")
Back to Journals » Journal of Inflammation Research » Volume 14
The Effects of Adoptively Transferred IL-23/IL-18-Polarized Neutrophils on Tumor and Collagen-Induced Arthritis in Mice
Authors Chen Y , Li Y, Guo H, Zhang Z, Zhang J, Dong X, Liu Y, Zhuang Y , Zhao Y
Received 15 July 2021
Accepted for publication 4 September 2021
Published 16 September 2021 Volume 2021:14 Pages 4669—4686
DOI https://doi.org/10.2147/JIR.S329528
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Yifang Chen,1,* Yang Li,1,* Han Guo,1,2,* Zhaoqi Zhang,1,2 Jiayu Zhang,1,2 Xue Dong,1 Yi Liu,3 Yuan Zhuang,3 Yong Zhao1,4
1Department of State Key Laboratory of Membrane Biology, Institute of Zoology, Chinese Academy of Sciences, Beijing, People’s Republic of China; 2Savaid Medical School, University of Chinese Academy of Sciences, Beijing, People’s Republic of China; 3Department of Blood Transfusion, First Medical Center of Chinese PLA General Hospital, Beijing, People’s Republic of China; 4Department of State Key Laboratory of Membrane Biology, Institute for Stem Cell and Regeneration, Chinese Academy of Science, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yong Zhao; Yuan Zhuang Email [email protected]; [email protected]
Background: Neutrophils present great diverse phenotypes in various microenvironments and play different immune regulatory functions. Neutrophils generally classified into inflammatory phenotype N1 and anti-informatory phenotype N2. Our recent studies showed that IL-23 alone stimulated neutrophils to express IL-17A, IL-17F and IL-22 and displayed a gene transcriptional profile similar to Th17 cells. In the present study, we tried to identify potential cytokines to promote IL-23-induced neutrophil polarization.
Methods: Mouse bone marrow-derived neutrophils and human peripheral blood neutrophils were treated with IL-23 (10 ng/mL) plus IL-18 (25 ng/mL) to induce Th17-like subset in vitro and detected by real-time PCR, flow cytometry, ELISA, immunofluorescence and RNA-seq assays. In vivo, collagen-induced arthritis (CIA) mouse model and EL4 tumor-bearing mouse model were used to characterize the potential roles of N(IL-23+IL-18) in inflammation and tumor.
Results: Real-time PCR, ELISA and flow cytometry assays showed that IL-18 could significantly enhance IL-23-induced IL-17A, IL-17F and IL-22 expressions in mouse and human neutrophils in a synergistic way, although IL-18 alone failed to induce these cytokines expression. RNA-seq and molecular studies showed that the polarization of N(IL-23+IL-18) is mainly mediated by the JNK/p38-STAT3-BATF signaling pathway. Adoptive transfer of the induced N(IL-23+IL-18) neutrophils significantly accelerated the tumor growth in EL4 tumor-bearing mice and enhanced disease progression in the CIA mouse model. IL-17A-deficient N(IL-23+IL-18) neutrophils failed to enhance the CIA pathogenesis in this model, suggesting that IL-17A may be involved in the N(IL-23+IL-18) neutrophils-promoted arthritis in mice.
Conclusion: The Th17-type subpopulation N(IL-23+IL-18) has pro-tumor and pro-inflammatory properties. Recognizing the different functional polarization of neutrophils would significantly help us to understand the distinctive protective/pathological roles of neutrophils in physiological and different pathological situations.
Keywords: IL-23, IL-18, neutrophils, polarization, tumor, arthritis
Introduction
Neutrophils are the primary defense line against pathogens and represent the main cell type in the innate immunity to regulate the initial inflammation and adaptive immunity.1 Accumulated evidence confirms that neutrophils have potential of functional plasticity and phenotypic diversity.2,3 It is generally recognized that neutrophils have different phenotypes due to the different location of settlement or microenvironments. In tumors, anti-tumor type I neutrophils (N1) display CD49d+CD11b− phenotype and pro-tumor type II neutrophils (N2) with CD49d−CD11b+ phenotype.4 Similarly, N2 can also be detected in both parasite infection and type II immune response.5 Unlike conventional neutrophils (Nc), spleen neutrophils (NBH) are divided into two subtypes, NBH1 and NBH2. CD15 and CD16 were highly expressed on Nc, moderately expressed on NBH1 and lowly expressed on NBH2. NBH1 highly expressed CD86, CD27, CD40L, CD95 and HLA-II, but lowly expressed CD24 compared with NBH2.6 The immature ly6Gint neutrophils that settled in spleens expanded rapidly and migrated out after pneumococcal infection, which reinforces the reserves of effective mature ly6Ghi neutrophils.7 These observations proved that neutrophils possess ability of diversity and plasticity.
Importantly, accumulating evidence indicated that neutrophils could secrete large amount of cytokines, which participate in the occurrence and progression of diseases and regulate the subsequent adaptive immunity.8 It is reported that neutrophils secreted IL-4, IL-5, IL-9 and other cytokines after IL-33 stimulation and significantly promotes the severity of the lung pathogenesis in an ovalbumin (OVA)-induced allergic asthma mouse model.9 Taylor et al suggested that IL-23 and IL-6-induced neutrophils secrete IL-17 and constitutively expressed retinoid-related orphan receptors (ROR)γt and this subset plays an important role in fungal infection model.10 Ramirez-Velazquez et al found that IL17+CD177+ neutrophils were increased in patients with allergic asthma and might play a role in the initial stage or progression of asthma disease by releasing IL-17.11 IL-17+ neutrophils were also found in arachidonic acid-induced psoriasis model as detected by immunohistochemistry (IHC), and suggested that neutrophils were the main effector cells to express IL-17 in this model rather than the traditional CD4+ T cells. Controversially, Tamassia et al detected IL-17+ neutrophils in the skin in healthy people and patients with psoriasis by IHC, but the IL-17 labeling AF-317-NA antibody did not show the corresponding molecular weight position of IL-17, so it was considered that the IL-17+ neutrophils detected by IHC might be a kind of false artifact.8 Importantly, human neutrophils can secrete IL-17 under the treatment of PMA and fMLF,12 ionomycin plus PMA,11 Mycobacterium,13 LPS and pPam3csk4 or L. pneumophila.14 We recently found that IL-23-induced mouse neutrophils secreted large amounts of IL-17A, IL-17F and IL-22, displayed distinctive gene expression profile, and aggravated colitis in mice after adoptive transfer.15 Wild-type p53-induced phosphatase 1 intrinsically controls IL-17 expression in neutrophils in a dextran sulphate sodium-induced colitis mouse model.16 However, the inducing cytokines, regulatory network, phenotypic characteristics and biological significance of the functionally polarized neutrophil subtypes need to be further explored.
In the present study, we identified that the combination of IL-23 plus IL-18 could synergistically promote neutrophils to secrete IL-17A, IL-17F and IL-22, named N(IL-23+IL-18). Adoptive transfer of N(IL-23+IL-18) neutrophils showed pro-tumor and pro-inflammatory functions in tumor-bearing and collagen-induced arthritis (CIA) mouse models, respectively.
Materials and Methods
Mice
Six-14 weeks old male C57BL/6 mice were purchased from SPF Biotechnology Co., Ltd. (Beijing, China). Six-14 weeks old male IL-17-GFP mice, IL-17A-KO mice and C57BL/6-GFP mice were purchased from Biosaitu Bioengineering Co., Ltd. (Beijing, China). All mice were raised in a specific pathogen-free surroundings, temperature 24 ± 2°C, relative humidity 55% ± 15%, 12h light/dark, eating self-selected diets. The number of laboratory animal certificate is SYXK (Jing) 2018–0021). All experiment operations were complying with the Institutional Guidelines for the Care and Use of Laboratory Animals, and approved by The Animal Care and Use Committee, Institute of Zoology, Chinese Academy of Sciences, Beijing, China.
Reagents
Anti-mCD11b-BV510, anti-mLy6G-FITC, anti-mF4/80-APC, anti-mCD45-BUV395, and K2 EDTA blood collection tubes were purchased from BD (San Diego, CA, USA). Immunomagnetic negative selection kits were purchased from Stemcell Technologies (Vancouver, Canada), anti-mCD11b-FITC, anti-mIL-17A-PE was purchased from Biolegend (San Diego, CA, USA), MojosortTM mouse neutral isolation kit purchased from Miltenyi Biotec (Bergisch Gladbach, Germany), BCA assay kit, LPS was purchased from Sigma-Aldrich (St Louis,MO, USA). JNK inhibitor (SP600125), A selective ERK inhibitor (PD98059), NF-ĸB inhibitor and P38 inhibitor (SB203580) was purchased from Cell Signaling Technology (Danvers, MA, USA), JNK activator (Anisomycin) and p38 activator (Dehydrocorydaline) was purchased from MedChemExpress (Monmouth Junction, NJ, USA). PVDF membranes (Millipore, Ternicula, CA, USA). SuperEnhanced chemiluminescence detection reagents were purchased from Applygen Technologies Inc. (Beijing, China). Stat3 (79D7) rabbit mAb, p-stat3 rabbit mAb, BATF rabbit mAb and β-actin mouse mAb was purchased from cell signaling technology (Boston, MA, USA). FICOLL PAQUE PREMIUM was purchased from GE Healthcare (Chicago, USA), Mouse IL-17A ELISA Kit, Mouse IL-17F ELISA Kit and IL-22 ELISA Kits were purchased from Biolegend (San Diego, CA, USA). RmIL-1β, rmIL-6, rmIL-2, rmIL-10, rmIL-12, rmIL-13, rmIL-17A, rmIFN-γ and rmTNF-α was purchased from PeproTech (Rocky Hill, NJ, USA). RmIL-4, rmIL-18, rmIL-21, rmIL-25, rmIL-33, rmIL-23, rmTGF-β1, and rmGM-CSF was purchased from R&D Systems (Minneapolis, MN, USA).
Mouse Bone Marrow Neutrophil Isolation
C57BL/6 mice were anaesthetized and decapitated in an aseptic environment, soak in 75% alcohol and sucked dry with sterile absorbent paper. The femur, tibia and iliac bone were separated and the bone marrow cavity was then washed thoroughly with PBS to obtain bone marrow cells. According to the instructions of mojosortTM mouse neutral isolation kit introduction, non-neutrophil cells were combined with antibodies, respective, then be discarded by magnetic activated cell sorting (MASC) separation system. Purity of neutrophils (CD11b+Ly6G+ cells) obtained with MASC system is >95% as detected by a flow cytometry. We also isolated neutrophils with flow cytometry separation system. The freshly isolated bone marrow cells were incubated with anti-mouse-CD45-BUV395, anti-mouse-CD11b-BV510, anti-mouse Ly6G FITC and anti-mouse-F4/80-APC in dark for 30 min at 4°C, then washing cells with sterile PBS for 2 times. The cells were then sorted by a flow cytometry and were re-suspended in RPMI-1640 complete medium with a density 1 × 108cells/mL. The purities of the sorted CD45+CD11b+Ly6G+F4/80− neutrophils were >99%.
Isolation of Human Peripheral Blood Neutrophils
Blood samples were from two healthy men volunteers (25 and 26 years old, respectively) after giving written informed consent. The study was approved by the ethics committee of the Chinese People’s Liberation Army General Hospital, in accordance with the Declaration of Helsinki. A total of 10 mL peripheral blood was collected from each participant by venipuncture using K2 EDTA blood collection tubes. Neutrophils are isolated by immunological negative selection kit followed by ficoll density gradient centrifugation using FICOLL PAQUE premium and lysis of red blood cells using ACK lysis buffer. The purity of human CD45+CD15+CD16+ neutrophils were >99% as confirmed by a FACSAria Fusion flow cytometry (BD, CA, USA).
Polarization of Neutrophils in vitro
The purified mouse or human neutrophils were adjusted to 1×106 cells/mL and added into 48 well plates. The cells were cultured with cytokines or inhibitors according to the following concentrations:17–24 rmIL-1β (100 ng/mL), rmIL-4 (10 ng/mL), rmIL-6 (50 ng/mL), rmIL-10 (20 ng/mL), rmIL-13 (50 ng/mL), rmIL-21 (50 ng/mL), rmIL-33 (100 ng/mL), LPS (100 ng/mL), rmIFN-γ (100 ng/mL), rmTNF-α (100 ng/mL), rmTGF-β1 (5 ng/mL), rmGM-CSF (40 ng/mL), rmG-CSF (100 ng/mL), rmIL-2 (10 ng/mL). rmIL-12 (5 ng/mL), IL-17A (100 ng/mL), rmIL-18 (25 ng/mL), rmIL-25 (20ng/mL), JNK inhibitor (SP600125, 10 or 20 μM), A selective ERK inhibitor (PD98059, 10 or 20 μM), NF-κB inhibitor (10 or 20 μM), and P38 inhibitor (SB203580, 10 or 20 μM). For the inhibitor assay, neutrophils were pretreated with each inhibitor for 0.5h before various cytokines added.
Immunofluorescent Staining
Neutrophils isolated from IL-17A-GFP reporter mice were cultured in glass bottom dishes with density 1×106 cells/mL, transferred to high speed confocal laser living cells imaging system (Andor Dragonfly, N. Ire, England) to record and take pictures continuously, set 9 observation fields, fluorescence capture thickness is 1 mm, which is divided into 12 layers, The instrument automatically takes fluorescent photos every 10min for 36h. We regard IL-17-GFP+ (Green fluorescence) as IL-17A+ neutrophils.
Real-Time PCR Analysis
Total RNA was extracted with TRIzol, RNA concentration determination with nanodrop 2000 (Thermo Scientific), and total RNA content > 400 ng/sample. cDNA synthesized with oligo-dT and AMV reverse transcription kit according to instructions. Real-time PCR was performed using SYBR Premix Ex Taq on real-time fluorescence quantitative PCR instrument (Roche, Basel, Switzerland), and the information and sequence of general primers reference Table 1. The mRNA expression of each gene calculated with 2 -ΔΔCT calculation, then relative mRNA expression was normalized to the housekeeping gene hypoxanthine guanine phosphoribosyl transferase (HPRT).
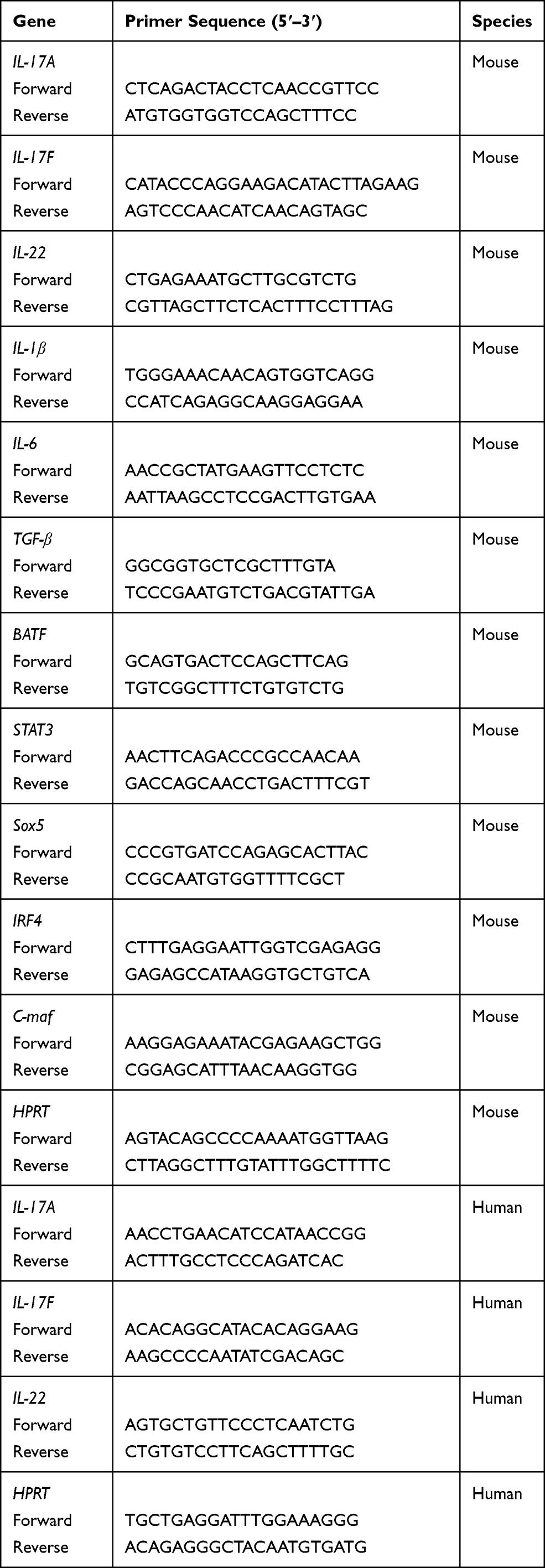 |
Table 1 Primers Used for Real-Time PCR Analysis |
ELISA Assays
ELISA assays were used to detect protein-level expression of cytokines IL-17A, IL-17F, and IL-22. This assay was performed following the manufacturer’s instructions. In short, 50 μL/well cell culture supernatant was added in antibody pre-coated 96-well, next horseradish peroxidase labeled detection antibody was added and culture in 37°C for 60 min, then wash plates 3 times and discard excessive moisture, culture with reactive substrate A and B in 37 oC for 15 min. The reaction was stopped with the stop solution determined absorbance at 450 nm using a microplate reader (Bio-red). Optical density value of each sample was converted into protein concentration by standard curve.
RNA-Seq
Total RNA was isolated by Trizol and the RNA purity was detected to ensure the concentration and integrity of RNA samples meet the quality requirements of transcriptome sequencing.25 Next, we construct the library, that main process is as follows: (1) Enrich eukaryotic mRNA with magnetic beads with Oligo-dT; (2) Random interruption of mRNA by adding fragment buffer; (3) The mRNA as a template to synthesize the first cDNA strand with six-base random hexamers, then the second cDNA strand was synthesized by adding buffer, dNTPs, RNase H and DNA polymerase I. The cDNA was purified by ampere XP beads; (4) The purified double-stranded cDNA is then repaired, A-tailed and connected to the sequencing adapter, and then AMPure XP beads are used for fragment size selection; (5) Finally, the cDNA library was obtained by PCR enrichment. Qubit 2.0 and Agilent 2100 are used to detect the concentration and insert size of the library, and the effective concentration of the library is accurately quantified using the Q-PCR method to ensure the quality of the library. The NovaSeq 6000 is used for high-throughput sequencing, and the sequencing read length is PE150. HISAT2 was used to align the reads to mice genome (mm10) and StringTIe was used to construct transcripts independently for each cell.26 DEseq2 was used to identify differentially expressed genes between each group.27 KEGG pathway enrichment was performed by KOBAS 3.0.28 Protein and protein interaction network were analyzed by String (https://string-db.org/) and visualized by Cytoscape.29
Western Blotting
Inhibitors or activators were added into the neutrophil culture system in which cells were induced with IL-23 or IL-23 plus IL-18 in 24-well plates for 12h. The stimulated neutrophils were washed with PBS and lysed in SDS lysis buffer for 10 min on ice. The concentrations of proteins were determined by BCA assay kit according to the instructions. Protein samples (20μg/test) were analyzed by SDS polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto PVDF membranes, and blocked with TBST-5% non-fat dried milk solution for 2h at room temperature, then incubated with primary antibodies overnight on a shaker at 4 °C. Primary antibodies diluted in TBST-5% non-fat dried milk solution, the dilution ratio as follow: β-actin (1:20,000), p-STAT3 (1:1000), STAT3 (1:1000), BATF (1:1000). PVDF membranes washed with TBST 3 times, and the cultured with HRP-conjugated anti-mouse or anti-rabbit secondary antibody for 2h at room temperature. The protein bands were visualized by enhanced chemiluminescence detection reagents and detected through a Tanon 1600R Gel Image System (Tanon Co., Ltd, Shanghai, China).
Flow Cytometry Assays
Flow cytometry assays were used to analyze mouse GFP+ neutrophils and human IL-17A+ neutrophils. GFP+ neutrophils were stained with anti-mCD45-BUV395, anti-mLy6G-FITC and anti-mCD11b-BV510. Human neutrophils were stained with anti-hCD45-FITC, anti-hCD16-Percp-cy5.5 and anti-hCD15-BV421, culture in dark at 4°C for 30min. Samples were washed by PBS containing 0.1% (w/v) BSA and 0.1% NaN3 for 3 times. For the detection of intracellular cytokines IL-17A, human neutrophils were fixed and permeabilized and stained with anti-hIL-17-PE.30 Flow cytometry data were acquired on a FACSCalibur (BD Biosciences) and analyzed with FlowJo Version 7.6.5 software.
EL4 Tumor Bearing Mouse Model
C57BL/6 mice, 6–8 weeks, the hair of the right leg was shaved with a shaver and skin sterilized with 75% alcohol. EL4 cells suspension density was adjusted to 2×106 cells/mL. 100 μL EL4 cells suspension hypodermics injected with 1 mL sterile injection syringe. 5×106 cells N0, N1, N2 and N(IL-23+IL-18) were adoptively transferred into mice on day 2, 6, and 10, respectively. To measure length, width and height of the tumor by vernier caliper every day, and calculated tumor size and the tumor incidence. The mice were executed on day 17, and the tumor tissue single-cell suspension was prepared.31 The mice were disinfected with 75% alcohol, and the skin was stripped with scissors and forceps to isolate the tumor. Trim the edge of the tumor tissue and break off the adhesive skin. The tumor tissue were cut into tissue particles, and 3mL digestive solution (digestive solution: RPMI-1640, add 2U DNase I and 5mM collagenase) was added to digest in 37°C shaker for 30 min. To stop digestion, sufficient PBS was added. Cells were washed twice and re-suspended with RPMI-1640.
Collagen Induced Arthritis (CIA) Mouse Model
100μL fully emulsified chicken type II collagen solution (2mg/mL) was injected into subcutaneous tissue of 14 weeks or older C57BL/6 mice twice (left and right points) with 1 mL syringe with 4.5 domestic syringe needle.32 On the 21st day of the first injection, the secondary immunization was carried out. The same volume of the above-mentioned chicken type II collagen solution was also used for immunizing mice. Sorted and induced WT N0, WT N(IL-23+IL-18) and IL-17AKO N(IL-23+IL-18) neutrophils (5 × 106 cells per mouse) were injected i.v. into these CIA mice on day 21, 25, 29 and 33. The onset of the disease usually started on about 28–35 days after the first immunization and reached the highest incidence rate of 50–70% to about 42–56 days. Three days before the second immunization, the comprehensive swelling degree and joint function of mice were detected, and clinical scores of mouse limbs were evaluated, and the data were recorded. To take the synovial fluid cells from the articular cavity, the mouse hind limbs were removed, the skin and muscle were stripped, and the synovial fluid in the articular cavity was repeatedly washed with 5 mL PBS, and centrifuged at 1700 rpm and 4°C for 5 min. After centrifugation, cells were sorted.
Statistical Analysis
All repeated experimental data are presented as the Mean ± SD, Two-way ANOVA analysis was used for comparison with SPSS 22.0 software. Student’s unpaired t-test for comparison of means was used to compare between two groups. The non-parametric Mann–Whitney U-test was used to calculated significant difference for multiple group comparison. P ≤ 0.05 was considered to be statistically significant.
Results
IL-18 Synergistically Promotes IL-23-Induced IL-17A, IL-17F and IL-22 Expressions in Mouse Neutrophils
We recently reported that IL-23 could induce IL-17A, IL-17F and IL-22 expression in mouse neutrophils. IL-17A+ neutrophils are present in the colons of dextran sulfate sodium-induced colitis mice and adoptive transfer of syngeneic IL-23-treated neutrophils greatly promoted the pathogenesis in this mouse model.15 To explore whether other cytokines could synergistically increase the expression of Th17-type cytokines in IL-23-treated neutrophils, we treated the freshly isolated neutrophils from mouse bone marrow with different cytokines and LPS combined with IL-23 for 12h and then detected the IL-17A, IL-17F and IL-22 mRNA expressions by real-time PCR. Among the 18 cytokines and LPS studied, IL-18 significantly promoted IL-23-induced IL-17A, IL-17F and IL-22 expression (P<0.01, Figure 1A), while other cytokines somewhat enhanced or decreased IL-17A, IL-17F and IL-22 expression compared with IL-23-treated neutrophils. In the following different doses of IL-23 and IL-18 stimulation assays, as low as 5ng/mL IL-23 could induce IL-17A, IL-17F and IL-22 expression, 10ng/mL or above of IL-23 showed synergistic effects with IL-18 to increase IL-17A, IL-17F and IL-22 expressions (P<0.01, Figure 1B). However, even high doses of IL-18 failed to induce IL-17A, IL-17F and IL-22 expression in neutrophils (Figure 1C). It is true that IL-18 increased IL-23-induced IL-17A, IL-17F and IL-22 expression (P<0.01, Figure 1C). Therefore, IL-18 can significantly promote IL-23-induced IL-17A, IL-17F and IL-22 expressions in neutrophils. The optimal dosages of IL-23 and IL-18 are 25 ng/mL and 10 ng/mL, respectively. The IL-23 plus IL-18 stimulated neutrophils were named as N(IL-23+IL-18) in the afterward.
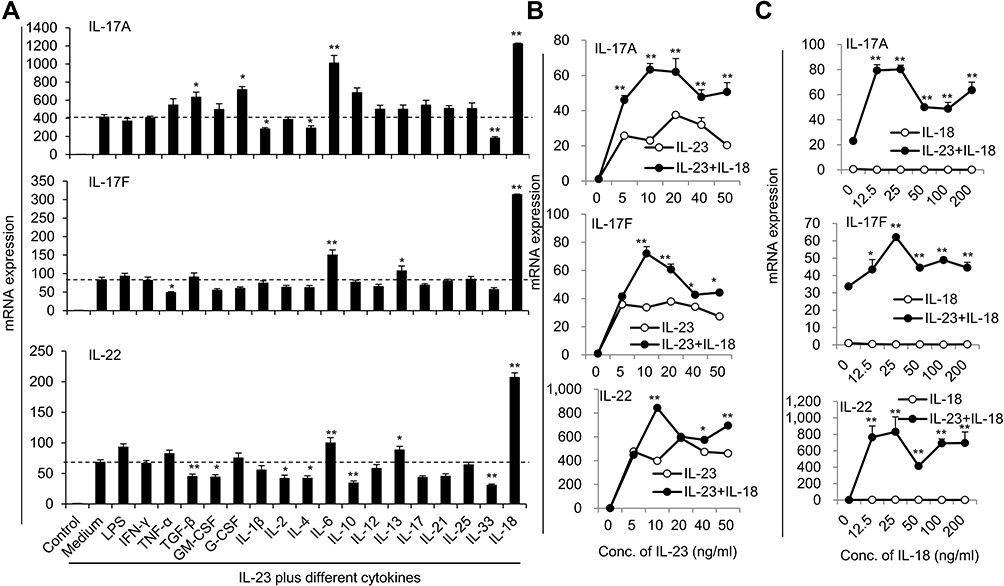 |
Figure 1 The expressions of IL-17A, IL-17F and IL-22 in neutrophils after stimulation with different cytokines combined with IL-23. (A) Freshly isolated bone marrow neutrophils treated with various cytokines plus 50ng/mL IL-23. The mRNA expression levels of Th17-type cytokines IL-17A, IL-17F and IL-22 were determined by real-time PCR. (B) Real-time PCR analysis of IL-17A, IL-17F and IL-22 mRNA expression in neutrophils treated with IL-18 (25 ng/mL) plus different concentrations of IL-23 (0–50 ng/mL) for 24 hr. (C) Real-time PCR analysis of IL-17A, IL-17F and IL-22 mRNA expression in neutrophils treated with IL-23 (10 ng/mL) plus different concentrations of IL-18 (0–200 ng/mL) for 24 h. All data were obtained from the average of repeated samples and shown as Mean±SD (n=3), *P<0.05, **P<0.01 compared with the control group or IL-23 or IL-18 alone treated cells. |
We also observed the expression kinetics of IL-17A, IL-17F and IL-22 in IL-23/IL-18-induced neutrophils. The results suggested that IL-23 plus IL-18 stimulated IL-17A, IL-17F and IL-22 expression by 6h after stimulation and reached a peak around 12h after stimulation as determined by real-time PCR (P<0.01, Figure 2A). Even importantly, certain levels of IL-17A, IL-17F and IL-22 in the cell culture supernatant of IL-23-stimulated neutrophils are detectable in a time-dependent manner as detected by ELISA assays. Significantly higher levels of IL-17A, IL-17F and IL-22 were observed in the cell culture supernatant of IL-23 plus IL-18 stimulated neutrophils compared with those of IL-23 alone treated neutrophils (P<0.01, Figure 2B). Furthermore, IL-18 or IL-18 plus IL-23 induced more IL-1β, IL-6 and TGF-β expression, but IL-23 did not do so (Figure 2C), which is in a line with the reported IL-18 function.33
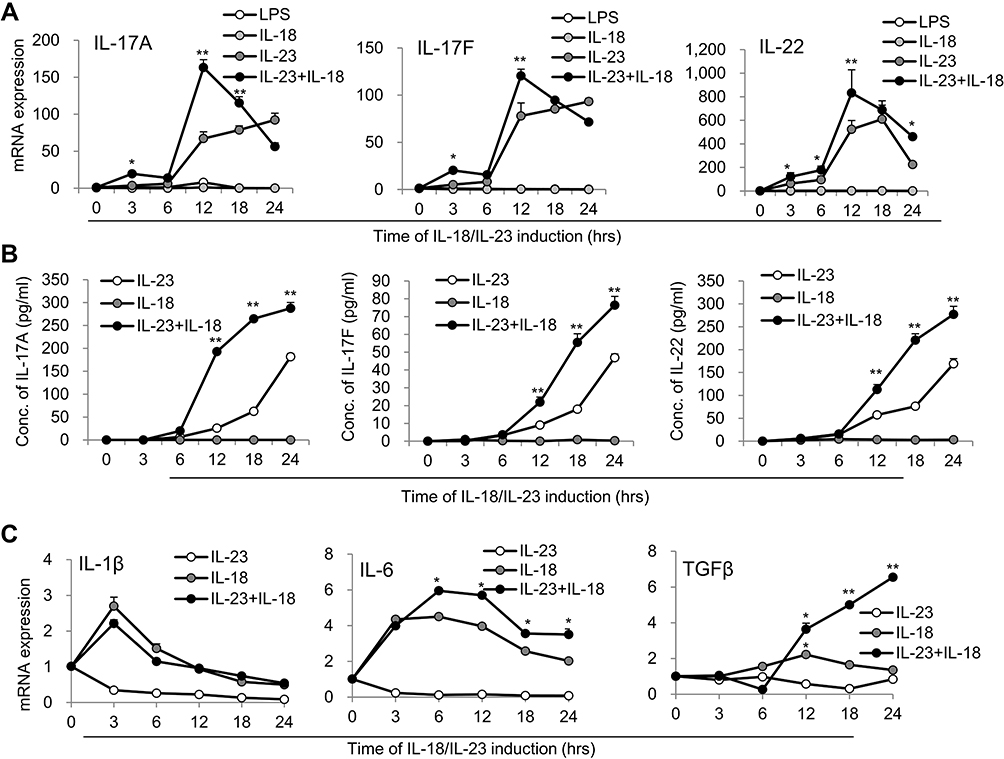 |
Figure 2 Time kinetics of cytokine expression in N(IL-23+IL-18). (A) Neutrophils treated with LPS (100 ng/mL), IL-23 (10 ng/mL), IL-18 (25 ng/mL) and IL-23 (10 ng/mL) plus IL-18 (25 ng/mL) for different time points (0–24h). Real-time PCR analysis of IL-17A, IL-17F and IL-22 mRNA expression in neutrophils were performed. (B) ELISA analysis of IL-17A, IL-17F and IL-22 protein levels in the supernatant of the treated neutrophils for different time (0–24h). (C) Real-time PCR analysis of IL-1β, IL-6 and TGF-β mRNA expression in neutrophils treated with IL-23 (10 ng/mL) and/or IL-18 (25 ng/mL) for different time (0–24h). All data were shown as Mean±SD (n=3), *P<0.05, **P<0.01 compared with IL-18 alone treatment group. |
To further exclude the potential impacts of the contaminated other cells on the cytokine expression of the treated neutrophils, we sorted the highly purified neutrophils (CD11b+ly6G+ cells, purity > 99%) from bone marrow used fluorescence-activated cell sorting system and treated these cells with IL-23 plus IL-18. Identical results were also observed that IL-18 promote IL-23-induced IL-17A, IL-17F and IL-22 expression in mRNA and protein levels by sorted neutrophils as detected by real-time PCR and ELISA assays (P<0.01, Figure 3A and B). In addition, we separated freshly resting bone marrow neutrophils from IL-17-GFP reporter mice, and the fluorescence intensity of GFP signal (indicating the synthesis of IL-17A) in cells was dynamically detected from 0–36h by high-speed laser confocal platform. Results showed that few green signal were detectable in freshly isolate neutrophils at 0h, but IL-17-GFP+ neutrophils gradually accumulated and increased with the IL-23 plus IL-18 stimulation (Figure 3C). Thus, all these results collectively suggested that N(IL-23+IL-18) express Th17-type cytokines in mRNA and protein levels in time- and dose-dependent manners.
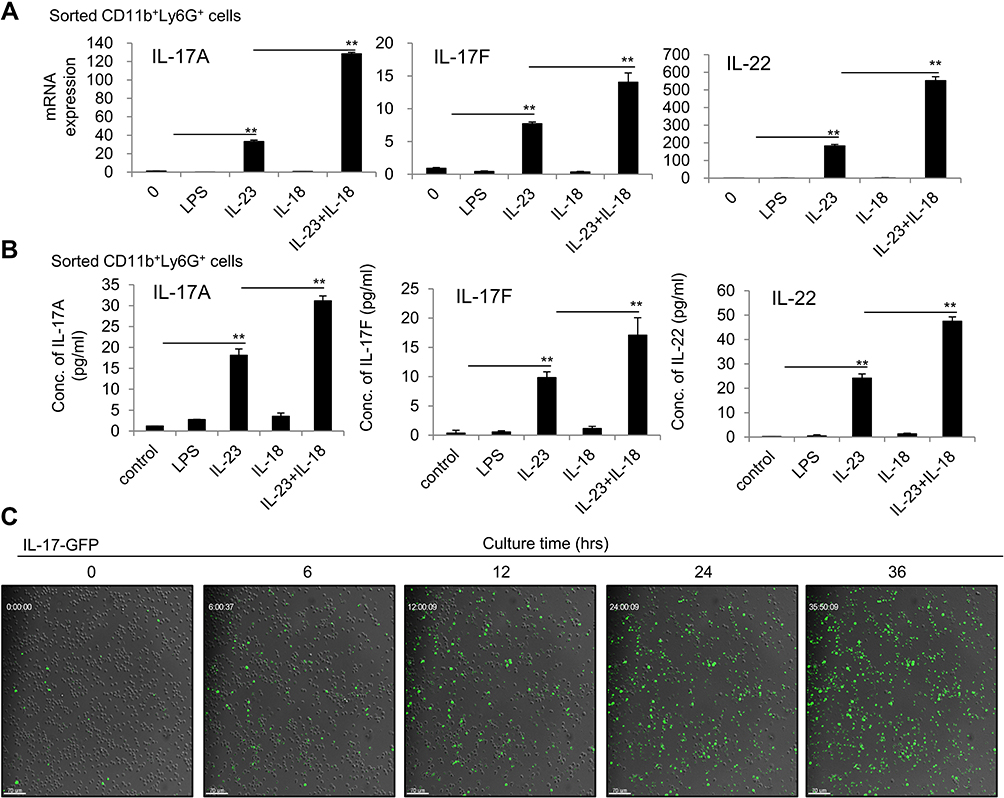 |
Figure 3 IL-23 plus IL-18 stimulate the highly purified mouse neutrophils to express IL-17A, IL-17F and IL-22. (A) The sorted mouse bone marrow neutrophils by a flow cytometry were treated with IL-23 (10 ng/mL) and/or IL-18 (25 ng/mL) for 24h. Real-time PCR analysis of IL-17A, IL-17F and IL-22 mRNA expressions in neutrophils. (B) Flow cytometry sorted mouse bone marrow neutrophils were treated with IL-23 (10 ng/mL) and/or IL-18 (25 ng/mL) for 24h. ELISA analysis of IL-17A, IL-17F and IL-22 protein levels in the supernatant of the treated neutrophils. (C) The kinetics of IL-17A-GFP expression in neutrophils treated with IL-23 (10 ng/mL) and IL-18 (25 ng/mL). All data were shown as Mean±SD (n=3), **P<0.01 compared with control group. |
IL-23 Plus IL-18 Induced Human Neutrophils to Express Th17-Type Cytokines
To verify whether IL-23 plus IL-18 can induce Th17-type cytokine expression in human neutrophils, we treated the isolated human peripheral blood neutrophils with IL-23 and/or IL-18. Indeed, we observed similar polarization characteristics in human peripheral blood neutrophils, IL-23 alone could induce significant levels of IL-17A, IL-17F and IL-22 expressions in human neutrophils compared with medium or LPS-stimulated cells (P<0.01, Figure 4A), but IL-18 alone did not show detectably increased expression of these cytokines. However, the co-stimulation of IL-23 and IL-18 significantly increased IL-17A, IL-17F and IL-22 expressions in human neutrophils compared with IL-23-treated neutrophils as detected by real-time PCR (P<0.01, Figure 4A), indicated that IL-18 had the ability to promote IL-23-induced Th17-type cytokines expression in human neutrophils. The IL-17A expression in IL-23 and IL-18-treated human neutrophils was further confirmed by IL-17A protein expression as detected by a flow cytometry (Figure 4B). Thus, IL-23 plus IL-18 could significantly induce human neutrophil functional polarization to express Th17-type cytokines.
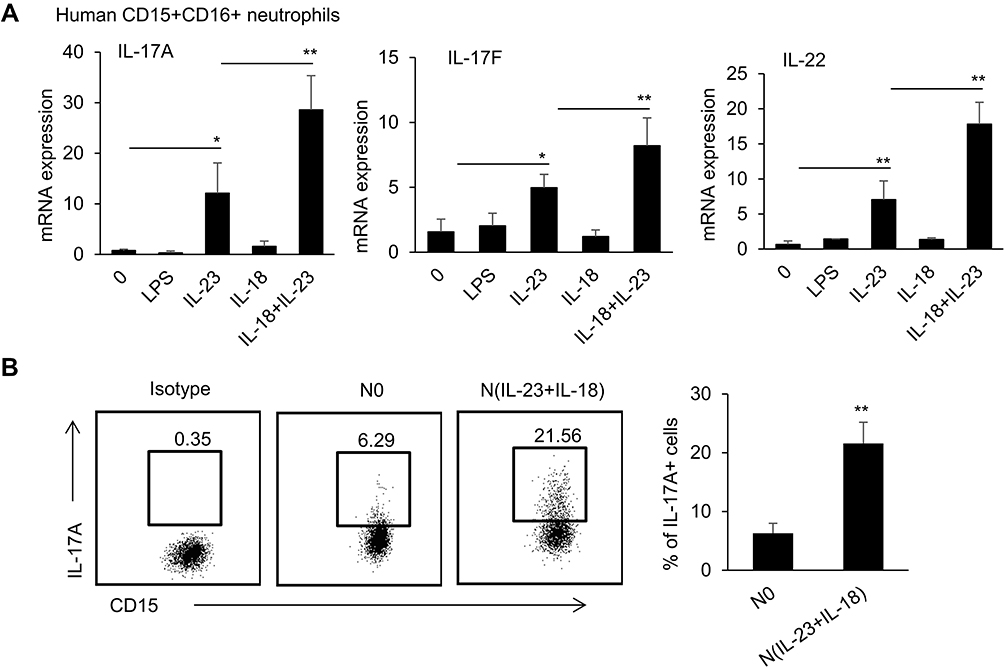 |
Figure 4 IL-23 plus IL-18 stimulate human peripheral neutrophils to express IL-17A, IL-17F and IL-22. (A) The sorted human peripheral blood neutrophils were treated with IL-23 (10 ng/mL) and/or IL-18 (25 ng/mL) for 12h. Real-time PCR analysis of IL-17A, IL-17F and IL-22 mRNA expressions in neutrophils was performed. (B) Flow cytometry analysis CD15+CD16+IL-17A+ neutrophils after the sorted human peripheral blood neutrophils were treated with IL-23 (10 ng/mL) and IL-18 (25 ng/mL) for 12h. All data were shown as Mean±SD (n=3). Experiments were repeated twice with identical results. *P<0.05, **P<0.01 compared with control group. |
IL-23 Plus IL-18 Induce Neutrophils to Express a Distinct Gene Transcriptional Profile
To see the gene transcriptional characteristics of N(IL-23+IL-18), we performed RNA-seq assays. The distance of N0, N(IL-18), N(IL-23) and N(IL-23+IL-18) were analyzed using principal component analysis (PCA) in three dimensions (Figure 5A). The PCA clustering reflects that N(IL-23) and N0 were closer while N(IL-18) and N(IL-23+IL-18) were closer. Based on P value < 0.05 and | fold change | > 2 conditions, we compared the feature genes of these cells compared with N0 control. The N(IL-18+IL-23) showed the characteristics of gene expression changes in both N(IL-23) and N(IL-18) (Figure 5B). KEGG pathway enrichment analysis showed that cell activation-related pathways were significantly up-regulated in neutrophils after stimulation with IL-23 and/or IL-18, in which that the metabolic pathways were enriched (Figure 5C). It is proposed that metabolic pathways and cytokine-cytokine receptor interaction pathways were crucial for Th17-type polarization. We analyzed the cytokine expression profiles of differently treated cells (Figure 5D). The result showed that Th17-type cytokines IL-17A, IL-17F and IL-22 were specifically expressed in N(IL-23) and N(IL-23+IL-18) but not in N0 and N(IL-18) cells, which indicated that IL-23 was crucial for neutrophils to polarize to Th17-type like cells and IL-18 displayed synergism with IL-23 to promote IL-17A, IL-17F and IL-22 expression (Figure 5D). In addition, more pro-inflammatory cytokine expressions in N(IL-23+IL-18) such as IL-6, IL-1f9, IL-1β, IL-12a, csf1, csf2, csf3 compared with N0 and N(IL-23) (Figure 5D). Moreover, the chemokine expression profile was also fully analyzed, N(IL-18+IL-23) highly expressed chemokines such as CCL9, CCL24, CXCL9 and CCL22, which was similar as N(IL-18) but was significantly higher than those in N0 and N(IL-23) (Figure 5E). Meanwhile, we tried to analyze the transcription factor changes in order to find the key factors that regulate the expression of these cytokines (Figure 5F). We downloaded the T transcription factors list from animal TFDB database for our analysis. We observed that IL-23 stimulation can only induce the activation of a few of transcription factors such as RORC, PPARGC1A, HOXA1, etc., but the combination of IL-23 and IL-18 can induce up-regulation of a cluster of transcription factors, suggesting that the synergistic effect of IL-18 with IL-23 may be generated through a wider range of transcriptional regulation.
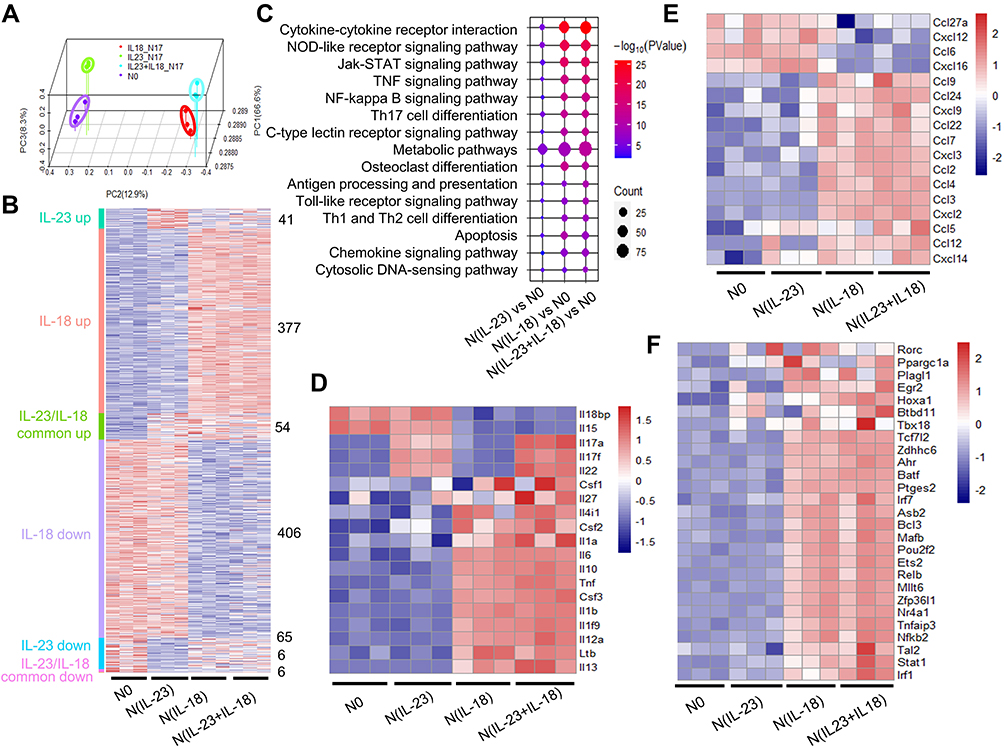 |
Figure 5 Gene transcriptional expression characteristics in IL-23 and IL-18-treated mouse neutrophils. (A) Principal component analysis of the four groups including neutrophils treated with none, IL-23 (10 ng/mL) and/or IL-18 (25 ng/mL). All expressed genes were used for the analysis. Each group contains three parallel samples labeled in the same color. (B) Heatmap of the whole gene expression profile in four group. Color and number represented that the genes obtained from the comparison of differential expressed genes between IL-23 group and IL-18 group. (C) bubble plot of KEGG pathway enrichment analysis. (D) Heatmap of the significantly changed cytokines in N(IL-23+IL-18). (E) Heatmap of the significantly changed chemokines in N(IL-23+IL-18). (F) Heatmap of the significantly changed transcriptional factors in N(IL-23+IL-18). |
We then used cellular signaling pathway network analysis to find pathways and regulatory molecules that regulate the expression of cytokines in N(IL-23+IL-18) (Figure 6A). We performed KEGG pathway enrichment analysis on the differential genes of N(IL-18+IL-23) vs N0 based on P-value<0.05 and obtained 200 up-regulated pathways. We then analyzed the pathways related to cytokine secretion in the top 30% up-regulated pathways, and found HIF-1 pathway, Th17 pathway, Jak-stat pathway, NLR pathway, TNF pathway and Pi3k-akt pathways significantly regulate the cytokines secretion of N(IL-18+IL-23) (Figure 6A). Subsequently, we use transcription factor network analysis to find the changes in transcription factors that regulate cytokine secretion pathways in N(IL-23+IL-18) (Figure 6B). We found that RORC and BATF was the top two transcription factors with the highest LogFC changes, which may play a crucial role in secretion of Th17-type cytokines in N(IL-23+IL-18) (Figure 6B). Furthermore, we analyzed the regulatory network of these transcription factors on upstream and downstream genes, and found that Irf1 was a significant transcription factor regulated by IL-18, which regulates pro-inflammatory cytokines expression, such as IL-12a, IL-12b and IL-18, and the transcription factor BATF and RORC regulated Th17-type gene expression in N(IL-23+IL-18), such as IL-23R, IL-17A, IL-17F and IL-21 (Figure 6C). This analysis indicated that N(IL-23+IL-18) neutrophils present a unique transcriptional profile, which induce neutrophil polarization into Th17-like cells.
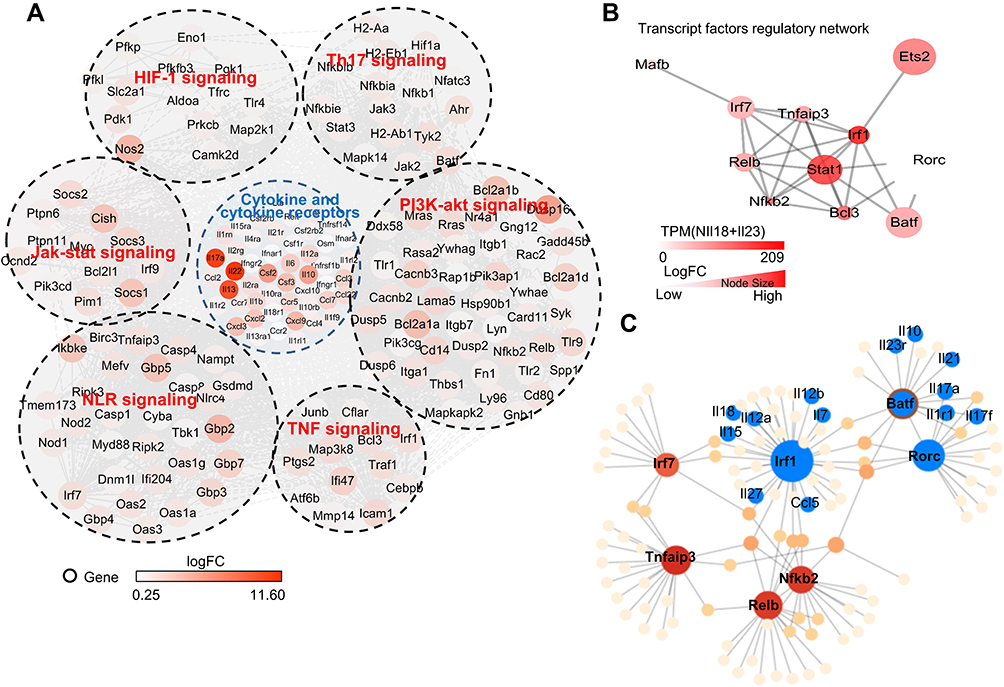 |
Figure 6 Transcription characteristics of IL-23 and IL-18 stimulated mouse neutrophils. (A) Network of cytokines and KEGG signaling regulating pathways of mouse N(IL-23+IL-18) vs N0. The color represented the Log Fold Change of differential expressing genes. (B) Network of up-regulated transcription factors in N(IL-23+IL-18). The color and size represented the TPM value and Log Fold Change respectively. (C) Transcription factors and genes regulatory network in mouse N(IL-23+IL-18). |
JNK/P38-STAT3-BATF Pathways Mediate N(IL-23+IL-18) Polarization
In order to determine the molecular mechanisms for IL-23 and IL-18-promoted Th17-type cytokine expression in neutrophils, we detected transcription factors and signaling pathways related to Th17-type polarization. Combined with the mentioned above RNA-Seq results, transcription factors BATF, STAT3, SOX5, IRF4 and c-Maf related to IL-17A expression were detected by real-time PCR. As expected, BATF, STAT3, SOX5, IRF4 and c-Maf in N(IL-23+IL-18) were significantly increased in a time-dependent manner (Figure 7A). JNK, p38, ERK and NF-κB signaling pathways are recognized as the downstream signaling pathway of IL-23 and/or IL-18,15,33 thus, we give different doses of JNK inhibitor (SP600125), p38 inhibitor (SB203580), ERK inhibitor (PD98059) and NF-κB inhibitor to verify the key pathway of IL-18 cooperating with IL-23 to regulate neutrophil polarization. Polarized phenotype of N(IL-23+IL-18) was significant inhibited by JNK and p38 inhibitors in a dose-dependent manner (Figure 7B and C, Supplement Figure 1A), but NF-κB inhibitor and ERK inhibitor (PD98059) treatment had no significant effect on the expressions of Th17-type cytokines by neutrophils even in high doses (Supplement Figure 2A and B). On the other hand, we give different doses of JNK activator (Anisomycin) and p38 activator (Dehydrocorydaline) in the N(IL-23+IL-18) induction system and found that these activators enhanced the IL-17A, IL-17F, IL-22, STAT3 and BATF expression as detected by real-time PCR (Figure 7D and E) and the IL-17A protein secretion as detected by ELISA assays (Supplement Figure 1B). Meanwhile, the inhibitors of JNK and p38 decreased transcription factor STAT3 and BATF protein expression and p-STAT3 expression (Figure 7F). In contrast, the activators of JNK and p38 enhanced transcription factor STAT3 and BATF protein expression and p-STAT3 expression (Figure 7G), supporting that JNK/p38 pathway regulated STAT3 expression and activation as well as BATF expression in neutrophils. Therefore, these data collectively indicate that JNK/P38-STAT3-BATF signaling pathway is likely taking the major role in the neutrophil polarization to Th17-type phenotype.
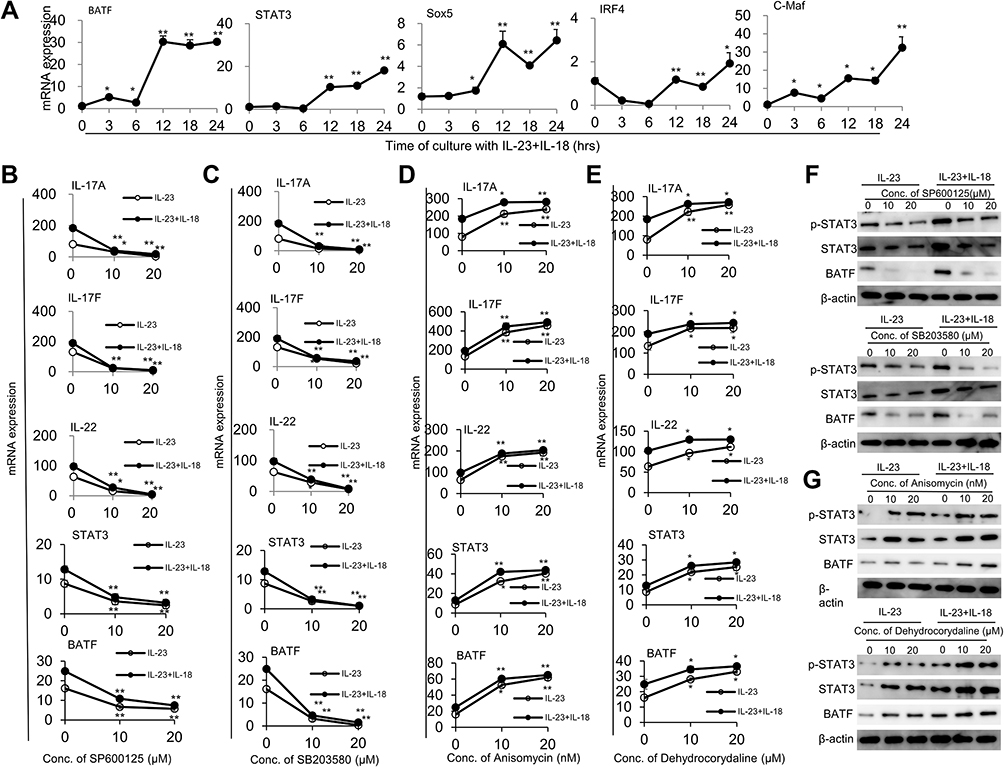 |
Figure 7 IL-17A, IL-17F and IL-22 expression in mouse N(IL-23+IL-18) cells are mediated by JNK/P38-STAT3-BATF signaling pathways. (A) The mRNA expression levels of BATF, STAT3, Sox5, IRF4, and C-Maf in mouse neutrophils treated with IL-23 (10 ng/mL) and IL-18 (25 ng/mL) for different time course were determined by real-time PCR. (B) N(IL-23+IL-18) treated with two concentrations of JNK inhibitor (SP600125) for 12h, real-time PCR was used to analysis IL-17A, IL-17F, IL-22, STAT3 and BATF mRNA expression levels. (C) N(IL-23+IL-18) treated with two concentrations of P38 inhibitor (SB203580) for 12h, real-time PCR was used to analysis IL-17A, IL-17F, IL-22, STAT3 and BATF mRNA expression. (D) Mouse N(IL-23+IL-18) neutrophils treated with two concentrations of JNK activator (Anisomycin) for 12h, real-time PCR was used to analyze IL-17A, IL-17F, IL-22, STAT3 and BATF mRNA expression. (E) Mouse N(IL-23+IL-18) neutrophils treated with two concentrations of p38 activator (Dehydrocorydaline) for 12h, real-time PCR was used to analysis IL-17A, IL-17F, IL-22, STAT3 and BATF mRNA expression. (F) Western blot assays were used to detect p-STAT3, STAT3, and BATF expression in mouse N(IL-23+IL-18) neutrophils treated with JNK/p38 inhibitors. (G) Western blot assays were used to detect p-STAT3, STAT3, and BATF expression in mouse N(IL-23+IL-18) neutrophils treated with JNK or p38 activator. All data were shown as Mean±SD (n=3), *P<0.05, **P<0.01 compared with control group. |
Pro-Tumor Effect of N(IL-23+IL-18) in EL4 Tumor-Bearing Mice
Different neutrophil subtypes play different roles in tumor progression, as anti-tumor N1 neutrophils perform negative roles on tumor pathogenesis, but tumor-promoting N2 present a positive effect on tumor growth.34 To identify the role of N(IL-23+IL-17) in the tumor pathogenesis, we adoptively transferred syngeneic N0, N1, N2 and N(IL-23+IL-18) into the EL4 lymphoma-bearing mice. Adoptive transfer of N1 neutrophils showed somehow inhibitory effects on tumor growth as indicated by the decreased tumor size, incidence, weight and cell number compared with those in mice received none or N0 cells (P<0.05, Figure 8A–F). In contrast, N(IL-23+IL-18) neutrophils showed promoting tumor growth effects as N2 cells as indicated by the increased tumor size, incidence, weight and cell number compared with those in mice received none, N0 or N1 cells (P<0.01, Figure 8A–F). By the end of the experiments, we detected the mRNA expressions of IL-17A, Il-17F, and IL-22 in tumor by real-time PCR. The expressions of IL-17A, Il-17F, and IL-22 were significantly up-regulated in tumor of mice received N(IL-23+IL-18) than tumor in mice received N0, N1 and no neutrophils (P<0.01, Figure 8G). Furthermore, we further explore whether the injected N(IL-23+IL-18) neutrophils infiltrated into tumor tissue to promote the pathogenesis of tumor by using N(IL-23+IL-18) neutrophils of C57BL/6-GFP mice. We detected the injected GFP+ neutrophils in mouse spleen, bone marrow, peripheral blood, lymph nodes and tumor by a flow cytometry at 0h, 4h, 12h and 24h after adoptive transfer of GFP+N(IL-23+IL-18) neutrophils. Certain levels of these GFP+ cells can be detected in tumor, spleen, bone marrow, peripheral blood and lymph nodes even as late as 24h after cell injection (Figure 8H). The adoptively transferred N(IL-23+IL-18) can survive and infiltrate into the tumor and immune system for at least 24h in mice.
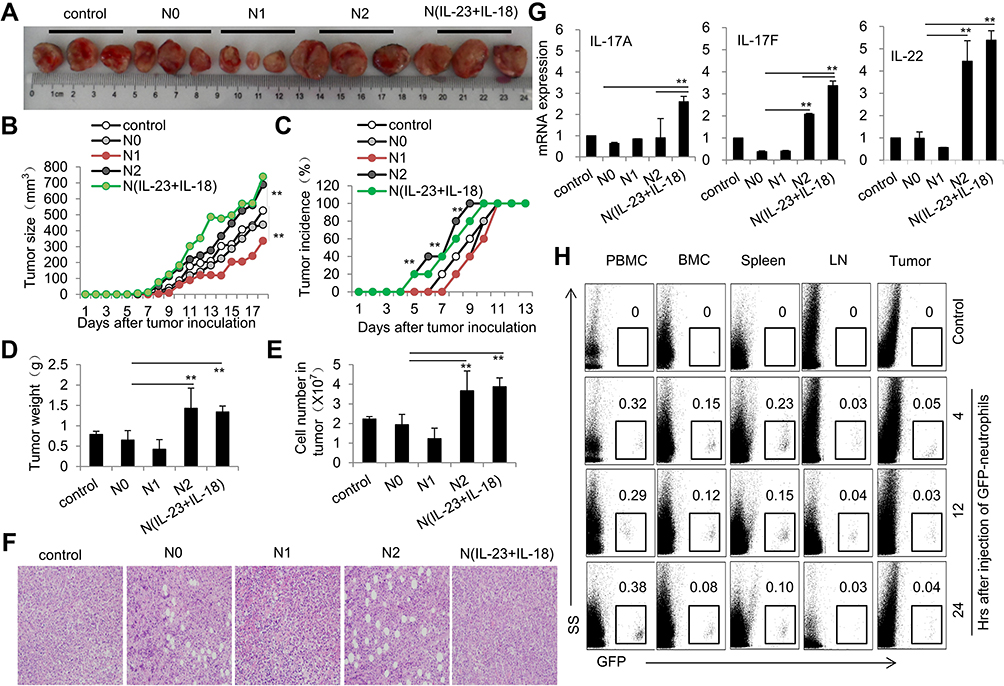 |
Figure 8 Adoptive transfer of N(IL-23+IL-18) promotes EL4 tumor growth in mice. (A) Adoptive transfer of medium, N0, N1, N2, and N(IL-23+IL-18) neutrophils (sorted CD11b+Ly6G+ neutrophils) to EL4 tumor-bearing mice on day 2, 6 and 10, respectively. Representative pictures of tumor on day 18. (B) Tumor size. (C) Tumor incidence. (D) Tumor weight. (E) Cell number in tumor. (F) HE staining of tumor tissue. (G) Real-time PCR analysis of IL-17A, IL-17F and IL-22 mRNA expressions in tumor tissue on day 17. (H) GFP+N(IL-23+IL-18) neutrophils were i.v. injected into syngeneic mice which were seeded EL4 tumor cells 10 days earlier. Flow cytometric analysis of the percentage of GFP+ neutrophils in PBMC, BMC, spleen, LNs and tumor at different times. All data were shown as Mean±SD, n=5, **P<0.01 compared with control group. |
Pro-Inflammatory Role of N(IL-23+IL-18) in CIA Mice
It is reported that IL-23 and IL-17 family cytokines are critical in the pathogenesis of CIA.35 We employed a chicken type II collagen-induced CIA mouse model, in which Th17-type cytokines, such as IL-17A and IL-17F, were significantly involved.36 We adoptively transferred bone marrow-derived N0, N(IL-23+IL-18) from either C57BL/6 mice or IL-17-KO mice into syngeneic recipient CIA mice, which aim to explore the role of N(IL-23+IL-18) in the pathogenesis of CIA and to verify whether the role of N(IL-23+IL-18) in CIA pathology is related to the secretion of IL-17A. The results showed that mice received WT-N (IL-23+IL-18) displayed severer CIA symptoms compared with mice received none or WT-N0 cells as indicated by the earlier occurrence, and higher incidence, increased arthritis score and hind paw thickness (P<0.01, Figure 9A–D). Meanwhile, mice received IL-17A-KO-N (IL-23+IL-18) displayed slower CIA incidence, lower arthritis score and decreased hind paw thickness than those in mice received WT-N (IL-23+IL-18) (P<0.01, Figure 9A–D), suggesting that IL-17A may be involved in the CIA-promoting effect of N(IL-23+IL-18) in this model. We also observed that more seriously synovial hyperplasia, cell infiltration, inflammation, bone erosion and joint ring breaking in the mice received WT-N (IL-23+IL-18) compared with control and N0 group, and those pathological indexes in the mice received IL-17A-KO-N (IL-23+IL-18) were less than those mice received WT-N (IL-23+IL-18) (Figure 9E). Consistently, the mRNA expressions of IL-17A, IL-17F, IL-22, IL-1β and TNF-α in synovial fluid cells of mice received WT-N (IL-23+IL-18) showed significantly higher than those in mice received none, N0, and IL-17A-KO-N (IL-23+IL-18), respectively, as detected by real-time PCR assays (P<0.01, Figure 9F). These results together indicated that adoptive transfer of N(IL-23+IL-18) neutrophils promoted severe pro-inflammatory response and CIA pathogenesis and IL-17A may be involved in this process.
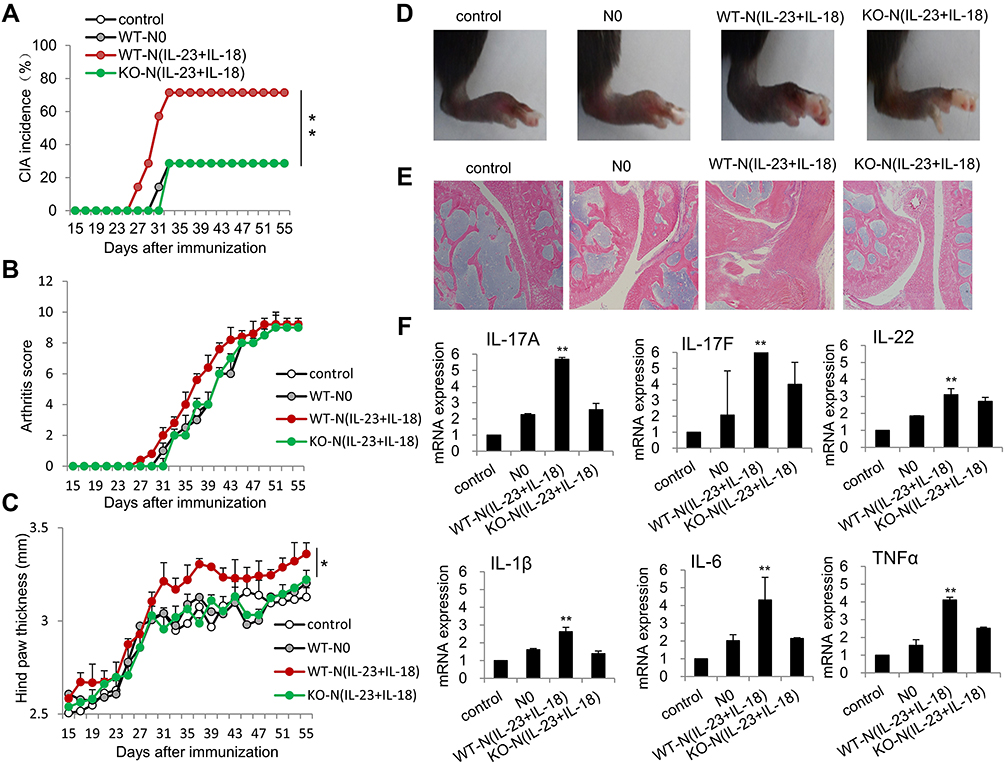 |
Figure 9 Adoptive transfer of N(IL-23+IL-18) promotes pathogenesis in the CIA mice. (A) Adoptive transfer of medium, WT-N0, WT-N (IL-23+IL-18), IL-17A-KO-N (IL-23+IL-18) (sorted CD11b+Ly6G+ neutrophils) to CIA mice, respectively, on day 21, 25, 29 and 33. CIA incidence were shown. All mice were sacrificed on day 56 and the following experiments were performed. (B) Arthritis score. (C) Hind paw thickness. (D) Representative pictures. (E) HE staining of the joint tissues. (F) The mRNA expression of IL-17A, IL-17F, IL-22, IL-1β, IL-6 and TNF-α in synovial fluid cells of mice. All data were shown as Mean±SD, n=7, *P<0.05, **P<0.01 compared with control group. |
Discussion
In this study, we found that IL-23 plus IL-18 can efficiently induce a group of neutrophils to express the Th17-type cytokine profile, providing new evidence for the existence of more wide and diverse polarization forms of neutrophils. Th17 family cytokines including IL-17 and IL-22 in Th cells are mainly induced by IL-23 and IL-6 stimulation.37 Increasing evidences showed that Th17-like cytokines are not only found in Th17 but also in many immune cells in different disease models.38 Th17 family cytokines are also synthesized and secreted in innate immune cells.39,40 We have confirmed our previous study that IL-23 alone can induce neutrophils to express IL-17A, IL-17F, and IL-22. In this study, after screening 17 cytokines, we found that IL-18 can cooperate with IL-23 to stimulate neutrophils to highly express IL-17A, IL-17F and IL-22. Importantly, IL-23 and IL-18 co-stimulation can induce high levels of IL-17A, IL-17F and IL-22 in human neutrophils.
IL-23 belongs to IL-12 cytokine family, which is related to the terminal differentiation of Th17 cells, promotes Th17 cell proliferation and effector functions, and stimulates activated T cells to secrete IL-22, but IL-23 alone could not induce naive CD4+ T cells differentiate into Th17 cells.41,42 The addition of TGF-β and IL-6 at the stage of T cell recognizing antigens promotes the differentiation of Th17 cells and the secretion of IL-17A and IL-17F.43 This phenomenon is different in innate immune cells. IL-23 alone induced IL-17A, IL-17F and IL-22 expression in mouse and human neutrophils, but IL-18 could not. However, IL-18 significantly promoted the expression of these pro-inflammatory cytokines in IL-23-treated neutrophils in a synergistic manner with IL-23. IL-23 activates JAK in T cells, leading to phosphorylation and activation of STAT3, promoting the transcription of IL-23R and Rorc, and stabilizing the expression of pro-inflammatory cytokines IL-17A, IL-17F, IL-22 and CSF2. RORγT, IRF7, IRF4, BATF, Sox5 and C-maf are the key transcription factors that induce Th17 cells. Our results showed that IL-23 induced Th17 cytokines expression in neutrophils through the classic STAT3-RORγT/BATF pathway. The synergistic effects of IL-18 on IL-23-induced IL-17A, IL-17F and IL-22 expressions are dependent on the JNK and P38 signaling pathways but not on the ERK and NF-κB signaling pathways.
The Th17 cytokines play an important role in the occurrence and development of tumors and many immune diseases.44,45 It is generally believed that the function of IL-17 in tumor microenvironments is to promote tumorigenesis mainly by increasing the angiogenesis of surrounding epithelial cells and fibroblasts.44 IL-17 induces IL-6, PGE2 and CAM-1 expression in fibroblasts, and plays an important role in blood vessel growth and tumor migration.46 IL-17 promotes the expression of IL-1β and TNF-α by macrophages, and increases chemokine expression to recruit neutrophils to infiltrate tumor tissues.47 IL-17 also induces the production of IL-8, which promotes the angiogenesis of endothelial cells, enhances the proliferation and survival of endothelial cells and tumor cells, and enhances the migration of tumor cells and infiltrating neutrophils.48 IL-17 enhances tumor cells and epithelial cells to produce chemokines like CXCX1, CXCL5, CXCL6, and CXCL8 that promote angiogenesis, and also inhibit secretion of anti-angiogenesis chemokines by fibroblasts.49,50 Therefore, IL-17 promotes angiogenesis in tumor tissue by multiple pathways.51 Although IL-17 mainly exhibits a tumor-promoting effect, there are quite a few of reports suggesting that IL-17 present an anti-tumor effect, mainly depending on the presence of IFN-γ. IL-17 has been proven to inhibit hematopoietic tumors by enhancing the cytotoxicity response.52 Similar to IL-17, IL-22 present both tumor-promoting and anti-tumor effects. Whether IL-17 has different effects in different tumor models needs to be clarified. Our results suggest that adoptive transfer of syngeneic N(IL-23+IL-18) neutrophils can significantly promote tumor growth in a EL4 tumor-bearing mouse model. Our research points out that N(IL-23+IL-18) express a wide range of the tumor-promoting cytokines and chemokines like IL-17A, IL-22, IL1f9, Csf1, Csf2, Csf3, etc., which can aggravate tumor development. But, the detailed mechanisms relevant to N(IL-23+IL-18) neutrophils-mediated tumor growth need to be studied in the future.
IL-17 is related to autoimmune arthritis.53 The levels of IL-17 mRNA and protein in the joints of patients with rheumatoid arthritis (RA) are higher than those in the normal control group, and IL-17 is positively correlated with disease severity indicators, such as erythrocyte sedimentation rate, C-reactive protein, rheumatoid factor.54 Overexpression of IL-17 aggravate CIA in mice, and IL-17 can stimulate fibroblasts-like synovial cells to produce vascular endothelial growth factor to aggravate inflammation.55–57 In addition, IL-17 induces synovial fibroblasts to secrete many inflammatory mediators and promotes the entire synovial tissue, synovial fibroblasts and cartilage to produce many matrix degrading enzymes, matrix metalloproteinases (MMP) such as MMP-1, −2, −9, −13. IL-17−/- mice were resistant to CIA compared with WT mice.58 IL-22 participates in RA pathological process.59–61 The level of IL-22 in the serum of patients with RA is related to erosive diseases, indicating that IL-22 has a role in the pathology of the disease.62 IL-22 has a pro-inflammatory effect in the CIA mouse model.63 However, the inflammation of the synovial membrane in IL-22-deficient mice is similar to that in WT mice.64,65 Thus, the roles of IL-22 in RA need to be clarified. Our results showed that adoptive transfer of N(IL-23+IL-18) neutrophils significantly promoted the occurrence and pathogenesis of arthritis in a CIA mouse model. IL-17-deficient N(IL-23+IL-18) neutrophils did not show this promoting effect, indicating that N(IL-23+IL-18) neutrophils act through an IL-17A-dependent pathway.
In a summary, IL-23 and IL-18 can synergistically induce IL-17A, IL-17F and IL-22 expression in mouse and human neutrophils mainly through JNK/P38- STAT3-RORγT/BATF pathway. Adoptive transfer of these N(IL-23+IL-18) neutrophils significantly promoted tumor growth and the pathogenesis of autoimmune arthritis in tumor bearing and CIA mouse models, respectively, indicating that N(IL-23+IL-18) neutrophils have pro-tumor and pro-inflammatory properties in these situations. We believe that identify of more and more neutrophil functional subpopulations would help us to more precisely understanding the detailed roles of neutrophils in physiological and pathological situations. Uncovering the different roles of different neutrophil subpopulations in various diseases might offer novel targeting neutrophil therapies to treat neutrophil-related immune disorders in the future.
Abbreviations
AMV, Avian myeoloblastosis virus reverse transcriptase; BATF, Basic leucine zipper ATF-like transcription factor; CIA, Collagen-induced arthritis; CSF, Colony-stimulating factor; EDTA, Ethylene diamine tetraacetic acid; EL4, Mouse lymphoma cells; ELISA, Enzyme-linked immunosorbent assay; ERK, Extracellular regulated protein kinases; fMLF, Formylmethionyl-leucyl-phenylalanine; GFP, Green fluorescent protein; HOXA1, Homeobox A1; IRF4, Interferon regulatory factor 4; JNK, C-Jun N-terminal kinases; KEGG, Kyoto encyclopedia of genes and genomes; LPS, Lipopolysaccharides; N1, Type 1 neutrophils; N2, Type 2 neutrophils; NBH, B cell-helper neutrophils; Nc, Conventional neutrophils; NF-κB, Nuclear factor kappa light chain enhancer of activated B cells; PBS, Phosphate buffered saline; PCA, Principal component analysis; PCR, Polymerase chain reaction; PGE, Prostaglandin E; PMA, Phorbol myristate acetate; PPARG, Peroxisome proliferator-activated receptor γ; RNase H, Ribonuclease H; RNA-seq, Messenger ribonucleic acid-sequencing; RORγ, Retinoic acid receptor-related orphan receptor γ; RPMI-1640, Roswell Park Memorial Institute-1640; Sox5, Sex determining region Y-box protein 5; STAT3, Signal transducers and activators of transcription 3.
Acknowledgments
We gratefully acknowledge Dr. Yinan Guo for his review on our manuscript and Mrs. Ling Li for her excellent lab management. This work was supported by grants from the National Natural Science Foundation for General and Key Programs (31930041, Y.Z), the National Key Research and Development Program of China (2017YFA0105002, 2017YFA0104402, Y.Z.), Knowledge Innovation Program of Chinese Academy of Sciences (XDA16030301, Y.Z.). Yifang Chen, Yang Li, and Han Guo are co-first authors.
Disclosure
The authors declare no conflict of interest.
References
1. Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol. 2011;12(11):1035–1044. doi:10.1038/ni.2109
2. Li Y, Wang W, Yang F, Xu Y, Feng C, Zhao Y. The regulatory roles of neutrophils in adaptive immunity. Cell Commun Signal. 2019;17(1):147. doi:10.1186/s12964-019-0471-y
3. Xu Y, Zhang Q, Zhao Y. The functional diversity of neutrophils and clustered polarization of immunity. Cell Mol Immunol. 2020;17(11):1212–1214. doi:10.1038/s41423-020-0378-y
4. Tsuda Y, Takahashi H, Kobayashi M, Hanafusa T, Herndon DN, Suzuki F. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity. 2004;21(2):215–226. doi:10.1016/j.immuni.2004.07.006
5. Chen F, Wu W, Millman A, et al. Neutrophils prime a long-lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat Immunol. 2014;15(10):938–946. doi:10.1038/ni.2984
6. Puga I, Cols M, Barra CM, et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol. 2011;13(2):170–180. doi:10.1038/ni.2194
7. Deniset JF, Surewaard BG, Lee WY, Kubes P. Splenic Ly6G(high) mature and Ly6G(int) immature neutrophils contribute to eradication of S. pneumoniae. J Exp Med. 2017;214(5):1333–1350. doi:10.1084/jem.20161621
8. Tamassia N, Bianchetto-Aguilera F, Arruda-Silva F, et al. Cytokine production by human neutrophils: revisiting the “dark side of the moon”. Eur J Clin Invest. 2018;48(Suppl 2):e12952. doi:10.1111/eci.12952
9. Sun B, Zhu L, Tao Y, et al. Characterization and allergic role of IL-33-induced neutrophil polarization. Cell Mol Immunol. 2018;15(8):782–793. doi:10.1038/cmi.2017.163
10. Taylor PR, Roy S, Leal SM
11. Ramirez-Velazquez CCE, Guido-Bayardo L, Ortiz-Navarrete V. IL-17-producing peripheral blood CD177+ neutrophils increase in allergic asthmatic subjects. Allergy Asthma Clin Immunol. 2013;9(1):23. doi:10.1186/1710-1492-9-23
12. Velden J, Paust HJ, Hoxha E, et al. Renal IL-17 expression in human ANCA-associated glomerulonephritis. Am J Physiol Renal Physiol. 2012;302(12):F1663–1673. doi:10.1152/ajprenal.00683.2011
13. Hu S, He W, Du X, et al. IL-17 production of neutrophils enhances antibacteria ability but promotes arthritis development during mycobacterium tuberculosis infection. EBioMedicine. 2017;23:88–99. doi:10.1016/j.ebiom.2017.08.001
14. Cai S, Batra S, Langohr I, Iwakura Y, Jeyaseelan S. IFN-gamma induction by neutrophil-derived IL-17A homodimer augments pulmonary antibacterial defense. Mucosal Immunol. 2016;9(3):718–729. doi:10.1038/mi.2015.95
15. Li Y, Zhu L, Chu Z, et al. Characterization and biological significance of IL-23-induced neutrophil polarization. Cell Mol Immunol. 2018;15(5):518–530. doi:10.1038/cmi.2017.39
16. Hu X, Wang P, Du J, et al. Phosphatase Wip1 masters IL-17-producing neutrophil-mediated colitis in mice. Inflamm Bowel Dis. 2016;22(6):1316–1325. doi:10.1097/MIB.0000000000000751
17. Ichii M, Oritani K, Toda J, et al. Signal-transducing adaptor protein-2 delays recovery of B lineage lymphocytes during hematopoietic stress. Haematologica. 2021;106(2):424–436. doi:10.3324/haematol.2019.225573
18. Chandrasekar B, Patel DN, Mummidi S, Kim JW, Clark RA, Valente AJ. Interleukin-18 suppresses adiponectin expression in 3T3-L1 adipocytes via a novel signal transduction pathway involving ERK1/2-dependent NFATc4 phosphorylation. J Biol Chem. 2008;283(7):4200–4209. doi:10.1074/jbc.M708142200
19. Singh A, Dashnyam M, Chim B, et al. Anxiolytic drug FGIN-1-27 ameliorates autoimmunity by metabolic reprogramming of pathogenic Th17 cells. Sci Rep. 2020;10(1):3766. doi:10.1038/s41598-020-60610-5
20. Shi H, Sun L, Wang Y, et al. N4BP1 negatively regulates NF-κB by binding and inhibiting NEMO oligomerization. Nat Commun. 2021;12(1):1379. doi:10.1038/s41467-021-21711-5
21. Lu Z, Miao Y, Muhammad I, et al. Colistin-induced autophagy and apoptosis involves the JNK-Bcl2-Bax signaling pathway and JNK-p53-ROS positive feedback loop in PC-12 cells. Chem Biol Interact. 2017;277:62–73. doi:10.1016/j.cbi.2017.08.011
22. Kong X, Chen Z, Xia Y, et al. Dehydrocorydaline accounts the majority of anti-inflammatory property of corydalis rhizoma in cultured macrophage. Evid Based Complement Alternat Med. 2020;2020:4181696. doi:10.1155/2020/4181696
23. Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351(Pt 1):95–105. doi:10.1042/bj3510095
24. Kim JA, Lee J, Margolis RL, Fotedar R. SP600125 suppresses Cdk1 and induces endoreplication directly from G2 phase, independent of JNK inhibition. Oncogene. 2010;29(11):1702–1716. doi:10.1038/onc.2009.464
25. Shi L, Tian H, Wang P, et al. Spaceflight and simulated microgravity suppresses macrophage development via altered RAS/ERK/NFkappaB and metabolic pathways. Cell Mol Immunol. 2021;18(6):1489–1502. doi:10.1038/s41423-019-0346-6
26. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11(9):1650–1667. doi:10.1038/nprot.2016.095
27. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8
28. Wu J, Mao X, Cai T, Luo J, Wei L. KOBAS server: a web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006;34(WebServer issue):W720–724. doi:10.1093/nar/gkl167
29. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
30. Chu Z, Sun C, Sun L, et al. Primed macrophages directly and specifically reject allografts. Cell Mol Immunol. 2020;17(3):237–246. doi:10.1038/s41423-019-0226-0
31. Wu T, Zhao Y, Wang H, et al. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci Rep. 2016;6(1):20250. doi:10.1038/srep20250
32. Hou Y, Lin H, Zhu L, et al. Lipopolysaccharide increases the incidence of collagen-induced arthritis in mice through induction of protease HTRA-1 expression. Arthritis Rheum. 2013;65(11):2835–2846. doi:10.1002/art.38124
33. Yoo JK, Kwon H, Khil LY, Zhang L, Jun HS, Yoon JW. IL-18 induces monocyte chemotactic protein-1 production in macrophages through the phosphatidylinositol 3-kinase/Akt and MEK/ERK1/2 pathways. J Immunol. 2005;175(12):8280–8286. doi:10.4049/jimmunol.175.12.8280
34. Furumaya C, Martinez-Sanz P, Bouti P, Kuijpers TW, Matlung HL. Plasticity in pro- and anti-tumor activity of neutrophils: shifting the balance. Front Immunol. 2020;11:2100. doi:10.3389/fimmu.2020.02100
35. Lubberts E. The IL-23-IL-17 axis in inflammatory arthritis. Nat Rev Rheumatol. 2015;11(7):415–429. doi:10.1038/nrrheum.2015.53
36. Vecellio M, Hake VX, Davidson C, Carena MC, Wordsworth BP, Selmi C. The IL-17/IL-23 axis and its genetic contribution to psoriatic arthritis. Front Immunol. 2020;11:596086. doi:10.3389/fimmu.2020.596086
37. Astry B, Venkatesha SH, Moudgil KD. Involvement of the IL-23/IL-17 axis and the Th17/Treg balance in the pathogenesis and control of autoimmune arthritis. Cytokine. 2015;74(1):54–61. doi:10.1016/j.cyto.2014.11.020
38. Ratsimandresy RA, Indramohan M, Dorfleutner A, Stehlik C. The AIM2 inflammasome is a central regulator of intestinal homeostasis through the IL-18/IL-22/STAT3 pathway. Cell Mol Immunol. 2017;14(1):127–142. doi:10.1038/cmi.2016.35
39. Hou Y, Zhu L, Tian H, et al. IL-23-induced macrophage polarization and its pathological roles in mice with imiquimod-induced psoriasis. Protein Cell. 2018;9(12):1027–1038. doi:10.1007/s13238-018-0505-z
40. Venken K, Jacques P, Mortier C, et al. RORgammat inhibition selectively targets IL-17 producing iNKT and gammadelta-T cells enriched in Spondyloarthritis patients. Nat Commun. 2019;10(1):9. doi:10.1038/s41467-018-07911-6
41. Mangan PR, Harrington LE, O’Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441(7090):231–234. doi:10.1038/nature04754
42. Hasan Z, Koizumi SI, Sasaki D, et al. JunB is essential for IL-23-dependent pathogenicity of Th17 cells. Nat Commun. 2017;8(1):15628. doi:10.1038/ncomms15628
43. Durant L, Watford WT, Ramos HL, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32(5):605–615. doi:10.1016/j.immuni.2010.05.003
44. Honorati MC, Neri S, Cattini L, Facchini A. Interleukin-17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthritis Cartilage. 2006;14(4):345–352. doi:10.1016/j.joca.2005.10.004
45. Li X, Bechara R, Zhao J, McGeachy MJ, Gaffen SL. IL-17 receptor-based signaling and implications for disease. Nat Immunol. 2019;20(12):1594–1602. doi:10.1038/s41590-019-0514-y
46. Li S, Cong X, Gao H, et al. Tumor-associated neutrophils induce EMT by IL-17a to promote migration and invasion in gastric cancer cells. J Exp Clin Cancer Res. 2019;38(1):6. doi:10.1186/s13046-018-1003-0
47. Jungnickel C, Schmidt LH, Bittigkoffer L, et al. IL-17C mediates the recruitment of tumor-associated neutrophils and lung tumor growth. Oncogene. 2017;36(29):4182–4190. doi:10.1038/onc.2017.28
48. Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14(21):6735–6741. doi:10.1158/1078-0432.CCR-07-4843
49. Lee JW, Wang P, Kattah MG, et al. Differential regulation of chemokines by IL-17 in colonic epithelial cells. J Immunol. 2008;181(9):6536–6545. doi:10.4049/jimmunol.181.9.6536
50. Numasaki M, Watanabe M, Suzuki T, et al. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol. 2005;175(9):6177–6189. doi:10.4049/jimmunol.175.9.6177
51. Li TJ, Jiang YM, Hu YF, et al. Interleukin-17-producing neutrophils link inflammatory stimuli to disease progression by promoting angiogenesis in gastric cancer. Clin Cancer Res. 2017;23(6):1575–1585. doi:10.1158/1078-0432.CCR-16-0617
52. Fabrice Benchetrit AC, Vives V, Warnier G, Gey A. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood. 2002;99(6):2114–2121. doi:10.1182/blood.V99.6.2114
53. Moran EM, Mullan R, McCormick J, et al. Human rheumatoid arthritis tissue production of IL-17A drives matrix and cartilage degradation: synergy with tumour necrosis factor-alpha, Oncostatin M and response to biologic therapies. Arthritis Res Ther. 2009;11(4):R113. doi:10.1186/ar2772
54. Rosu AMC, Stepan A, Musetescu A, Ene M. IL-17 patterns in synovium, serum and synovial fluid from treatment-naive, early rheumatoid arthritis patients. Rom J Morphol Embryol. 2012;53(1):73–80.
55. Pickens SR, Volin MV, Mandelin AM
56. Chabaud MLE, Joosten L, van Den Berg W, Miossec P. IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res. 2001;3(3):168–177. doi:10.1186/ar294
57. Dudler J, Renggli-Zulliger N, Busso N, Lotz M, So A. Effect of interleukin 17 on proteoglycan degradation in murine knee joints. Ann Rheum Dis. 2000;59(7):529–532. doi:10.1136/ard.59.7.529
58. Nakae SSS, Horai R, Sudo K, Mori S, Iwakura Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci U S A. 2003;100(10):5986–5990. doi:10.1073/pnas.1035999100
59. Liang SC, Tan XY, Luxenberg DP, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203(10):2271–2279. doi:10.1084/jem.20061308
60. Zheng Y, Valdez PA, Danilenko DM, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14(3):282–289. doi:10.1038/nm1720
61. Miyazaki Y, Nakayamada S, Kubo S, et al. Th22 cells promote osteoclast differentiation via production of IL-22 in rheumatoid arthritis. Front Immunol. 2018;9:2901. doi:10.3389/fimmu.2018.02901
62. Zhao LJZ, Jiang Y, Ma N, Zhang Y, Feng L, Wang K. IL-22+CD4+T cells in patients with rheumatoid arthritis. Int J Rheum Dis. 2013;16(5):518–526. doi:10.1111/1756-185X.12099
63. Leipe J, Schramm MA, Grunke M, et al. Interleukin 22 serum levels are associated with radiographic progression in rheumatoid arthritis. Ann Rheum Dis. 2011;70(8):1453–1457. doi:10.1136/ard.2011.152074
64. Corneth OBJ, Reijmers RM, Mus AMC, et al. Loss of IL-22 inhibits autoantibody formation in collagen-induced arthritis in mice. Eur J Immunol. 2016;46(6):1404–1414. doi:10.1002/eji.201546241
65. van Hamburg JP, Corneth OB, Paulissen SM, et al. IL-17/Th17 mediated synovial inflammation is IL-22 independent. Ann Rheum Dis. 2013;72(10):1700–1707. doi:10.1136/annrheumdis-2012-202373
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.