")
Back to Journals » Journal of Pain Research » Volume 10
The efficacy and safety of S-flurbiprofen plaster in the treatment of knee osteoarthritis: a phase II, randomized, double-blind, placebo-controlled, dose-finding study
Authors Yataba I, Otsuka N , Matsushita I, Matsumoto H, Hoshino Y
Received 26 January 2017
Accepted for publication 3 March 2017
Published 11 April 2017 Volume 2017:10 Pages 867—880
DOI https://doi.org/10.2147/JPR.S131779
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Katherine Hanlon
Ikuko Yataba,1 Noboru Otsuka,1 Isao Matsushita,1 Hideo Matsumoto,2 Yuichi Hoshino3
1Taisho Pharmaceutical Co, Ltd, 2Institute for Integrated Sports Medicine, School of Medicine, Keio University, Tokyo, 3Department of Orthopedics Surgery, School of Medicine, Jichi Medical University, Tochigi, Japan
Background: Nonsteroidal anti-inflammatory drug (NSAID) patches are convenient for use and show much less gastrointestinal side effects than oral NSAIDs, whereas its percutaneous absorption is not sufficient for the expression of clinical efficacy at satisfactory level. S-flurbiprofen plaster (SFPP) has shown dramatic improvement in percutaneous absorption results from animal and clinical studies. In this study, the efficacy and safety of SFPP were compared with placebo in patients with knee osteoarthritis (OA) to determine its optimal dose. This was a multicenter, randomized, double-blind, parallel-group comparative study.
Patients and methods: Enrolled 509 knee OA patients were treated with placebo or SFPP at 10, 20, or 40 mg applied on the affected site once daily for 2 weeks. The primary endpoint for efficacy was improvement in knee pain on rising from the chair assessed by visual analog scale (VAS). The other endpoints were clinical symptoms, pain on walking, and global assessment by both investigator and patient. Safety was evaluated by observing adverse events (AEs).
Results: VAS change in knee pain from baseline to trial end was dose-dependent, least squares mean was 29.5, 31.5, 32.0, and 35.6 mm in placebo and SFPP 10, 20, and 40 mg, respectively. A significant difference was observed between placebo and SFPP 40 mg (P=0.001). In contrast, the effect of SFPP at a dose ≤20 mg was not significantly different from that of placebo. The proportion of the patients who achieved 50% pain relief was 72.4% in 40 mg and 51.2% in placebo (P<0.001). In all other endpoints, SFPP 40 mg showed significant improvement compared with placebo. The incidence of AEs was not different across all four groups, and no severe AEs were observed.
Conclusion: Clinically relevant pain relief was observed in all groups including placebo. Especially 40 mg showed remarkable pain relief in not only primary endpoint but also all the other endpoint with significant differences over placebo. The safety profile of SFPP 40 mg was not different from that of placebo. Therefore, 40 mg was determined as the optimal tested dose.
Keywords: nonsteroidal anti-inflammatory drugs, patch, double-blind, visual analog scale, topical, optimal dose, randomized controlled trial
Introduction
Osteoarthritis (OA) is a disease characterized by chronic pain, inflammation, and impaired overall functioning, significantly affecting the quality of life.1,2 Incidence of OA is increasing by rising aged population, and it is highly prevalent.3,4
Among the pharmacological treatments for symptomatic OA, nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used.5–7 However, NSAIDs are known to have a risk of adverse reactions related to class effects of NSAIDs such as gastrointestinal disorders.8–10 On the contrary, topical administration of NSAIDs leads to lower risk of gastrointestinal disorder6 because they generally indicate lower systemic concentration and avoid the direct action to gastrointestinal mucosa.11
Therefore, it was considered that it would be beneficial for OA therapy to employ the favorable characteristics of topical NSAIDs and develop a formulation with better efficacy. This study developed S-flurbiprofen plaster (SFPP), which is a novel topical NSAID patch formulation. To maximize the efficacy of the topical formulation, the potent cyclooxygenase inhibitory NSAID S-flurbiprofen was selected12,13 and the SFPP formulation was designed to allow sufficient deep-tissue penetration of S-flurbiprofen. To improve penetration of S-flurbiprofen into deeper tissues, a tape-type patch with superior percutaneous absorption was selected as the dosage form, along with additional efforts to maximize the formulation performance. Drug products containing racemic flurbiprofen are commercially available as oral formulations,14 patches,15 and injectable solution16 and are used widely and globally.
SFPP has been shown to be superior to existing NSAID patch products in terms of percutaneous absorption, analgesic, and anti-inflammatory effects in in vivo studies.17,18 In a study conducted in healthy adult males, SFPP provided sustained and stable plasma levels of S-flurbiprofen.19 Furthermore, a study in knee OA patients demonstrated higher synovial transfer of S-flurbiprofen compared with that of an existing flurbiprofen patch formulation.19 The results from these studies support the promising and high efficacy of SFPP in OA therapy. This study was conducted in patients with symptomatic knee OA, randomized into four groups of 10, 20, 40 mg of SFPP treatment and placebo to examine the efficacy of SFPP, verify the dose response, and determine the optimal dose.
Patients and methods
The study was conducted in Japan according to the Declaration of Helsinki and Good Clinical Practice guidelines. In the planning of the study method, we referred to the guidelines on clinical study proposals of the European Medicines Agency and of the US Food and Drug Administration.20,21 The study was conducted in a randomized, placebo-controlled, double-blind, parallel-group comparison design in collaboration with 59 study sites from February 2009 to August 2010. The study protocol and the informed consent form were approved by relevant institutional review boards (IRB) (Sapporo Dermatology Clinic IRB, Yokohama Minoru Clinic IRB, Oikawa Clinic IRB, Sendai IRB, Nakajou Orthopaedic Clinic IRB, Maebashi Hirosegawa Clinic IRB, NS Clinic IRB, Irahara Primary Care Hospital IRB, Meiwa Hospital IRB, Sinagawa East One Medical Clinic IRB, Akane Family Clinic IRB, Fussa Hospital IRB, Edogawa Hospital IRB, Kita Shinagawa 3rd. Hospital IRB, Yokohama Shinmidori General Hospital IRB, Hiratsuka Kyosai Hospital IRB, Tana Orthopedic IRB, Aichi Medical Association IRB, Fukuoka Shin Mizumaki Hospital IRB, and Nagata Orthopedic Hospital IRB). The investigators obtained written informed consent from all patients prior to study participation.
Patients
The study enrolled knee OA patients aged ≥20 years who had pain symptoms and were classified as Kellgren–Lawrence grade22 II or III (grade of knee for evaluation is the same or heavier than the grade of knee not for evaluation) in the central assessment of X-ray findings. Each patient visited the study site weekly for a total of five times during the study period (Figure 1).
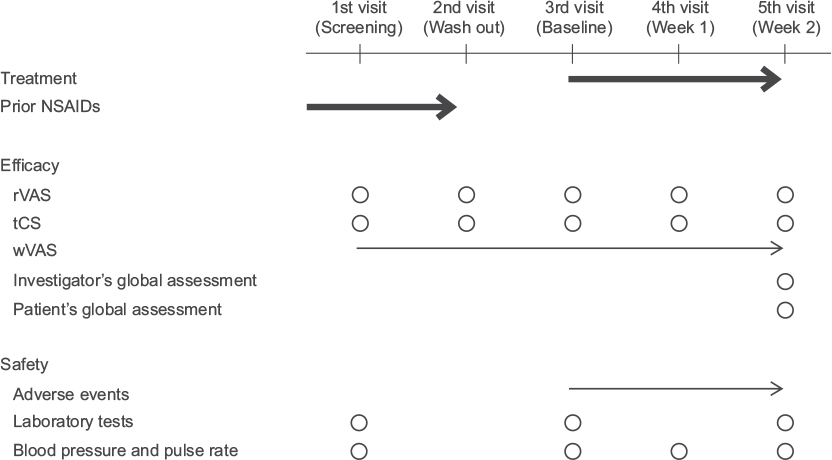 | Figure 1 Study schedule. Notes: Each patient made weekly visits. At the first visit (screening), the investigator verified the eligibility for study participation. At the second visit (wash out), any ongoing NSAIDs treatment was discontinued to exclude its potential effect on the study. At the third visit (baseline), the investigator verified the conformity with the inclusion criteria and initiated the assigned study treatment. The investigator assessed the efficacy and safety variables at the fourth and fifth visits (week 1 and 2) and completed the observation on the fifth visit unless there were ongoing adverse events to be followed up. Thick arrow, treatment; thin arrow, daily evaluations; circle, evaluations at the time of visit. Abbreviations: NSAIDs, nonsteroidal anti-inflammatory drugs; rVAS, pain on rising from the chair as determined by visual analogue scale; tCS, total clinical symptoms score; wVAS, pain on walking as determined by visual analog scale. |
To achieve the accurate evaluation of the efficacy, those patients who had unilateral knee affected or who had much less pain in not evaluated knee than evaluated knee or patients who did not need treatment for not evaluated knee were enrolled. Furthermore, less pain in not evaluated knee than evaluated knee throughout the observation period (from first visit to third visit) was confirmed using the diary written by patients.
The patients were eligible if they had been using any oral and/or topical NSAIDs continuously to treat their evaluated knee for at least 3 weeks prior to the screening visit (first visit). In addition, the following inclusion criteria were used for pain on rising from the chair as determined by the visual analog scale (rVAS) (a 100-mm VAS, see the “Efficacy assessments” section) to select patients who were suitable for the NSAID efficacy evaluation based on published study protocols.23–25
- rVAS before wash-out of prior NSAIDs: <80 mm (at second visit)
- rVAS after wash-out: ≥40 mm (at third visit)
- Difference of rVAS before and after wash-out: ≥15 mm (ie, worsening of pain)
Furthermore, worsening of the pain on ascending or descending stairs by at least 1 point (on a 4-point scale) by the investigator’s assessment through wash-out was also added to the inclusion criteria (see the “Efficacy assessments” section).
Patients were excluded from the study if any of the following criteria were met:
- Complication of rheumatoid arthritis
- History of knee surgery
- Complication due to a malignant tumor, neuropsychiatric disease, serious cardiac disease, renal disease, or hepatic disease
- Taking oral steroid, administrating intra-articular steroid or needle puncture and aspiration within 13 days prior to the first visit
- Administering intra-articular hyaluronic acid or local anesthetics within 6 days prior to the first visit
Treatment
Four patches containing 0 (ie, placebo), 10, 20, or 40 mg of S-flurbiprofen (10×14 cm, from Tokuhon Corporation, Tokyo, Japan) were used. Each patch contained 36.2–37.0 mg of peppermint oil. The formulations were indistinguishable from each other. Patients whose eligibility was confirmed at the baseline visit (third visit) were randomly assigned to treatment with one of placebo or SFPP 10, 20, or 40 mg in a ratio of 1:1:1:1. Randomization was performed by the permuted block method with block size of four and the block size was also blinded. The third person prepared randomization codes in an environment separated from the persons/parties concerned using random numbers from PLAN Procedure of SAS®9.1.3. Randomization codes were concealed until database lock. Each patient applied one patch of the assigned study drug to cover their tender point of evaluated knee and replaced the patch once daily for 2 weeks.20,26 All concerned parties in the study were blinded throughout the study period.
No rescue medication was allowed during the treatment period. The following treatments were not allowed during the treatment period:
- Systemic and topical NSAIDs
- Opioid analgesics
- Paracetamol
- Corticosteroids, except topical corticosteroids
- Intra-articular knee injections (hyaluronic acid injection, local anesthetics)
- Psychotropic drug
- Antiulcer and gastrointestinal agents
- Agents known to interact with flurbiprofen, such as lithium
- Surgical therapy
- Needle puncture and aspiration
Exercise and physical therapy of lower leg and hip were continued if they were started at least 2 weeks prior to the screening visit (first visit). Those patients who started the exercise or physical therapy within 13 days prior to the first visit were excluded. Patients were not allowed to start the exercise and physical therapy after study had been started.
Efficacy assessments
The primary efficacy endpoint was the change in rVAS (ΔrVAS) from baseline to the end of study which was at the fifth visit or the visit when the application was stopped. For the selection of primary endpoint, we set the first priority on minimizing the deviation among the study sites and the reproducibility. The knee pain on rising is one of the symptoms seen most frequently in knee OA, and it occurs from early to late stage in knee OA. Therefore, the evaluation of knee pain on rising from the chair was selected, and the whole procedure for assessment was standardized throughout the study sites. All study sites used same chairs and adjusted the seat height for a sitting knee angle of 110° according to the patient’s height. Each patient was instructed to sit on the chair, rest for 5 minutes, and then rise up without any support. The knee pain on rising from the chair was rated on a 100-mm VAS. The pain assessment was performed in the presence of the clinical research coordinators (each study site had their own) following the standardized procedure at all study sites.
The secondary efficacy endpoint was the total clinical symptoms score (the change from baseline; ΔtCS). The clinical symptoms were assessed by the investigator at all visits for the following nine components on a 4-point scale (none, mild, moderate, or severe).27,28
- Components of pain: passive exercise, ascending or descending stairs, rest, and tenderness
- Components of inflammation: swelling and ballottement of the patella
- Components of disability of active daily living: sitting and rising up motion, and walking
The other endpoints included the change in knee pain on walking assessed by VAS (ΔwVAS), investigator’s global assessment, and patient’s global assessment. For the assessment of wVAS, each patient was instructed to self-assess the knee pain during walking by VAS daily using a diary. The investigator’s and patient’s global assessments were performed by the investigator and the patient, respectively, at the end of the study using a 5-point scale (marked, moderate, mild, no change, or worse).28 A good correlation between ΔrVAS and investigator’s global assessment had been observed in the previous clinical knee OA study.
Safety assessments
The investigator examined the adverse events (AEs) throughout the study period (Figure 1). The investigator also determined the causal relationship of each AE to the study drug. In addition, the investigator rated the severity of each AE on a 3-point scale (mild = treatment not required or daily living not affected, moderate = some treatment required, and severe = particular emergency treatment required).
In order to evaluate safety of SFPP and influence on the results of test, the investigator performed laboratory testing and measured blood pressure and pulse rate according to the schedule shown in Figure 1. Parameters of laboratory testing were hematology (red blood cell count, hemoglobin, hematocrit, white blood cell count, white blood cell classification, and platelets), blood chemistry (aspartate aminotransferase, alanine aminotransferase, γ-glutamyltransferase, alkaline phosphatase, total bilirubin, lactate dehydrogenase, blood urea nitrogen (BUN), creatinine, total protein, sodium, potassium, chloride, and calcium), and urinalysis (protein, glucose, urobilinogen, and hemoglobin). The investigator assessed each parameter for the presence of abnormal variations by considering whether the change was clinically significant. If an abnormal variation was found, it was reported as an AE.
Statistical methods
The mean (standard deviation) of ΔrVAS was 30.1 (17.9) and 39.2 (16.6) mm in placebo and SFPP 40 mg group in the previous clinical knee OA study. Then 123 patients per treatment group would be necessary to confirm the superiority of SFPP to placebo with a difference in ΔrVAS of 9.1 mm between the groups, at a significance level of 2.5% (one-sided) and with a power of 90%, based on the double confidence limits method, that is, sample size estimation which used the lower confidence limit of mean and the upper confidence limit of standard deviation. Because of the assumption that some patients will not complete the study, 125 patients were needed in each treatment group.
All the analyses were carried out according to the pre-specified statistical analysis plan by using SAS®9.1.3. Significance levels were 2.5% (one-sided) at the primary analysis and 15% (two-sided) at the comparison of baseline characteristics. Adjustment for multiplicity in the primary outcome was carried out by using the fixed-sequence testing method, which compared the higher SFPP dose groups and placebo group in turn. Multiplicity in other outcomes was not adjusted because these analyses were exploratory. Missing data at the end of the study were imputed by using the last observation carried forward method; missing data at other time-points were not imputed. Normal probability plot and Shapiro–Wilk test were used to evaluate whether the assumptions of normality and homogeneity of variance were reasonable. No interim analysis was performed in this study.
For efficacy analyses, the primary population was the full analysis set (FAS; it is also called modified intention-to-treat, mITT, population), which comprised all patients who had applied the study drug at least once, and for whom one efficacy data had been obtained at least after the application. The means of primary outcome and other continuous outcomes were analyzed with analysis of covariance (ANCOVA) by using the treatment group as a fixed effect and the baseline as a covariate. The medians of primary outcome were analyzed by using Wilcoxon’s rank-sum test. Analyses for rVAS were performed by using two efficacy criteria: a moderate (30% pain relief) and substantial (50% pain relief) reduction in pain. Categorical outcomes were analyzed using Wilcoxon’s rank-sum test or χ2 test. In addition, the dose–response was evaluated by using contrasts for continuous outcomes.
Safety analyses were based on the safety population, which comprised all patients who had applied the study drug at least once. The number and percentage of patients who had an AE were summarized by using the Medical Dictionary for Regulatory Activities (MedDRA/J ver.13.0) terminology (preferred term). The incidence proportions of AEs and treatment-related AEs were analyzed by using the χ2 test without continuity correction or Fisher’s exact test when appropriate. Since SFPP is a topical formulation, AEs were assessed separately for local AEs at the application sites (skin symptoms) and systemic AEs. Continuous outcomes in the laboratory tests, blood pressure, and pulse rate were analyzed by using Student’s t-test.
Results
Patients
A total of 765 patients underwent screening, 509 of whom were randomly assigned to four groups and received the assigned treatment; 498 (97.8%) completed and 11 (2.2%) discontinued the study (Figure 2). All 509 randomized patients were included in the primary outcome analysis set. The baseline characteristics of the patients in the analysis set showed no statistical significant differences across the treatment groups (Table 1).
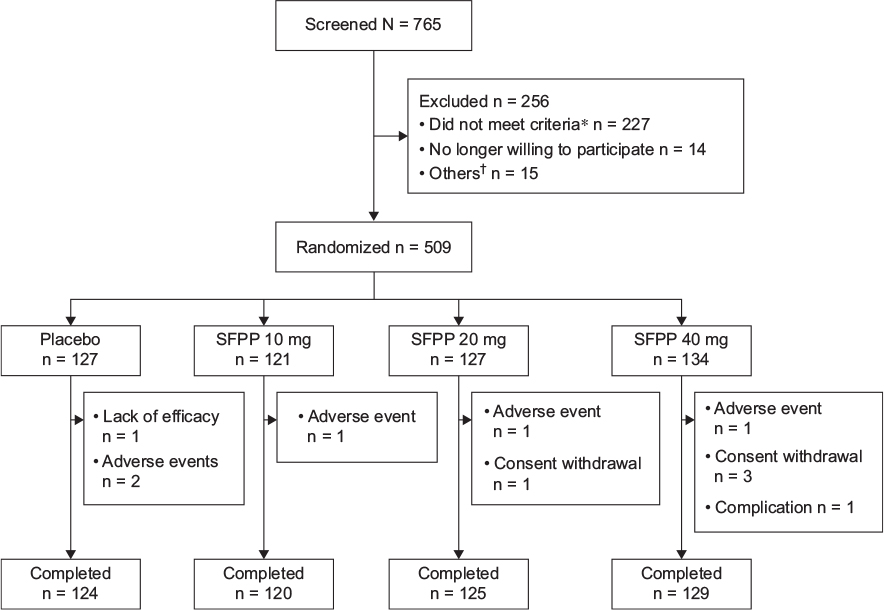 | Figure 2 Flowchart representing the patients throughout the study. Notes: *Inadequate rVAS, KL grade, complications, concomitant drugs, and others. †Patients who could not perform pain assessment according to the standard procedure and who had inadequate laboratory test results at the baseline, and others. Abbreviations: SFPP, S-flurbiprofen plaster; rVAS, pain on rising from the chair as determined by visual analogue scale; KL grade, Kellgren–Lawrence X-ray grade. |
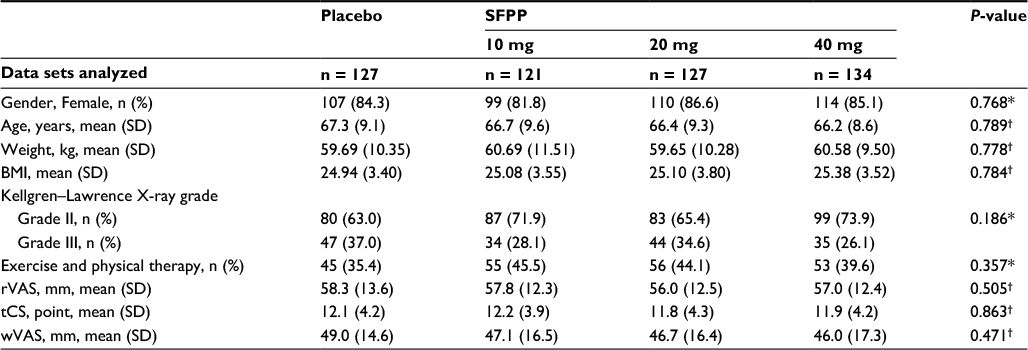 | Table 1 Baseline characteristics of the study population (FAS) Notes: *χ2 test. †Analysis of variance. The baseline characteristics of the study population showed no statistically significant differences across the treatment groups. Abbreviations: FAS, full analysis set; SFPP, S-flurbiprofen plaster; SD, standard deviation; BMI, body mass index; rVAS, pain on rising from the chair as determined by the visual analog scale; tCS, total clinical symptoms score; wVAS, pain on walking as determined by the visual analog scale. |
Efficacy
At the end of the study, the least squares mean (95% confidence interval) of ΔrVAS was 29.5 (26.7–32.4) mm and 35.6 (32.9–38.4) mm in placebo and SFPP 40 mg group (Table 2). A significant difference was observed between placebo and SFPP 40 mg group (P=0.001, Figure 3A and B). In an exploratory dose–response analysis using the ΔrVAS comparison, (−3 −1 1 3) afforded a high goodness of fit at the end of the study (P=0.004). The proportion of patients who achieved substantial (50% pain relief) pain reduction was 51.2% (65/127) in placebo and 72.4% (97/134) in SFPP 40 mg with significant difference (P<0.001) (Table 3). Dose dependency was observed in the substantial pain reduction. On the contrary, dose dependency was not observed in the proportion of patients who achieved moderate reduction in pain.
 | Table 2 Overview of the pain on rising from the chair assessed by visual analog scale Notes: *Mean ± standard error. †Least squares mean (95% confidence interval). ‡Analysis of covariance with the treatment group as a fixed effect and the baseline as a covariate (significance levels = 0.025 [one-sided]). §Median (normal approximation) (95% confidence interval). ||Wilcoxon’s rank-sum test (significance levels = 0.025 [one-sided]). ¶One subject in placebo group was excluded because of lack of data. End of the study means each patient’s last visit, which was the 5th visit or the visit when the application was stopped. Abbreviations: SFPP, S-flurbiprofen plaster; rVAS, pain on rising from the chair as determined by visual analog scale; ΔrVAS, change in rVAS from baseline; NT, not tested. |
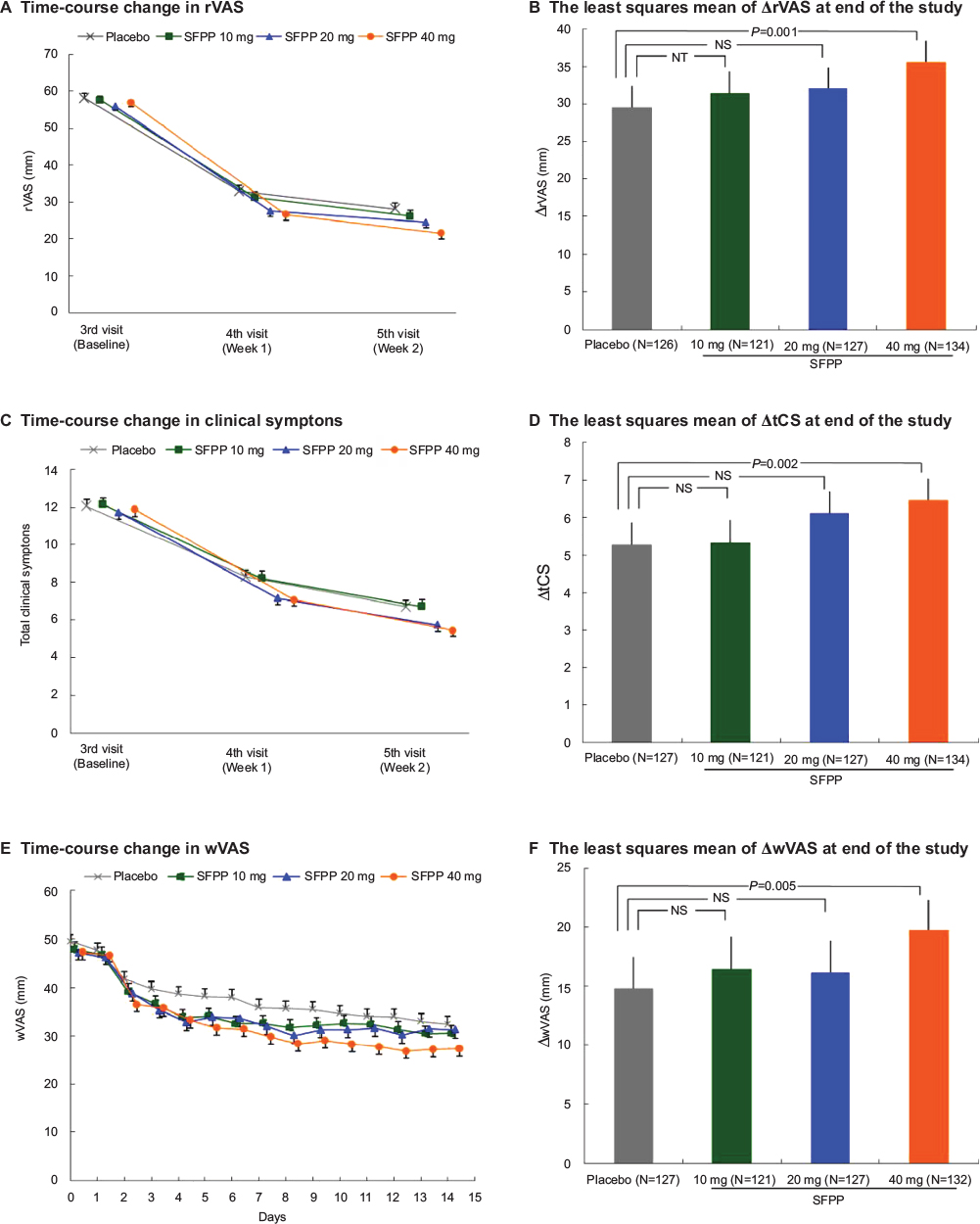 | Figure 3 SFPP 40 mg showed significant improvement compared with placebo in rVAS, tCS, and wVAS. Notes: (A) FAS; time-course change in rVAS (mean and standard error). (B) FAS; the least squares mean (95% confidence interval) of ΔrVAS at the end of the study. One subject of placebo group was excluded because of lack of data. (C) FAS; time-course change in clinical symptoms (mean and standard error). (D) FAS; the least squares mean (95% confidence interval) of ΔtCS at the end of the study. (E) FAS; time-course change in wVAS. Day 1 is the day of initiation of the study drug treatment (mean and standard error). (F) FAS; the least squares mean (95% confidence interval) of ΔwVAS at the end of the study. Two subjects of SFPP 40 mg group were excluded because of lack of data. (B), (D), and (F) The outcomes were ANCOVA with the treatment group as a fixed effect and the baseline as a covariate (significance levels = 0.025 [one-sided]). Abbreviations: rVAS, pain on rising from the chair as determined by visual analog scale; tCS, total clinical symptoms score; wVAS, pain on walking as determined by the visual analog scale; FAS, full analysis set; ANCOVA, analysis of covariance; NT, not tested; NS, not significant; SFPP, S-flurbiprofen plaster. |
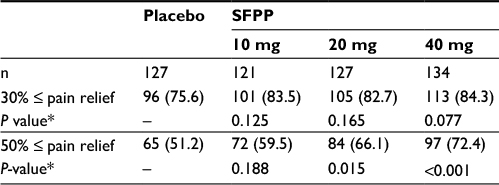 | Table 3 Number of patients who achieved 30% or 50% pain relief Notes: Values are shown as n (%). *Comparisons with placebo, χ2 test (significance levels = 0.05 [two-sided]). Abbreviation: SFPP, S-flurbiprofen plaster. |
At the end of the study, the least squares mean (95% confidence interval) of ΔtCS of SFPP 40 mg group was 6.5 (5.9–7.0), which was significantly larger than that of placebo group 5.3 (4.7–5.9) (P=0.002, Table 4, Figure 3C and D).
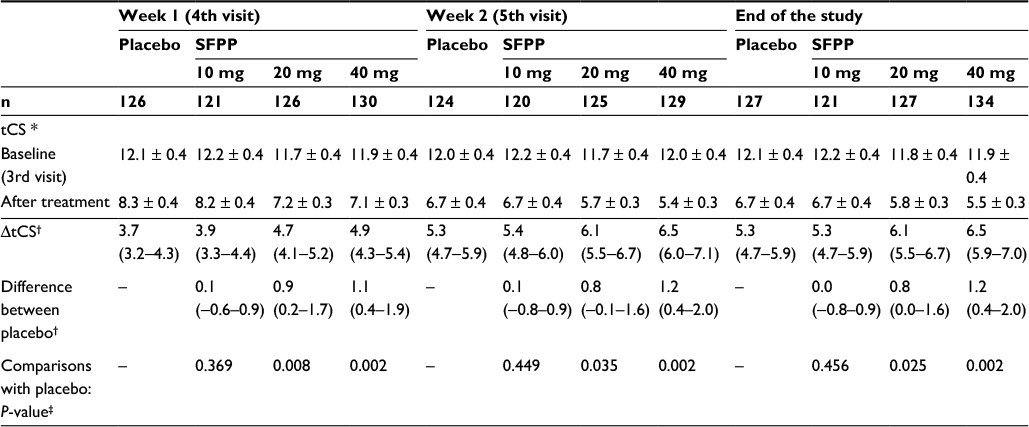 | Table 4 Overview of the total clinical symptoms score Notes: *Mean ± standard error. †Least squares mean (95% confidence interval). ‡Analysis of covariance with the treatment group as a fixed effect and the baseline as a covariate (significance levels = 0.025 [one-sided]). End of the study means each patient’s last visit, which was the 5th visit or the visit when the application was stopped. Abbreviations: SFPP, S-flurbiprofen plaster; tCS, total clinical symptoms score; ΔtCS, change in tCS from baseline. |
At the end of the study, the least squares mean (95% confidence interval) of ΔwVAS of SFPP 40 mg group was 19.7 (17.1–22.3) mm which was significantly larger than that of placebo group 14.8 (12.2–17.5) mm (P=0.005, Table 5, Figure 3E and F). In addition, SFPP 40 mg group showed significantly different ΔwVAS from that of placebo group at the day after treatment initiation (P=0.014).
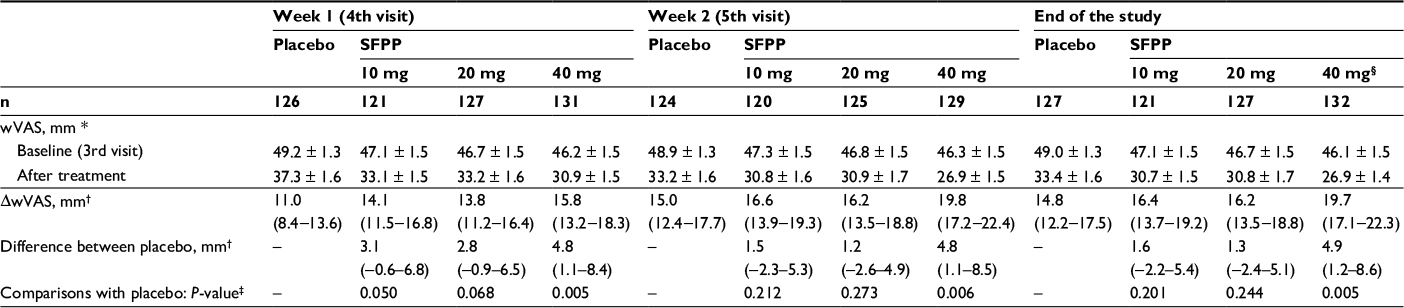 | Table 5 Overview of the pain on walking assessed by visual analog scale Notes: *Mean ± standard error. †Least squares mean (95% confidence interval). ‡Analysis of covariance with the treatment group as a fixed effect and the baseline as a covariate (significance levels = 0.025 [one-sided]). §Two subjects in SFPP 40 mg group were excluded because of lack of data. End of the study means each patient’s last visit, which was the 5th visit or the visit when the application was stopped. Abbreviations: SFPP, S-flurbiprofen plaster; wVAS, pain on walking as determined by the visual analog scale; ΔwVAS, change in wVAS from baseline. |
In the investigator’s global assessment, a significant difference was only detected between SFPP 40 mg and placebo group (P=0.003) in the intergroup comparisons (Figure 4A). In the patient’s global assessment, a significant difference was detected for SFPP 40 mg versus placebo group and SFPP 20 mg versus placebo group (P=0.003 and 0.045, respectively) in the intergroup comparisons (Figure 4B).
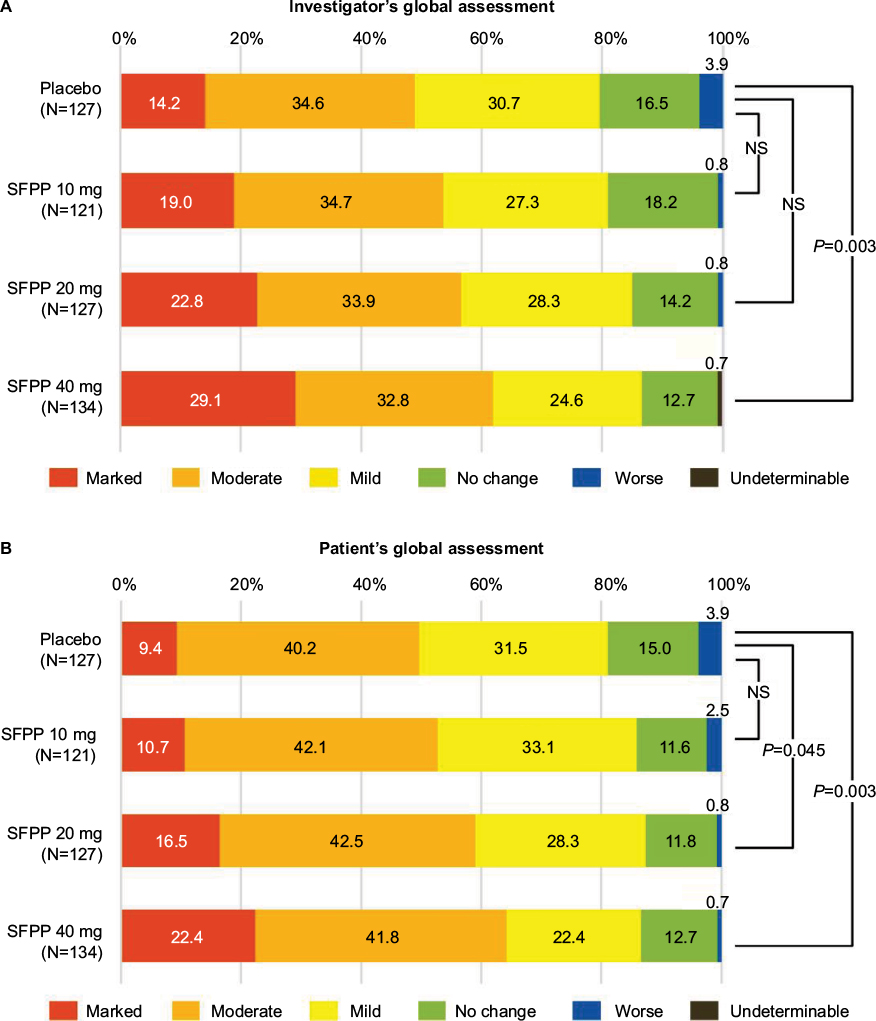 | Figure 4 SFPP 40 mg showed significant improvement compared with placebo in investigator’s and patient’s global assessments. Notes: (A) FAS; investigator’s global assessment. (B) FAS; patient’s global assessment. The outcomes were analyzed by using Wilcoxon’s rank-sum test (significance levels = 0.05 [two-sided]). Abbreviations: FAS, full analysis set; NS, not significant; SFPP, S-flurbiprofen plaster. |
Results of efficacy from the data set, which consists of the patients who completely followed the study protocol, were similar to FAS results mentioned earlier (data not shown).
Safety
The AEs at the application site were reported in all groups, details were 5.5%, 9.9%, 3.9%, and 10.4% in placebo, SFPP 10, 20, and 40 mg, respectively (Table 6). AE incidence rate at the application site was not significantly different across the treatment groups and indicated the absence of dose dependency. All AEs at application site were drug-related. A total of 35 of 38 AEs were rated as mild, and none of those was rated as severe. AEs leading to discontinuation of study treatment were reported in one subject in each group.
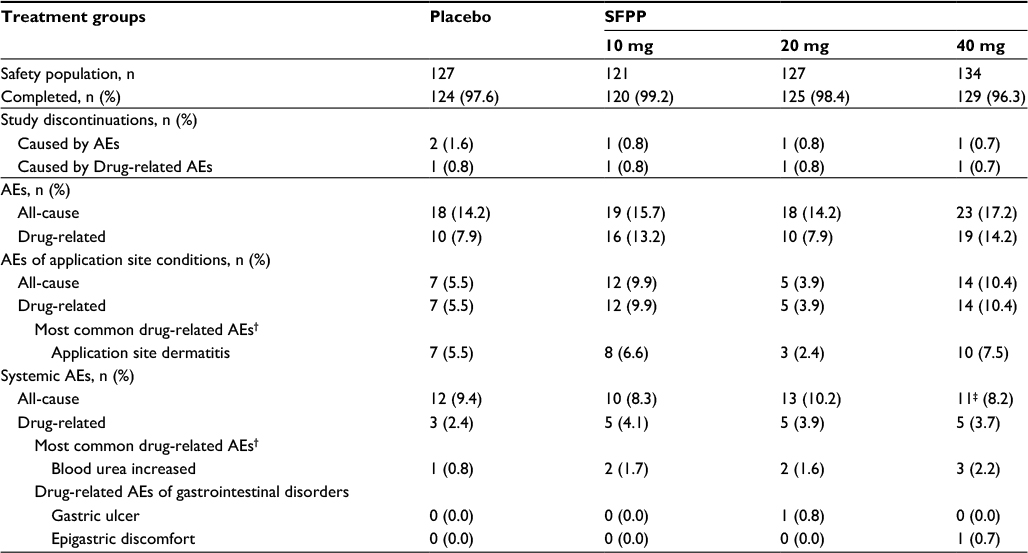 | Table 6 The incidence of adverse events per treatment group* Notes: *The same patient may be listed under different adverse events. †More than 2% in any of the groups. ‡An AE that led to hospitalization (vertigo) was observed in one subject. The investigator ruled out the causal relationship to the study treatment. Abbreviations: SFPP, S-flurbiprofen plaster; AEs, adverse events. |
The incidence rate of systemic and drug-related AEs of which the causal relationships were determined by the investigators before key code breaking were 2.4%, 4.1%, 3.9%, and 3.7% in placebo, SFPP 10, 20, and 40 mg, respectively (Table 6), statistical intergroup difference was not noted in these rates, and dose dependency was absent. There were 27 systemic drug-related AEs in 18 subjects in all groups, 25 and 2 of which were mild and moderate in severity, respectively. About two moderate AEs, glucose urine present in SFPP 40 mg and gastric ulcer in 20 mg were reported. Gastric ulcer with Helicobacter pylori infection was observed in a 65-year-old female, but did not require inpatient treatment. The subject was anemic prior to the initiation of the study treatment. The investigator thought that she might have had preexisting gastric ulcer, but the investigator did not rule out a causal relationship because the study treatment could have been an exacerbating factor.
Significant differences were detected in some laboratory test parameters, blood pressure, and pulse rate data at the end of the study as determined in the intergroup comparisons by the Student’s t-test (Table 7).
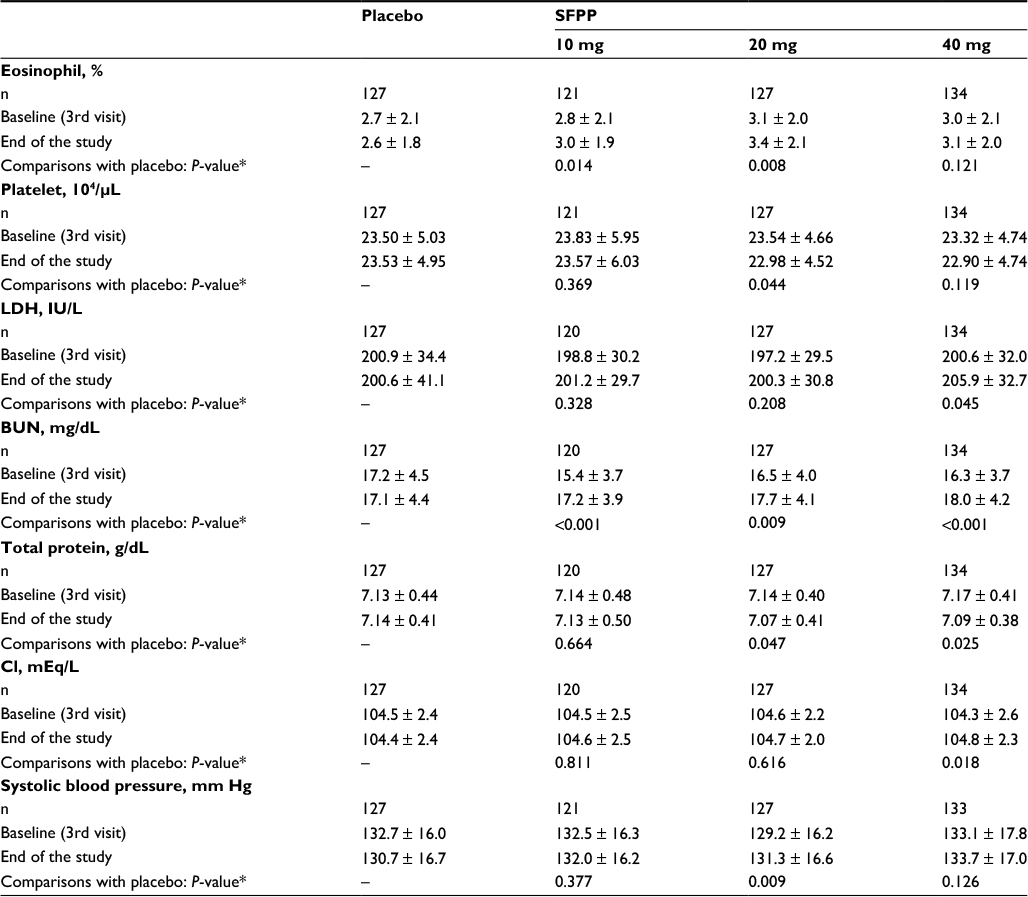 | Table 7 Laboratory test parameters, blood pressure, and pulse rate data with significant differences in the intergroup comparisons Notes: Values “at baseline” and “at the end of the study” are the mean ± standard deviation. *Student’s t-test (significance levels = 0.05 [two-sided]). Abbreviations: SFPP, S-flurbiprofen plaster; LDH, Lactate dehydrogenase; BUN, Blood urea nitrogen; Cl, Chloride; SFPP, S-flurbiprofen plaster. |
Discussion
In the present study, clinically relevant pain relief (the change of rVAS: 29.5–35.6 mm; the change rate: 51.15%-66.89%) was observed in all groups including placebo.29–31 Especially 40 mg showed remarkable pain relief, and a statistical significance was observed versus placebo. Although a 6.1 mm difference in ΔrVAS between SFPP 40 mg and placebo was smaller than the estimated value of 9.1 mm at the planning stage, statistical significance was detected. This difference was also smaller than those reported in the results of two comparative studies of diclofenac patch and placebo.26,32 Basically, the same inclusion criteria for the pain severity was adopted in the present study and the diclofenac studies, and the mean values of VAS at baseline were almost the same, which ranged from 56.0 to 58.4 mm. However, change of rVAS in placebo in the present study (29.5 mm) was larger than those in diclofenac studies (17 mm32 and 22.1 mm26). We speculated that the reason for the difference was partly due to the relatively high content of peppermint oil in placebo of the present study. Peppermint oil has “minty” smell and contains >30% l-menthol, which has been used widely in medicinal preparations for pain relief in arthritis and other painful conditions.33,34 So peppermint oil is not suitable for the study of analgesic effect of SFP for its analgesic property. However, it is indispensable for the improved transdermal absorption of SFP. Therefore, peppermint oil was added to active drugs and placebo as well to keep the blindness among four groups.
The proportion of the patients who achieved moderate improvement (achieved 30% pain reduction) was very high (75.6%–84.3%). Placebo of topical treatment tends to show high efficacy.35,36 Furthermore peppermint oil in placebo in the present study must have contributed. On the contrary, the proportion achieved substantial improvement (50% pain reduction) showed apparent dose-dependency and only 40 mg exceeded 70%. The difference of 21.2% between SFPP 40 mg and placebo also indicates the clinically relevant improvement.
In addition, all efficacy endpoints including the assessments by patients (rVAS, wVAS, and patient’s global assessment) and investigators (clinical symptoms and investigator’s global assessment) also revealed a statistically significant difference between SFPP 40 mg and placebo, indicating the robust efficacy of SFPP 40 mg.
Test drugs were applied for 2 weeks in this study in consideration of EMA guideline20 for clinical investigation of topical NSAIDs, which recommends 1–3 weeks. On the contrary, practical treatment with NSAIDs tends to be longer than 2 or 3 weeks. It is a limitation in terms of generalizability of the studies for topical NSAIDs.
In the safety assessment, the incidence of AEs and the discontinuation status indicated that SFPP was well tolerated. No intergroup difference was noted in the AE incidence rate, and most AEs reported in SFPP groups were mild in severity. No AEs were rated as severe. All local AEs were drug-related, but no intergroup differences were found. These AEs reported might not have been causally related to S-flurbiprofen because no consistent trend depending on doses was observed. These AEs might have been associated with physical stimuli, such as irritation upon patch removal.37
Although topical NSAIDs have a lower risk of gastrointestinal disorder compared with oral NSAIDs,6 the use of SFPP might increase the risk due to its high percutaneous absorption and systemic exposure.19 Nevertheless, the study results demonstrated low incidence of gastrointestinal disorder in SFPP groups (0%, 0.8%, and 0.7% in SFPP 10, 20, and 40 mg, respectively), indicating that SFPP had a similarly low risk as that of existing topical NSAIDs.
In other systemic AEs, the increments in BUN in all SFPP groups were significantly higher compared with those in placebo group, and no significant variation was observed in the blood creatinine levels. An increase in BUN may lead to renal disorder. Because a long-term administration of NSAIDs may result in renal injury as a class effect and BUN is one of the biomarkers of renal function, the BUN variation needs to be investigated in a long-term treatment.
Photosensitivity has been associated with some NSAID products,38,39 but no subjects in this study experienced photosensitivity. The nonclinical data indicate that SFPP does not cause photosensitivity (unpublished), and this is consistent with the results of the present study.
Based on the efficacy and safety results from this study, the optimal tested dose of SFPP was determined to be 40 mg. SFPP is provided in a size (10×14 cm) that is commonly used worldwide such as Flector® Patch and TransAct® patch. Because this formulation size can contain a maximum of 40 mg of S-flurbiprofen in a dispersed and soluble state, any higher dose could not be evaluated.
In future studies, establishing the clinical positioning of SFPP by conducting comparative studies to evaluate its efficacy against existing NSAID patch products is required. In addition, the evaluation of efficacy and safety for the long-term treatment of SFPP is also required.
Conclusion
Clinically relevant pain relief was observed in all groups including placebo. Especially 40 mg showed remarkable pain relief in not only primary endpoint but also all the other endpoints with significant differences over placebo. The safety profile of SFPP 40 mg was not different from that of placebo. Therefore, 40 mg was determined as the optimal tested dose.
Acknowledgments
The authors would like to thank the SFPP investigators of this study (during the clinical trial): Daisuke Kawamura, MD, Hirotaka Azuma, MD, Masahito Harada, MD, Mitsugu Takahashi, MD, Susumu Asano, MD, and Yoshimitsu Aoki, MD (Hokkaido); Sadafumi Kato, MD (Iwate); Satoru Nakajo, MD, Takashi Sato, MD, Takato Kanabuchi, MD, Tetsuro Sato, MD, and Tomomaro Kawamata, MD (Miyagi); Kazushi Kishi, MD, Kunio Kamatani, MD, Susumu Maehara, MD, and Yasuo Kobuna, MD (Gunma); Akira Kobayashi, MD, Masaki Iwahashi, MD, Ruriko Ozawa, MD, and Ryoichi Yamazaki, MD (Saitama); Aiichiro Yamamoto, MD, Masashi Kimoto, MD, Masashi Shiraishi, MD, Minoru Irahara, MD, and Yoshinori Nakata, MD (Chiba); Akira Takiguchi, MD, Eijiro Okumura, MD, Fumitoshi Omura, MD, Ko Matsumoto, MD, Koh Maruyama, MD, Koichi Tanaka, MD, Masahiro Shibasaki, MD, Ryosuke Kanda, MD, Ryuji Ikeda, MD, Sanshiro Hashimoto, MD, Shojiro Kato, MD, Takashi Yokoyama, MD, Takeshi Inoue, MD, Yu Miyazaki, MD, and Yutaka Suzuki, MD (Tokyo); Akihito Tomonaga, MD, Hideyuki Miki, MD, Hiroaki Shibata, MD, Kuniaki Katayama, MD, Masato Kasuga, MD, and Ryuji Suto, MD (Kanagawa); Motoaki Fujimori, MD, and Nobuaki Hachisuka, MD (Yamanashi); Chisato Kato, MD (Aichi); Daisuke Matsunaga, MD, Hiroshi Nomiyama, MD, Ichiro Hoshiko, MD, Koichi Kamihirakawa, MD, Nobuyuki Matsuguchi, MD, Takaaki Morooka, MD, Takaaki Yoshimoto, MD, Takahide Kozuma, MD, Takahiko Sannomiya, MD, Yosuke Kanazawa, MD, and Yuzoh Fukahori, MD (Fukuoka).
SFPP was kindly gifted by Tokuhon Corporation, Saitama, Japan. The authors would like to thank Yukikazu Kamada of Taisho Pharmaceutical Co, Ltd, for assistance with drafting and revising this manuscript.
The current affiliation of Yuichi Hoshino is Tochigi Rehabilitation Center, Tochigi, Japan.
Author contributions
All the authors were involved in drafting the article or revising it critically for important intellectual content and approved the final version for publication. Ikuko Yataba and Noboru Otsuka had full access to all of the study data and take responsibility for the integrity of the data and the accuracy of the data analysis. Ikuko Yataba, Noboru Otsuka, Isao Matsushita, Hideo Matsumoto, and Yuichi Hoshino were involved in study conception and design. Ikuko Yataba was involved in data acquisition. Ikuko Yataba, Noboru Otsuka, Isao Matsushita, Hideo Matsumoto, and Yuichi Hoshino were involved in data analysis and interpretation.
Disclosure
Taisho Pharmaceutical funded this clinical trial. Taisho Pharmaceutical was involved in the design of the study, its conduct, and the data analysis. Ikuko Yataba, Noboru Otsuka, and Isao Matsushita are employees of Taisho Pharmaceutical. Hideo Matsumoto has received consultancy fees from Taisho Pharmaceutical. Yuichi Hoshino had received consultancy fees from Taisho Pharmaceutical while in a previous affiliation. The authors report no other conflicts of interest in this work.
References
Muraki S, Akune T, Oka H, et al. Association of radiographic and symptomatic knee osteoarthritis with health-related quality of life in a population-based cohort study in Japan: the ROAD study. Osteoarthritis Cartilage. 2010;18:1227–1234. | ||
Peat G, McCarney R, Croft P. Knee pain and osteoarthritis in older adults: a review of community burden and current use of primary health care. Ann Rheum Dis. 2001;60:91–97. | ||
Fransen M, Bridgett L, March L, Hoy D, Penserga E, Brooks P. The epidemiology of osteoarthritis in Asia. Int J Rheum Dis. 2011;14:113–121. | ||
Wittenauer R, Smith L, Aden K. Priority medicines for Europe and the world update report, 2013. Priority medicines for Europe and the world “A public health approach to innovation. Background paper 6.12 osteoarthritis. World Health Organization. Available from: http://www.who.int/medicines/areas/priority_medicines/BP6_12Osteo.pdf. Accessed March 23, 2017. | ||
American Academy of Orthopaedic Surgeons. Treatment of Osteoarthritis of the Knee, Evidence-Based Guideline. 2nd edn; 2013. Available from: http://www.aaos.org/cc_files/aaosorg/research/guidelines/treatmentofosteoarthritisofthekneeguideline.pdf. Accessed March 23, 2017. | ||
McAlindon TE, Bannuru RR, Sullivan MC, et al. OARSI guidelines for the non-surgical management of knee osteoarthritis. Osteoarthritis Cartilage. 2014;22:363–388. | ||
National Clinical Guideline Centre. Osteoarthritis: Care and Management in Adults. 2014. Available from: https://www.nice.org.uk/guidance/cg177/evidence/full-guideline-191761309. Accessed March 23, 2017. | ||
Huang JQ, Sridhar S, Hunt RH. Role of Helicobacter pylori infection and non-steroidal anti-inflammatory drugs in peptic-ulcer disease: a meta-analysis. Lancet. 2002;359:14–22. | ||
Sakamoto C, Sugano K, Ota S, et al. Case-control study on the association of upper gastrointestinal bleeding and nonsteroidal anti-inflammatory drugs in Japan. Eur J Clin Pharmacol. 2006;62:765–772. | ||
Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med. 1999;340:1888–1899. | ||
Sinha M, Gautam L, Shukla PK, Kaur P, Sharma S, Singh TP. Current perspectives in NSAID-induced gastropathy. Mediators Inflamm. 2013;2013:258209. | ||
Carabaza A, Cabré F, Rotllan E, et al. Stereoselective inhibition of inducible cyclooxygenase by chiral nonsteroidal antiinflammatory drugs. J Clin Pharmacol. 1996;36:505–512. | ||
Peskar BM, Kluge S, Peskar BA, Soglowek SM, Brune K. Effects of pure enantiomers of flurbiprofen in comparison to racemic flurbiprofen on eicosanoid release from various rat organs ex vivo. Prostaglandins 1991;42:515–531. | ||
Froben 100mg Tablets (flurbiprofen) [package insert]. Maidenhead, Berkshire, UK: BGP Products, Ltd.; 2015. | ||
Memeo A, Garofoli F, Peretti G. Evaluation of the efficacy and tolerability of a new topical formulation of flurbiprofen in acute soft tissue injuries. Drug Invest. 1992;4:441–449. | ||
Tanaka S, Sonoda H, Nakabayashi K, Namiki A. [Preoperative flurbiprofen provides pain relief after laparopscopic cholecystectomy]. Masui. 1997;46:679–683. Japanese. | ||
Sugimoto M, Toda Y, Hori M, et al. Analgesic effect of the newly developed S(+)-flurbiprofen plaster (SFPP) on inflammatory pain in a rat adjuvant-induced arthritis model. Drug Dev Res. 2016;77:20–28. | ||
Sugimoto M, Toda Y, Hori M et al. Topical anti-inflammatory and analgesic effects of multiple applications of S(+)-flurbiprofen plaster (SFPP) in a rat adjuvant-induced arthritis model. Drug Dev Res. 2016;77:206–211. | ||
Yataba I, Otsuka N, Matsushita I, et al. Plasma pharmacokinetics and synovial concentrations of S-flurbiprofen plaster in humans. Eur J Clin Pharmacol. 2016;72:53–59. | ||
European Medicines Agency. Committee for medicinal products for human use, guideline on clinical investigation of medicinal products used in the treatment of osteoarthritis, Draft; 2009. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003443.pdf. Accessed March 23, 2017. | ||
U.S. Department of Health and Human Services, Food and Drug Administration. Guidance for industry: clinical development programs for drugs, devices, and biological products intended for the treatment of osteoarthritis (OA), draft guidance; 1999. Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM071577.pdf. Accessed March 23, 2017. | ||
Kellgren JH, Lawrence JS. Radiological assessment of osteoarthrosis. Ann Rheum Dis. 1957;16:494–502. | ||
Day R, Morrison B, Luza A, et al. A randomized trial of the efficacy and tolerability of the COX-2 inhibitor rofecoxib vs ibuprofen in patients with osteoarthritis. Rofecoxib/ibuprofen comparator study group. Arch Intern Med. 2000;160:1781–1787. | ||
Geba GP, Weaver AL, Polis AB, Dixon ME, Schnitzer TJ; Vioxx, Acetaminophen, Celecoxib Trial (VACT) Group. Efficacy of rofecoxib, celecoxib, and acetaminophen in osteoarthritis of the knee: a randomized trial. JAMA. 2002;287:64–71. | ||
Hiraga K, Ohashi Y. [Efficacy evaluation of analgesic agents used for cancer pain management by visual analogue scale]. Pain Res. 1999;14:9–19. Japanese (English abstract). | ||
Direiser RL, Tisne-Camus M. DHEP plasters as a topical treatment of knee osteoarthritis – a double-blind placebo-controlled study. Drugs Exp Clin Res. 1993;19:117–123. | ||
Sugawara S, Kuroki Y, Tateishi A, et al. [The effects of LOXONIN PAP 100mg (hydrogel patch containing loxoprofen sodium) for the treatment of patients with osteoarthritis of the knee – randomized, double-blind clinical study with loxoprofen sodium tablets]. J Clin Ther Med. 2006;22:393–409. Japanese (English abstract). | ||
Nagaya I, Niwa S, Matsui N, et al. [Study for defining the adequate concentration of the active ingredient of KPT-220 – double-blind study in gonarthrosis deformans]. Clin Rep. 1993;27:245–269. Japanese (English abstract). | ||
Busse JW, Bartlett SJ, Dougados M, et al. Optimal strategies for reporting pain in clinical trials and systematic reviews: recommendations from an OMERACT 12 workshop. J Rheumatol. 2015;42:1962–1975. | ||
Tubach F, Ravaud P, Baron G, et al. Evaluation of clinically relevant changes in patient reported outcomes in knee and hip osteoarthritis: the minimal clinically important improvement. Ann Rheum Dis. 2005;64:29–33. | ||
Tubach F, Ravaud P, Baron G, et al. Evaluation of clinically relevant states in patient reported outcomes in knee and hip osteoarthritis: the patient acceptable symptom state. Ann Rheum Dis. 2005;64:34–37. | ||
Brühlmann P, Michel BA. Topical diclofenac patch in patients with knee osteoarthritis: a randomized, double-blind, controlled clinical trial. Clin Exp Rheumatol. 2003;21:193–198. | ||
Eccles, R. Menthol and related cooling compounds. J Pharm Pharmacol. 1994;46:618–630. | ||
Johar P, Grover V, Topp R, Behm DG. A comparison of topical menthol to ice on pain, evoked tetanic and voluntary force during delayed onset muscle soreness. Int J Sports Phys Ther. 2012;7:314–322. | ||
Bannuru RR, McAlindon TE, Sullivan MC, Wong JB, Kent DM, Schmid CH. Effectiveness and implications of alternative placebo treatments: a systematic review and network meta-analysis of osteoarthritis trials. Ann Intern Med. 2015;163:365–372. | ||
Zhang W, Robertson J, Jones AC, Dieppe PA, Doherty M. The placebo effect and its determinants in osteoarthritis: meta-analysis of randomised controlled trials. Ann Rheum Dis. 2008;67:1716–1723. | ||
Kawamura N, Shinkai N, Yamauchi H, Takayama S. Comparison of poultice-type and tape-type patches containing ketoprofen on human skin irritation. J Toxicol Sci. 2003;28:415–425. | ||
European Medicines Agency. Assessment report for ketoprofen containing medicinal products (topical formulations); 2010. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/Ketaprofen_107/WC500117251.pdf. Accessed March 23, 2017. | ||
Guy RH, Kuma H, Nakanishi M. Serious photocontact dermatitis induced by topical ketoprofen depends on the formulation. Eur J Dermatol. 2014;24:365–371. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.