redacted in the CSRs, the CDP for the autologous CD34+ enriched cell fraction containing CD34+ cells transduced with retroviral vector that encodes for the human ADA cDNA sequence had the overall timeline data in the clinical overview. The CDP timelines could not be fully reconstructed in three additional IMAs (pandemic influenza vaccine H5N1, drisapersen, and saxagliptin + dapagliflozin). In the remaining 23 CDPs, timelines could be reconstructed for analysis. Eight of these CDPs (albutrepenonacog alfa, eftrenonacog alfa, human coagulation factor X, rociletinib, daratumumab, osimertinib, infliximab, and idarucizumab) were clinical developments, in which either Phase I, II and/or III studies were not part of the pathway; they were in the non-core set and excluded from the timeline analyses. The 55 IMAs were categorized into: a) a core CDP set of 15 CDPs for a detailed time analysis; b) a non-core CDP set of 18 CDPs with limited information from the clinical overview or CSRs; c) a set of nine CDPs with redacted CSRs; and d) 13 CDPs without CSRs (Table 1).
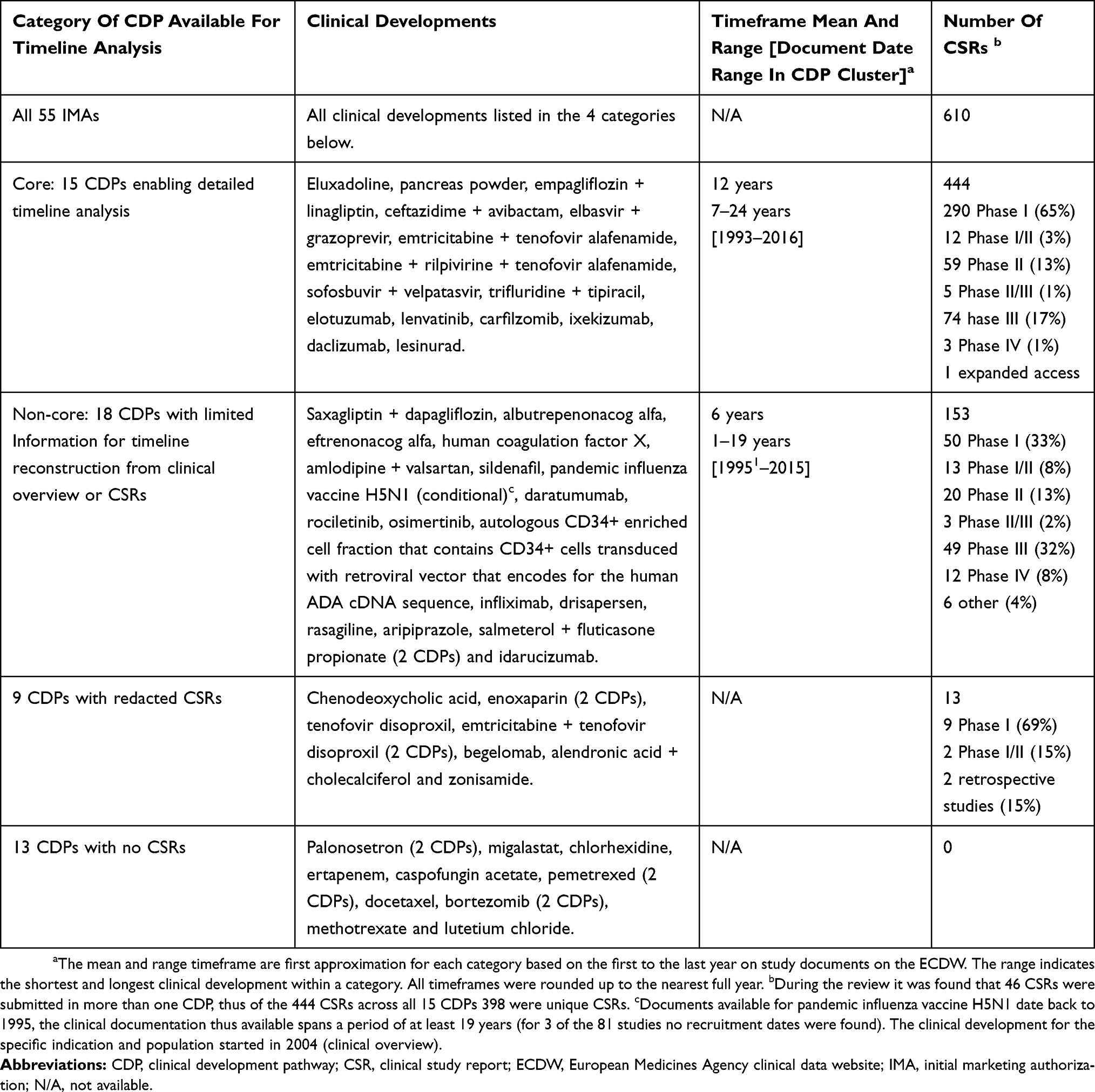 |
Table 1 Categories And Data Availability Of Clinical Development Pathways For Timeline Analysis |
The 18 non-core CDPs differed from the 15 CDPs in the core set (Figure 2). Although the proportion of Phase II studies was similar that of Phase I studies was higher in the core CDP set. The non-core set had proportionately more Phase III clinical studies than the core CDP set.
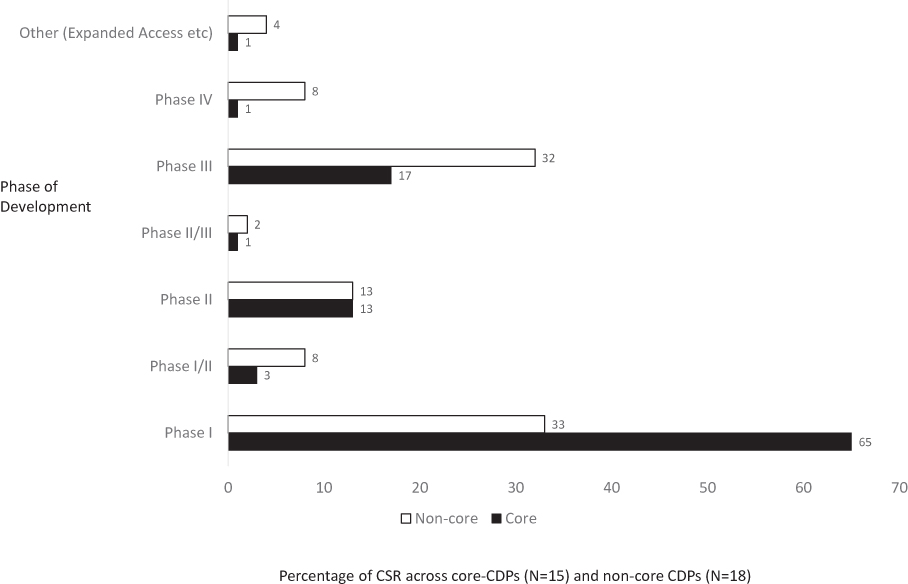 |
Figure 2 Comparison of document composition in the core and non-core CDPs. Percentage of clinical study reports according to the phase of clinical development. Note: There were 444 CSRs from the 15 CDPs in the core set and 153 CSRs from the 18 CDPs in the non-core set. Abbreviations: CDP, clinical development pathway; CSR, clinical study report; EMA, European Medicines Agency. |
Therapeutic indication (ATC), breakdown by eCTD leaf structure and by phase of clinical development, and the diversity of the core CDPs
The 15 core CDPs analyzed cover eight high-level ATC categories (Figure 2). For five of the ATC categories, there was only one submission. These five CDPs with 105 publicly available CSRs were antibacterial for systemic use (fixed-dose combination of ceftazidime + avibactam with 26 CSRs), antidiarrheal (eluxadoline with 14 CSRs), antigout (lesinurad with 45 CSRs), blood glucose-lowering drugs (empagliflozin + linagliptin [EMPA+LINA] with 13 CSRs), and enzyme preparations (pancreas powder with seven CSRs). CDPs in multiple myeloma (L04X), hepatitis C (J01A), and HIV (J05AR) had more than one CDP available for timeline analysis (Figure 3). These three ATC categories included two immunosuppressant CDPs (ixekizumab and daclizumab with 13 and 11 CSRs, respectively), four antineoplastic (fixed-dose combination of trifluridine + tipiracil [FTD+TPI], elotuzumab, lenvatinib, and carfilzomib with 15, 9, 25, and 13 CSRs, respectively), and four direct-acting antiviral CDPs (all four fixed-dose combination developments: elbasvir + grazoprevir [EBR+GZR], emtricitabine + tenofovir alafenamide [F/TAF], emtricitabine + rilpivirine + tenofovir alafenamide [FTC+RFV+TAF], sofosbuvir + velpatasvir [SOF+VEL]). Each of the four direct-acting antiviral CDPs available for timeline analysis had >50 CSRs. The clinical study reports in this ATC cluster accounted for 57% of the 444 CSRs in the core set. The two CDPs with the emtricitabine + tenofovir alafenamide combination (FTC+RFV+TAF and F/TAF) had 38 CSRs in common.
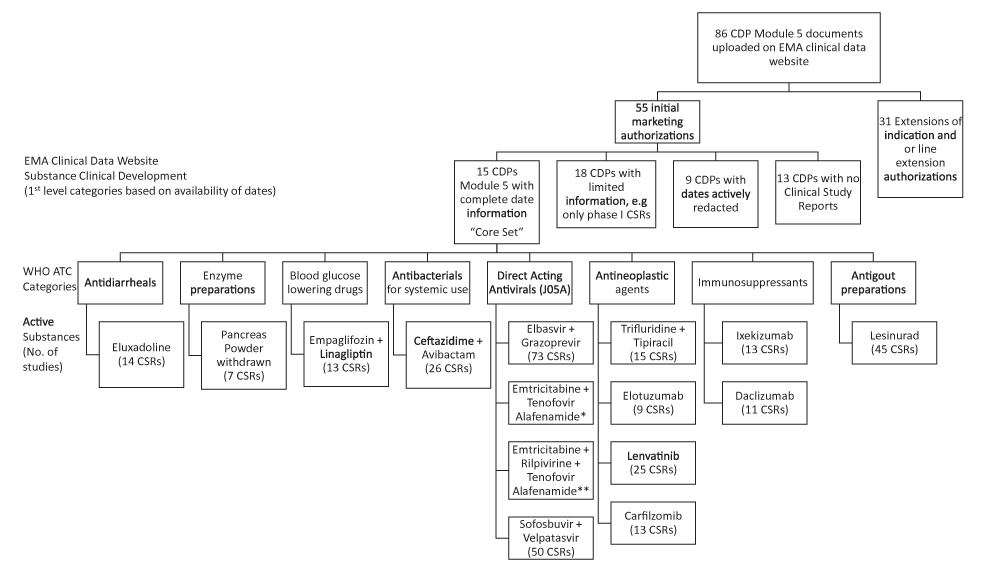 |
Figure 3 Clinical development documents available on the clinical data website of the EMA up to April 1, 2018. Note: Breakdown to show the documents in Module 5.3 that could be used to evaluate project management timelines. Abbreviations: CDP, clinical development pathway; CSR, clinical study report; No, number. |
Table 2 lists the categorization of CSRs based on the M.5.3 leaf structure required by ICH M4. The most common documents in the 15 core CDPs, based on the high-level regulatory leaf structure (Table 2), were 212 CSRs of “human pharmacokinetic studies,” followed by 77 reports of “CCSPCI,” and 44 CSR “reports of biopharmaceutic studies.” Each of the sections M.5.3.5.2 “Study reports of uncontrolled clinical studies” and M.5.3.5.4 “Other study reports” had 40 CSRs. The smallest section with five documents in the 15 CDPs pertained to “reports of studies pertinent to pharmacokinetics using human biomaterials” (Table 2).
 |
Table 2 CSR Categorization Based On The eCTD M.5.3 Leaf Structure And Phase Of Clinical Development – Core CDPs |
Timeline Analyses
The core CDP set comprised 444 studies: 290 Phase I, 59 Phase II, 74 Phase III, three Phase IV, 17 multiple-phase, and one expanded access program. The dates extracted from the 15 core CDPs enabled us to perform seven different timeline analyses: CDT, DST, TSRT, time to Phase II, time to Phase III, the duration of the Phase III part of the CDP, and the duration of CCSPCI.
The Clinical Development Timeline (CDT)
The median CDT in the 15 core CDPs was 9.3 years (3397 days; interquartile range [IQR]: 2922–4026 days). The number of subjects ranged from 272 subjects in the CDP for pancreas powder to 17,022 subjects in the EMPA+LINA CDP (Table 3). The CDT for the pancreas powder CDP was 9.3 years (3397 days), despite being the core CDP with the lowest number of subjects. The CDT of the EMPA+LINA CDP was 1.8 years longer: 11.1 years (4052 days; Table 3). Nine of the 15 CDPs had a CDT <10 years (Table 3).
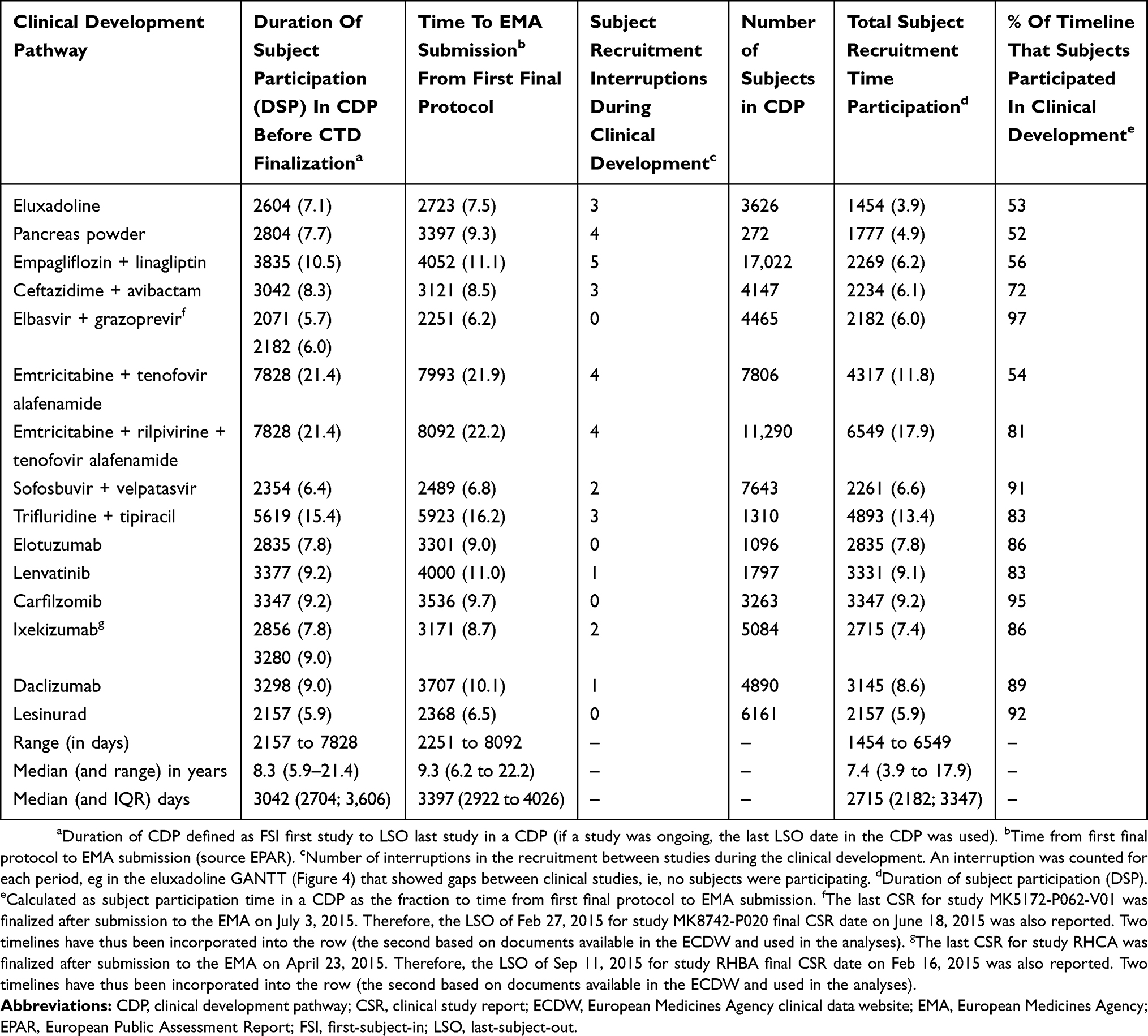 |
Table 3 Clinical Development Observations With Respect To Timelines In Days (And Years) |
Total Subject Recruitment Time (TSRT) In The Core CDP Set
The TSRT is the timespan from FSI to LSO in the CDP in the documents submitted to the EMA (Table 4). The TSRT ranged from 5.9 years (2157 days) in the lesinurad CDP, to 21.4 years (7828 days) in the F/TAF and FTC+RFV+TAF CDPs (Table 3). The 143-001 and GS-US-311-1387 studies were the first and last, respectively, in both the FTC+RFV+TAF and F/TAF CDPs. The median TSRT was 7.4 years (2715 days; IQR: 2182–3347).
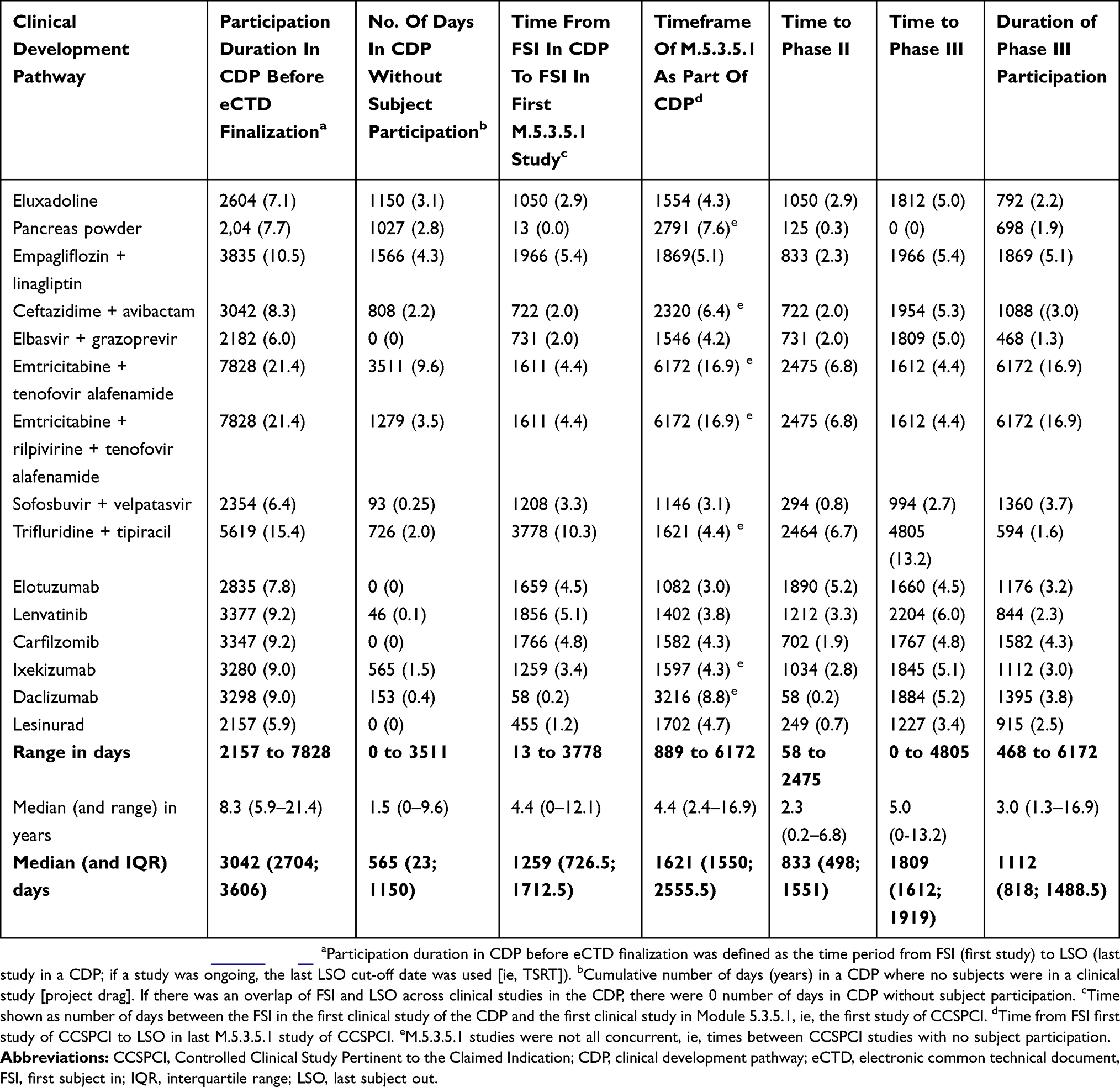 |
Table 4 Project Evaluation And Review In Exploring CDP Critical Paths In Days (And Years) |
The Duration Of Subject Participation (DSP) In The Core CDPs
The DSP is the TSRT including the days in a CDP during which no subjects were participating in a clinical study. The CDP with the longest period without subjects in a clinical study (ie, project drag27) was 9.6 years (3511 days) in the F/TAF CDP (Table 4). The F/TAP CDP had four periods in which no subjects were participating in any of the 54 clinical studies (Table 3). In the 15 core CDPs, 79,872 subjects participated in CDPs with a median DSP of 8.3 years (3042 days; IQR: 2704–3606), ranging from 5.9 years in the lesinurad to 21.4 years in the FTC+RFV+TAF CDP (Table 3).
Interruptions In Subject Participation In The Core CDPs
Eleven of the 15 clinical developments had interruptions in subject participation across the clinical studies in a CDP (Table 3). Interruptions are indicative of sequential project work packages in a CDP. Theoretically, projects with sequential elements have a longer overall timeline.25 Four CDPs (EBR+GZR, elotuzumab, carfilzomib, and lesinurad) did not have any interruptions (ie, at least one study was actively recruiting or following-up subjects during the TSRT period) (Table 3). These four CDPs were considered to be concurrent clinical developments, with sequential aspects in individual studies. The EMPA+LINA CDP had the highest number of interruptions (5), where subjects did not participate in a clinical study during the development. In addition, the F/TAF CDP had the longest period (9.6 years) in a CDP without subject participation (Table 4). The F/TAF CDP had many CSRs that were also submitted in the FTC+RFV+TAF CDP. The FTC+RFV+TAF CDP had four interruptions and no subject participation totaling 3.5 years. The EMPA+LINA CDP had the second-longest period (4.3 years) in a CDP without subject participation. In the nine CDPs with a CDT <10 years, no associations (eg, fixed-dose combination, duration of Phase III) were observed with respect to the number of interruptions.
Time To Phase II In The Core CDPs
The median time from the FSI in the first Phase I study to the FSI in the Phase II study was 2.3 years (833 days; IQR: 498–1551 days) for the 15 core CDPs (Table 4). The observed range spanned from daclizumab, where the FSI of the first Phase II clinical study was 58 days after the FSI in the CDP, to >6.7 years in 3 CDPs (F/TAF, FTC+RFV+TAF, and FTD+TPI).
Time To Phase III In The Core CDPs
The median time from the FSI in the first Phase I study to the FSI in Phase III study was 5.0 years (1809 days; IQR: 1612–1919 days) (Table 4). The earliest study to enroll subjects based on the CSRs in the pancreas powder CDP was a Phase III clinical study. The longest time prior to the initiation of the Phase III program in the 15 CDPs was observed in the FTD+TPI CDP. The FSI in the Phase III study was 13.2 years (4805 days) after the FSI in the first clinical study of the FTD+TPI CDP.
Duration Of The Phase III Core CDPs
The median time from the FSI in the first Phase III study to the LSO in the last Phase III study was 3.0 years (1112 days; IQR: 818–1488.5 days) (Table 4). The shortest (1.3 years) Phase III program was noted in the EBR+GZR CDP. The longest (16.9 years) Phase III programs were reported in the F/TAF and FTC+RFV+TAF CDPs.
Timeframe Of CCSPCI In The Core CDP Set
Documents in Module 5.3.5.1 are “Controlled Clinical Studies Pertinent to the Claimed indication,” and were available in all core and non-core CDPs. However, timeline analysis was not possible in the non-core CDP set. In the core CDP set, the median time from the FSI in the first study to the LSO in the regulatory label “controlled clinical studies pertinent to the claimed indication” was 4.4 years (1621 days; IQR: 1550–2555.5 days) (Table 4). The shortest (2.4 years) CCSPCI program was reported in the SOF+VEL CDP. The longest (16.9 years) CCSPCI programs were noted in the FTC+RFV+TAF and F/TAF CDPs. It was observed that, in the six CDPs with a Phase III duration <1000 days (ie, pancreas powder, eluxadoline, EBR+GZR, FTD+TPI, lenvatinib, and lesinurad), the CCSPCI timeframe was longer than the Phase III duration (Table 4). Phase II studies (eg, RDEA594-203 in the lesinurad CDP and IBS-2001 in the eluxadoline CDP) were categorized as M.5.3.5.1 clinical studies (ie, pertinent to the claimed label). In the non-core CDP, there were programs that did not have Phase III studies (eg, osimertinib and daratumumab) (Supplemental Table S3). The Phase III reports in the osimertinib and daratumumab CDPs are listed as post-marketing requirements in the corresponding EPARs.
Subject Participation In The Core CDP Set
It was estimated that subject participation in the F/TAF CDP accounted for 54% of the CDT (ie, from the date of the first final protocol to EMA submission). The subject participation in 10 CDPs (EBR+GZR, FTC+RFV+TAF, FTD+TPI, SOF+VEL, elotuzumab, lenvatinib, carfilzomib, ixekizumab, daclizumab, and lesinurad) accounted for >80% of the CDT. The estimated contribution was highest in the EBR+GZR CDP (Table 3).
Study Amendments In The Core CDP Set
Among the 444 clinical study protocols, 310 (70%) had a total of 888 amendments (Supplemental Table S3). All core CDPs had clinical studies with protocol amendments. It was found that 46 CSRs were submitted in more than one CDP; thus, 398 of the 444 CSRs across all 15 CDPs were unique. These unique CSR protocols had a total of 803 amendments. Protocol amendments were present in all phases of clinical development. The protocol amendments were as follows: 323 in 265 Phase I (range: 1–13), 177 in 53 Phase II (range: 1–14), 226 in 63 Phase III (range: 1–21), seven in three Phase IV (range: 2–5), 55 in 10 Phase I/II (range: 3–10), and 15 in four Phase II/III (range: 3–5) clinical studies. A total of 108 Phase I studies (41%) did not have protocol amendments. In the later phases of clinical development, most studies had protocol amendments. Only three Phase II studies (6%), three Phase III studies (5%) and one Phase IV study (33%) did not have protocol amendments. All Phases I/II and II/III studies had amendments.
Modeling Of The Core CDPs
The visualization of a CDP was initially performed by producing simplified GANTT charts. The 14 clinical studies (11 Phase I, one Phase II, and two Phase III) in the eluxadoline CDP covered a time period of 7.5 years (2007–2015). In the CDP, the FSI was in a PK dose-escalation study (EDI-1001), while the FSI in the IBS-2001 study was 1051 days later, heralding the initiation of the Phase II part of the clinical development (Figure 4). The FSI in the mass balance study (EDI-1003) was after the FSI in the Phase II study. The CCSPCI part of the eCTD spanned 4.3 years (1554 days) and included the Phase II IBS-2001, as well as the two Phase III IBS-3001 and IBS-3002 clinical studies. There were three periods without subject participation, totaling 1150 days (3.1 years).
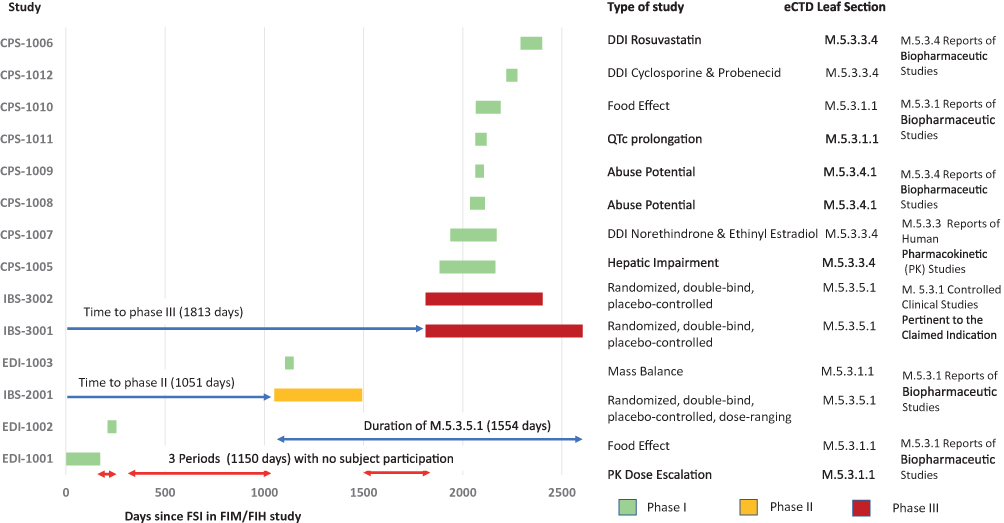 |
Figure 4 GANTT chart of the clinical development of eluxadoline based on the FSI to LSO, according to phase and regulatory eCTD leaf structure with project timelines. Note: The metrics shown are listed in Supplemental Table S2 and S3 for the 15 core CDPs (CPS, EDI & IBS are part of the study number). Abbreviations: CDP, clinical development pathway; DDI, drug–drug interaction; eCTD, electronic common technical document; FIH, first in human study; FIM, first in man study; FSI, first-subject-in; GANTT, generalized activity normalization time table; LSO, last-subject-out; PK, pharmacokinetic; QTc, thorough QT/QTc prolongation/QTc-like study. |
A more detailed approach to visualizing a CDP is through the generation of a network analysis diagram based on the FSI dates. The lesinurad clinical development had a total of 43 milestones, based on 42 clinical studies and 45 CSRs uploaded on the ECDW. The first study and, therefore, first-time defined event in the CDP was a Phase I PK study; the final event was the submission of the eCTD to the EMA in this top-line process. The first Phase III clinical study was the 23rd event in the CDP. Phase III enrolment was initiated in the second half of the lesinurad CDP (Figure 5). Twenty-four clinical studies had longer subject participation times than planned for a single subject or all cohorts. Eighteen clinical studies had shorter study periods (FSI to LSO) than the planned study duration, including one Phase II study (RDEA594-202) and one Phase III study (RDEA594-307). Five clinical studies had a shorter duration than planned, as the optional cohorts were included in the planned study duration and did not participate. The 13 other studies had protocol amendments. A simplified process flow was fitted to the lesinurad CDP to assess the inclusion of special Phase I clinical studies (Figure 6). In this example for lesinurad, the pictogram shows that the time from the FSI in the RDEA594-101 to the FSI in the Phase II study was 249 days. The FSI in the last lesinurad clinical study (RDEA594-132) was 5.7 years (2092 days) after the FSI in RDEA594-101. There were no Phase III clinical studies before the renal impairment clinical study. However, five Phase I and Phase II studies had initiated subject enrollment. Similarly, the mass balance and QT-prolongation clinical studies initiated enrollment prior to the first Phase III study. Phase I clinical studies of special interest did not impact the lesinurad CDT, as there were no interruptions. An overview of Phase I clinical studies of special interest in the core CDPs is shown in Table 5. In the 15 core CDPs, 53 (18%) of the 290 Phase I studies were studies of special interest. This clearly differed from the non-core set (ie, three Phase I studies of special interest). The three non-core CDPs were for osimertinib and rociletinib (both had a single mass balance CSR), and idarucizumab with a renal impairment CSR.
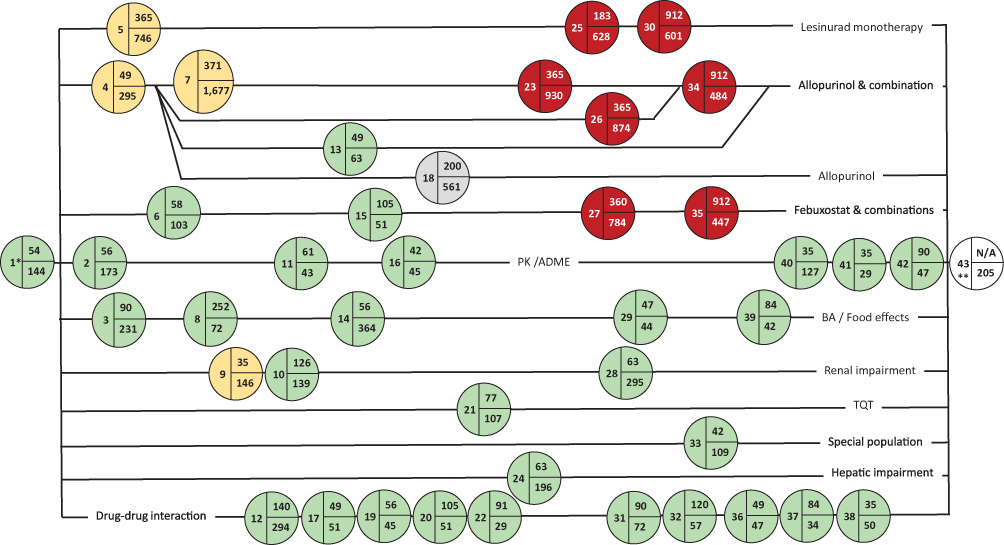 |
Figure 5 Network analysis of the lesinurad CDP based on an ICH E8 categorization and the FSI in each clinical study. Notes: There were 45 CSRs for 42 clinical studies, the 43rd milestone was the submission of the eCTD to the EMA. Color coding: phase I (green), phase II (yellow), phase III (red), phase IV (grey). Abbreviations: ADME, absorption, distribution, metabolism and elimination study; BA, bioavailability study; CDP, clinical development pathway; CSR, clinical study report; eCTD, electronic common technical document; EMA, European Medicines Agency; ICH, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; FSI, first-subject-in; PK, pharmacokinetic; TQT, thorough QT/QTc prolongation/QTc-like study. |
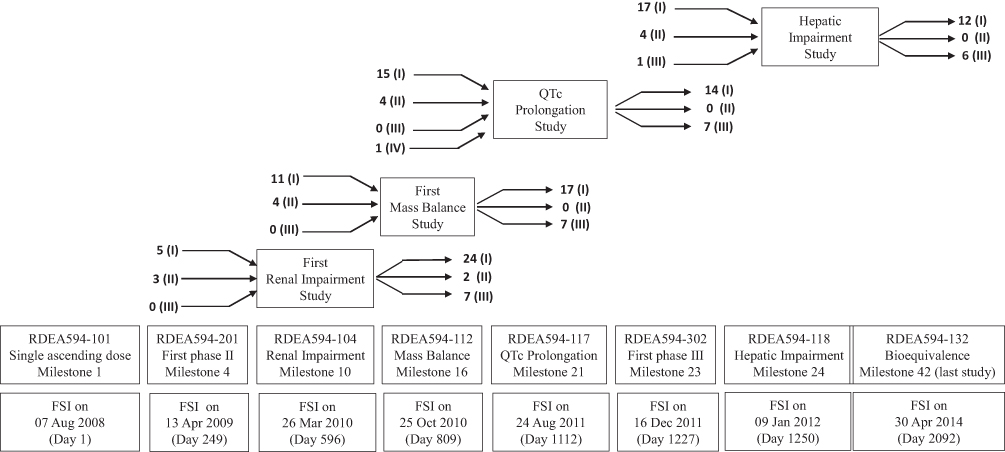 |
Figure 6 Pictogram of the lesinurad CDP with a focus on Phase I studies of special interest in relation to the initiation of enrolment to the Phase II and Phase III studies. The Phase IV study is only partially shown (prior to the QTc prolongation). Note: Numbers indicate the studies preceding the milestone/event and those initiated subsequent to the milestone/event. Abbreviations: (I) Phase I; (II) Phase II; (III) Phase III; (IV) Phase IV; CDP, clinical development pathway; CSR, clinical study report; EMA European Medicines Agency; FSI, first-subject-in; MB, mass balance study, PK, pharmacokinetic; QTc, thorough QT/QTc prolongation/QTc-like study. |
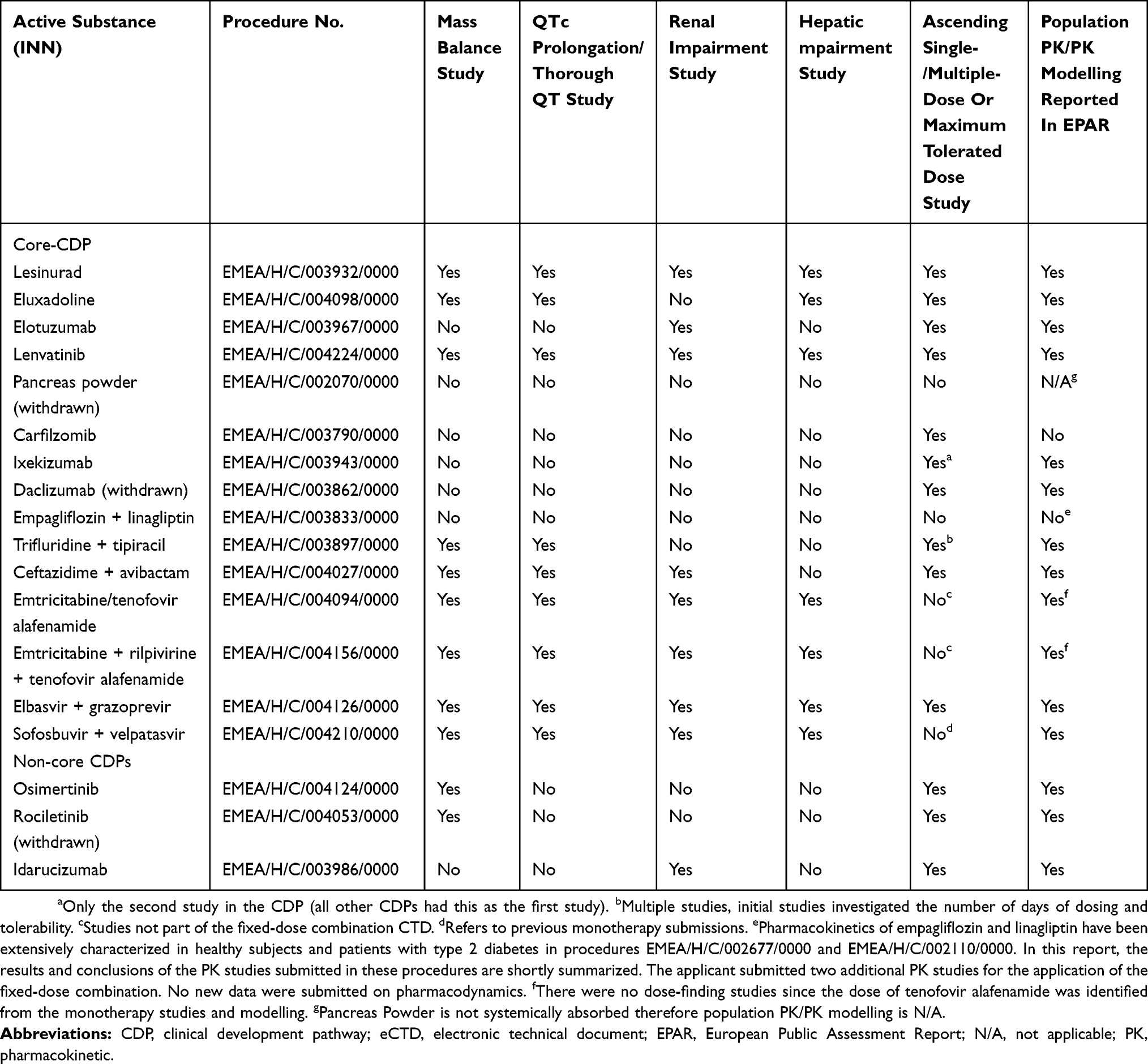 |
Table 5 Overview Of Specific Phase I Studies Or Measures That Have The Potential To Affect Clinical Development Timelines |
The diversity of the strategies used in the CDPs, even within a therapeutic indication, is shown in Figure 7. The 3 antineoplastic (ATC code L01X) CDPs (lenvatinib [core CDP]), and two non-core CDPs (rociletinib and osimertinib) showed that the CDTs were significantly different and dependent on the methodology used to cluster CDPs.
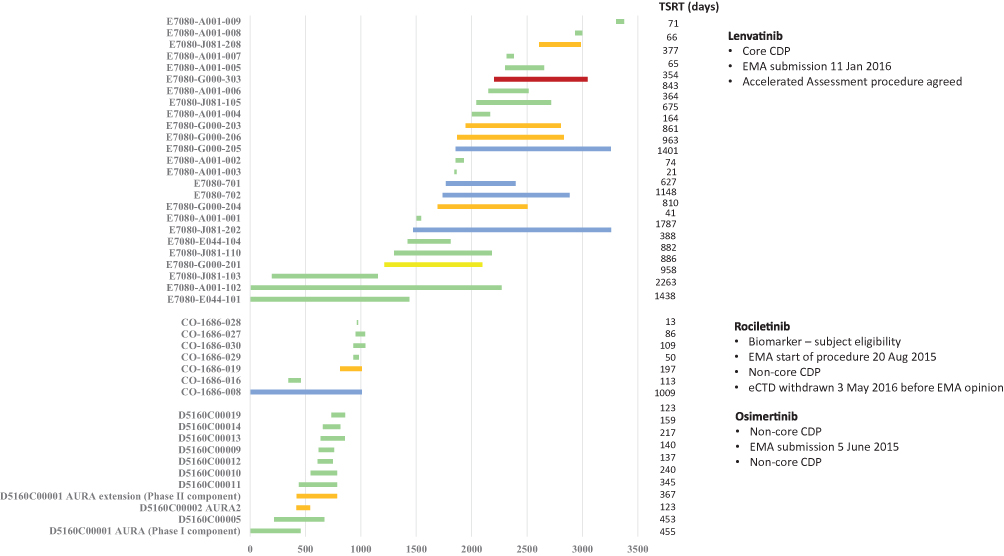 |
Figure 7 A comparison of the lenvatinib [core CDP], rociletinib, and osimertinib [non-core CDPs] total subject recruitment times (based on the FSI to LSO) in the clinical study reports submitted and on the ECDW. Notes: The metrics shown are listed in Supplemental Table S3 for the 15 core CDPs (the study identification numbers for each CDP are shown on the y-axis). Color coding: phase I (green), phase II (yellow), phase I/II (blue), phase III (red). Abbreviations: CDP, clinical development pathway, eCTD, electronic common technical document, EMA, European Medicines Agency, FSI, first-subject-in, LSO, last-subject-out; TSRT, total subject recruitment times. |
In summary (core CDP analyses), an ECDW CDP with a new active ingredient had a median of 30 clinical studies (range: 7–76 studies) with a median CDT of 9.3 years (range: 6.2–22.2 years). A median of four Phase II (range: 1–9) and five Phase III (range: 1–14) studies was observed. The greatest variation was noted with respect to the number of Phase I clinical studies (range: 1–59 studies) and the sequence of the Phase I studies in a CDP. The median time to FSI in a CCSPCI and Phase III study was 4.4 years and 5.0 years, respectively (Table 4). Phase I/II and Phase II/III studies accounted for 3% and 1% of all clinical studies, respectively. Four CDPs (EBR+GZR, F/TAF, FTC+RFV+TAF, and albutrepenonacog alfa) had Phase II/III clinical studies. The albutrepenonacog alfa CDP had a Phase I/II and a Phase II/III clinical study, but not a Phase II study. However, the albutrepenonacog alfa CDP had two additional Phase III clinical studies. Phase IV (eg, lesinurad) and other clinical studies (eg, expanded access in the carfilzomib CDP) were exceptions. The Phase IV study in the lesinurad CDP had no effect on the overall CDT.
Observations On CDP Project Crashing
The strategy that sponsors implemented to potentially reduce the clinical development timelines in the 15 core CDPs is summarized in Figure 8. An IMA CDP based on well-characterized drugs (eg, the fixed-dose combination FTC+RFV+TAF) does not exhibit the shortest development timeline. A more universal approach appeared to be submissions with ongoing clinical studies, based on interim reports (eg, lesinurad). Concurrent subject enrolment in a CDP was observed in four CDPs. A single CCSPCI was sufficient in the elotuzumab CDP, as well as in the non-core rociletinib CDP (ie, the CDP with the shortest CDT). The fixed-dose combination EBR+GZR CDP had the shortest core CDP CDT (6.2 years). The CDPs of three fixed-dose combinations (FTD+TPI, F/TAF, and FTC+RFV+TAF) had the longest CDTs (16.2, 21.9, and 22.2 years, respectively) (Figure 8).
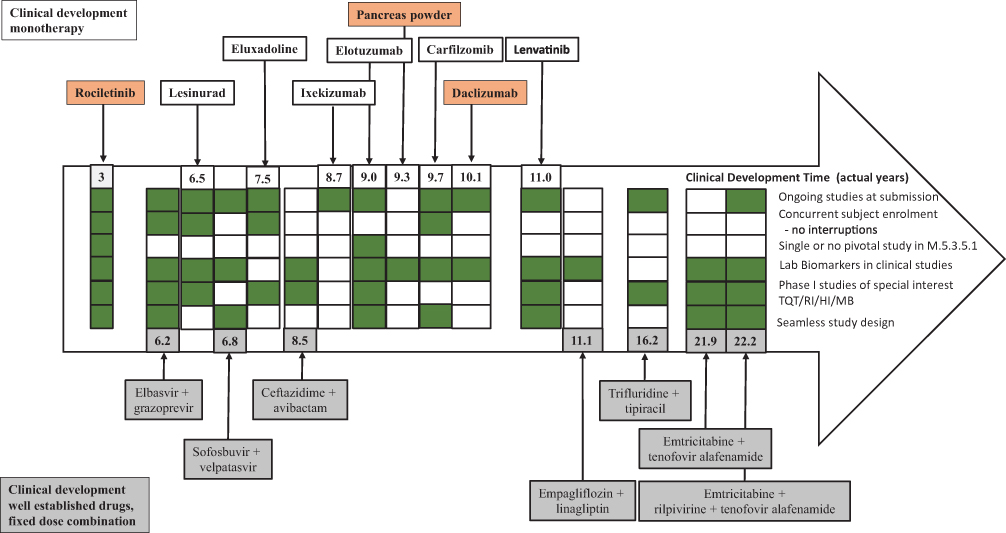 |
Figure 8 Overview of different strategies applied in the clinical development of the core CDPs (monotherapy and fixed-dose combination) compared with the shortest CDT, ie, rociletinib (non-core CDP, with breakthrough designation). Note: The numbers indicate the development time of the indicated CDP. Green boxes indicate that the strategy was implemented in a CDP. Empty white boxes indicate that the strategy was not implemented. Rociletinib, pancreas powder, and daclizumab are color-coded in orange to indicate withdrawal. None of the core fixed-dose combination CDPs (in grey) were withdrawn. Abbreviations: CDP, clinical development pathway; HI, hepatic impairment study, MB, mass balance study; QTc, thorough QT/QTc prolongation/QTc-like study; RI, renal impairment study. |
Discussion
The EMA clinical data website offers an overview of regulatory documents spanning all EU central marketing eCTD submissions. The diversity of the CDPs analyzed ranged from generic (eg, salmeterol + fluticasone fixed-dose combination), monoclonal (eg, daclizumab and elotuzumab), biosimilar (eg, infliximab), orphan (eg, albutrepenonacog alfa), small molecules (eg, lesinurad), advanced therapy (eg, drisapersen) to medicines for use outside the EU (ie, Article 58 procedure for chlorhexidine).
Considering that documents date back to 1993, the publication of documents by the EMA should be analyzed under the perspective that these documents were produced at different times targeting various audiences. Moreover, as the wealth of knowledge increases, it will impact the way that clinical study protocols develop in the future. The number of protocol amendments may reflect the increasing amount of data becoming available, and it is unlikely that amendments to protocols can be eliminated.28–30 The finding that 310 of the 444 clinical study protocols in the 15 core CDPs had amendments appears high (Supplemental Table S3). However, it reflects the uncertainties faced and external information that becomes available and needs to be incorporated into clinical development.
The analysis of the diversity of CDPs was an important initial step to determine whether it would be possible to perform comparative analyses between clinical developments within a single therapeutic area. Based on the available data, it was decided that such analyses would be premature. The number of generics classified by the EMA as IMAs reduced the number of CDPs available for timeline analysis of innovative monotherapies. However, the observation that one-third of the IMAs (ie, 19 of the 55 CDPs) were generics is consistent with the increasing number of drugs reaching the end of their patent.31
Timeline analyses of the available data offer insight into the amount of time invested by sponsors and the amount of time dedicated by clinical study subjects in the clinical development of a medicinal product for marketing authorization. Learning about time optimization from sponsors outside of one’s own organization is also possible when considering a clinical development in a new therapeutic indication.
In the analysis and modeling of the CDP timelines, there were numerous observations that sponsors attempted project crashing to accelerate the release of the therapy to the market. The withdrawal of rociletinib from the market highlights the high risk associated with marketing approval in the absence of a Phase III study. Nevertheless, market authorization without completion of the Phase III part of clinical development is potentially efficient, considering that it may reduce the development time by ≥1.3 years. This observation triggered the analysis of a potential significant difference between the duration of Phase III clinical development and the duration of the CCSPCI timeline in a CDP. It was observed that a CDP with a relatively short duration of Phase III clinical development (ie, <1000 days) coincided with a longer CCSPCI duration. For example, the CCSPCI duration in the EBR+GZR CDP was 3.3-fold longer than that reported for the Phase III program. Thus, a shorter Phase III program appeared to be associated with additional regulatory documents from clinical studies other than Phase III. Other elements of project crashing (eg, integration of seamless Phase I/II and Phase II/III clinical studies into the CDP) were observed. However, their effectiveness can only be assessed once there is an adequate number of published CDPs for a medical condition in similar patient populations. Additional data, such as the date of final protocol, FSI, LSO, dates of scientific advice (available in the clinical overviews but not included in our analyses), dates of implementation of substantial amendments, phase of development, regulatory leaf structure, and planned duration of a clinical study (including cohorts and wash-out periods) are required for a fair comparison of CDPs.
An aspect of data utility is lost in the CSRs (non-core CDPs) in which dates are redacted to reduce the chance of subject reidentification. In the absence of data regarding the FSI and LSO dates, it is not possible to categorize a clinical study into a period with corresponding regulatory guidelines for a clinical development or the prevailing medical practice. This complicates the placement of the clinical study into context. It is also impossible to assess whether a regulatory strategy (eg, Breakthrough or Sakigake Designation32) leads to accelerated approval, and the appropriate timing for the inclusion of CSRs and regulatory documents into systematic reviews.33
The relevance of a clinical study can be easily determined by performing a literature search in peer-reviewed journals, or by analyzing the results available in a freely accessible WHO primary registry.34 Currently, accessing the clinical data in the ECDW is not the first choice, as some documents have limited search functionality. Moreover, during our analyses, multiple sources (eg, the FDA website,35 ClinicalTrials.gov, PubMed, etc.) had to be consulted for a product. Therefore, all possible sources of data should be consulted to construct a comprehensive database for evidence-based medicine reviews. In the case of rociletinib, the CDT was reduced to approximately 3 years. Even if all development strategies could be employed and earlier marketing approval obtained, the development lifecycle for rociletinib continued after approval. Thus, the CDT appears to be artificially shifted in some IMAs as the marketing authorization date was earlier. However, additional development costs and risks (eg, withdrawal from the market) remain.36
High-level timelines have been previously published for USA marketing approvals,4 and were found to be in line with the findings of the present analysis. Prior to this, it was reported that an average of 12 years was necessary for an experimental drug to progress from bench to market.37 The incorporation of a biomarker-based strategy in drug development timelines for new oncology drugs has been reported to reduce the time to approval.38 Unfortunately, this could not be analyzed as the non-personalized carfilzomib was the only common CDP. Innovative concepts related to the Phase I program have also been debated but not analyzed.39 With the publication of CSRs by the EMA as per Policy 0070, detailed analysis has become possible even with respect to study participation at the individual clinical study level, as shown in the lesinurad network diagram. There are limitations in the reconstruction of timelines, particularly regarding the impact of non-clinical activities on the clinical development strategies. The discontinued preclinical tetrahydrobiopterin (BH4) synthesis target, which was identified as a key modulator of both neuropathic and inflammatory pain in the periphery, is a recent example of the amount of work invested and the interrelations of the non-clinical and clinical developments.40 Thus, although preclinical data are included in the EPAR, the impact of this inclusion on clinical timelines could not be assessed. An overview of the limitations of this study is provided in Supplemental Table S4. The information available on the ECDW demonstrates the time invested by participants (ie, healthy volunteers and patients), and enhances the transparency of CDP decisions. This has positive and negative impacts on future clinical development projects. The positive aspect is that future clinical development strategies can be developed based on previous scientific argumentation. A potential negative aspect is that more resources may be necessary to analyze previous CDPs.
Conclusions
The publication of CSR documents by the EMA has offered an insight into the timelines and project management aspects of the clinical development of a medicinal product. Although the EMA timelines are published in the initial EPAR with respect to the regulatory review milestones, clinical development milestones are not published. In this article, we highlight that the ECDW permits the implementation of project management best practices. The findings obtained from the ECDW need to be put into context with the global regulatory framework, as well as other commercial considerations. This can be analyzed by consulting sponsor press releases and financial statements. There are a few areas in which sponsors could reduce the clinical development time, such as avoiding sequential project management (eg, F/TAF). Phase III studies with short durations appeared to be associated with longer CCSPCI durations. Marketing approval without completion of the Phase III clinical development was linked to a post-marketing risk (eg, the withdrawal of pancreas powder, drisapersen, and rociletinib from the market). The CDPs available on the ECDW for the first 18 months represent a diverse spectrum. Understanding the uniqueness of each CDP requires a varying degree of specialist knowledge to permit analysis. Visual techniques assisted in understanding the interrelationship between clinical studies in a CDP, and following strategic decision-making. The significant time that subjects contribute has been quantified and found to vary from 52% to 97% of the CDT. Implementation of the Phase 2 of the EMA Policy 0070 in the future17 and the continuously increasing number of publicly available CDPs would permit a more detailed analysis to ascertain the potential time reduction.
The clinical development approach (ie, the type of information required by the regulators for a submission) will continue to be driven to a large extent by the type of medicinal product under development and the medical need. Based on the currently available documents and the resources available to an organization, it is unlikely that a significant decrease in the clinical development timeframe can be consistently achieved.
Abbreviations
ADME, absorption, distribution, metabolism and elimination study; ATC, anatomical therapeutic chemical classification system; CCSPCI, controlled clinical study pertinent to the claimed indication; CDP, clinical development pathway; CDT, clinical development timeline; CO, clinical overview; CSR, clinical study report; CTR, (EU) clinical trials register; EBR+GZR, elbasvir + grazoprevir; ECDW, European Medicines Agency clinical data website; eCTD, electronic common technical document; EMA, European Medicines Agency; EMPA+LINA, empagliflozin + linagliptin; EPAR, European Public Assessment Report; FTC+RFV+TAF, emtricitabine + rilpivirine + tenofovir alafenamide; F/TAF, emtricitabine + tenofovir alafenamide; FIH/FIM, first-in-human/first-in-man; FSI, first-subject-in; GANTT, generalized activity normalization time table; HI, hepatic impairment study; ICH, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; IMA, initial marketing authorization; IQR, interquartile range; LSO, last-subject-out; MB, mass balance; PK, pharmacokinetic; RI, renal impairment; SOF+VEL, sofosbuvir + velpatasvir; FTD+TPI, trifluridine + tipiracil; TPP, target product profile; TQT, Thorough QT/QTc prolongation/QTc-like; WHO, World Health Organization.
Acknowledgments
The authors would like to thank Liz Roberts and Elke Grosselindemann for their review and comments.
Data Sharing Statement
All source documents used in this project are freely accessible on the clinical data website of the European Medicines Agency.
Disclosure
Sarah Lehmann performed part of this work as part of her MSc at the TH Koeln – University of Applied Sciences. René Allard was an employee of Grünenthal GmbH. The authors report no other conflicts of interest in this work.
References
1. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. General considerations for clinical trials; 1997. Available from: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E8/Step4/E8_Guideline.pdf.
2. Food and Drug Administration. Guidance for Industry and Review Staff Target Product Profile – A Strategic Development Process Tool. Available from: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm080593.pdf.
3. Food and Drug Administration. Critical path initiative. Available from: https://www.fda.gov/science-research/science-and-research-special-topics/critical-path-initiative.
4. Hwang TJ, Darrow JJ, Kesselheim AS. The FDA’s expedited programs and clinical development times for novel therapeutics, 2012-2016. JAMA. 2017;318(21):2137–2138. doi:10.1001/jama.2017.14896
5. The European Parliament and the Council of the European Union. Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 Laying Down Community Procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency. Available from: http://eurlex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2004:136:0001:0033:en:PDF.
6. Garvin DA. Building a learning organization. Harv Bus Rev. 1993;71(4):78–79. Available from: https://hbr.org/1993/07/building-a-learning-organization. Accessed March 10, 2019.
7. Porter ME. Competitive Advantage: Creating and Sustaining Superior Performance. New York: Free Press; 1985.
8. Porter ME. Towards a dynamic theory of strategy. Strategic Manag J. 1991;12:95–117. doi:10.1002/(ISSN)1097-0266.
9. Papathanasiou P, Brassart L, Blake P, et al. Transparency in drug regulation: public assessment reports in Europe and Australia. Drug Discov Today. 2016;21:1806–1813.
10. Van Luijn JC, Stolk P, Gribnau FW, et al. Gap in publication of comparative information on new medicines. Br J Clin Pharmacol. 2008;65(5):716–722. doi:10.1111/j.1365-2125.2007.03092.x
11. Eichler HG, Abadie E, Breckenridge A, et al. Open clinical trial data for all? A view from regulators. PLoS Med. 2012;9(4):e1001202. doi:10.1371/journal.pmed.1001202
12. European Medicines Agency. Access to clinical-trial data and transparency. Workshop report. Available from: https://www.ema.europa.eu/en/documents/report/access-clinical-trial-data-transparency-workshop-report_en.pdf.
13. European Medicines Agency. European Medicines Agency Policy on Publication of Clinical Data for Medicinal Products for Human Use. Available from: https://www.ema.europa.eu/en/documents/other/european-medicines-agency-policy-publication-clinical-data-medicinal-products-human-use_en.pdf.
14. European Medicines Agency. Background to Clinical Data Publication Policy. Available from: https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/clinical-data-publication/background-clinical-data-publication-policy.
15. European Medicines Agency. Clinical Data Publication. Available from: https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/clinical-data-publication.
16. European Medicines Agency. Clinical Data. Available from: https://clinicaldata.ema.europa.eu/web/cdp/home.
17. European Medicines Agency. External Guidance on the Implementation of the European Medicines Agency Policy on the Publication of Clinical Data for Medicinal Products for Human Use: Version 1.1. Available from: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/external-guidance-implementation-european-medicines-agency-policy-publication-clinical-data_en-2.pdf.
18. European Medicines Agency. External Guidance on the Implementation of the European Medicines Agency policy on the publication of clinical data for medicinal products for human use: version 1.2. Available from: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/external-guidance-implementation-european-medicines-agency-policy-publication-clinical-data_en.pdf.
19. European Medicines Agency. External Guidance on the Implementation of the European Medicines Agency Policy on the Publication of Clinical Data for Medicinal Products For Human Use: Version 1.3. Available from: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/external-guidance-implementation-european-medicines-agency-policy-publication-clinical-data_en-1.pdf.
20. Henry-Eude SA. European Medicines Agency. EMA update on Clinical Data Publication (CDP); 2018 Available from: https://www.ema.europa.eu/en/documents/presentation/presentation-european-medicines-agency-update-clinical-data-publication-cdp-anne-sophie-henry-eude_en.pdf.
21. Ross JS, Waldstreicher J, Bamford S, et al. Overview and experience of the YODA Project with clinical trial data sharing after 5 years. Sci Data. 2018;5:180268. doi:10.1038/sdata.2018.187
22. So D, Knoppers BM. Ethics approval in applications for open-access clinical trial data: an analysis of researcher statements to clinicalstudydatarequest.com. PLoS One. 2017;12(9):e0184491. doi:10.1371/journal.pone.0184491
23. Billiones R, Schneider A. Navigating the EMA clinical data website. Med Writing. 2018;27(2):19–21.
24. Moen RD, Norman CL; Circling back. Clearing up myths about the Deming cycle and seeing how it keeps evolving. Qual Prog. 2010. Available from: http://www.apiweb.org/circling-back.pdf.
25. Maylor H. Project Management.
26. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Revision of M4E guideline on enhancing the format and structure of benefit-risk information in ICH Efficacy -– E4E(R2); 2016. Available from: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/CTD/M4E_R2_Efficacy/M4E_R2__Step_4.pdf.
27. Devaux SA. The drag efficient. The missing quantification of time on the critical path. Defense AT&L. 2012, Available from: https://web.archive.org/web/20130313022307/http://www.dau.mil/pubscats/ATL%20Docs/Jan_Feb_2012/Devaux.pdf.
28. European Medicines Agency. EMA/CHMP/ICH/309348/2008 ICH guideline E2F on development safety update report September 2011. Available from https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-26.pdf.
29. Ananthakrishnan R, Gona P. Pharmacological modeling and biostatistical analysis of a new drug. Open Access J Clin Trials. 2010;2:59–82. doi:10.2147/OAJCT.S9290
30. Hung H. Considerations in adapting clinical trial design. J Formos Med Assoc. 2008;107(12 Suppl):14–18. doi:10.1016/S0929-6646(09)60004-8
31. Burgess LJ, Terblanche M. The future of the pharmaceutical, biological and medical device industry. Open Access J Clin Trials. 2011;3:45–50.
32. Kondo H, Hata T, Ito K, et al. The current status of Sakigake designation in Japan, PRIME in the European Union, and breakthrough therapy designation in the United States. Ther Innov Regul Sci. 2017;51(1):51–54.
33. Vu HH, Pariser AR. Pre-Investigational new drug meetings with the FDA: evaluation of meeting content and characteristics of applications for new drug and biologic products. Ther Innov Regul Sci. 2015;49(3):434–442.
34. WHO Primary Registries. Available from: http://www.who.int/ictrp/network/primary/en/.
35. Drugs@FDA: FDA approved drug products. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/.
36. van Der Steen N, Caparello C, Rolfo C, et al. New developments in the management of non-small-cell lung cancer, focus on rociletinib: what went wrong. Onco Targets Ther. 2016;9:6065–6074. doi:10.2147/OTT.S97644
37. Kraljevic S, Stambrook PJ, Pavelic K. Accelerating drug discovery. EMBO Rep. 2004. doi:10.1038/sj.embor.7400236.
38. Hobbs BP, Barata PC, Kanjanapan Y, et al. Seamless designs: current practice and considerations for early-phase drug development in oncology. J Natl Cancer Inst. 2019;111(2):118–128. doi:10.1093/jnci/djy196
39. Miseta E. Acceptable risk reduces clinical development time. Clin Leader. 2018, Available from: https://www.clinicalleader.com/doc/acceptable-risk-reduces-clinical-development-time-0001.
40. Booth B. Painful truth: the successful failure of a biotech startup. Forbes. 2017, Available from: https://www.forbes.com/sites/brucebooth/2017/11/17/painful-truth-successful-failure-of-a-biotech-startup/#619f89b3d025.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.