")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
The effects of repeated Toll-like receptors 2 and 4 stimulation in COPD alveolar macrophages
Authors Lea SR, Reynolds SL, Kaur M, Simpson KD, Hall SR, Hessel EM , Singh D
Received 24 September 2015
Accepted for publication 7 December 2017
Published 2 March 2018 Volume 2018:13 Pages 771—780
DOI https://doi.org/10.2147/COPD.S97071
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Simon R Lea,1,* Sophie L Reynolds,1,* Manminder Kaur,1 Karen D Simpson,2 Simon R Hall,2 Edith M Hessel,2 Dave Singh1
1Division of Infection, Immunity and Respiratory Medicine, School of Biological Sciences, Faculty of Biology, Medicine and Health, Manchester Academic Health Science Centre, The University of Manchester, Manchester University NHS Foundation Trust, Manchester, 2Refractory Respiratory Inflammation DPU, GlaxoSmithKline Medicines Research Centre, Stevenage, UK
*These authors contributed equally to this work
Background: COPD is a progressive inflammatory airway disease characterized by increased numbers of alveolar macrophages in the lungs. Bacterial colonization of the lungs is a common feature in COPD and can promote inflammation through continual and repeated Toll-like receptor (TLR) stimulation. We have studied the response of COPD alveolar macrophages to repetitive stimulation with TLR2 and TLR4 ligands. We investigated the effect of sequential stimulation with different ligands to determine whether this results in tolerance or amplification of the immune response.
Methods: We stimulated alveolar macrophages from COPD patients (n=9) and smokers (n=8) with the TLR4 agonist lipopolysaccharide (LPS) or the TLR2 agonist Pam3CSK4 for 24 hours before restimulating again for 24 hours. Cytokine protein release and gene expression were investigated.
Results: Repetitive stimulation of COPD and smokers macrophages with LPS for both 24-hour periods caused a reduction in tumor necrosis factor α, CCL5, and IL-10 production compared to cells that were not exposed initially to LPS. IL-6 and CXCL8 production were not significantly altered following repetitive LPS stimulation. The same pattern was observed for repeated stimulation with Pam3CSK4. Using COPD macrophages, LPS followed by Pam3CSK4 stimulation increased the levels of all cytokines compared to media followed by Pam3CSK4.
Conclusion: TLR tolerance in COPD alveolar macrophages occurs after repetitive stimulation with the same TLR ligand, but this only occurs for selected cytokines. CXCL8 production is not reduced after repetitive TLR stimulation with the same ligand; this may be an important mechanism for the increased CXCL8 levels that have been observed in COPD. We showed that TLR4 stimulation followed by TLR2 stimulation does not cause tolerance, but enhances cytokine production. This may be a relevant mechanism by which bacteria cause excessive inflammation in COPD patients.
Keywords: alveolar macrophages, tolerance, COPD
Introduction
COPD is characterized by an increased inflammatory response to the inhalation of noxious particles, such as cigarette smoke.1 There are increased numbers of macrophages, neutrophils, and lymphocytes in the lungs of COPD patients,2 with these cells releasing a broad range of inflammatory mediators.
Bacterial colonization of the airways is a feature of COPD.3,4 There are a variety of possible underlying causes for this, including defective phagocytosis by COPD macrophages allowing persistence of bacteria.5 Bacterial colonization may lead to chronic stimulation of immune cells through Toll-like receptors (TLRs) which sense pathogen-associated molecular patterns (PAMPs).6 The bacterial PAMPs lipopolysaccharide (LPS) and lipopeptide signal through TLR4 and TLR2, respectively, promoting activation of signaling pathways such as mitogen-activated protein kinases and nuclear factor kB7,8 that increase the secretion of pro-inflammatory mediators.7,9
Endotoxin tolerance is the development of refractoriness to LPS, where repeat exposure to LPS results in a limited inflammatory response characterized by reduced production of cytokines such as tumor necrosis factor α (TNFα).10–12 The mechanisms proposed to be responsible for endotoxin tolerance include downregulation of TLRs13 or increased expression of negative regulators of TLR signaling.14–16 The behavior of some inflammatory mediators following repeat LPS exposure tolerance may be different to TNFα, as it has been reported that the neutrophil chemoattractant CXCL8 showed tolerance in one study12 but not in another.11 CXCL8 levels are increased in the lungs of COPD patients, and its production by alveolar macrophages is poorly suppressed by corticosteroids.17–21 CXCL8 appears to be an important driver of corticosteroid-insensitive neutrophilic inflammation in COPD.
Tolerance to repeated TLR stimulation has been described in various macrophage models,10–12 but this phenomenon has not been investigated in COPD lung macrophages. The phenotype and behavior of COPD macrophages differ from other types of macrophages; for example, the response to LPS is lower than healthy alveolar macrophages17 and they display less pro-inflammatory potential compared to healthy cells.20,22,23 The response of COPD macrophages to repetitive TLR stimulation is clinically relevant, as these cells are exposed to chronic stimulation by bacteria in the lungs which may contribute to the frequency and severity of exacerbations.
The bacterial flora within the lungs of COPD patients often contains multiple pathogens capable of stimulation through different or multiple TLRs.24 COPD alveolar macrophages may therefore be exposed to sequential stimulation with different PAMPs. The inflammatory response of alveolar macrophages to such sequential stimulation has not been studied, and it is not known if a tolerized or an increased response will occur in this situation.
The aim of this study was to characterize the response of COPD alveolar macrophages to repetitive stimulation with TLR2 and TLR4 ligands. The novelty of this study is that we have used COPD alveolar macrophages to investigate the development of tolerance after repeated TLR stimulation. Furthermore, we investigated the production of inflammatory mediators after sequential stimulation with different ligands to study whether tolerance or an increased inflammatory response occurs.
Methods
Subjects
Patients undergoing surgical resection for suspected or confirmed lung cancer (Table 1) were recruited. COPD was diagnosed based on a history of smoking for >10 pack-years, typical symptoms, and airflow obstruction, defined as forced expiratory volume in 1 second/forced vital capacity ratio <0.7. COPD patients and smokers without airflow obstruction included current and ex-smokers who had ceased smoking for 1 year. All subjects gave written informed consent and the study was approved by the local ethics committee.
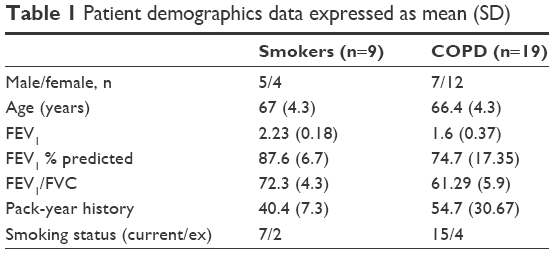 | Table 1 Patient demographics data expressed as mean (SD) |
Alveolar macrophage isolation
Alveolar macrophages were isolated as previously described.17 Briefly, COPD patients and smokers without airflow obstruction undergoing lung surgery were recruited. Lung tissue distant from the tumor site was removed before perfusing with 0.1 M NaCl for macrophage isolation. Flushed fluid was centrifuged and the cell pellet was resuspended in RPMI-1640 (Sigma-Aldrich Co, St Louis, MO, USA). The cell suspension was floated over a Ficoll gradient, centrifuged, and viable macrophages were isolated and counted using trypan blue exclusion.
Cell culture
Alveolar macrophages in supplemented RPMI-1640 media were plated out in 96- and 24-well plates at a concentration of 1×106/mL. Cells were incubated with LPS (Sigma-Aldrich, serotype O26:B6; 1 μg/mL) or Pam3CSK4 (Sigma-Aldrich; 0.1 μg/mL) for a 24-hour period before removing supernatants and washing 3 times with media. Cells were then stimulated again with LPS (1 μg/mL) or Pam3CSK4 (0.1 μg/mL) for 24 hours or as indicated; these concentrations have been shown to give optimal stimulation of alveolar macrophages at 24 hours.17,25 For TLR mRNA analysis, cells were stimulated with LPS (1 μg/mL), Pam3CSK4 (0.1 μg/mL), or ultrapure Escherichia coli O111:B4 LPS (UPLPS; InvivoGen, San Diego, CA, USA; 0.1 μg/mL) for 4, 6, 24, and 48 hours. Where necessary, supernatants were removed and frozen for future cytokine analysis.
Cytokine and chemokine quantification
Cell culture supernatants were harvested and enzyme-linked immunosorbent assays (ELISA) were used according to manufacturers’ instructions (R&D Systems, Inc, Minneapolis, MN, USA) to quantify levels of CXCL8 and TNFα. Lower limits of detection were 31.25 and 15.625 pg/mL, respectively. IL-6 and IL-10 (lower limits of detection 0.17 pg/mL) were measured using the Luminex 100 System. CCL5 was measured by both ELISA (15.625 pg/mL) and Luminex (0.17 pg/mL); these results were comparable.
Analysis of mRNA levels
Samples harvested in TRIzol were homogenized using a 21-gauge needle. Chloroform was added to the TRIzol samples (1 in 5 v/v) and separation was achieved following centrifugation at 12,000 g for 15 minutes. The upper aqueous RNA phase was purified using DNase I and an RNeasy Mini Kit (Qiagen NV, Venlo, the Netherlands) according to the manufacturers’ instructions.
cDNA was synthesized from 50 ng RNA using the Verso™ 2-step qRT-PCR kit (Thermo Fisher Scientific, Waltham, MA, USA). Real-time PCR was performed using the Stratagene machine (Agilent Technologies, Santa Clara, CA, USA). cDNA (1 μL) was added to ABsolute Blue qPCR mix (Thermo Fisher Scientific) according to the manufacturers’ instructions in the presence of 6-carboxyfluorescein (FAM)-labeled primers for CXCL8, TNFa, TLR1, TLR2, TLR4, or GAPDH (Thermo Fisher Scientific). Amplification conditions were 95°C for 15 minutes, 95°C for 15 minutes before cooling to 60°C for 1 minute for 40 cycles. Levels of CXCL8, TNFα, TLR1, TLR2, and TLR4 were normalized to housekeeping gene GAPDH to quantify mRNA levels.
Statistical analysis
Data normality was measured using the Kolmogorov–Smirnov test. For comparison of 2 parametrically distributed data sets a Student’s paired t-test was used. For comparison of 2 nonparametrically distributed data sets, Wilcoxon matched pairs (paired data) or Mann–Whitney (unpaired) tests were used. In tolerance experiments with multiple conditions, pairwise tests were used to analyze a priori determined selected conditions only, and not comparisons between all conditions. These a priori determined comparisons are stated in the “Results” section. All statistical analysis was carried out using GraphPad InStat version 3.00 (GraphPad Software, Inc, La Jolla, CA, USA).
Results
Repeated LPS stimulation
Alveolar macrophages from smokers (n=7) and COPD patients (n=9) were incubated with media (M) or LPS for 24 hours. Supernatants were removed, and cells from both conditions were subsequently incubated with either M or LPS for 24 hours. Inflammatory mediators secreted in the latter 24-hour cell culture period were measured. Cells exposed to M and then LPS (ML) showed significant increases in inflammatory mediator production compared to cells not exposed to LPS (MM), confirming that cells left initially in M culture were subsequently responsive to LPS stimulation. There were no differences in cytokine levels between smoking controls and COPD patients for either MM or ML conditions (p>0.05 for all inflammatory mediators; Figure S1).
Repetitive stimulation with LPS for both 24-hour cell culture periods (LL) caused a statistically significant reduction in TNFα compared to cells that were not exposed initially to LPS (ML) in both smokers and COPD patients (p<0.05; Figure 1). LL treatment also significantly reduced CCL5 and IL-10 production compared to ML treatment in COPD patients (p<0.05 for each cytokine). In smokers, there were numerical decreases in CCL5 and IL-10 levels after LL compared to ML treatment, but these were not statistically significant (p=0.3 and p=0.15, respectively). Contrastingly, the levels of CXCL8 and IL-6 production were not significantly altered following LL compared to ML treatment in either smokers or COPD patients. These results demonstrate tolerance to LPS stimulation for selected inflammatory mediators (TNFα, CCL5, and IL-10). Overall, the pattern of results was similar between smokers and COPD patients.
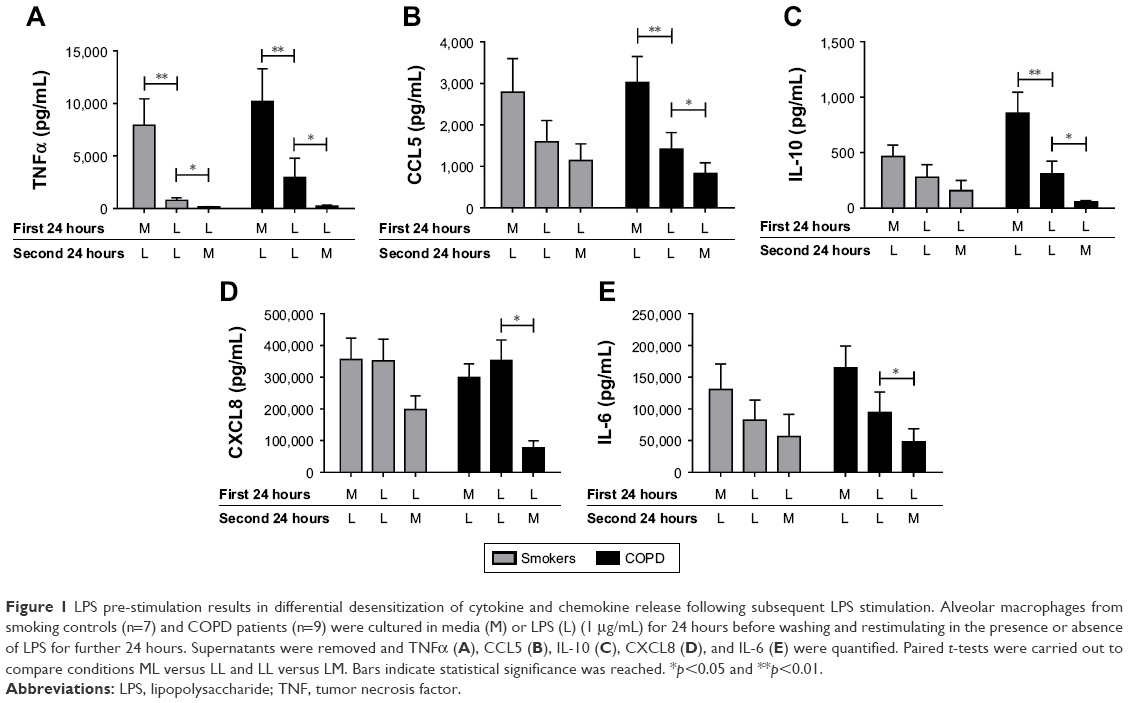 | Figure 1 LPS pre-stimulation results in differential desensitization of cytokine and chemokine release following subsequent LPS stimulation. Alveolar macrophages from smoking controls (n=7) and COPD patients (n=9) were cultured in media (M) or LPS (L) (1 μg/mL) for 24 hours before washing and restimulating in the presence or absence of LPS for further 24 hours. Supernatants were removed and TNFα (A), CCL5 (B), IL-10 (C), CXCL8 (D), and IL-6 (E) were quantified. Paired t-tests were carried out to compare conditions ML versus LL and LL versus LM. Bars indicate statistical significance was reached. *p<0.05 and **p<0.01. |
We investigated whether inflammatory mediator release in the 24-hour period after a second LPS exposure (LL) was greater than the natural time course observed after a single LPS exposure followed by media (LM). Inflammatory mediator production after LL was significantly greater (p<0.05) than LM in all experiments apart from IL-6 in smokers. This demonstrates that even though tolerance occurred for TNFα, CCL5, and IL-10, LL stimulation still increased inflammatory mediator production above the levels caused by a single LPS stimulation (LM; Figure 1). These results again showed a generally similar pattern between smokers and COPD cells.
Repeated Pam3CSK4 stimulation
COPD alveolar macrophages (n=8) incubated with M for 24 hours followed by stimulation with Pam3CSK4 (P) for 24 hours produced significantly increased levels of TNFα, CCL5, IL-10, CXCL8, and IL-6 compared to cells incubated in M for 48 hours (p<0.05 for each inflammatory mediator; Figure S2).
Following 2 consecutive Pam3CSK4 stimulations (PP), TNFα, CCL5, and IL-10 release were significantly reduced (p<0.05) compared to MP treatment. CXCL8 and IL-6 production after PP and MP treatment were similar (Figure 2).
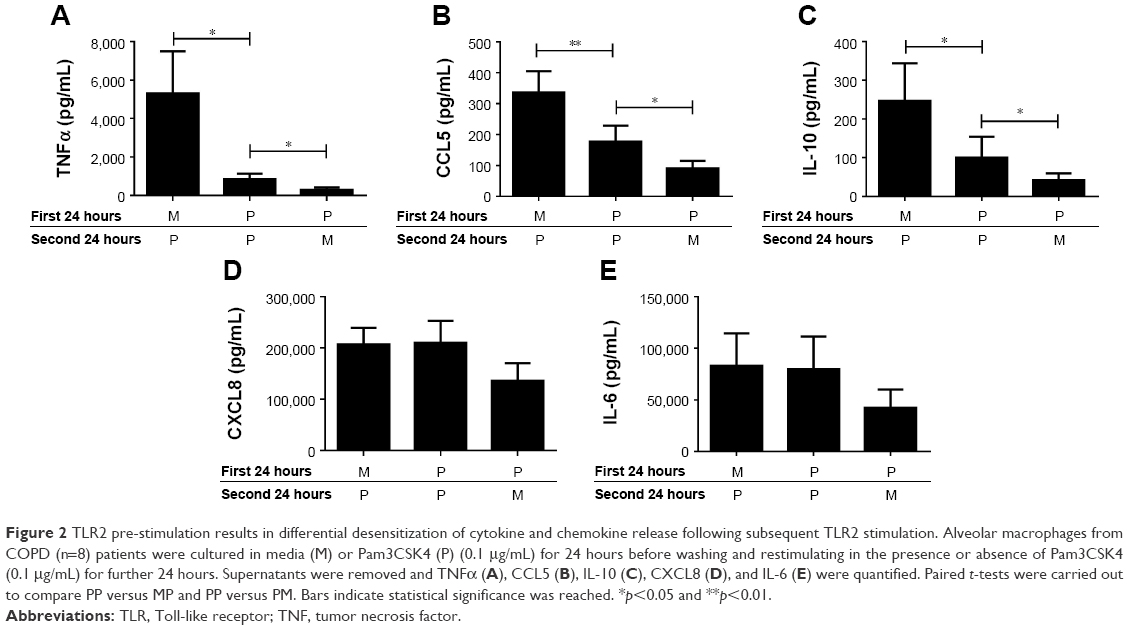 | Figure 2 TLR2 pre-stimulation results in differential desensitization of cytokine and chemokine release following subsequent TLR2 stimulation. Alveolar macrophages from COPD (n=8) patients were cultured in media (M) or Pam3CSK4 (P) (0.1 μg/mL) for 24 hours before washing and restimulating in the presence or absence of Pam3CSK4 (0.1 μg/mL) for further 24 hours. Supernatants were removed and TNFα (A), CCL5 (B), IL-10 (C), CXCL8 (D), and IL-6 (E) were quantified. Paired t-tests were carried out to compare PP versus MP and PP versus PM. Bars indicate statistical significance was reached. *p<0.05 and **p<0.01. |
We investigated whether inflammatory mediator release in the 24-hour period after a second Pam3CSK4 exposure (PP) was greater than the natural time course after a single P exposure followed by M (PM). Inflammatory mediator release after PP was numerically greater compared to PM, reaching significance for IL-10 and CCL5. This demonstrates that even though tolerance occurred for IL-10 and CCL5, PP stimulation still increased mediator production above the levels that would have been caused by a single Pam3CSK4 stimulation (PM; Figure 2).
TNFα and CXCL8 gene expression
The effects of repeated LPS stimulation on TNFα and CXCL8 gene expression were assessed. Alveolar macrophages from COPD patients (n=6) were cultured with media or LPS for 24 hours followed by LPS for 4 or 24 hours. TNFα and CXCL8 gene expression were increased at 4 and 24 hours after ML; TNFα production peaked at 4 hours, while CXCL8 production was greater at 24 hours (Figure 3). Twenty-four-hour pre-treatment with LPS reduced the effect of the second LPS stimulation on TNFα, with mRNA levels reduced at both 4- and 24-hour time points. Contrastingly, CXCL8 gene expression was not reduced by LL treatment compared to ML.
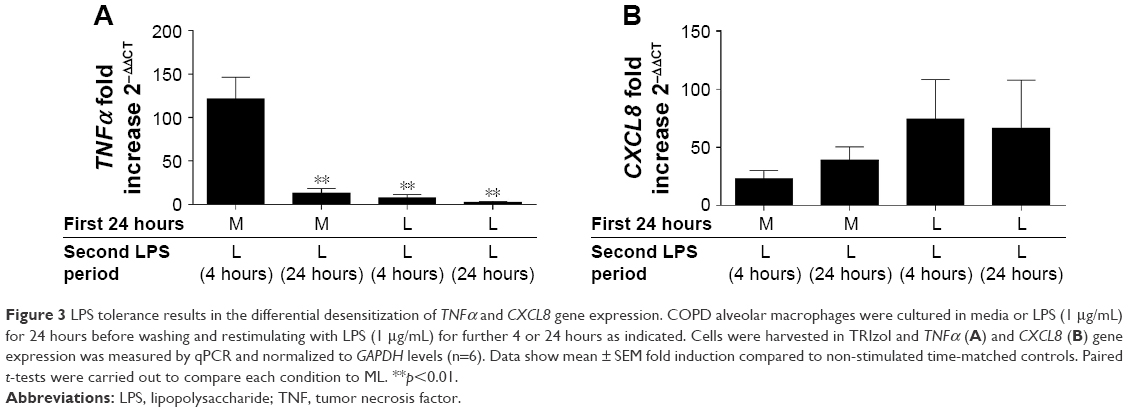 | Figure 3 LPS tolerance results in the differential desensitization of TNFα and CXCL8 gene expression. COPD alveolar macrophages were cultured in media or LPS (1 μg/mL) for 24 hours before washing and restimulating with LPS (1 μg/mL) for further 4 or 24 hours as indicated. Cells were harvested in TRIzol and TNFα (A) and CXCL8 (B) gene expression was measured by qPCR and normalized to GAPDH levels (n=6). Data show mean ± SEM fold induction compared to non-stimulated time-matched controls. Paired t-tests were carried out to compare each condition to ML. **p<0.01. |
LPS stimulation of alveolar macrophages increases TLR2 gene expression
TLR2 and TLR4 gene expression levels were measured following LPS stimulation (n=6; Figure 4). TLR2 mRNA levels were significantly increased at 6, 24, and 48 hours after LPS stimulation (p<0.05 at each time point). LPS did not change TLR4 gene expression at any time point (Figure 4A).
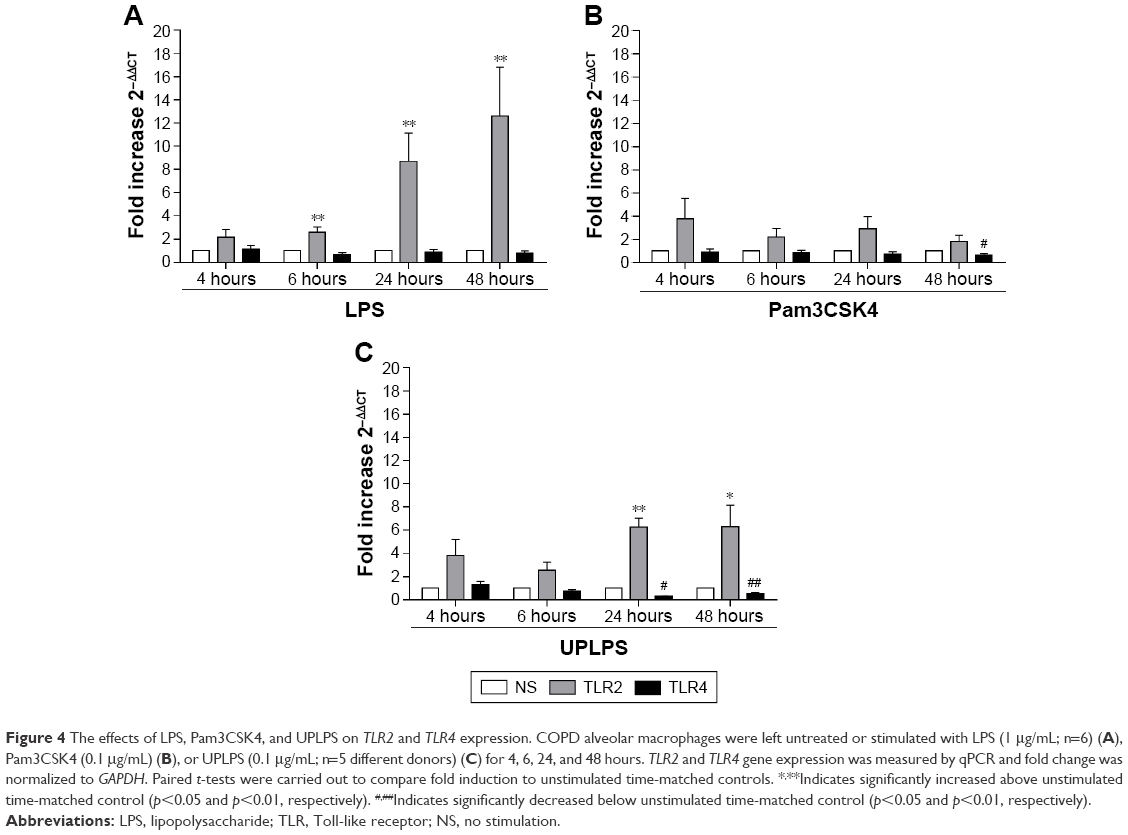 | Figure 4 The effects of LPS, Pam3CSK4, and UPLPS on TLR2 and TLR4 expression. COPD alveolar macrophages were left untreated or stimulated with LPS (1 μg/mL; n=6) (A), Pam3CSK4 (0.1 μg/mL) (B), or UPLPS (0.1 μg/mL; n=5 different donors) (C) for 4, 6, 24, and 48 hours. TLR2 and TLR4 gene expression was measured by qPCR and fold change was normalized to GAPDH. Paired t-tests were carried out to compare fold induction to unstimulated time-matched controls. *,**Indicates significantly increased above unstimulated time-matched control (p<0.05 and p<0.01, respectively). #,##Indicates significantly decreased below unstimulated time-matched control (p<0.05 and p<0.01, respectively). |
Alveolar macrophages from 4 further COPD patients were treated with either TLR2-specific ligand Pam3CSK4 (Figure 4B) or TLR4-specific ligand UPLPS (Figure 4C). TLR1, TLR2, and TLR4 gene expression levels were measured; TLR2 mRNA levels were significantly increased at 24 and 48 hours by UPLPS stimulation but were not changed by Pam3CSK stimulation. TLR4 mRNA was significantly reduced by UPLPS at 24 and 48 hours and Pam3CSK4 at 48 hours. TLR1 mRNA levels were unchanged by either UPLPS or Pam3CSK4 stimulation at any time point (Figure S3).
LPS and subsequent Pam3CSK4 stimulation
As LPS increased TLR2 expression, we investigated the effect of treatment with LPS or M followed by stimulation with Pam3CSK4 using COPD alveolar macrophages (n=8; Figure 5).
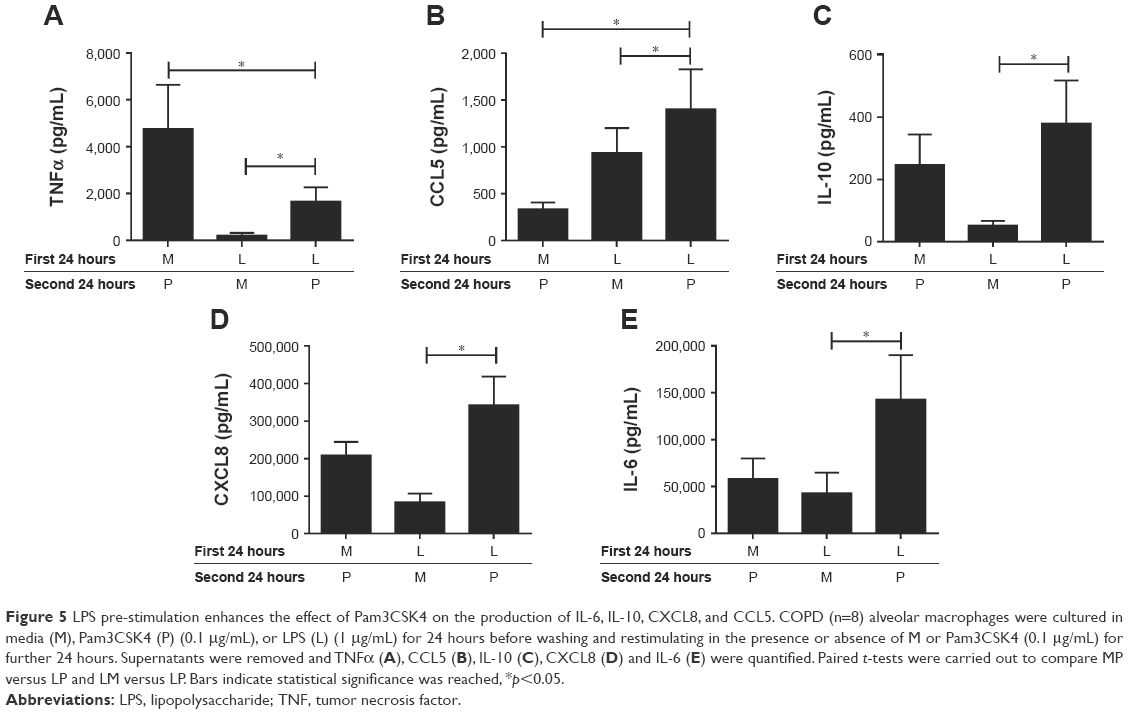 | Figure 5 LPS pre-stimulation enhances the effect of Pam3CSK4 on the production of IL-6, IL-10, CXCL8, and CCL5. COPD (n=8) alveolar macrophages were cultured in media (M), Pam3CSK4 (P) (0.1 μg/mL), or LPS (L) (1 μg/mL) for 24 hours before washing and restimulating in the presence or absence of M or Pam3CSK4 (0.1 μg/mL) for further 24 hours. Supernatants were removed and TNFα (A), CCL5 (B), IL-10 (C), CXCL8 (D) and IL-6 (E) were quantified. Paired t-tests were carried out to compare MP versus LP and LM versus LP. Bars indicate statistical significance was reached, *p<0.05. |
LP treatment caused significantly greater production of all 5 cytokines compared to LM treatment (p<0.05), demonstrating an additional stimulatory effect of Pam3CSK4 after LPS treatment. There were numerical increases in the levels of IL-6, IL-10, CCL5, and CXCL8 production after LP treatment compared to MP that reached significance for CCL5 (p=0.016). LP treatment resulted in lower TNFα release compared to MP-treated cells (p=0.016).
Discussion
COPD alveolar macrophages develop tolerance to repetitive LPS stimulation, but this only occurs for selected cytokines such as TNFα, CCL5, and IL-10. In contrast, tolerance was not observed for CXCL8 and IL-6. There was similar cytokine selective tolerance after repetitive TLR2 stimulation. This difference between cytokine responses after repetitive TLR stimulation may be relevant to the pathophysiology of COPD as a mechanism by which selected cytokines are produced at persistently high levels, thus promoting inflammation. LPS upregulated TLR2 gene expression, potentially enhancing the sensitivity of these cells to TLR2 ligands. We tested this possibility, and found that LPS followed by Pam3CSK4 stimulation (LP) increased the levels of all cytokines measured compared to media followed by Pam3CSK4 (MP). This was a different pattern of results in comparison to stimulation with the same TLR ligand. There was reduced production of selected cytokines when comparing PP to MP. This upregulation of cytokine production after different TLR stimuli may be of clinical relevance in COPD patients who are exposed to more than one bacterial strain and hence exposed to different TLR ligands. This may occur in some patients in the stable state, or during an acute exacerbation.
This is the first study of TLR tolerance using COPD alveolar macrophages. COPD alveolar macrophages have unique characteristics17,20,22,23 and are very different to other sources of macrophages such as those derived from cell lines or blood-derived monocytes. Nevertheless, our results for repetitive LPS or Pam3CSK4 stimulation of macrophages agree with previous reports concerning TNFα and CCL5, which develop tolerance10,11,13,26,27 and for CXCL8, which does not.11 Our results for IL-6, which did not develop tolerance, differ from previous reports for both LPS and Pam3CSK4 stimulation.10,11 The reasons for this are not known, but are likely to be a reflection of the different phenotype and function of human lung macrophages from smokers and COPD patients. The majority of our experiments were performed in COPD patients, but we did observe a similar tolerance pattern in smokers. Lung cancer surgery is usually performed on patients with a history of smoking. Never smokers are a minority,28 so samples from such patients were unavailable. This limited the scope of the study. However, smokers without airflow obstruction allowed changes due to COPD rather than chronic smoking to be assessed.
CXCL8 is a neutrophil chemoattractant present at higher levels in the lungs of COPD patients compared to controls.29–32 CXCL8 is produced by different cell types including epithelial cells, macrophages, and smooth muscle cells. The overproduction of CXCL8 in the lungs of COPD patients could be partly due to bacterial stimulation of lung cells, as it is known that lung CXCL8 levels and neutrophil numbers are higher in COPD patients with bacterial colonization.33 We showed here a mechanism by which this may occur; TLR2 or TLR4 repetitive stimulation causes persistent CXCL8 secretion, which may lead to enhanced neutrophilic lung inflammation. The reasons for the differences between cytokines in the development of tolerance are unclear. The time course experiment showed different patterns between TNFα and CXCL8 gene expression after TLR4 activation, with CXCL8 showing a slower induction and longer duration of expression upregulation. The transcriptional control mechanisms responsible for these different profiles after a single TLR activation are likely to dictate the response to a repeated activation, with prolonged promotor activation for CXCL8 being associated with a lack of tolerance. For cytokines such as TNFα, there may be a natural promotor activation shutdown mechanism34 after a single TLR activation that reduces the effect of a repeated identical TLR activation, also highlighting the fact that there is differential transcriptional regulation for different cytokines. The regulation of other inflammatory cytokines not reported in this manuscript such as IL-1β may also provide interesting results within the context of COPD pathophysiology.
It has previously been shown using mouse macrophages that TLR4 stimulation increases TLR2 gene expression,35 and that TLR2 stimulation after TLR4 stimulation causes increased cytokine production compared to repetitive TLR4 stimulation.13 We now report the same phenomenon within human lung macrophages, confirmed by using a specific TLR4 agonist. The upregulation of TLR2 expression, caused by LPS, appears to enhance the sensitivity to subsequent TLR2 activation, thereby altering downstream cell signaling events capable of upregulating cytokine production.
The exact mechanisms underpinning TLR tolerance have not been elucidated. In our experiments, there was downregulation of TLR4 gene expression after using ultrapure LPS, which targets TLR4 only, but not LPS which also shows some activity against TLR2. The reason for this difference is unclear. The experiments measuring cytokines were performed using LPS, so TLR4 gene expression downregulation was not the reason for tolerance. It has been suggested that impaired activation of kinases such as interleukin receptor-associated kinase 1,16 or altered tyrosine activation of TLR435 could be contributing to this phenomenon. Impaired activation of specific signaling pathways could lead to the differences observed here between inflammatory mediators.
In conclusion, we described TLR tolerance in COPD alveolar macrophages. Interestingly, CXCL8 production is not reduced after repetitive TLR stimulation with the same ligand; this may be an important mechanism causing persistent CXCL8 secretion in COPD. Furthermore, we showed that TLR4 stimulation followed by TLR2 stimulation does not cause tolerance, but enhances cytokine production; this again may be a relevant mechanism by which inflammation occurs in the airways of COPD patients.
Disclosure
SRL, SLR and MK have no conflicts of interest. KDS, SRH and EMH are employees of GSK. DS has received sponsorship to attend international meetings, honoraria for lecturing or attending advisory boards and research grants from various pharmaceutical companies including Apellis, AstraZeneca, Boehringer Ingelheim, Chiesi, Cipla, Genentech, GlaxoSmithKline, Glenmark, Johnson and Johnson, Menarini, Mundipharma, Novartis, Peptinnovate Pfizer, Pulmatrix, Skypharma, Teva, Therevance and Verona. The authors report no other conflicts of interest in this work.
References
Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med. 2017;195(5):557–582. | ||
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | ||
Desai H, Eschberger K, Wrona C, et al. Bacterial colonization increases daily symptoms in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2014;11(3):303–309. | ||
Patel IS, Seemungal TA, Wilks M, Lloyd-Owen SJ, Donaldson GC, Wedzicha JA. Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax. 2002;57(9):759–764. | ||
Taylor AE, Finney-Hayward TK, Quint JK, et al. Defective macrophage phagocytosis of bacteria in COPD. Eur Respir J. 2010;35(5):1039–1047. | ||
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11(5):373–384. | ||
Foster SL, Medzhitov R. Gene-specific control of the TLR-induced inflammatory response. Clin Immunol. 2009;130(1):7–15. | ||
Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. | ||
Bhattacharyya S, Gutti U, Mercado J, Moore C, Pollard HB, Biswas R. MAPK signaling pathways regulate IL-8 mRNA stability and IL-8 protein expression in cystic fibrosis lung epithelial cell lines. Am J Physiol Lung Cell Mol Physiol. 2011;300(1):15. | ||
Wang JH, Doyle M, Manning BJ, Di Wu Q, Blankson S, Redmond HP. Induction of bacterial lipoprotein tolerance is associated with suppression of toll-like receptor 2 expression. J Biol Chem. 2002;277(39):36068–36075. | ||
Kaufmann A, Gemsa D, Sprenger H. Differential desensitization of lipopolysaccharide-inducible chemokine gene expression in human monocytes and macrophages. Eur J Immunol. 2000;30(6):1562–1567. | ||
Mendes ME, Baggio-Zappia GL, Brunialti MK, Fernandes Mda L, Rapozo MM, Salomao R. Differential expression of toll-like receptor signaling cascades in LPS-tolerant human peripheral blood mononuclear cells. Immunobiology. 2011;216(3):285–295. | ||
Sato S, Nomura F, Kawai T, et al. Synergy and cross-tolerance between toll-like receptor (TLR)2- and TLR4-mediated signaling pathways. J Immunol. 2000;165(12):7096–7101. | ||
Kobayashi K, Hernandez LD, Galan JE, Janeway CA Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110(2):191–202. | ||
Piao W, Song C, Chen H, et al. Endotoxin tolerance dysregulates MyD88- and Toll/IL-1R domain-containing adapter inducing IFN-beta-dependent pathways and increases expression of negative regulators of TLR signaling. J Leukoc Biol. 2009;86(4):863–875. | ||
Xiong Y, Qiu F, Piao W, Song C, Wahl LM, Medvedev AE. Endotoxin tolerance impairs IL-1 receptor-associated kinase (IRAK) 4 and TGF-beta-activated kinase 1 activation, K63-linked polyubiquitination and assembly of IRAK1, TNF receptor-associated factor 6, and IkappaB kinase gamma and increases A20 expression. J Biol Chem. 2011;286(10):7905–7916. | ||
Armstrong J, Sargent C, Singh D. Glucocorticoid sensitivity of lipopolysaccharide-stimulated chronic obstructive pulmonary disease alveolar macrophages. Clin Exp Immunol. 2009;158(1):74–83. | ||
Higham A, Lea S, Ray D, Singh D. Corticosteroid effects on COPD alveolar macrophages: dependency on cell culture methodology. J Immunol Methods. 2014;405:144–153. | ||
Kent L, Smyth L, Clayton C, et al. Cigarette smoke extract induced cytokine and chemokine gene expression changes in COPD macrophages. Cytokine. 2008;42(2):205–216. | ||
Kent LM, Fox SM, Farrow SN, Singh D. The effects of dexamethasone on cigarette smoke induced gene expression changes in COPD macrophages. Int Immunopharmacol. 2010;10(1):57–64. | ||
Lea S, Plumb J, Metcalfe H, et al. The effect of peroxisome proliferator-activated receptor-gamma ligands on in vitro and in vivo models of COPD. Eur Respir J. 2014;43(2):409–420. | ||
Kaku Y, Imaoka H, Morimatsu Y, et al. Overexpression of CD163, CD204 and CD206 on alveolar macrophages in the lungs of patients with severe chronic obstructive pulmonary disease. PLoS One. 2014;9(1):e87400. | ||
Vlahos R, Bozinovski S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol. 2014;5:435. | ||
Cullen L, McClean S. Bacterial adaptation during chronic respiratory infections. Pathogens. 2015;4(1):66–89. | ||
Metcalfe HJ, Lea S, Hughes D, Khalaf R, Abbott-Banner K, Singh D. Effects of cigarette smoke on Toll-like receptor (TLR) activation of chronic obstructive pulmonary disease (COPD) macrophages. Clin Exp Immunol. 2014;176(3):461–472. | ||
Nimah M, Zhao B, Denenberg AG, et al. Contribution of MKP-1 regulation of p38 to endotoxin tolerance. Shock. 2005;23(1):80–87. | ||
Dalpke AH, Lehner MD, Hartung T, Heeg K. Differential effects of CpG-DNA in Toll-like receptor-2/-4/-9 tolerance and cross-tolerance. Immunology. 2005;116(2):203–212. | ||
Samet JM, Avila-Tang E, Boffetta P, et al. Lung cancer in never smokers: clinical epidemiology and environmental risk factors. Clin Cancer Res. 2009;15(18):5626–5645. | ||
Yamamoto C, Yoneda T, Yoshikawa M, et al. Airway inflammation in COPD assessed by sputum levels of interleukin-8. Chest. 1997;112(2):505–510. | ||
Caramori G, Adcock IM, Di Stefano A, Chung KF. Cytokine inhibition in the treatment of COPD. Int J Chron Obstruct Pulmon Dis. 2014;9:397–412. | ||
Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 1996;153(2):530–534. | ||
Culpitt SV, Rogers DF, Fenwick PS, et al. Inhibition by red wine extract, resveratrol, of cytokine release by alveolar macrophages in COPD. Thorax. 2003;58(11):942–946. | ||
Soler N, Ewig S, Torres A, Filella X, Gonzalez J, Zaubet A. Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary disease. Eur Respir J. 1999;14(5):1015–1022. | ||
Falvo JV, Tsytsykova AV, Goldfeld AE. Transcriptional control of the TNF gene. Curr Dir Autoimmun. 2010;11:27–60. | ||
Medvedev AE, Sabroe I, Hasday JD, Vogel SN. Tolerance to microbial TLR ligands: molecular mechanisms and relevance to disease. J Endotoxin Res. 2006;12(3):133–150. |
Supplementary materials
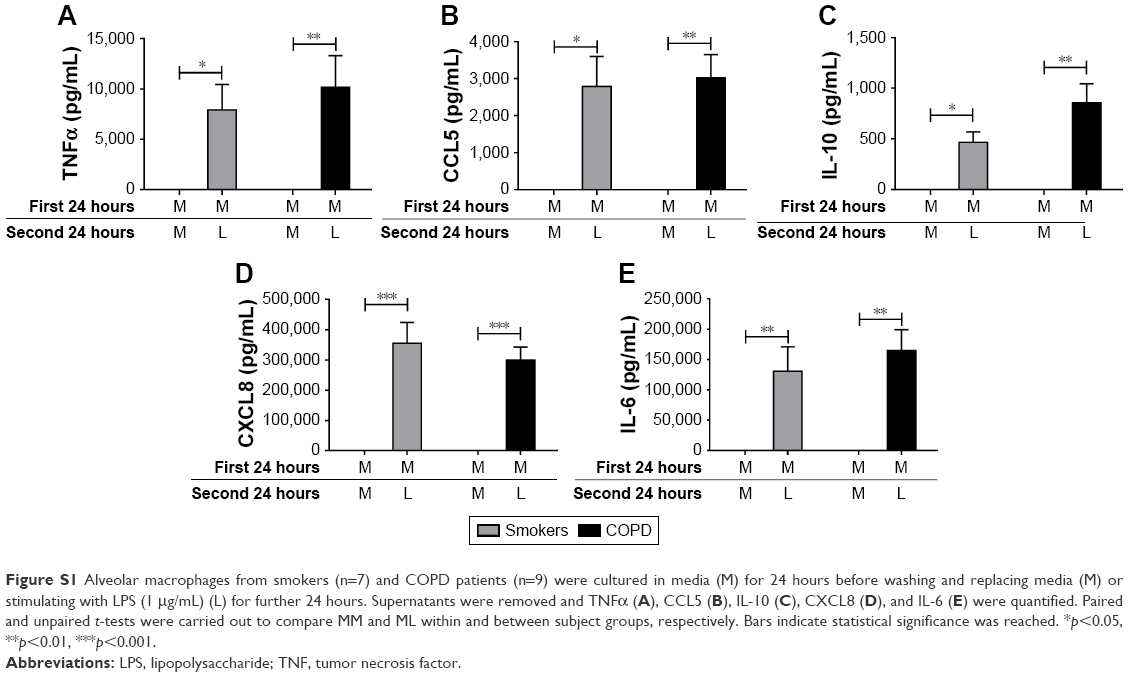 | Figure S1 Alveolar macrophages from smokers (n=7) and COPD patients (n=9) were cultured in media (M) for 24 hours before washing and replacing media (M) or stimulating with LPS (1 μg/mL) (L) for further 24 hours. Supernatants were removed and TNFα (A), CCL5 (B), IL-10 (C), CXCL8 (D), and IL-6 (E) were quantified. Paired and unpaired t-tests were carried out to compare MM and ML within and between subject groups, respectively. Bars indicate statistical significance was reached. *p<0.05, **p<0.01, ***p<0.001. |
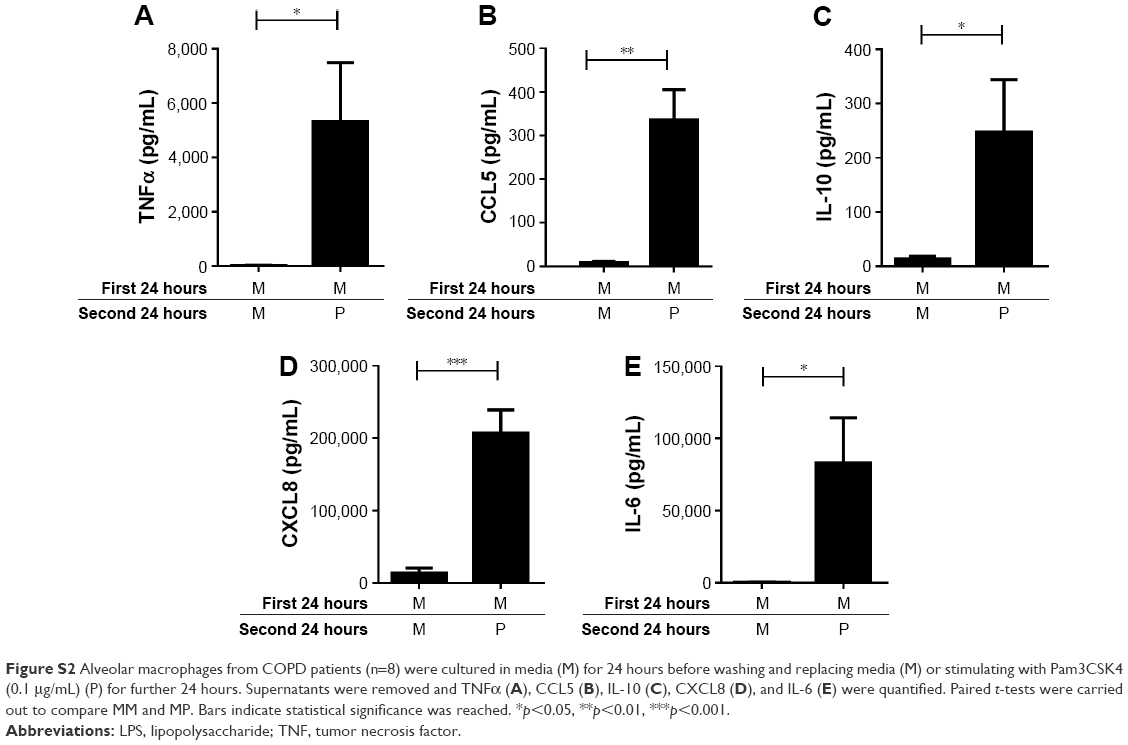 | Figure S2 Alveolar macrophages from COPD patients (n=8) were cultured in media (M) for 24 hours before washing and replacing media (M) or stimulating with Pam3CSK4 (0.1 μg/mL) (P) for further 24 hours. Supernatants were removed and TNFα (A), CCL5 (B), IL-10 (C), CXCL8 (D), and IL-6 (E) were quantified. Paired t-tests were carried out to compare MM and MP. Bars indicate statistical significance was reached. *p<0.05, **p<0.01, ***p<0.001. |
 | Figure S3 The effects of Pam3CSK4 and UPLPS on TLR1 expression. COPD alveolar macrophages were left untreated or stimulated with Pam3CSK4 (0.1 μg/mL) (A) or UPLPS (0.1 μg/mL) (B) (n=5) for 4, 6, 24, and 48 hours. TLR1 gene expression was measured by qPCR and fold change was normalized to GAPDH. Paired t-tests were carried out to compare fold induction to unstimulated time-matched controls. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.