")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
The effect of electronic cigarette and tobacco smoke exposure on COPD bronchial epithelial cell inflammatory responses
Authors Higham A, Bostock D, Booth G , Dungwa JV, Singh D
Received 21 November 2017
Accepted for publication 7 February 2018
Published 23 March 2018 Volume 2018:13 Pages 989—1000
DOI https://doi.org/10.2147/COPD.S157728
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Andrew Higham,1,2 Declan Bostock,1 George Booth,2 Josiah V Dungwa,2 Dave Singh1,2
1Division of Infection, Immunity and Respiratory Medicine, School of Biological Sciences, Faculty of Biology, Medicine and Health, Manchester Academic Health Science Centre, The University of Manchester and University Hospital of South Manchester, NHS Foundation Trust, Manchester, UK; 2Medicines Evaluation Unit, University Hospital of South Manchester, Manchester, UK
Background: Electronic cigarettes (e-cigs) are used to help smoking cessation. However, these devices contain harmful chemicals, and there are safety concerns. We have investigated the effects of e-cigs on the inflammatory response and viability of COPD bronchial epithelial cells (BECs).
Methods: BECs from COPD patients and controls were exposed to e-cig vapor extract (ECVE) and the levels of interleukin (IL)-6, C-X-C motif ligand 8 (CXCL8), and lactate dehydrogenase release were measured. We also examined the effect of ECVE pretreatment on polyinosinic:polycytidylic acid (poly I:C)-stimulated cytokine release from BECs. Parallel experiments using Calu-3 cells were performed. Comparisons were made with cigarette smoke extract (CSE).
Results: ECVE and CSE caused an increase in the release of IL-6 and CXCL8 from Calu-3 cells. ECVE only caused toxicity in BECs and Calu-3 cells. Furthermore, ECVE and CSE dampened poly I:C-stimulated C-X-C motif ligand 10 release from both cell culture models, reaching statistical significance for CSE at an optical density of 0.3.
Conclusion: ECVE caused toxicity and reduced the antiviral response to poly I:C. This raises concerns over the safety of e-cig use.
Keywords: e-cigs, epithelial cells, COPD, air–liquid interface, cigarette smoke
Introduction
Electronic cigarettes (e-cigs) are nicotine delivery systems used to help reduce or stop tobacco smoking.1 e-cig vapor contains toxic chemicals including acrolein, styrene, and formaldehyde,2,3 raising questions about the safety of these devices. Emerging evidence suggests that there is long-term risk associated with the use of e-cigs.4 At present, it is difficult to quantify this risk due to the lack of longitudinal studies, especially in relation to tobacco smoking; it is assumed that this risk is lower due to the lack of tar and potentially lower levels of other harmful chemicals in e-cig vapor.
The respiratory system is delicately poised in a state of homeostasis, maintained through the interaction between the airway epithelium, immune cells, and the external environment. The chronic inhalation of particulate matter, including tobacco smoke and biomass fuels, can interfere with this balance, causing chronic inflammation and tissue remodeling.5 This can lead to airflow limitation which is a cardinal feature of COPD. The effect of long-term exposure to e-cig vapor is unknown, but there are concerns that it may cause chronic pulmonary inflammation.
We have previously shown that neutrophils exposed to e-cigs become activated and secrete higher levels of matrix metalloproteinase-9 and C-X-C motif ligand 8 (CXCL8).2 There is also evidence that the human airway epithelial cell line, H292, secretes higher levels of CXCL8 and interleukin-6 (IL-6) when exposed to e-cig vapor.6 In two separate studies, exposure of mice to e-cig vapors caused increased pulmonary inflammation.7 These studies underscore the potential for e-cigs to cause pulmonary inflammation in humans.
COPD patients are susceptible to viral and bacterial infections, including Streptococcus pneumoniae and non-typeable Haemophilus influenzae8 which can cause exacerbations of symptoms,5 which are associated with hospitalizations and increased mortality.9 Bacteria and viruses activate toll-like receptors which are expressed on pulmonary immune cells and bronchial epithelial cells (BECs). Toll-like receptor 3 (TLR3) is activated by viral double stranded RNA; the viral mimetic polyinosinic:polycytidylic acid (poly I:C) is a useful tool to study antiviral responses in vitro.
Cigarette smoke reduces the antimicrobial response of the lungs. For example, cigarette smoke extract (CSE) exposure attenuates the induction of the antimicrobial peptide human β-defensin-2 from BECs following infection with human rhinovirus or Pseudomonas aeruginosa.10,11 Furthermore, cigarette smoke dampens the production of antiviral cytokines, including interferon (IFN)-β, IFN-λ, and C-X-C motif ligand 10 (CXCL10) from BECs.12,13 CSE also dampens the production of proinflammatory cytokines from COPD lung macrophages exposed to bacteria.14,15 There is also accumulating evidence that e-cigs alter the host response to pulmonary infection. For example, e-cig vapor reduces macrophage and neutrophil killing of methicillin-resistant Staphylococcus aureus in vitro.16 Moreover, mice exposed to e-cig vapor are more susceptible to viral and bacterial infections.17 e-cig users have suppressed expression of immune-related genes in nasal epithelial cells, with greater suppression compared to tobacco smokers.18
The effects of e-cigs on COPD BECs have not been studied. The aim of the current study was to evaluate the effects of e-cig exposure on human airway epithelial cells air–liquid interface (ALI) models. Calu-3 cells and BECs obtained from COPD patients and controls were used. We examined the effects of e-cig vapor extract (ECVE) and CSE on cytokine production, cell viability, and the response to TLR3 stimulation which mimics viral infection.
Methods
Study subjects
Three healthy nonsmokers and five COPD patients were recruited for bronchoscopy to obtain bronchial brushings for primary BEC culture. Demographics are presented in Table 1. COPD was diagnosed based on ≥10 pack years smoking history, typical symptoms, and airflow obstruction. All subjects were nonatopic, with no history of respiratory illness or antibiotic use within 6 weeks of the study. The study was approved by NRES Committee North West – Greater Manchester Central (ref code 06/Q1403/156) and participants provided written and informed consent.
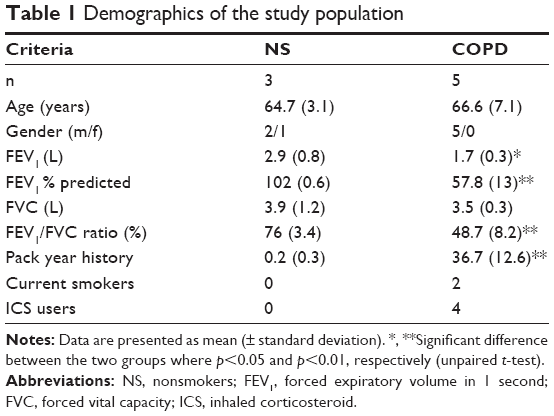 | Table 1 Demographics of the study population |
Isolation and culture of BECs
BECs were obtained from bronchial brushings and culture at the ALI until fully differentiated. Full details of isolation and culture of BECs can be found in the Supplementary materials.
Culture of Calu-3 cells at ALI
Calu-3 adenocarcinoma cells were a gift from Dr Peter West (The University of Manchester). Details of Calu-3 culture can be found in the Supplementary materials.
Transepithelial electrical resistance (TEER)
TEER was used to confirm tight junction formation. Full details can be found in the Supplementary materials.
ECVE vapor and CSE preparation
Details of ECVE and CSE preparation can be found in the Supplementary materials. ECVE was generated using a VIP® 1,100 mAh battery with a V5/CE5 clearomizer containing USA tobacco flavor e-liquid with a nicotine strength of 24 mg.
ECVE and CSE treatment of primary BECs and Calu-3 cells
For experiments examining the effect of ECVE or CSE only, cells were incubated apically with ECVE (0.01–0.3 optical density [OD]) or CSE (0.1–0.3 OD) and incubated at 37°C and 5% CO2 for 24 hours. For experiments examining the effect of ECVE or CSE pretreatment prior to poly I:C (Invivogen, San Diego, CA, USA) stimulation, cells were incubated apically with ECVE (0.01–0.1 OD) or CSE (0.1–0.3 OD) for 1 hour prior to stimulation with 10 μg/mL of poly I:C and incubated at 37°C and 5% CO2 for 24 hours.
Apical and basolateral supernatants were removed and analyzed for lactate dehydrogenase (LDH) by the LDH cytotoxicity assay kit (Fisher Scientific, Hampton, NH, USA), and IL-6, CXCL8, C-C motif ligand 5 (CCL5), and CXCL10 by enzyme-linked immunosorbent assay (ELISA; R&D Systems, Abingdon, UK) according to the manufacturer’s instructions. Fresh medium was then applied to the apical and basolateral surfaces and TERR was measured after 30 minutes.
Inflammatory pathway signaling
For experiments examining the effect of ECVE on p38 mitogen-activated protein kinase (MAPK) phosphorylation, cells were treated with ECVE (0.1 OD) and incubated at 37°C and 5% CO2 for 5–240 minutes. Cells were then lysed in RIPA buffer (10 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.25%) containing phosphatase (Sigma-Aldrich Co, St Louis, MO, USA) and protease inhibitors (Merck Millipore, Billerica, MA, USA) and samples were prepared for Western blot.
Western blot
Details for Western blot can be found in the Supplementary materials.
Mucin-5AC (MUC5AC) ELISA
An ELISA to detect MUC5AC in the supernatants from the apical wash of ALI cultures was developed in house. Full details can be found in the Supplementary materials.
Embedding and processing of Transwell membranes
Transwell membranes were embedded and processed for immunohistochemistry and histology. Details can be found in the Supplementary materials.
Immunohistochemistry and histology
Embedded Transwell membranes were stained for markers to confirm cellular differentiation at ALI. Full details can be found in the Supplementary materials.
Data analysis
Statistical analysis was performed using GraphPad InStat (GraphPad Software Inc, La Jolla, CA, USA). Data were analyzed by two-way analysis of variance followed by Tukey’s post hoc test for group data or unpaired t-test for direct comparisons between controls and COPD patients where p<0.05 was considered significant.
Results
TEER and LDH release
Calu-3 cells
LDH is a cytosolic enzyme which is released upon damage to the plasma membrane, and can be measured in the supernatant as an indicator of cellular toxicity.19 TEER is a measure of tight junction integrity. Cellular damage reduces TEER levels.20 There was a significant reduction in the TEER of Calu-3 cells exposed to 0.3 ECVE (Figure 1; p<0.01). In addition, LDH release into the apical supernatants of cells exposed to 0.3 ECVE was increased (Figure 1; p<0.01). CSE had no effect on TEER or LDH at any concentration.
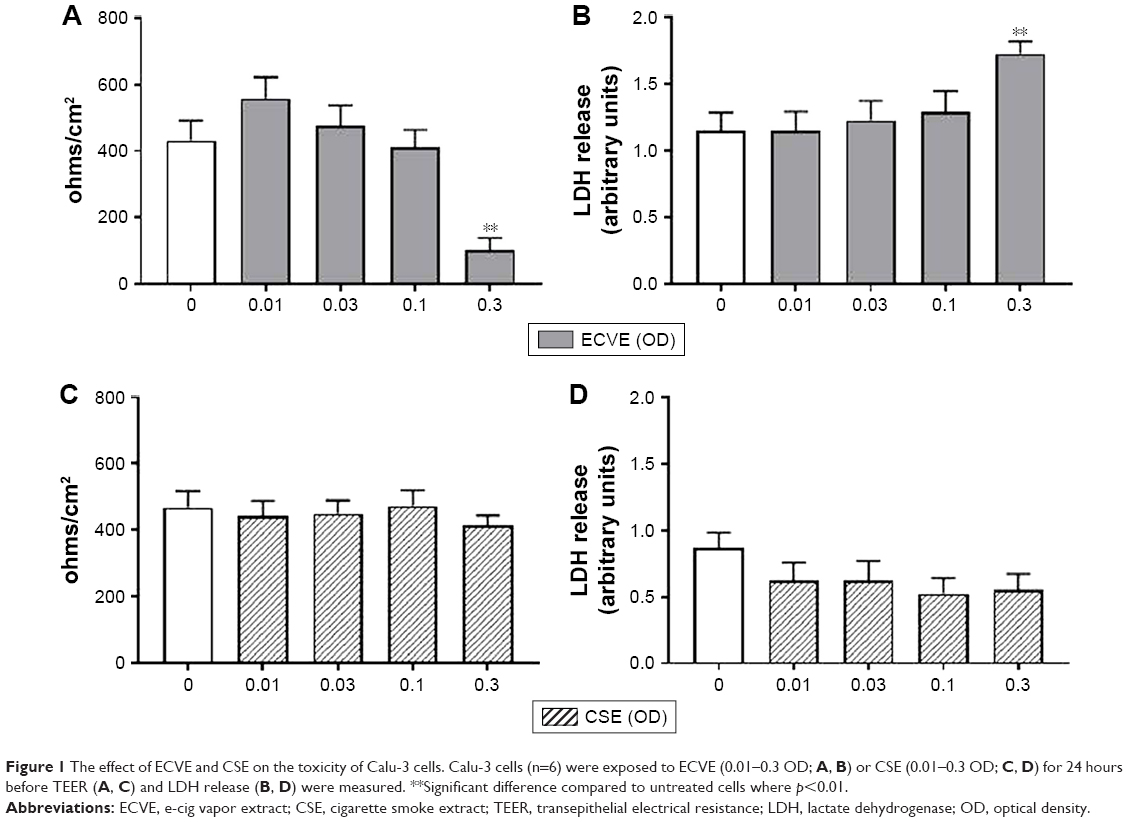 | Figure 1 The effect of ECVE and CSE on the toxicity of Calu-3 cells. Calu-3 cells (n=6) were exposed to ECVE (0.01–0.3 OD; A, B) or CSE (0.01–0.3 OD; C, D) for 24 hours before TEER (A, C) and LDH release (B, D) were measured. **Significant difference compared to untreated cells where p<0.01. |
Primary BECs
Differentiation of BECs at the ALI was confirmed by several parameters including tight junction formation, mucous production, and the presence of cilia (Figure S1). In primary BECs from controls and COPD patients exposed to ECVE, there was a numerical reduction in TEER at 0.3 ECVE in controls and 0.1 and 0.3 ECVE in COPD patients but this did not reach statistical significance (Figure 2). However, there was a significant increase in LDH release from controls and COPD BECs into the apical supernatants after exposure to 0.1 and 0.3 ECVE (p<0.01), and 0.03 in COPD BECs only.
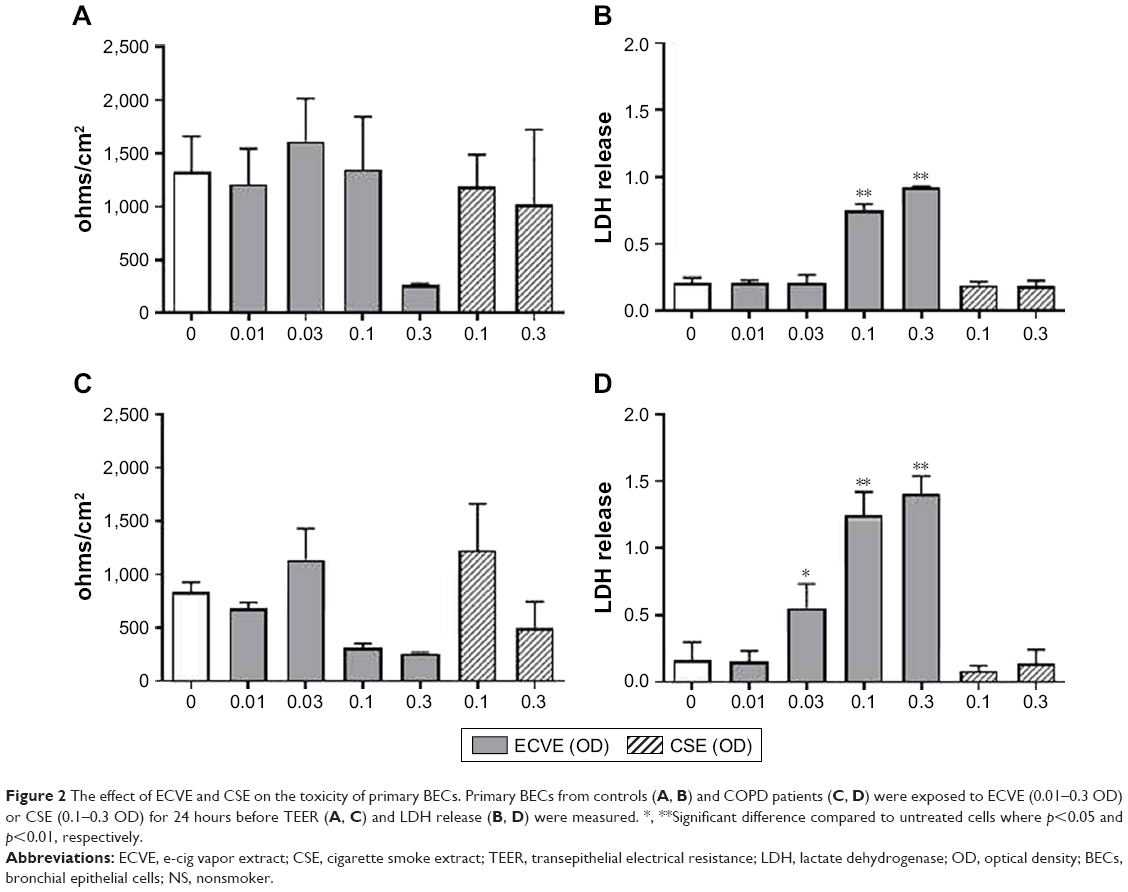 | Figure 2 The effect of ECVE and CSE on the toxicity of primary BECs. Primary BECs from controls (A, B) and COPD patients (C, D) were exposed to ECVE (0.01–0.3 OD) or CSE (0.1–0.3 OD) for 24 hours before TEER (A, C) and LDH release (B, D) were measured. *, **Significant difference compared to untreated cells where p<0.05 and p<0.01, respectively. |
Cytokine release
Calu-3 cells
ECVE exposure increased apical and basolateral IL-6 release and basolateral release of CXCL8 compared to untreated cells (n=6; Figure 3). Maximal release was observed at 0.1 ECVE (p<0.05). There was a significant decrease in CXCL8 levels at 0.3 ECVE in the apical supernatants (p<0.01).
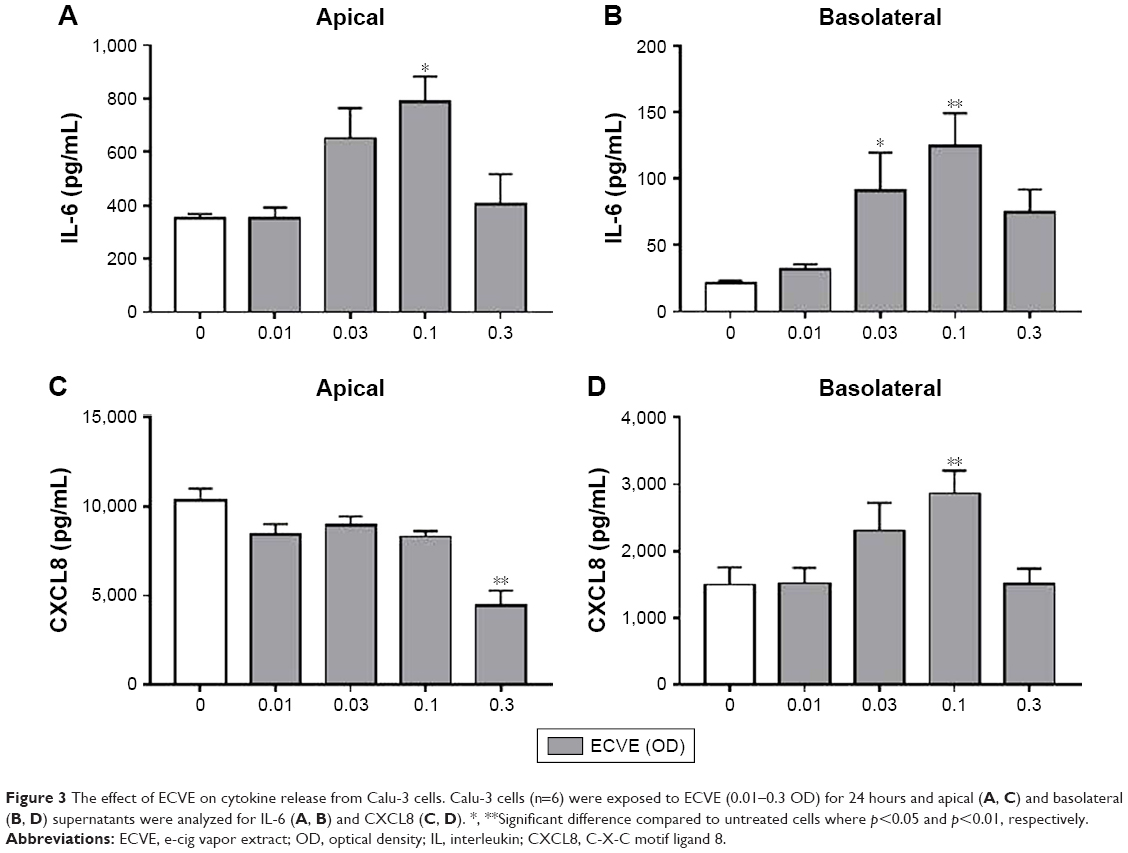 | Figure 3 The effect of ECVE on cytokine release from Calu-3 cells. Calu-3 cells (n=6) were exposed to ECVE (0.01–0.3 OD) for 24 hours and apical (A, C) and basolateral (B, D) supernatants were analyzed for IL-6 (A, B) and CXCL8 (C, D). *, **Significant difference compared to untreated cells where p<0.05 and p<0.01, respectively. |
Similarly, CSE (0.3 OD only) exposure increased apical and basolateral IL-6 release and basolateral release of CXCL8 compared to untreated cells (p<0.01; Figure 4). There was no change in the levels of CXCL8 in the basolateral supernatants. The levels of CCL5 and CXCL10 were below the lower limit of detection for all conditions (data not shown).
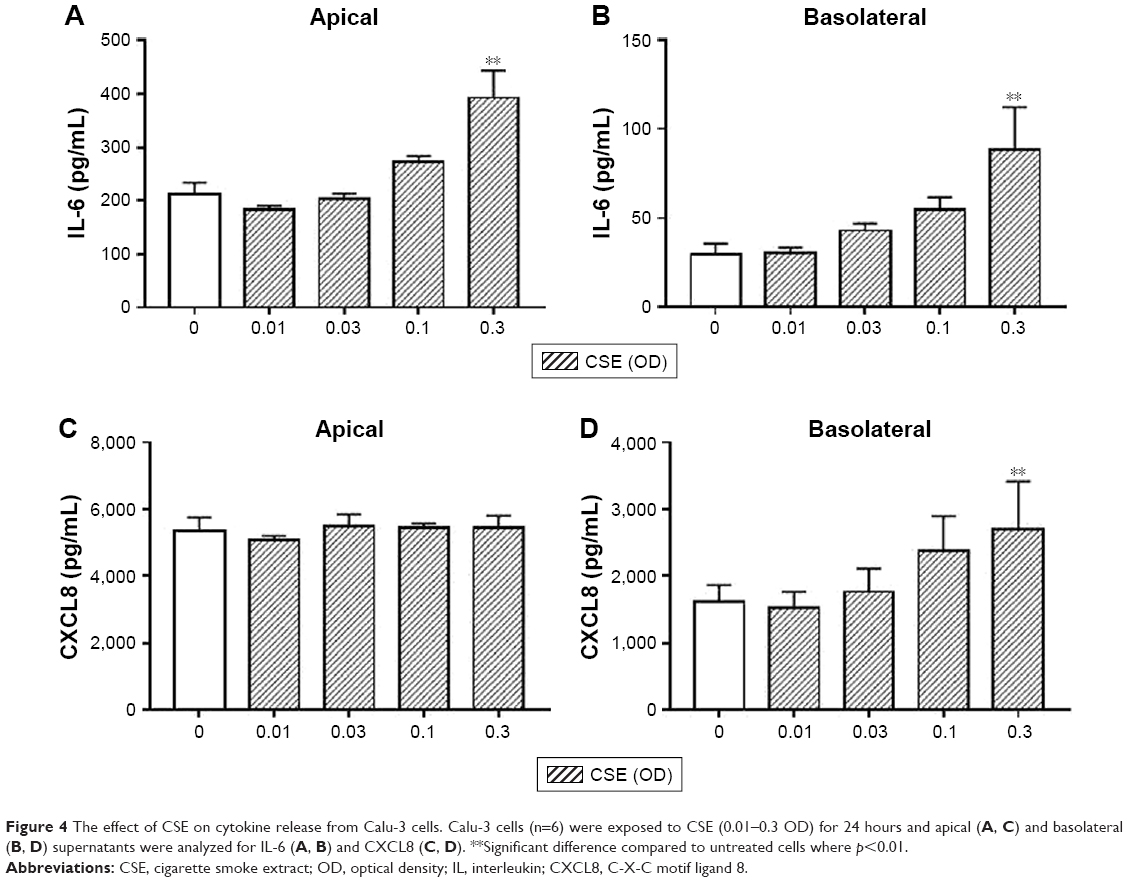 | Figure 4 The effect of CSE on cytokine release from Calu-3 cells. Calu-3 cells (n=6) were exposed to CSE (0.01–0.3 OD) for 24 hours and apical (A, C) and basolateral (B, D) supernatants were analyzed for IL-6 (A, B) and CXCL8 (C, D). **Significant difference compared to untreated cells where p<0.01. |
Primary BECs
We also compared the baseline release of IL-6 and CXCL8 between controls and COPD BECs; there was a trend for greater IL-6 and CXCL8 secretion by COPD compared to control BECs, but statistical significance was reached only for basolateral IL-6 secretion (Figure S2).
Exposure of BECs from controls and COPD patients to ECVE did not significantly change apical or basolateral release of IL-6 or CXCL8 (Figure 5). There was a reduction in the IL-6 and CXCL8 secretion at 0.3 ECVE, but this did not reach statistical significance. There was no effect of CSE on apical and basolateral IL-6 or CXCL8 release in control and COPD BECs (Figure 5).
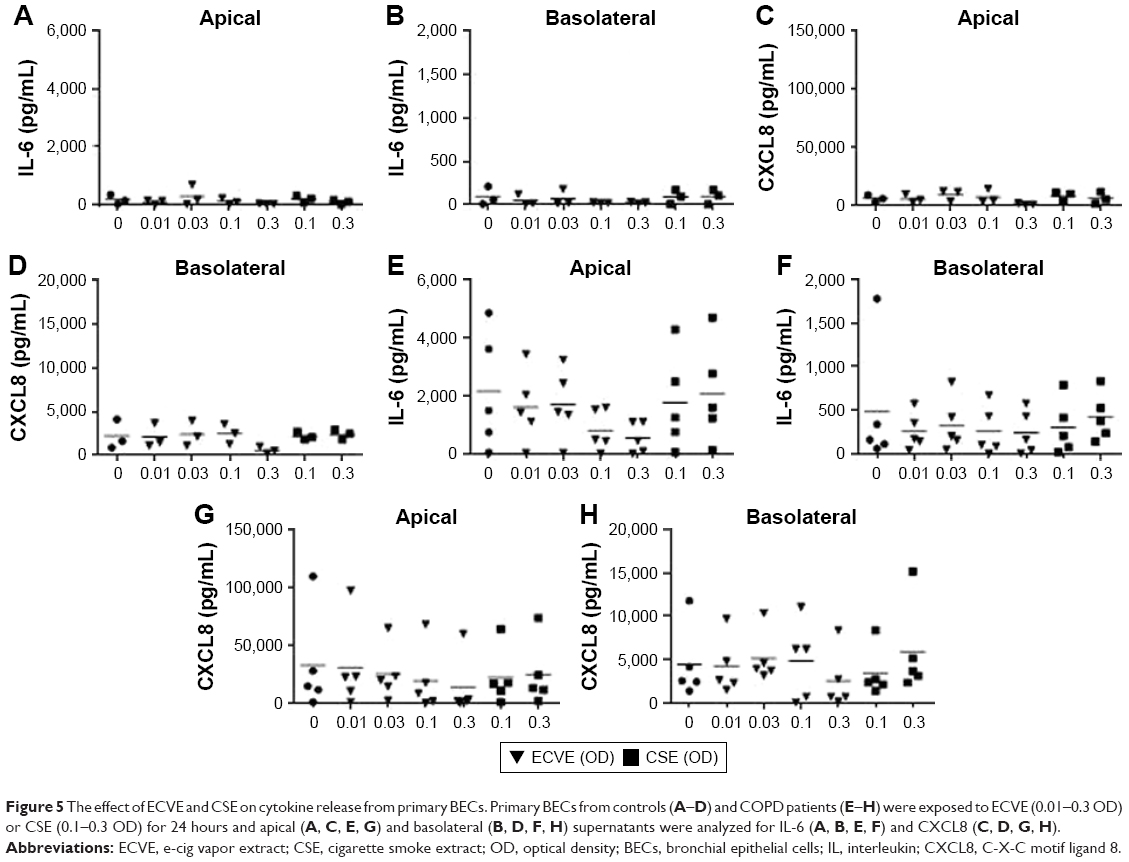 | Figure 5 The effect of ECVE and CSE on cytokine release from primary BECs. Primary BECs from controls (A–D) and COPD patients (E–H) were exposed to ECVE (0.01–0.3 OD) or CSE (0.1–0.3 OD) for 24 hours and apical (A, C, E, G) and basolateral (B, D, F, H) supernatants were analyzed for IL-6 (A, B, E, F) and CXCL8 (C, D, G, H). |
Inflammatory pathway activation
We observed a maximal increase in IL-6 and CXCL8 release at 0.1 ECVE in Calu-3 cells, and so we examined the effect on p38 MAPK phosphorylation at this concentration (0.1 ECVE). There was a time-dependent increase in the phosphorylation of p38 MAPK with maximal phosphorylation at 60 minutes (Figure 6; p<0.001). 0.3 CSE had a similar effect on p38 MAPK phosphorylation (Figure S3) in-line with previous publications.15,21–23
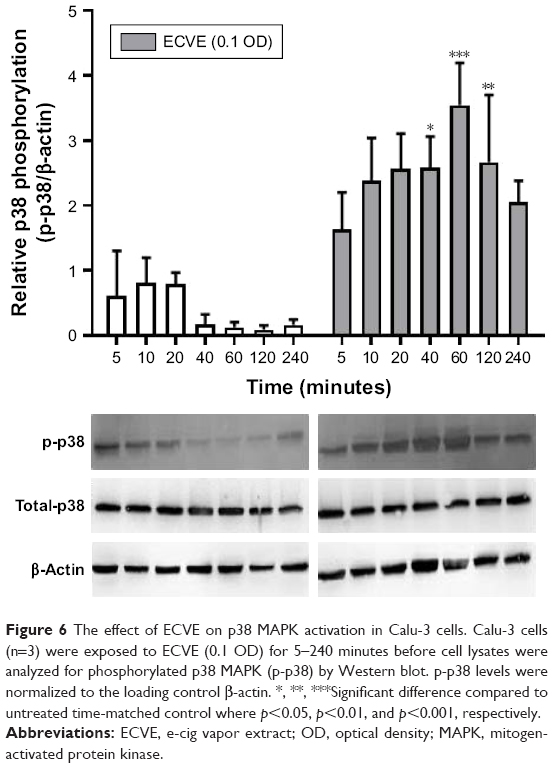 | Figure 6 The effect of ECVE on p38 MAPK activation in Calu-3 cells. Calu-3 cells (n=3) were exposed to ECVE (0.1 OD) for 5–240 minutes before cell lysates were analyzed for phosphorylated p38 MAPK (p-p38) by Western blot. p-p38 levels were normalized to the loading control β-actin. *, **, ***Significant difference compared to untreated time-matched control where p<0.05, p<0.01, and p<0.001, respectively. |
Poly I:C stimulation
Calu-3 cells
Poly I:C stimulation caused a significant increase in cytokine release compared to untreated cells (n=3; Table S1). Pretreatment of Calu-3 cells with ECVE or CSE for 1 hour prior to poly I:C stimulation (10 μg/mL) caused a reduction in the apical release of CXCL10 (Figure 7); for ECVE, this reached ~50% inhibition at 0.1 OD, but this did not reach statistical significance, while for CSE at 0.3 OD there was ~80% inhibition (p<0.05). Pretreatment with either ECVE or CSE did not significantly change CCL5, IL-6, or CXCL8 release.
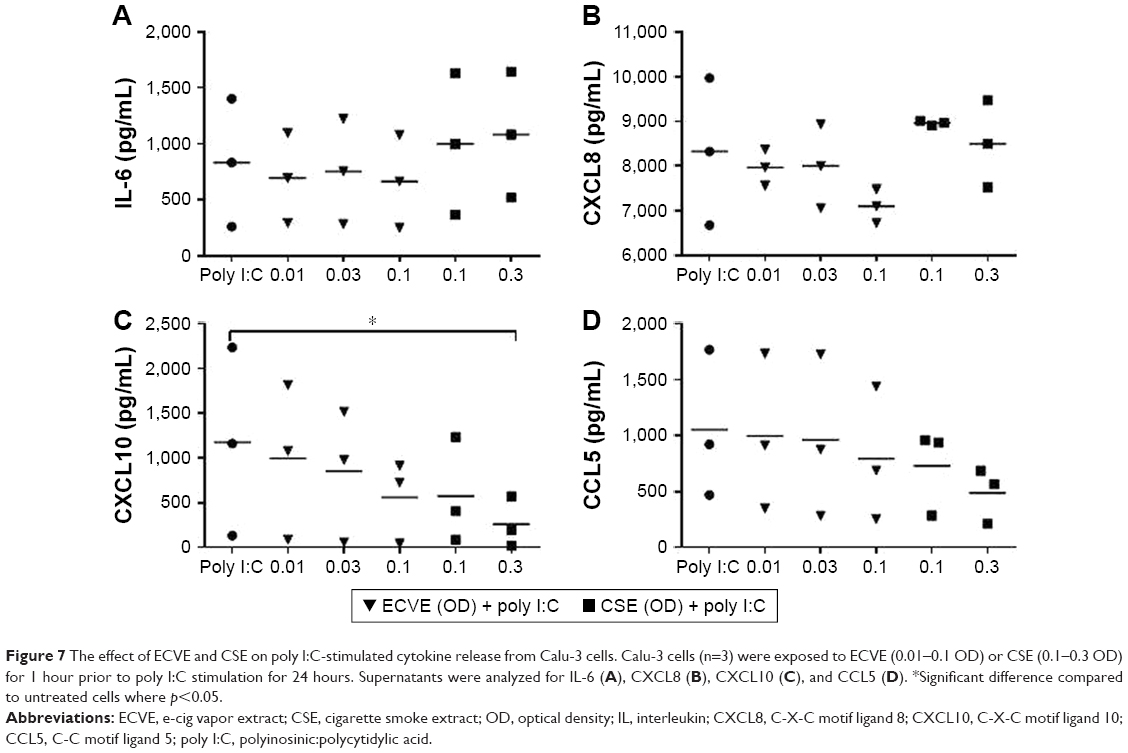 | Figure 7 The effect of ECVE and CSE on poly I:C-stimulated cytokine release from Calu-3 cells. Calu-3 cells (n=3) were exposed to ECVE (0.01–0.1 OD) or CSE (0.1–0.3 OD) for 1 hour prior to poly I:C stimulation for 24 hours. Supernatants were analyzed for IL-6 (A), CXCL8 (B), CXCL10 (C), and CCL5 (D). *Significant difference compared to untreated cells where p<0.05. |
Primary BECs
COPD BECs stimulated with poly I:C released significantly greater amounts of basolateral IL-6 compared to controls (Figure S4). There was a trend for increased amounts of apical and basolateral CXCL8 release in COPD compared to control cells, but this did not reach statistical significance (Figure S4C and D; p=0.09 and p=0.06, respectively). There was no difference in the release of CXCL10 or CCL5.
Control and COPD BECs were pretreated for 1 hour with ECVE or CSE prior to poly I:C stimulation. ECVE and CSE caused numerical reductions in IL-6, CXCL10, and CCL5 secretion (Figures 8 and 9), which reached significance for apical IL-6 release in controls at 0.1 ECVE where ~90% reduction was observed (Figure 8; p<0.01), and apical CCL5 production in controls with 0.3 CSE (p<0.05).
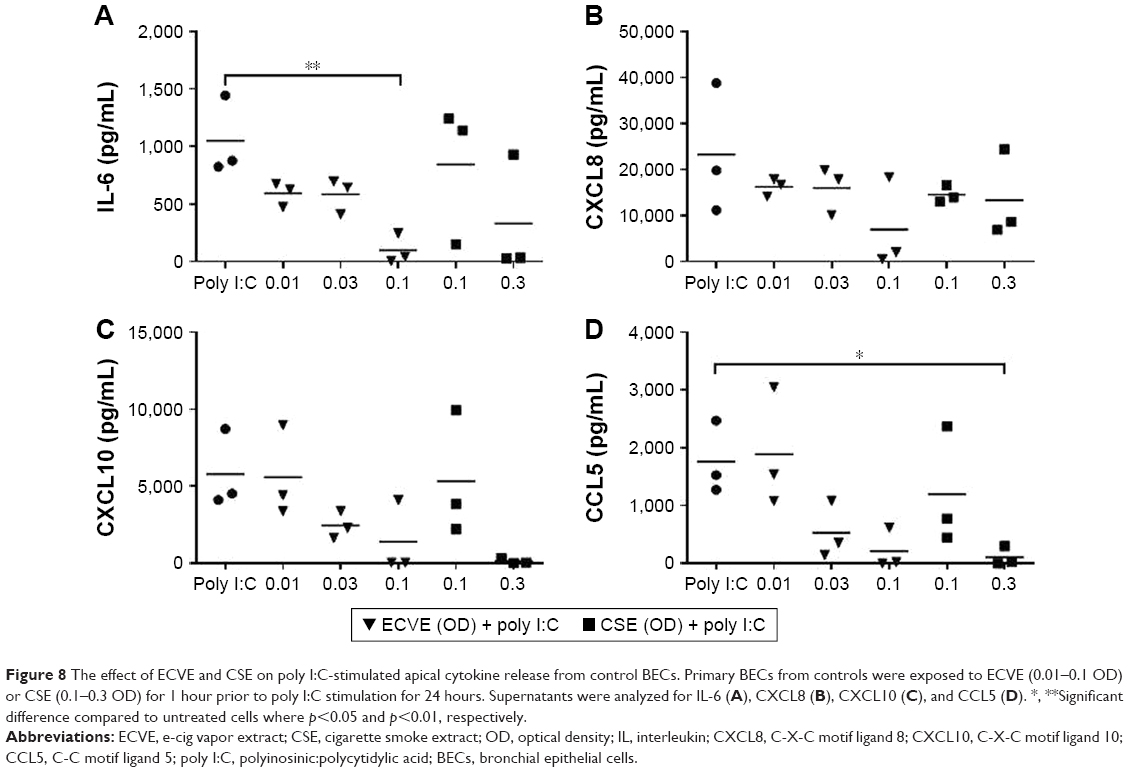 | Figure 8 The effect of ECVE and CSE on poly I:C-stimulated apical cytokine release from control BECs. Primary BECs from controls were exposed to ECVE (0.01–0.1 OD) or CSE (0.1–0.3 OD) for 1 hour prior to poly I:C stimulation for 24 hours. Supernatants were analyzed for IL-6 (A), CXCL8 (B), CXCL10 (C), and CCL5 (D). *, **Significant difference compared to untreated cells where p<0.05 and p<0.01, respectively. |
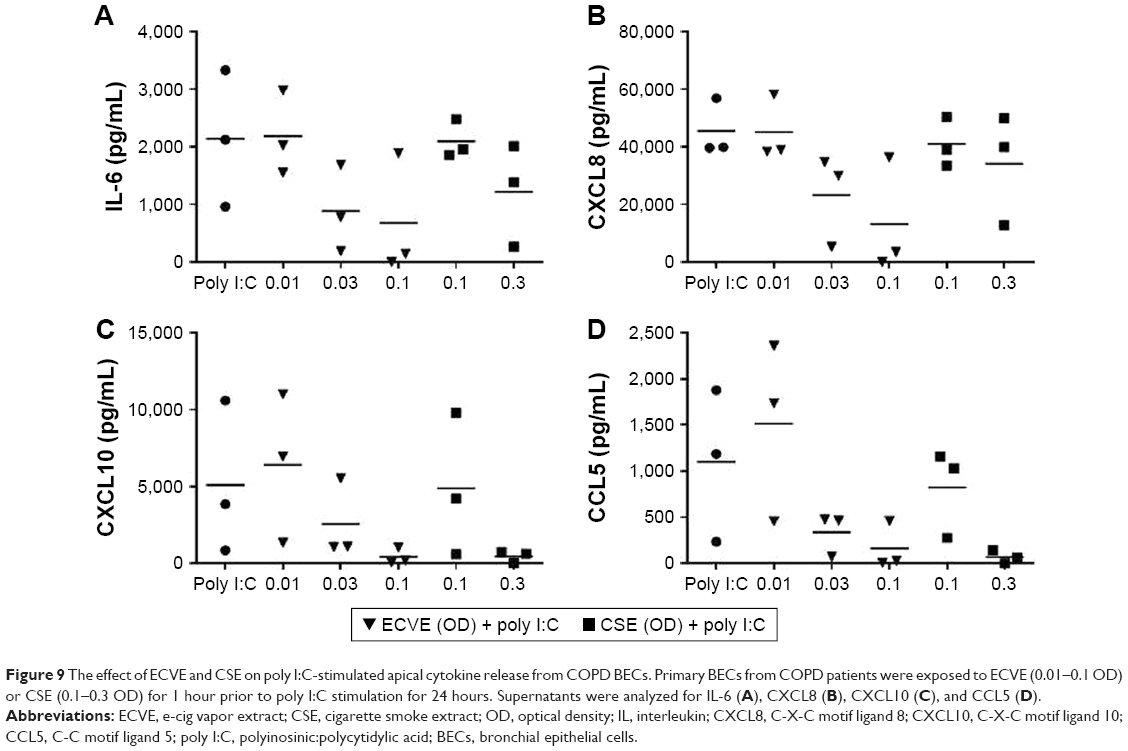 | Figure 9 The effect of ECVE and CSE on poly I:C-stimulated apical cytokine release from COPD BECs. Primary BECs from COPD patients were exposed to ECVE (0.01–0.1 OD) or CSE (0.1–0.3 OD) for 1 hour prior to poly I:C stimulation for 24 hours. Supernatants were analyzed for IL-6 (A), CXCL8 (B), CXCL10 (C), and CCL5 (D). |
The levels of LDH release were higher in apical supernatants from cells exposed to ECVE and poly I:C compared to ECVE alone (Figure S5), being significant at 0.03 ECVE (p<0.05). The levels of LDH release were not different between cells exposed to CSE alone compared to cells exposed to CSE and poly I:C.
Discussion
We show that e-cig exposure causes toxicity in ALI models using Calu-3 cells and primary BECs from controls and COPD patients, while CSE had no toxic effect. There were some differences between the ALI models; ECVE and CSE increased IL-6 and CXCL8 release from Calu-3 cells but not from primary BECs. ECVE and CSE dampened the immune response to TLR3 stimulation in both ALI models.
Despite growing evidence which questions the safety of e-cigs, these devices are recommended as an alternative to combustible cigarettes. It is well known that smoking tobacco cigarettes damages the airway epithelium, which forms an integral part of the first line of defence against inhaled particles. There is also mounting evidence that e-cigs may also damage the airway epithelium.6,16,24 We showed that ECVE is toxic to BECs from controls and COPD patients. This was also demonstrated in Calu-3 cells. In general terms, ECVE increased some inflammatory responses at lower concentrations but had the potential to cause toxicity at higher concentrations. This toxicity caused a reduction in inflammatory responses, which we believe may have relevance to the possible effects of e-cigs on the lungs in real life.
Immortalized BEC lines are often used as surrogates to primary epithelial cells as the latter are difficult to obtain. Another advantage is that cell lines such as Calu-3 cells can produce robust ALI cultures in a short amount of time. This provides data to guide experiments using primary cells which are a more limited resource. ECVE induced toxicity in both ALI models and both ECVE and CSE dampened the antiviral response. However, Calu-3 cells produced greater amounts of IL-6 and CXCL8 in response to ECVE and CSE while primary BECs did not. This suggests that there are differences in the way these cells behave and highlights the need to confirm any findings using cell lines with primary cells from patients with the disease. Calu-3 cells are derived from lung adenocarcinoma. They have limited ability to fully differentiate into bronchial epithelium at ALI as evidenced by their lack of mature cilia and reduced expression of tight junction protein 1.25 This results in a culture system which may behave differently to primary cells at ALI, as we demonstrated.
There are mixed reports regarding the effect of cigarette smoke on cytokine release from BEC lines and primary BECs.26–28 This is likely due to the differential methodology used in each of the studies, including the use of CSE or cigarette smoke, time of exposure, and concentration of CSE used.29 We did not observe an increase in cytokine release following exposure to CSE in primary BECs from controls and COPD patients. Nevertheless, using this method of producing CSE, we have previously demonstrated an increase in the release of CXCL8 from lung macrophages, monocyte-derived macrophages, and neutrophils.7,15,30 This shows the variability in the effect of CSE on different cell types.
A consistent finding in this study is the dampening of the response to poly I:C stimulation by ECVE in both ALI models. This was due to the toxic effects of ECVE demonstrated by increased LDH release. CXCL10 and CCL5 contribute to the antiviral response by recruiting and activating lymphocytes.31–33 A reduction in the levels of CXCL10 and CCL5 will impact the antiviral response of the bronchial epithelium which is potentially damaging to the host by increasing susceptibility to viral infection. Viruses are a major cause of COPD exacerbations.34 Tobacco cigarettes also dampen the antiviral response in primary BECs12,35,36 and we now confirm these findings. However, unlike ECVE, CSE did not induce toxicity, as there was no change in the LDH release. This could be due to a difference in the mechanism of action or methodology of extract preparation.
A limitation of this study is the small size of primary BEC samples assessed, partly due to the failure of some samples to successfully grow to maturity. Nevertheless, some of the (nonsignificant) numerical reductions in primary BECs were significant when using Calu-3 cells, such as TEER. During the study, the batch of e-liquid used to generate ECVE ran out, and a different batch would have been used for further experiments to increase sample size. The manufacture of e-liquid is not conducted to fulfil research standards and thus batch to batch variability could not be controlled for. Nevertheless, despite a lack of statistical significance in some experiments, our data clearly show a negative impact of e-cigs on BEC biology. In 1968, the University of Kentucky answered the call of the US Scientific Advisory Board of the Council for Tobacco Research to produce an international research standard cigarette. We believe a similar measure is necessary to assess the harm of e-cigs and allow more accurate and reliable data comparisons between different laboratories.
In addition to the variability between e-cig models and e-liquids, puffing topography and frequency of use also vary widely among users.37 This makes it difficult to accurately replicate real-world user profiles in the laboratory environment. For example, it is difficult to judge the relevant duration of total exposure time in vitro. The cells in this study were exposed to ECVE and CSE for 24 hours prepared using a manual flow rate of 238 mL/min; such a study design has been widely used for investigating the effects of tobacco cigarettes in vitro.15,38–40 While these study designs clearly differ from the intermittent use of e-cigs and tobacco cigarettes in real-life, these experiments are a recognized way to gain insights into the biological effects of the chemicals within these products.
Recently, it has been reported that COPD patients who reduce tobacco cigarette use experience fewer exacerbations, despite using e-cigs.41 The relationship between current cigarette smoking and COPD exacerbations is unclear, as large studies with multivariate analyses suggest that current tobacco cigarette smoking is not a predictor of COPD exacerbations.42–44 However, it is possible that current cigarette smoking or e-cig use may dampen the antiviral response, as we have shown, and thereby impact the severity of exacerbations in susceptible individuals rather than the total number of events.
This is the first study to use BECs from COPD patients and compare the effect of e-cigs and tobacco cigarettes on these cells. There is evidence that e-cig use is associated with increased rates of chronic bronchitis symptoms in adolescents.4 Moreover, a recent study has shown that e-cig use is associated with an increase in aberrant neutrophil responses and altered mucin secretion.45 We now demonstrate a reduced antiviral response following exposure to ECVE. These findings point to a potential negative impact on COPD patients who use e-cigs.
Acknowledgments
This work was funded by the North West Lung Centre Charity. This report is an independent research supported by National Institute for Health Research South Manchester Respiratory and Allergy Clinical Research Facility at the University Hospital of South Manchester, NHS Foundation Trust. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
DS has received sponsorship to attend international meetings, honoraria for lecturing or attending advisory boards, and research grants from various pharmaceutical companies including Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi, Genentech, GlaxoSmithKline, Glenmark, Johnson and Johnson, Merck, NAPP, Novartis, Pfizer, Skyepharma, Takeda, Teva, Theravance, and Verona. The other authors report no conflicts of interest in this work.
References
Etter JF, Bullen C. Electronic cigarette: users profile, utilization, satisfaction and perceived efficacy. Addiction. 2011;106(11):2017–2028. | ||
Higham A, Rattray NJ, Dewhurst JA, et al. Electronic cigarette exposure triggers neutrophil inflammatory responses. Respir Res. 2016;17(1):56. | ||
Jensen RP, Luo W, Pankow JF, Strongin RM, Peyton DH. Hidden formaldehyde in e-cigarette aerosols. N Engl J Med. 2015;372(4):392–394. | ||
McConnell R, Barrington-Trimis JL, Wang K, et al. Electronic cigarette use and respiratory symptoms in adolescents. Am J Respir Crit Care Med. 2017;195(8):1043–1049. | ||
Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report: GOLD executive summary. Eur Respir J. 2017;49(3):1700214. | ||
Lerner CA, Sundar IK, Yao H, et al. Vapors produced by electronic cigarettes and e-juices with flavorings induce toxicity, oxidative stress, and inflammatory response in lung epithelial cells and in mouse lung. PLoS One. 2015;10(2):e0116732. | ||
Garcia-Arcos I, Geraghty P, Baumlin N, et al. Chronic electronic cigarette exposure in mice induces features of COPD in a nicotine-dependent manner. Thorax. 2016;71(12):1119–1129. | ||
Beasley V, Joshi PV, Singanayagam A, Molyneaux PL, Johnston SL, Mallia P. Lung microbiology and exacerbations in COPD. Int J Chron Obstruct Pulmon Dis. 2012;7:555–569. | ||
Soler-Cataluna JJ, Martinez-Garcia MA, Roman Sanchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax. 2005;60(11):925–931. | ||
Arnason JW, Murphy JC, Kooi C, et al. Human beta-defensin-2 production upon viral and bacterial co-infection is attenuated in COPD. PLoS One. 2017;12(5):e0175963. | ||
Herr C, Beisswenger C, Hess C, et al. Suppression of pulmonary innate host defence in smokers. Thorax. 2009;64(2):144–149. | ||
Wu W, Zhang W, Booth JL, et al. Human primary airway epithelial cells isolated from active smokers have epigenetically impaired antiviral responses. Respir Res. 2016;17(1):111. | ||
Hudy MH, Traves SL, Proud D. Transcriptional and epigenetic modulation of human rhinovirus-induced CXCL10 production by cigarette smoke. Am J Respir Cell Mol Biol. 2014;50(3):571–582. | ||
Kulkarni R, Rampersaud R, Aguilar JL, Randis TM, Kreindler JL, Ratner AJ. Cigarette smoke inhibits airway epithelial cell innate immune responses to bacteria. Infect Immun. 2010;78(5):2146–2152. | ||
Metcalfe HJ, Lea S, Hughes D, Khalaf R, Abbott-Banner K, Singh D. Effects of cigarette smoke on Toll-like receptor (TLR) activation of chronic obstructive pulmonary disease (COPD) macrophages. Clin Exp Immunol. 2014;176(3):461–472. | ||
Hwang JH, Lyes M, Sladewski K, et al. Electronic cigarette inhalation alters innate immunity and airway cytokines while increasing the virulence of colonizing bacteria. J Mol Med. 2016;94(6):667–679. | ||
Sussan TE, Gajghate S, Thimmulappa RK, et al. Exposure to electronic cigarettes impairs pulmonary anti-bacterial and anti-viral defenses in a mouse model. PLoS One. 2015;10(2):e0116861. | ||
Martin EM, Clapp PW, Rebuli ME, et al. E-cigarette use results in suppression of immune and inflammatory-response genes in nasal epithelial cells similar to cigarette smoke. Am J Physiol Lung Cell Mol Physiol. 2016;311(1):L135–L144. | ||
Danpure CJ. Lactate dehydrogenase and cell injury. Cell Biochem Funct. 1984;2(3):144–148. | ||
Narai A, Arai S, Shimizu M. Rapid decrease in transepithelial electrical resistance of human intestinal Caco-2 cell monolayers by cytotoxic membrane perturbents. Toxicol In Vitro. 1997;11(4):347–354. | ||
Moretto N, Bertolini S, Iadicicco C, et al. Cigarette smoke and its component acrolein augment IL-8/CXCL8 mRNA stability via p38 MAPK/MK2 signaling in human pulmonary cells. Am J Physiol Lung Cell Mol Physiol. 2012;303(10):L929–L938. | ||
Low B, Liang M, Fu J. p38 mitogen-activated protein kinase mediates sidestream cigarette smoke-induced endothelial permeability. J Pharmacol Sci. 2007;104(3):225–231. | ||
Sailland J, Grosche A, Baumlin N, et al. Role of Smad3 and p38 signalling in cigarette smoke-induced CFTR and BK dysfunction in primary human bronchial airway epithelial cells. Sci Rep. 2017;7(1):10506. | ||
Sundar IK, Javed F, Romanos GE, Rahman I. E-cigarettes and flavorings induce inflammatory and pro-senescence responses in oral epithelial cells and periodontal fibroblasts. Oncotarget. 2016;7(47):77196–77204. | ||
Stewart CE, Torr EE, Mohd Jamili NH, Bosquillon C, Sayers I. Evaluation of differentiated human bronchial epithelial cell culture systems for asthma research. J Allergy. 2012;2012:943982. | ||
Kode A, Yang SR, Rahman I. Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir Res. 2006;7:132. | ||
Phillips J, Kluss B, Richter A, Massey E. Exposure of bronchial epithelial cells to whole cigarette smoke: assessment of cellular responses. Altern Lab Anim. 2005;33(3):239–248. | ||
Pickett G, Seagrave J, Boggs S, Polzin G, Richter P, Tesfaigzi Y. Effects of 10 cigarette smoke condensates on primary human airway epithelial cells by comparative gene and cytokine expression studies. Toxicol Sci. 2010;114(1):79–89. | ||
Comer DM, Elborn JS, Ennis M. Cigarette smoke, airway epithelial cells and host defence. Inflamm Cell Signal. 2014;1:e203. | ||
Kent LM, Fox SM, Farrow SN, Singh D. The effects of dexamethasone on cigarette smoke induced gene expression changes in COPD macrophages. Int Immunopharmacol. 2010;10(1):57–64. | ||
Yuan J, Liu Z, Lim T, et al. CXCL10 inhibits viral replication through recruitment of natural killer cells in coxsackievirus B3-induced myocarditis. Circ Res. 2009;104(5):628–638. | ||
Liu J, Li F, Ping Y, et al. Local production of the chemokines CCL5 and CXCL10 attracts CD8+ T lymphocytes into esophageal squamous cell carcinoma. Oncotarget. 2015;6(28):24978–24989. | ||
Olsnes AM, Motorin D, Ryningen A, Zaritskey AY, Bruserud O. T lymphocyte chemotactic chemokines in acute myelogenous leukemia (AML): local release by native human AML blasts and systemic levels of CXCL10 (IP-10), CCL5 (RANTES) and CCL17 (TARC). Cancer Immunol Immunother. 2006;55(7):830–840. | ||
Rohde G, Wiethege A, Borg I, et al. Respiratory viruses in exacerbations of chronic obstructive pulmonary disease requiring hospitalisation: a case-control study. Thorax. 2003;58(1):37–42. | ||
Bauer CM, Dewitte-Orr SJ, Hornby KR, et al. Cigarette smoke suppresses type I interferon-mediated antiviral immunity in lung fibroblast and epithelial cells. J Interferon Cytokine Res. 2008;28(3):167–179. | ||
Eddleston J, Lee RU, Doerner AM, Herschbach J, Zuraw BL. Cigarette smoke decreases innate responses of epithelial cells to rhinovirus infection. Am J Respir Cell Mol Biol. 2011;44(1):118–126. | ||
Robinson RJ, Hensel EC, Morabito PN, Roundtree KA. Electronic cigarette topography in the natural environment. PLoS One. 2015;10(6):e0129296. | ||
Bourgeois JS, Jacob J, Garewal A, Ndahayo R, Paxson J. The bioavailability of soluble cigarette smoke extract is reduced through interactions with cells and affects the cellular response to CSE exposure. PLoS One. 2016;11(9):e0163182. | ||
Chen M, Yang T, Meng X, Sun T. Azithromycin attenuates cigarette smoke extract-induced oxidative stress injury in human alveolar epithelial cells. Mol Med Rep. 2015;11(5):3414–3422. | ||
Wahl EA, Schenck TL, Machens HG, Egana JT. Acute stimulation of mesenchymal stem cells with cigarette smoke extract affects their migration, differentiation, and paracrine potential. Sci Rep. 2016;6:22957. | ||
Polosa R, Morjaria JB, Caponnetto P, et al. Evidence for harm reduction in COPD smokers who switch to electronic cigarettes. Respir Res. 2016;17(1):166. | ||
Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363(12):1128–1138. | ||
Han MK, Quibrera PM, Carretta EE, et al. Frequency of exacerbations in patients with chronic obstructive pulmonary disease: an analysis of the SPIROMICS cohort. Lancet Respir Med. 2017;5(8):619–626. | ||
Keene JD, Jacobson S, Kechris K, et al. Biomarkers predictive of exacerbations in the SPIROMICS and COPDGene cohorts. Am J Respir Crit Care Med. 2017;195(4):473–481. | ||
Reidel B, Radicioni G, Clapp P, et al. E-cigarette use causes a unique innate immune response in the lung involving increased neutrophilic activation and altered mucin secretion. Am J Respir Crit Care Med. 2018;197(4):492–501. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.