")
Back to Journals » International Journal of Nanomedicine » Volume 11
The effect of dual-functional hyaluronic acid-vitamin E succinate micelles on targeting delivery of doxorubicin
Authors Wang J, Ma W, Guo Q, Li Y, Hu Z, Zhu Z, Wang X, Zhao Y, Chai X, Tu P
Received 30 May 2016
Accepted for publication 5 September 2016
Published 7 November 2016 Volume 2016:11 Pages 5851—5870
DOI https://doi.org/10.2147/IJN.S113882
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Jinling Wang,1 Wenzhuan Ma,1,2 Qiang Guo,1,2 Ying Li,1,2 Zhongdong Hu,1 Zhixiang Zhu,1 Xiaohui Wang,1 Yunfang Zhao,1 Xingyun Chai,1 Pengfei Tu1
1Modern Research Center for Traditional Chinese Medicine, 2School of Chinese Materia Medica, Beijing University of Chinese Medicine, Beijing, People’s Republic of China
Abstract: Tumor-targeted delivery system has been developed as an attractive strategy for effective tumor therapy. In this study, in order to enhance the antitumor effects of doxorubicin (DOX), amphiphilic hyaluronic acid (HA)-conjugated vitamin E succinate (VES) copolymers (HA-VES) with different degrees of substitution (DS) were prepared with synergistic antitumor effects and active targeting activities, and utilized as nanocarriers for the efficient delivery of DOX. DOX-loaded HA-VES polymeric micelles (HA-VES/DOX) self-assembled from dual-functional HA-VES copolymer and exhibited excellent loading efficiency and superior colloidal stability. In vitro, HA-VES/DOX displayed enhanced cytotoxicity with synergistic anticancer effects of HA-VES copolymer, high apoptosis-inducing activities of tumor cells, and reversal effects of DOX on multidrug resistance, in comparison with DOX. Also, in vitro cellular uptake and subcellular localization studies revealed that HA-VES/DOX could more efficiently internalize into cancer cells and selectively release DOX within lysosomes, thereby enhancing the distribution of DOX in nuclei and facilitating its interactions with DNA. Specifically, HA-VES/DOX decreased the activity of CD44 mRNA and improved the targeting efficiency on MCF-7 cells, based on the active recognition between HA and CD44 receptor. More importantly, HA-VES/DOX displayed better tumor accumulation and targeting, and enhanced antitumor efficacy with reduced systemic toxicity in 4T1 tumor-bearing mice. In summary, the developed HA-VES–based drug delivery system, which increased drug targeting on the tumor site and exhibited preferable anticancer activity, could hold great potential as an effective and promising strategy for efficient tumor therapy.
Keywords: hyaluronic acid, self-assembled micelles, tumor targeting, doxorubicin, vitamin E succinate
Introduction
Most of the chemotherapeutic drugs such as doxorubicin (DOX) fail in effective cancer therapy, mainly due to nonselective tumor accumulation, severe systemic toxicity, and multidrug resistance (MDR). Nanotechnology-based drug delivery system acts as an effective strategy to circumvent MDR by bypassing P-glycoprotein (P-gp) efflux, improving anticancer efficacy, and reducing the side effects of chemotherapeutic drugs.1,2 In particular, self-assembled polymeric micelles (PMs) have emerged as promising vehicles for drug delivery in cancer therapy. PMs have a typical core–shell structure, where hydrophobic drugs can be solubilized and stabilized in the hydrophobic inner core of the micelles, and the hydrophilic outer shell can extend the circulation time and improve the steric stabilization by reducing opsonization in blood circulation.3
In general, PMs could selectively and effectively accumulate in tumor through enhanced permeability and retention (EPR) effect, thereby enhancing the therapeutic effects of chemotherapeutic drugs.4 Nonetheless, passive targeting through EPR effect is still limited in tumor therapy, due to the recognition and clearance by reticuloendothelial system (RES), systemic toxicity, and low delivery efficiency.5,6 Therefore, active tumor-targeted PMs, which could be internalized by specific receptor-mediated endocytosis in tumor cells, and achieve a higher chemotherapy efficacy and are beneficial in reducing the side effects in normal tissues, have been extensively focused toward cancer treatment.7,8 To achieve active targeting, various specific targeting ligands, such as folic acid,9,10 peptide,11–13 hyaluronic acid,14–16 and other ligands,17 have been grafted onto the surface of PMs, and then the PMs specifically interact with the receptor expressed on target cancer cells.
Hyaluronic acid (HA), a natural glycosaminoglycan composed of repeating disaccharide units of D-glucuronic acid and (1-β-3)-N-acetyl-D-glucosamine, has been extensively studied and applied for pharmaceutical and biomedical delivery, due to its biological functions including modulating cell motility and mediating cell proliferation.18–21 Moreover, HA reinforces its potential as a targeted ligand that could specifically bind onto CD44 receptors. CD44 receptors are overexpressed on the surface of various tumor cells and play an important role in cancer cell proliferation, migration, and invasion.8,22 Apart from this, HA has a functional group for chemical modification, and many studies have proved that conjugation HA with cytotoxic agent could significantly enhanced the efficiency of anticancer agents.20 Therefore, based on the above features, HA-based polymers have been applied as promising active targeting vehicles for cancer therapy, due to their biocompatibility, biodegradability, and specific CD44 receptor-mediated active targeting on tumor cells.19,23,24
HA is hydrophilic and could self-assemble in aqueous medium to form micelles after conjugating with hydrophobic polymer. α-Tocopheryl succinate (vitamin E succinate [VES]) is a succinyl derivative of vitamin E, which is different from other vitamin E derivatives in that VES cannot act as an antioxidant.25,26 VES, not other vitamin E derivatives, has anticancer properties against multiple cancer cells and nontoxic side effects on normal cells, due to increased generation of reactive oxygen species inducing membrane-destabilizing properties in the tumor cells by VES.26–28 Based on the hydrophobicity and synergistic antitumor effects of VES, a functional copolymer, with VES as the hydrophobic group and HA as the hydrophilic part, has been designed which acts as a nanocarrier for the delivery of antitumor drugs.29
In this study, to integrate CD44 receptor-mediated active targeting effects of HA and synergistic antitumor effects of VES, the copolymers of HA-ethylenediamine–VES (called HA-VES) with different degrees of substitution (DS) were synthesized. PMs based on HA-VES conjugates were developed for targeting delivery and improving the anticancer efficacy of DOX. The reversal MDR effects, endocytosis mechanism, and the subcellular distribution of DOX-loaded HA-VES micelles (HA-VES/DOX) were investigated in MCF-7/Adr cells. Moreover, to confirm the CD44 receptor-mediated active targeting effects of HA-VES, the expression of CD44 and HA competitive inhibition test were also evaluated in MCF-7 cells by real-time polymerase chain reaction (PCR), and confocal microscopy and flow cytometric analysis, respectively. In vivo, Sprague Dawley rats and 4T1-tumor bearing mice were used to investigate pharmacokinetic behaviors, biodistribution, antitumor effects, and systemic toxicity of HA-VES/DOX micelles.
Materials and methods
Materials
Doxorubicin hydrochloride (DOX·HCl) was purchased from Beijing HuaFeng Co. Ltd (Beijing, People’s Republic of China). VES was obtained from Wuhan Yuancheng Gongchuang Technology Co. Ltd (Hubei, People’s Republic of China). Sodium hyaluronate with an average molecular weight of 7 kDa was purchased from Frida Biological Engineering Co. Ltd (Shandong, People’s Republic of China). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI), N-hydroxysuccinimide (NHS), and 1-hydroxybenzotriazole (HoBt) were obtained from Zhenjiang Pukang Pharm Co. Ltd (Zhejiang, People’s Republic of China). RPMI 1640 medium and fetal bovine serum were provided by Thermo Fisher Scientific (Waltham, MA, USA). ProteoExtract™ Subcellular Proteome Extraction Kit was purchased from EMD Millipore (Billerica, MA, USA). Apoptosis assay kit and Annexin V-FITC kit were provided by Beyotime Institute of Biotechnology (Shanghai, People’s Republic of China). The near-infrared (NIR) dye Cy7 was obtained from Fanbo Biochemical (Beijing, People’s Republic of China). Pyrene and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Creatine kinase (CK), creatine kinase MB (CKMB), lactate dehydrogenase (LDH), and aspartate transaminase (AST) were obtained from Beijing Leadman Biochemistry Co. Ltd (Beijing, People’s Republic of China). All solvents were of high-performance liquid chromatography (HPLC) grade.
Synthesis and characterization of HA-VES
HA-VES conjugate was synthesized in two steps. Details of the synthesis are as follows. 1) VES (5 mmol) was made to react with EA (5 mmol) in the presence of EDCI (10 mmol) and HoBt (5 mmol) in 20 mL anhydrous dichloromethane under nitrogen atmosphere at 37°C. The coarse product obtained after the reaction was purified by silica column chromatography to obtain a yellow powder of VES-EA-NH2 (VES-NH2), and the yield of VES-NH2 was about 80%. 2) HA (0.36 mmol, moles of carboxyl group) was dissolved in 4 mL anhydrous formamide, and stirred with EDCI (0.5 mmol) and NHS (0.5 mmol) under ice bath for 2 hours. Anhydrous formamide solution (10 mL) of VES-NH2 was added dropwise to the mixture of HA, with different molar ratios between the amino group of VES-NH2 and the carboxyl groups of HA, that is, 5:1, 10:1, and 20:1, respectively. The reaction mixture was stirred for another 24 hours at 60°C under nitrogen atmosphere. The resulting mixture was purified by dialysis against excess amount of water–ethanol (1:1, v/v) and distilled water, respectively, for 2 days. The polymers of HA-VES with different DS were obtained as white powder after freeze-drying, with a general yield of 45%. The gross yield of the reaction was about 34%.
The chemical structure of HA-VES was characterized by 1H-nuclear magnetic resonance (1H-NMR) spectrometry (500 Hz; Varian, USA) and Fourier transform infrared spectroscopy (ANTARIS, USA). The DS of VES was defined as the number of VES molecules per 100 sugar residues of HA.
The critical micelle concentrations (CMC) of HA-VES copolymers were determined using pyrene as the fluorescence probe, as described before.14 In brief, aliquots of pyrene solution (3×10−6 mol/L in acetone) were transferred into 10 mL volumetric flasks and the acetone was evaporated under nitrogen gas streams. Different DS of HA-VES copolymers with a series of concentration ranging from 5×10−4 to 1×10−1 mg/mL were added into each flask, with the final concentration of pyrene being 6×10−7 mol/L. The mixed solutions had ultrasound 4 hours and equilibrated for another 12 hours. The fluorescence spectrum was recorded on a fluorescence spectrophotometer (F-4500; Hitachi Ltd., Tokyo, Japan). The excitation (λex) was 336 nm, and the emission wavelengths (λem) were 373 nm and 384 nm.
The hemolytic activity of HA-VES with different DS was investigated using rat red blood cells (RBCs). RBCs were separated from plasma by centrifugation at 3,000 rpm for 5 minutes and washed three times with normal saline to collect erythrocytes. The erythrocytes were diluted to 2% RBC suspension with normal saline, and then used for hemolysis assay within 24 hours. Then 0.5 mL of RBC suspension was mixed with 0.5 mL copolymer to obtain final concentrations (0.1, 0.5, 1, 2, 4, 8, 10 mg/mL) of HA-VES4, HA-VES7, HA-VES12, and Tween 80, respectively. The mixture was incubated in an incubator shaker at 37°C for 4 hours and then centrifuged at 3,000 rpm for 5 minutes. The supernatant was transferred into a 96-well plate, and the released hemoglobin was determined by a microplate reader at 540 nm. RBC suspensions treated with 2% Triton X-100 and normal saline were considered to have 100% and 0% hemolysis, respectively. Hemolysis percentage was calculated using the following equation:
Hemolysis (%) = (As − A0)/(A100 − A0) ×100 |
where As, A0, and A100 are the absorbance values of samples, normal saline, and 2% Triton, respectively.
Preparation and characterization of HA-VES/DOX
HA-VES/DOX was prepared by probe ultrasonication method. Briefly, DOX·HCl (5 mg) was made to react with 2 M excess of triethylamine in methanol to obtain DOX base. Then, 20 mg of HA-VES copolymer was dispersed in 10 mL deionized water, and 4 mg DOX was dissolved in 0.5 mL acetone. The DOX solution was slowly added to HA-VES aqueous solution with stirring. The mixture was stirred for another 24 hours to evaporate the organic solvent. Then, HA-VES/DOX was developed after probe ultrasonication under an ice bath for 20 minutes at 200 W (active every 2 seconds with an interval of 3 seconds; Vernon Hills Co. Ltd, USA). The final micelles were centrifuged at 3,500 rpm for 30 minutes and filtered through 0.45 μm microfiltration membrane to remove unencapsulated DOX. Blank micelles were prepared in the same way without DOX.
Size distribution and zeta potential of micelles were determined by dynamic light scattering using a Zetasizer (Nano ZS 90; Malvern Instruments, Malvern, UK). Measurements were repeated in triplicate. The morphology of blank and HA-VES/DOX micelles was evaluated by transmission electron microscopy (TEM) (Tecnai 20 200 kV; FEI). The diluted micelles were placed on a copper grid and negative stained with 2% phosphotungstic acid before observation by TEM.
For the measurement of encapsulation efficiency (EE) and drug loading (DL) content of micelles, HA-VES/DOX micelles and lyophilized HA-VES/DOX micelles were disrupted in methanol and sonicated for 10 minutes, and the concentration of DOX in micelles was analyzed by HPLC (Agilent 1260; Agilent Technologies, Santa Clara, CA, USA). The chromatographic separations were performed on an Eclipse XDB-C18 column (4.6×250 mm, 5 μm; Agilent Technologies) with the mobile phase of methanol–3 mmol/L monopotassium phosphate–acetic acid (58:42:0.5, v/v/v) at a detection wavelength of 227 nm. The EE and DL were calculated as the amount of drug encapsulated in micelles to the amount of drug added and the weight of micelles, respectively. All the measurements were performed in triplicate.
The colloidal stability of HA-VES/DOX based on HA-VES12/DOX was evaluated to measure the changes in size and zeta potential after incubation with 1%, 5%, and 10% fetal bovine serum (FBS) at 37°C for 24 hours.
The release studies were carried out by dialysis method with a dialysis bag (molecular weight 14,000). Briefly, 2 mL of HA-VES/DOX micelles was sealed in dialysis bags. Then, the bags were immersed in 50 mL of phosphate buffer (pH 7.4, 6.5, and 5.5) or acetate buffer (pH 4.5), respectively, with constant shaking at 100 rpm at 37°C. At predetermined time intervals, 1 mL of release mediums was sampled and replenished by the same volume of fresh medium. The concentration of released DOX was analyzed by HPLC as described before. The in vitro release experiments were carried out in triplicate.
Cell culture and animals
MCF-7/Adr (DOX resistant),25 MCF-7 (DOX sensitive and relatively high CD44 receptor expression),30,31 and HepG2 (relatively low CD44 receptor expression)32 cells were obtained from Nanjing Kaiji Biotech. Ltd Co. (Jiangsu, People’s Republic of China). MCF-7/Adr and MCF-7 cells were cultured in RPMI 1640 medium and HepG2 cells were incubated with high glucose Dulbecco’s Modified Eagle’s Medium (DMEM) at 37°C in the presence of 5% CO2. These two media contain 10% (v/v) FBS and 1% penicillin–streptomycin. The cells were trypsinized when 80%–90% confluence was reached.
Sprague Dawley rats weighing 220±20 g and female Balb/c mice aged 5 weeks and weighing about 20 g were purchased from Beijing Vita River Company (ethical approval number was SCXK2012-0001). All animal experiments were performed in accordance with the Guidelines for the Use and Care of Animals approved by the Beijing University of Chinese Medicine Committee of Ethics. The animals were maintained in the animal care facility for at least 3 days with fresh diet and water daily before the experiments as per the Chinese Medicine Committee Guidelines of Ethical Animal Care.
In vitro cytotoxicity
The in vitro cytotoxicity of HA-VES copolymers with different DS, free DOX-Sol, HA-VES/DOX, and the mixture of DOX-Sol and HA-VES polymer was evaluated against MCF-7 and MCF-7/Adr cells by MTT method. Briefly, MCF-7 and MCF-7/Adr cells were seeded in 96-well plates at a density of 5×104 cells/well, and cultured in 0.1 mL of 1640 culture medium and attached for 24 hours. Then, the MCF-7 and MCF-7/Adr cells were incubated in multiple concentrations of HA-VES copolymers with different DS and various equivalent DOX concentrations of DOX-Sol, HA-VES/DOX, and a mixture of DOX and the corresponding concentration of HA-VES copolymer for 48, 72, and 96 hours, respectively. The cells incubated with medium only were utilized as control. Then, the cell growth inhibition rate was calculated by MTT assay.
Half-maximal inhibitory concentration (IC50) was defined as the drug concentration at 50% inhibition efficiency. The resistant index (RI) and reversal factor (RF) were studied to evaluate the MDR reversal effects of nanomicelles.33 Importantly, RI was calculated as the IC50 of samples in MCF-7/Adr cells compared with that in MCF-7 cells, and RF was evaluated by comparing the IC50 values of DOX-Sol with the IC50 values of other samples in MCF-7/Adr cells.
Apoptosis
MCF-7/Adr cells were seeded in 24-well plates at a density of 1×105 cells/well and incubated for 24 hours. Then, the cells were incubated with DOX-Sol, HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX (8 μg/mL DOX) for 48 hours. For qualitative observation, the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) and the cellular and nuclear morphology was observed with a fluorescence inversion microscope (Olympus Corporation, Tokyo, Japan). For quantitative measurement, the cells were trypsinized, washed with cold phosphate-buffered saline (PBS) twice, harvested, and stained with Annexin V-FITC and propidium iodide for 10 minutes at room temperature in the dark. Thereafter, the binding buffers were added and the samples were analyzed by fluorescence-activated cell sorting (FACS).
Western blot
The effects of different DOX formulations on the apoptotic protein caspase 3 and poly(ADP-ribose) polymerase (PARP) were assessed by Western blot analysis. In brief, MCF-7/Adr cells were cultured in six-well plates at 5×105 cells/well for 24 hours. Then, the cells were treated with DOX-Sol, HA-VES4/DOX, HA-VES7/DOX, HA-VES12/DOX, and the corresponding HA-VES12 nanocarrier (at concentration of 8 μg/mL DOX) for 48 hours. After this, the cells were washed with PBS and harvested with lysis buffer. Protein was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis before being transferred to nitrocellulose membranes. These membranes were blocked by milk and then incubated with caspase 3 and PARP antibody at 4°C overnight. The β-actin antibody was probed as loading control. The membranes were washed, and secondary antibodies were added and incubated for 1 hour at 37°C. Finally, the membranes were washed three times and the protein was visualized by enhanced chemiluminescence.
Quantitative real-time PCR analysis of CD44 expression
The targeted efficiency of HA-VES micelles on CD44 and the expression level of CD44 were further quantified by quantitative real-time PCR (qRT-PCR). Briefly, MCF-7 cells (1×106 cells/well) were seeded in six-well plates for 24 hours and then incubated with DOX-Sol, HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX (DOX concentration of 8 μg/mL) for 2 hours. Then, the cells were washed with cold PBS twice. Total RNA was extracted from the MCF-7 cells using EZNA. total RNA kit (Omega) according to the manufacturer’s instructions. Total RNA was reverse transcribed and analyzed by qRT-PCR as described previously.34,35 β-actin was used as the control for normalization. qRT-PCR for CD44 mRNA was performed with CD44 mRNA primers and the primer sequences were as follows:
Forward: 5′-GCAACCCTACTGATGATGAC-3′
Reverse: 5′-CCACCTTCTTGACTCCCAT-3′.
Cellular uptake and efflux
The cellular uptake of DOX formulations was investigated quantitatively by flow cytometry (FACS) and qualitatively by confocal laser scanning microscopy (CLSM) in MCF-7/Adr, MCF-7, and HepG2 cells. For FACS, the cells were seeded in 24-well plates at a density of 1×105 cells/well for 24 hours. Then, DOX-Sol, HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX at an equivalent DOX concentration of 8 μg/mL DOX were incubated in MCF-7 and HepG2 cells for 2 hours, and cultured in MCF-7/Adr cells for 1, 2, and 4 hours, respectively. Then, the cells were washed with cold PBS three times, trypsinized, and harvested in PBS. The fluorescent intensity of DOX in cells was measured by FACS.
For CLSM, the cells were seeded onto cover slips for 24 hours preincubation and then co-cultured with DOX-Sol, HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX for 2 hours in MCF-7 and HepG2 cells, and for 1 hour and 2 hours in MCF-7/Adr cells, respectively. Then, the cells were washed with cold PBS twice, fixed with 4% paraformaldehyde for 10 minutes, and counterstained with DAPI to mark the cell nucleus and with LysoTracker Green to mark the lysosome. The cells were visualized by confocal laser scanning microscope (Olympus Corporation).
For competitive inhibition studies, HA was used as the competitive inhibitor. Briefly, MCF-7 and HepG2 cells were preincubated with HA (10 mg/mL) for 1 hour and then incubated with HA-VES/DOX micelles with different DS for another 2 hours. Then, the cells were analyzed by FACS and CLSM as described above.
To investigate the efflux effects of DOX formulations, MCF-7/Adr cells were cultured with DOX-Sol, HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX (8 μg/mL DOX) for 2 hours. Then, the cells were washed with PBS twice and incubated with fresh medium for another 1 hour and 2 hours. After that, the cells were trypsinized and harvested. The intracellular fluorescent intensity of DOX was analyzed by FACS.
Endocytosis mechanism in MCF-7/Adr cells
The endocytic pathway of HA-VES/DOX was investigated with specific endocytosis inhibitors and detected by FACS in MCF-7/Adr cells based on HA-VES12/DOX. First, MCF-7/Adr cells were seeded in 24-well plates and cultured for 24 hours. Then, the cells were preincubated with chlorpromazine (10 μg/mL), sodium azide (3 μg/mL), quercetin (6 μg/mL), indomethacin (6 μg/mL), amiloride (8 μg/mL), and β-cyclodextrin (1 mg/mL) for 1 hour at 37°C. Thereafter, the cellular monolayers were incubated with a combination of HA-VES12/DOX and corresponding endocytosis inhibitors for an additional 1 hour at 37°C. After co-incubation, the MCF-7/Adr cells were washed, digested, harvested, and detected by FACS.
Subcellular distribution of DOX in MCF-7/Adr cells
Subcellular protein fractions (cytosol, membrane/organelle, nucleus, cytoskeleton) were extracted from MCF-7/Adr cells based on different solubilities of the subcellular fractions using ProteoExtract™ Subcellular Proteome Extraction Kit.36 Briefly, MCF-7/Adr cells were seeded at a density of 5×106 cells in Petri dishes for 2 days, and then the cells were incubated with DOX-Sol and HA-VES12/DOX at an equivalent DOX concentration of 8 μg/mL for 0.5, 1, 2, and 4 hours, respectively. Subcellular protein fractions were sequentially extracted according to the manufacturer’s instructions. Cytosolic fraction, membrane/organelle, nuclear fraction, and cytoskeletal fraction were sequentially prepared by incubating the cells with 0.4 mL extraction buffer I–IV for 10 minutes at 4°C, respectively, washing with wash buffer, and transferring the supernatant to an Eppendorf tube. The fractions were ultrasonic, and then the samples (100 μL) were mixed with 300 μL methanol and 100 μL glibenclamide (internal standard, IS) and centrifuged at 12,000 rpm for 10 min. An aliquot of 5 μL upper organic layer was injected into LC-MS/MS for analysis. Protein in the fractions was determined using BCA Protein Assay Kit.
Pharmacokinetics in vivo
Twenty-four male Sprague Dawley rats were randomly divided into four groups (n=6). The rats were fasted for 12 hours with free access to water before dosing. DOX-Sol, HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX were injected into the rats through the tail vein at a dose of 5 mg/kg DOX, respectively. Briefly, at the end of the experiment, the blood samples were extracted by puncturing the reto-orbital sinus and then were centrifuged to obtain serum. Plasma samples were obtained after centrifugation at 4,000× g for 10 minutes immediately and analyzed by LC-MS/MS after precipitation with methanol.
In vivo tumor targeting effects
To evaluate in vivo tumor targeting and biodistribution of HA-VES/DOX micelles, FX Pro in vivo imaging system (Carestream Health) was used for NIR fluorescence imaging in 4T1-bearing Balb/c mice after they are treated with NIR dye Cy7-loaded HA-VES micelles. The preparation of Cy7-loaded HA-VES micelles (HA-VES/Cy7) involved the same procedure as that of HA-VES/DOX, except for substituting DOX for Cy7. When the tumor volume of 4T1-bearing mice reached approximately 150 mm3, 4T1-bearing mice were intravenously injected with HA-VES4/CY7, HA-VES7/CY7, and HA-VES12/CY7, respectively, through the tail vein. Then, at 2 hours and 4 hours postadministration, the mice were sacrificed, and the tumor, heart, liver, spleen, lung, and kidney were excised, washed with saline, and imaged with FX Pro in vivo imaging system. Moreover, to further observe the distribution of DOX formulations in the tumor tissue, the tumors were sectioned at 20 μm, fixed with 4% formalin for 10 minutes, and the nuclei stained with DAPI and observed by CLSM.
For quantitative analysis, after 4 hours of administering HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX, the tissues including tumor, heart, liver, spleen, lung, and kidney were excised, weighed, homogenized with methanol, and quantified by LC-MS/MS.
In vivo antitumor effects
Female Balb/c mice bearing 4T1 tumor were randomly divided into five groups (n=10). When the tumors reached about 100 mm3, the mice were intravenously injected every 2 days with DOX-Sol, HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX at a dose of DOX 10 mg/kg, respectively, and the group administered saline was used as a control. Body weights and tumor volumes (V = ab2/2, where “a” was the major axis and “b” the minor axis measured by slide caliper) were measured every 2 days after the administration. At the end of the experiment, mice were sacrificed, and tumors were excised, weighed, and photographed.
The side effects of DOX formulations in vivo were evaluated using enzyme-linked immunosorbent assay (ELISA) kit. Briefly, at the end of the experiment, the blood of each mouse was extracted by eyeballs and the serum separated. Then, the activities of CK, CKMB, LDH, and AST were evaluated in serum to investigate the organ toxicity.
In addition, at day 12, the mice were sacrificed and their major organs including heart, liver, spleen, lung, kidney, and tumors were collected, fixed in 4% formalin, and embedded in paraffin. The paraffin tissues, except for tumors, were stained with hematoxylin and eosin, and the tumors were stained with terminal deoxynucleotidyl transferase dUTP nick end labeling for microscopic observation and pathological study.
Statistical analysis
Results were expressed as mean ± standard deviation (SD). A Student’s t-test or one-way analysis of variance was applied in the experiments to test for significance. Statistical differences were considered significant at P<0.05 and highly significant at P<0.01.
Results and discussion
Synthesis and characterization of HA-VES copolymers
The HA-VES copolymer was synthesized by grafting VES onto HA with the linker EA. The scheme showing synthesis of HA-VES is presented in Supplementary materials. Specifically, VES was made to react with EA in the presence of EDCI and HoBt to form VES-NH2. HA was activated in the presence of EDCI and NHS, and then amidated with VES-NH2. The structure of HA-VES was confirmed by 1H-NMR spectrum (shown in Supplementary materials). Intense peaks for the methyl group (0.8 ppm) of VES, the N-acyl group (6.42 ppm), and the methylene group (3.34 and 2.47 ppm) of EA were observed in the 1H-NMR of VES-NH2, indicating that VES-NH2 was successfully synthesized (Supplementary materials). Meanwhile, the characteristic peaks for the N-acetyl group (1.8 ppm) of HA and the methyl group (0.8 ppm) of VES-NH2 indicated that VES was grafted onto HA (Supplementary materials). In Fourier transform infrared spectrum, the stretching vibration (3,431.34 cm−1) of hydroxyl groups in HA-VES was strengthened after grafting VES onto HA, which is because VES has only one hydroxyl carboxylic acid (3,445.35 cm−1), but many hydroxyl groups exist in the side chain of HA (Supplementary materials). The peaks of –NHCO- and –COOH in HA overlapped in 1,616.04 cm−1. However, in HA-VES, these two peaks appeared in 1,558.11 cm−1 and 1,653.18 cm−1, respectively, and the peak of –NHCO- was strengthened after VES-NH2 conjugated with HA due to more –NHCO- groups in HA-VES. In conclusion, these results demonstrated that HA-VES was successfully synthesized.
The DS of VES in HA-VES, controlled by adjusting the molar ratio of VES to HA, was calculated by the relative intensity ratio between the representative peaks of N-acetyl peak in HA at 1.8 ppm and the methyl group in VES at 0.8 ppm in the 1H-NMR of HA-VES (Supplementary materials). When the molar ratios of VES to HA were 5:1, 10:1, and 20:1, the DS of VES in HA-VES copolymer was 4%, 7%, and 12%, respectively, and the copolymers of HA-VES were named as HA-VES4, HA-VES7, and HA-VES12, respectively. The DS of VES increased with the increase in molar ratio of VES to HA from 5:1 to 20:1, because the amino group of VES-NH2 could react with the carboxyl group of HA more completely.
To reflect the self-assembling behavior of HA-VES and the stability of micelles, the CMC values of HA-VES with different DS were determined by spectrofluorometry using pyrene as fluorescence probe.25 The curve of fluorescence intensity ratio (I373/I384), as a function of logarithm of HA-VES concentration, was plotted, and the interaction point of the curve slope was the CMC value (shown in Supplementary materials). The CMC values of HA-VES were 43.3, 21.3, and 9.4 μg/mL, respectively, when the corresponding DS was 4%, 7%, and 12%. The CMC values decreased with increase of DS, that is, stronger hydrophobic domain of VES could encapsulate more pyrene molecules and result in the aggregation of pyrene at a lower concentration. Moreover, such low CMC value of HA-VES is conductive for the structural stability of micelles, which avoids the rapid release of encapsulated drugs from micelles in the systemic circulation before they reached the tumor sites.37
The potential hemolytic activity of HA-VES micelles is one of the most important issues and needs to be determined to evaluate the biocompatibility of copolymers before they are injected into the vein. Hemolysis of HA-VES with different DS and Tween 80 was compared with that of 2% Triton X-100, a surfactant that is considered to have 100% hemolysis (shown in Supplementary materials). Tween 80, a surfactant used for intravenous injection, was used as a positive contrast. Significant hemolysis was observed after the RBCs were exposed to Tween 80, and the hemolysis caused by Tween 80 at a concentration of 0.5 mg/mL reached 81.01%±1.66%. In contrast, HA-VES4, HA-VES7, and HA-VES12 all caused no obvious hemolysis, and they exhibited less than 2.10% hemolysis at a concentration of 4 mg/mL and not more than 8.60% even at a concentration of 10 mg/mL. The very low hemolysis of HA-VES indicated that HA-VES copolymers with different DS exhibited excellent hemocompatibility and could be safe for intravenous administration.
Preparation and characterization of HA-VES/DOX
HA-VES/DOX was prepared by ultrasonic method with HA-VES copolymers having different DS. HA-VES copolymers could self-assemble in aqueous medium to form core/shell nanoassemblies with hydrophobic VES as the inner core and hydrophilic HA as the outer shell. As shown in Figure 1A, HA-VES12/DOX exhibited a particle size of 196.60±2.96 nm and a negative zeta potential of −9.78±0.32 mV, which could be helpful for avoiding the clearance of RES and improving the stability of micelles. The TEM images shown in Figure 1B reveal that blank and HA-VES12/DOX had relatively spherical morphology with uniform size distribution. Supplementary materials summarizes the detailed physicochemical characterization of different HA-VES/DOX micelles, which ranged from 199.30 nm to 253.20 nm in size and had polydispersity index below 0.28. Meanwhile, with an increase of DS values, the average sizes and zeta potentials of HA-VES micelles correspondingly decreased, which was probably because the increased hydrophobic force made the micelles pack more tightly and the reduced carboxyl groups of HA on the surface led to a decrease in particle size and zeta potentials of micelles with higher DS. Moreover, due to the carboxyl groups of HA on the micellar surface, HA-VES/DOX had negative zeta potential, which could protect the micelles from aggregation by means of electrostatic repulsion and maintain the stability of micelles. The EE and DL slightly increased with an increase in DS of micelles, which could be attributed to the better hydrophobic interactions between the hydrophobic DOX and the stronger hydrophobic core in HA-VES polymer with higher DS.
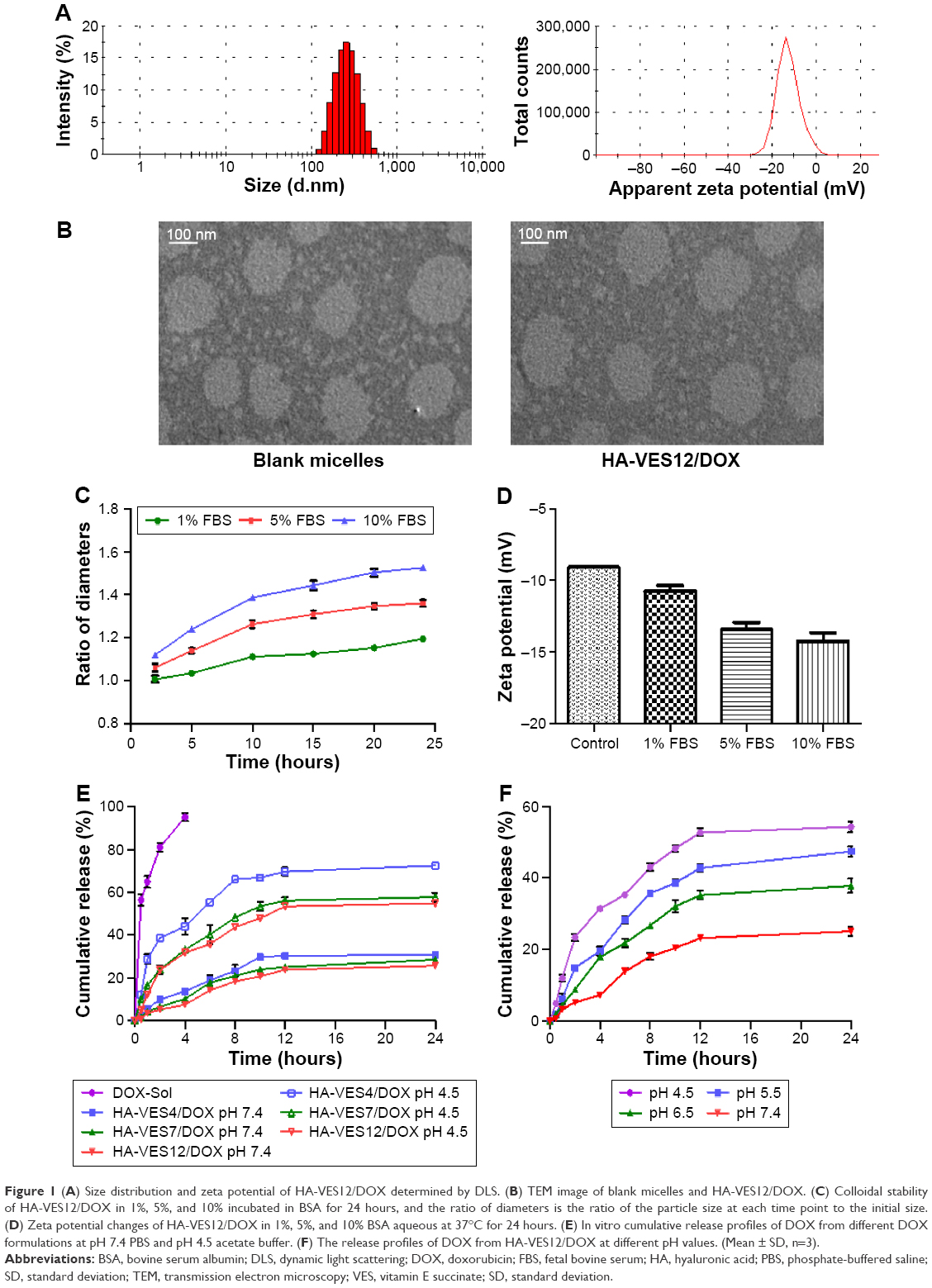 | Figure 1 (A) Size distribution and zeta potential of HA-VES12/DOX determined by DLS. (B) TEM image of blank micelles and HA-VES12/DOX. (C) Colloidal stability of HA-VES12/DOX in 1%, 5%, and 10% incubated in BSA for 24 hours, and the ratio of diameters is the ratio of the particle size at each time point to the initial size. (D) Zeta potential changes of HA-VES12/DOX in 1%, 5%, and 10% BSA aqueous at 37°C for 24 hours. (E) In vitro cumulative release profiles of DOX from different DOX formulations at pH 7.4 PBS and pH 4.5 acetate buffer. (F) The release profiles of DOX from HA-VES12/DOX at different pH values. (Mean ± SD, n=3). |
The colloidal stability of HA-VES12/DOX was investigated in different concentrations of FBS aqueous at 37°C. As shown in Figure 1C and D, the particle sizes were slightly increased with increase in FBS concentrations and incubation times. The size of the micelles in physiological concentration of 5% FBS aqueous increased 1.3-fold of the original size at 24 hours, but still with good dispersibility.38,39 Moreover, even when the concentration of FBS reached 10%, the size of the micelles increased 1.5-fold of the original size after 24-hour incubation, but no visible precipitation of micelles was observed. Due to the slight negative charge of FBS,40 the zeta potentials of HA-VES12/DOX decreased with an increase in FBS concentrations, which could further reduce the adsorption of FBS on the surface of HA-VES micelles by means of charge repulsion and maintain micellar structure integrity in the blood circulation.
The release of DOX from HA-VES/DOX with different DS is shown in Figure 1E, which was rapid at a lysosomal pH (4.5) and slow at a physiological pH (7.4). Furthermore, the release of DOX from HA-VES/DOX was decreased with an increase of copolymer DS under the same pH conditions, due to the stronger hydrophobic force and the structural stability of micelles with higher DS. Moreover, the release behaviors of DOX from HA-VES12/DOX were investigated at pH 7.4 (physiological pH), pH 6.5 (tumor extracellular pH), pH 5.5 (endosomal pH), and pH 4.5 (lysosomal pH), respectively, as shown in Figure 1F. The release of DOX from HA-VES12/DOX was increased with a reduction in pH. Less than 27% DOX was released from HA-VES12/DOX at pH 7.4, indicating that micelles could maintain micellar structure integrity in physiological condition before reaching the tumor sites. In contrast, DOX was rapidly released at pH 5.5 and pH 4.5 within 24 hours, which indicates that DOX could be released more rapidly in endosome and lysosome and has more effective antitumor effects on tumor cells.
In vitro cytotoxicity
Cytotoxicity of HA-VES copolymers
The cytotoxicity of HA-VES copolymers with different DS was evaluated against MCF-7 and MCF-7/Adr cells. As shown in Supplementary materials, the inhibition rates of HA-VES copolymers with different DS increased with the increase in concentrations and incubation times in both MCF-7 and MCF-7/Adr cells, indicating that all HA-VES copolymers have obvious cytotoxicity. The cytotoxicity of HA-VES copolymers could be attributed to the function of VES, which is to selectively induce apoptosis and inhibit the proliferation of tumor cells without any cytotoxic effects on normal cells.25,28 Moreover, enhanced cytotoxicity was observed with increase of DS, especially of HA-VES12, due to more VES existing in high DS of HA-VES12. Therefore, synthetic copolymers of HA-VES with different DS have selective antitumor abilities and could exert antitumor effects.
Cytotoxicity of HA-VES/DOX micelles
In vitro cytotoxicity of DOX-Sol, the mixture of DOX-Sol and HA-VES4 polymer, and HA-VES/DOX was investigated in DOX-sensitive MCF-7 and DOX-resistant MCF-7/Adr cells by MTT assay and the IC50 values were also calculated. As shown in Supplementary materials, the inhibition rates of DOX in all formulations were also time- and concentration dependent in both cells. The cytotoxicity of the mixture of DOX-Sol and HA-VES4 copolymer was higher than that of DOX-Sol or HA-VES4 copolymer alone in both cell lines, and HA-VES4 polymer had synergistic effects on DOX. In MCF-7 cells, due to the low expression of P-gp, DOX was quickly internalized into the cells by passive diffusion, resulting in no obvious differences in cytotoxicity with HA-VES/DOX and the mixture of HA-VES copolymer and DOX. However, in MCF-7/Adr cells, the cytotoxicity of DOX was obviously enhanced in the cells treated with HA-VES/DOX compared with that of DOX-Sol group. Meanwhile, the IC50 value of DOX in the mixture of HA-VES4 copolymer and DOX (30.40 μg/mL) was lower than that of DOX (38.04 μg/mL) and HA-VES4 copolymer (189.71 μg/mL) after 96 hours of incubation in MCF-7/Adr cells, which once again proves that HA-VES copolymer has synergistic antitumor effect. The IC50 values of DOX in HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX micelles were respectively 9.83-, 12.27-, and 13.74-fold lower than that of DOX-Sol in MCF-7/Adr cells after incubation for 96 hours. Moreover, the IC50 value of DOX in HA-VES4/DOX micelles was 49.04- and 7.85-times lower than that of HA-VES4 copolymer only and the mixture of DOX and HA-VES copolymer, respectively, after 96 hours of incubation in MCF-7/Adr cells. Therefore, HA-VES/DOX could enhance the sensitivity of resistant cells and overcome MDR, which can be attributed to the synergistic antitumor effect of the released HA-VES copolymer and the escape of P-gp recognition by encapsulating DOX in hydrophobic core.
As shown in Supplementary materials, RI and RF values were calculated to evaluate MDR reversal effects of HA-VES/DOX micelles after 96 hours of incubation. The RI value of DOX-Sol was 88.57, indicating that MCF-7/Adr cells have strong resistant effects on DOX and could be selected as a good model for MDR investigation. However, the RI values of HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX micelles decreased to 9.56, 8.47, and 8.55, respectively, which were 9.26-, 10.46-, and 10.36-fold lower than that of DOX-Sol. These results revealed that the sensitivity against resistant cells was significantly enhanced and MDR was partly overcome by encapsulating DOX into HA-VES micelles. Meanwhile, the RF values of HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX micelles were 9.83, 12.27, and 13.74, respectively, which once again proved that MDR was partly overcome by micelles. Specifically, based on the smaller size of micelles and the cytotoxicity of the released copolymer with high DS, the MDR reversal effects of micelles were augmented with increase of DS.
Apoptosis
HA-VES/DOX could enhance cytotoxicity and overcome MDR of DOX in P-gp–overexpressed MCF-7/Adr cells. Therefore, the effects of micelles on promoting cell apoptosis were evaluated in MCF-7/Adr cells. As shown in Figure 2A, most of the cells apeared to have apoptotic morphology after incubation with DOX formulation, such as membrane association and chromatin marginalization. Furthermore, apoptotic cells were quantified by FACS (Figure 2B) and it was found that 10.8% of cell apoptosis was induced after incubation with DOX for 48 hours. However, HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX induced 32.1%, 50%, and 48.7% cell apoptosis, resulting in 2.97-, 4.63-, and 4.51-fold increase in apoptotic cells, respectively, compared with DOX-Sol treated cells. Therefore, DOX encapsulated in HA-VES micelles was more effective in promoting cell apoptosis. Specifically, apoptotic cells in HA-VES7/DOX and HA-VES12/DOX treated group were nearly 1.56-fold higher than those in HA-VES4/DOX treated group, which was due to the stronger synergistic anticancer effects of the polymer and the smaller particle size of micelles with high DS. This was consistent with the results of in vitro cytotoxicity.
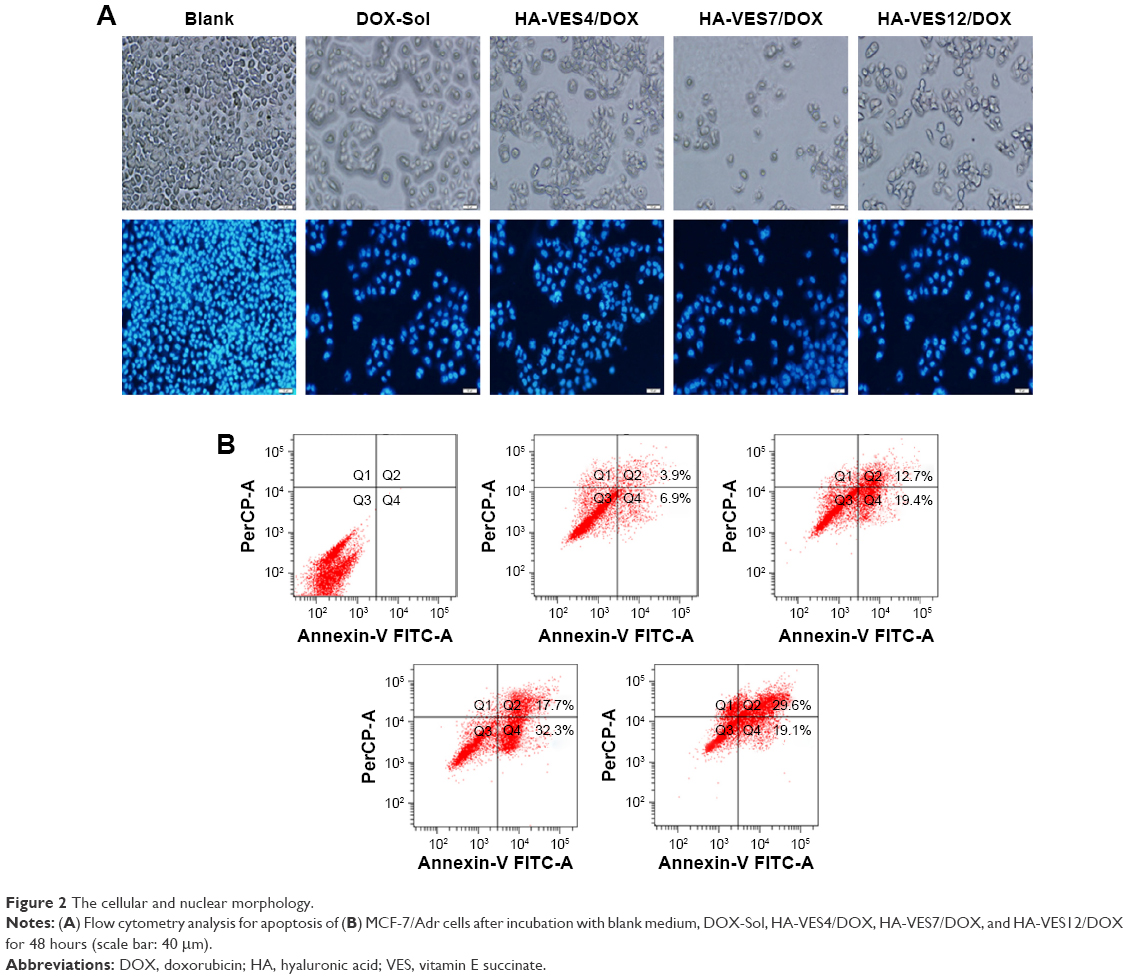 | Figure 2 The cellular and nuclear morphology. |
Apoptotic pathways in MCF-7/Adr cells
Caspase activation and PARP cleavage are the key markers of cells undergoing apoptosis. Therefore, after DOX formulation was pretreated for 48 hours, caspase 3 and PARP activation were detected by Western blot analysis. As shown in the results in Supplementary materials, caspase 3 was not activated. However, treatment of MCF-7/Adr cells with HA-VES12 polymer slightly upregulated the expression of PARP compared with the control group, but this was significantly lower when compared to the different DOX groups. Moreover, it is expected that PARP expression would be markedly higher for the HA-VES/DOX micelles compared with the DOX-Sol group, but showed no significant difference among the three HA-VES/DOX groups. Accordingly, HA-VES/DOX micelles activated the apoptotic pathway through the activation of PARP in MCF-7/Adr cells.
CD44 competitive inhibition assay
To evaluate the CD44-targeting efficiency of HA-VES/DOX micelles in tumor cells, the intracellular uptake of various DOX formulations was studied quantitatively and qualitatively by CLSM and FACS in MCF-7 and HepG2 cells. As shown in the CLSM images in Figure 3A and B, enhanced fluorescence intensity of DOX was observed in both MCF-7 and HepG2 cells treated with HA-VES/DOX, in comparison with DOX-Sol treated group. Even more remarkable is the finding that the fluorescence of HA-VES/DOX in MCF-7 cells was significantly higher than that in HepG2 cells, since CD44 receptor is expressed more in MCF-7 cells compared to HepG2 cells. Moreover, after competitive incubation with HA, the fluorescence of DOX in HA-VES12/DOX + HA treated group was significantly decreased in comparison with HA-VES12/DOX treated group in MCF-7 cells, whereas no changes were observed in HepG2 cells. Above all, these results suggested that the cellular uptake of HA-VES/DOX was mediated by CD44 receptor targeting based on HA.
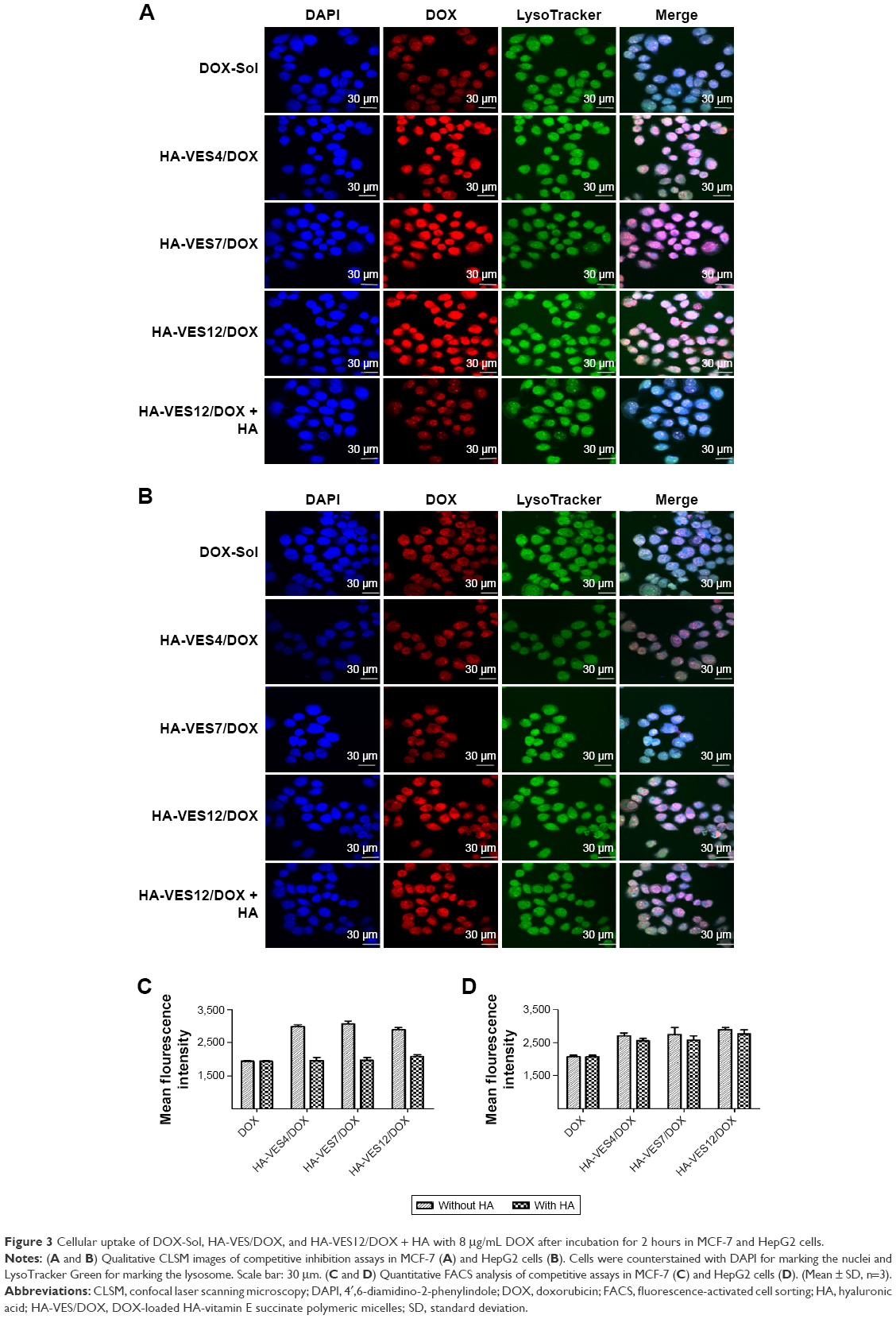 | Figure 3 Cellular uptake of DOX-Sol, HA-VES/DOX, and HA-VES12/DOX + HA with 8 μg/mL DOX after incubation for 2 hours in MCF-7 and HepG2 cells. |
The cellular uptake of HA-VES/DOX micelles was further quantitatively measured by FACS (Figure 3C and D). Free HA has no effect on the cellular uptake of DOX-Sol in both MCF-7 and HepG2 cells. Moreover, the mean fluorescence intensity of HA-VES/DOX in MCF-7 cells was higher than that in HepG2 cells. More importantly, after preincubation with HA, the mean fluorescence intensity of HA-VES/DOX was significantly reduced in MCF-7 cells, whereas no obvious changes were observed in HepG2 cells, which was in accordance with the CLSM results. In conclusion, both CLSM and FACS results revealed that HA-VES/DOX could enhance the cellular uptake of DOX in a CD44 receptor-mediated endocytosis on CD44-overexpressing cells.
qRT-PCR analysis of CD44 expression
To further determine the targeted efficiency of HA-VES micelles on CD44, the expression of CD44 mRNA was quantified by qRT-PCR. As shown in Figure 4, compared with DOX-Sol treated group, the expression of CD44 was significantly decreased in MCF-7 cells after incubation with HA-VES/DOX (P<0.05), indicating that HA-VES micelles could specifically conjugate with CD44 and then decrease CD44 expression. Therefore, HA-VES/DOX has CD44-targeting ability. Specifically, compared with HA-VES12/DOX, HA decreased the expression of CD44, which further confirmed that the CD44-targeting effects of HA-VES micelles were mainly derived from HA. Moreover, compared with HA-VES12/DOX treated group, the expression of CD44 was lower in cells treated with HA-VES4/DOX and HA-VES7/DOX. It demonstrated that the micelles with low DS could better identify CD44 receptors and then decrease CD44 expression, which is in agreement with a previous finding that the carboxylic groups of HA were the main CD44 target sites. So, the micelles with high DS exhibited little lower CD44 targeting.
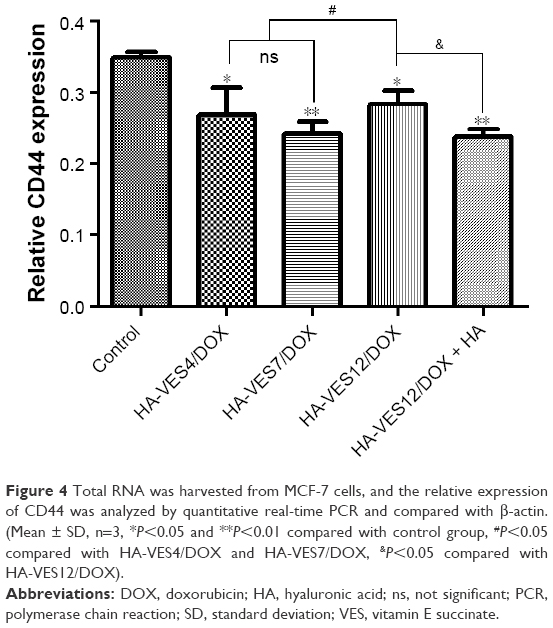 | Figure 4 Total RNA was harvested from MCF-7 cells, and the relative expression of CD44 was analyzed by quantitative real-time PCR and compared with β-actin. (Mean ± SD, n=3, *P<0.05 and **P<0.01 compared with control group, #P<0.05 compared with HA-VES4/DOX and HA-VES7/DOX, &P<0.05 compared with HA-VES12/DOX). |
Cellular uptake and efflux in MCF-7/Adr cells
To further evaluate the effects of HA-VES/DOX on overcoming MDR, cellular uptake and efflux assays were performed in MCF-7/Adr cells. For quantitative study, as shown in Figure 5A, the mean fluorescence intensity of DOX in all formulations increased with the extension of time. The intracellular accumulation of DOX-Sol was relatively lower due to P-gp efflux, whereas HA-VES/DOX significantly enhanced the uptake of DOX in MCF-7/Adr cells through avoiding P-gp efflux by endocytosis. Moreover, due to the smaller size of micelles and their easier penetration into resistant cells, HA-VES/DOX micelles with higher DS led to higher cellular accumulation of DOX in MCF-7/Adr cells, resulting in enhanced cytotoxicity.
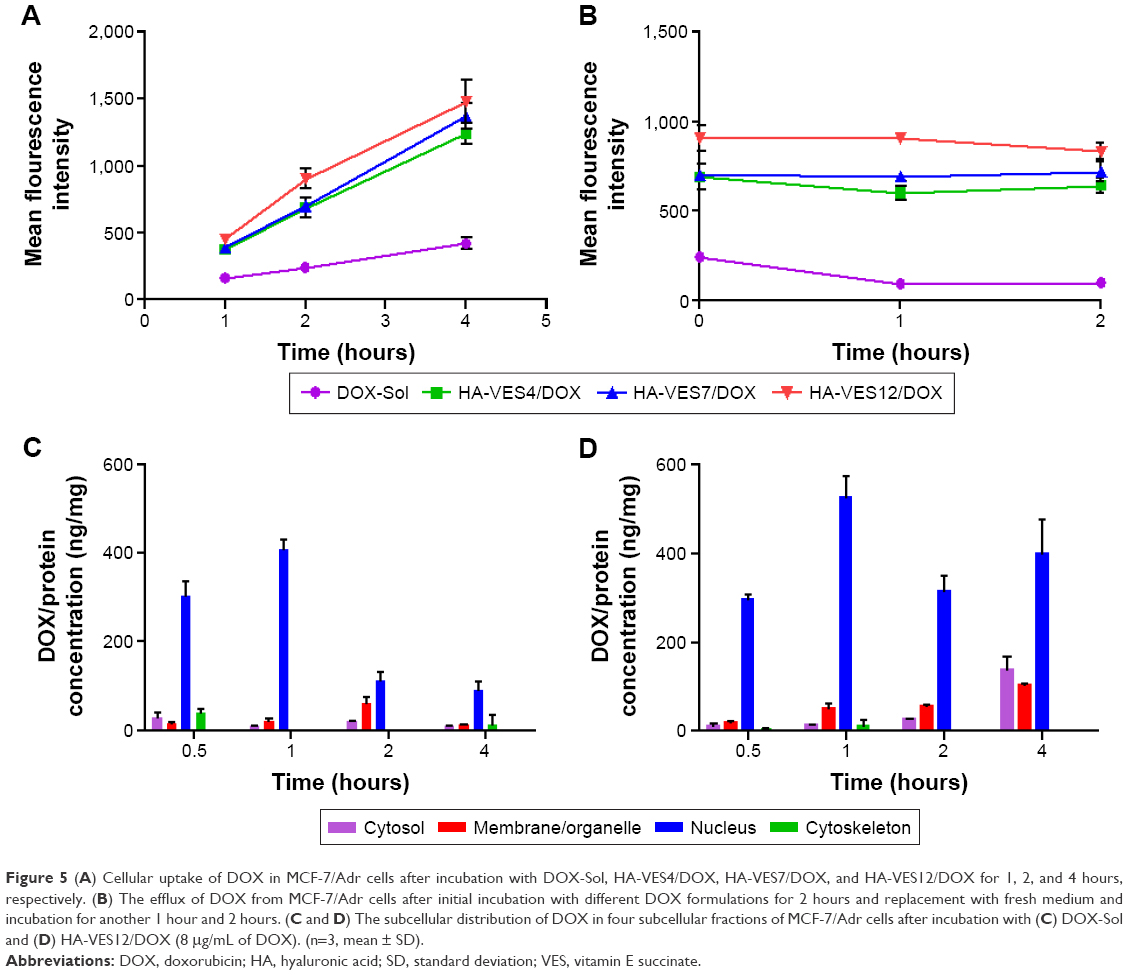 | Figure 5 (A) Cellular uptake of DOX in MCF-7/Adr cells after incubation with DOX-Sol, HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX for 1, 2, and 4 hours, respectively. (B) The efflux of DOX from MCF-7/Adr cells after initial incubation with different DOX formulations for 2 hours and replacement with fresh medium and incubation for another 1 hour and 2 hours. (C and D) The subcellular distribution of DOX in four subcellular fractions of MCF-7/Adr cells after incubation with (C) DOX-Sol and (D) HA-VES12/DOX (8 μg/mL of DOX). (n=3, mean ± SD). |
In cellular efflux study, the intracellular amount of DOX retained in MCF-7/Adr cells gradually decreased with incubation time prolongation in DOX-Sol group, as a result of P-gp efflux (Figure 5B). As expected, the intracellular amounts of DOX were still higher in cells treated with HA-VES/DOX and almost with no downward trend, indicating that DOX encapsulated in HA-VES/DOX could overcome P-gp–mediated drug efflux and reverse the MDR.
Endocytosis mechanism in MCF-7/Adr cells
Endocytic pathways were involved in the primary cellular uptake mechanism of nanoparticle.41,42 Endocytosis mechanism of HA-VES/DOX based on HA-VES12/DOX was investigated with various specific endocytosis inhibitors in MCF-7/Adr cells. As shown in Supplementary materials, compared with HA-VES12/DOX, no changes in the uptake of DOX were observed after incubation with a clathrin-mediated endocytosis inhibitor of chlorpromazine,43 and a caveolae- and clathrin-independent endocytosis inhibitor of quercetin,25 indicating that clathrin-mediated endocytosis and caveolae- and clathrin-independent endocytosis were not involved in the endocytosis pathways of HA-VES12/DOX. Interestingly, intracellular uptake of HA-VES12/DOX was significantly decreased after co-incubation with sodium azide (energy inhibitor) (P<0.01), β-cyclodextrin (inhibitor of lipid raft and caveolae-dependent endocytosis)44 (P<0.01), indomethacin (inhibitor of cyclooxygenase and caveolae-mediated endocytosis)45 (P<0.01), and amiloride (the inhibitor of Na+/H+ pump and micropinocytosis)45 (P<0.05). Accordingly, these results demonstrated that the endocytosis pathways of HA-VES12/DOX were energy-dependent, caveolae-mediated endocytosis and micropinocytosis.
Subcellular fraction distribution of DOX in MCF-7/Adr cells
Four subcellular fractions were investigated to study the subcellular distribution of DOX-Sol and HA-VES12/DOX in MCF-7/Adr cells. As shown in Figure 5C and D, DOX was mainly distributed in the nucleus at each time point in the cells treated with either DOX-Sol group or HA-VES12/DOX. The results were consistent with the mechanism of action of DOX, which works by interfering with DNA and RNA biosynthesis to induce tumor cell death. Notably, enhanced accumulation in the nucleus was observed in HA-VES12/DOX treated group compared with DOX-Sol treated group, which led to obvious cytotoxicity. This was in accordance with the results of subcellular localization discussed below.
Subcellular localization
Furthermore, subcellular localization of DOX-Sol and HA-VES/DOX with different DS was also visualized using CLSM in MCF-7/Adr cells. As shown in Supplementary materials, the red fluorescence of DOX in all DOX formulations was increased with the extension of time. Less red fluorescence of DOX was observed in cells treated with DOX-Sol, while HA-VES/DOX significantly enhanced the fluorescence intensity and cellular uptake of DOX compared with DOX-Sol after either 1 hour or 2 hours of co-incubation, which was in accordance with the results of FACS. Meanwhile, after 1 hour of co-incubation, strong red fluorescence of DOX in HA-VES/DOX groups was co-localized with the green fluorescence of lysosomes to generate large yellow fluorescence in merged images, and weak red fluorescence was observed near or in nuclei, indicating that HA-VES/DOX micelles were initially taken up into lysosome and then they gradually escaped from the lysosome into the nucleus. By contrast, most of the DOX was dispersed in cell nuclei after incubation for 2 hours with HA-VES/DOX, demonstrating that DOX was released from HA-VES/DOX in lysosome, followed by its successful escape from the lysosome and entry into the nuclei by simple diffusion to exert its effects.
To sum up, HA-VES/DOX enhanced cytotoxicity and overcame P-gp–mediated MDR by the following pathways: (1) CD44 receptor-mediated active target improved the accumulation of HA-VES/DOX on tumor cells; (2) HA-VES/DOX was internalized into P-gp–overexpressed cells by endocytosis and it improved DOX uptake by reducing the recognition of P-gp and escaping the P-gp efflux; and (3) the released HA-VES copolymer had a synergistic anticancer effect.
In vivo pharmacokinetics
The mean plasma concentration–time curves of DOX in rats after intravenous administration of DOX-Sol, HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX are shown in Figure 6A, and the main pharmacokinetic parameters are summarized in Supplementary materials. DOX-loaded micelles significantly enhanced the plasma concentration of DOX and changed the main pharmacokinetic parameters of area under the curve (AUC)0–t, t1/2, and MRT0–t, compared with DOX-Sol. The AUC0–t for HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX was 2.92-, 3.31-, and 4.05-fold higher, t1/2 was 3.31-, 2.40-, and 3.85-times more, and MRT0–t was 3.91-, 3.56-, and 4.42-fold higher, respectively, than those of DOX-Sol. The results implied that HA-VES/DOX, especially based on high DS of HA-VES micelles, could significantly improve the concentration in blood and prolong the blood circulation of DOX.
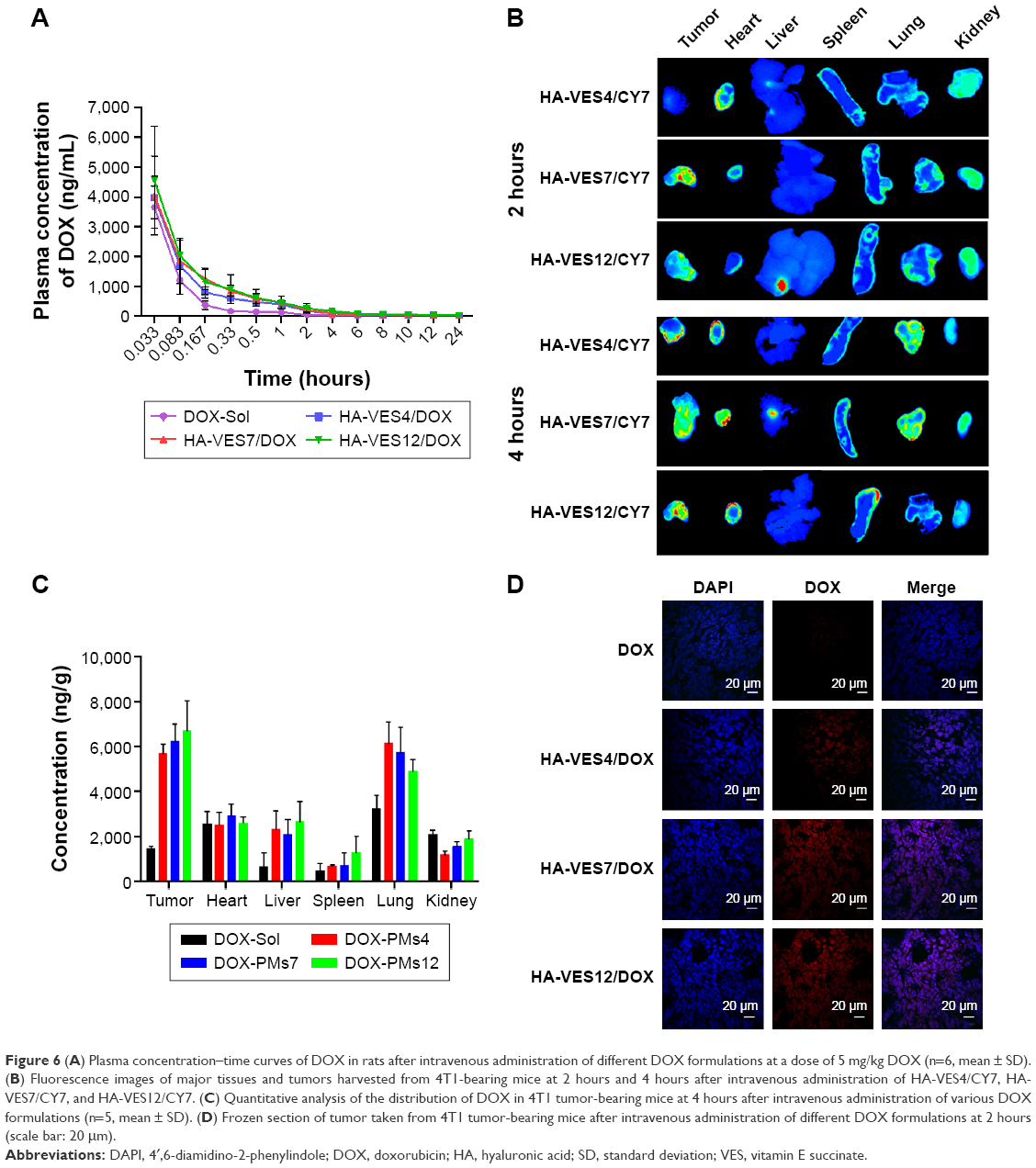 | Figure 6 (A) Plasma concentration–time curves of DOX in rats after intravenous administration of different DOX formulations at a dose of 5 mg/kg DOX (n=6, mean ± SD). (B) Fluorescence images of major tissues and tumors harvested from 4T1-bearing mice at 2 hours and 4 hours after intravenous administration of HA-VES4/CY7, HA-VES7/CY7, and HA-VES12/CY7. (C) Quantitative analysis of the distribution of DOX in 4T1 tumor-bearing mice at 4 hours after intravenous administration of various DOX formulations (n=5, mean ± SD). (D) Frozen section of tumor taken from 4T1 tumor-bearing mice after intravenous administration of different DOX formulations at 2 hours (scale bar: 20 μm). |
In vivo tumor targeting effects
As shown in Figure 6B, the ex vivo fluorescent images of excised organs and tumors confirmed that most of the micelles accumulated in the tumor, and some were distributed in heart, lung, and kidney because of the unavoidable uptake by RES. Meanwhile, Cy7-loaded micelles exhibited tumor accumulation, and the fluorescence intensity in tumor increased with the prolongation of time, mainly due to the EPR effects and CD44-targeted effects of HA-VES micelles. Moreover, in excised tumors, higher fluorescence intensity was found in the group treated with HA-VES7/Cy7 and HA-VES12/Cy7 compared with HA-VES4/Cy7, which was consistent with the results of cellular uptake in vitro and further confirmed that high tumor accumulation was observed in the micelles with higher DS.
The biodistribution of DOX in DOX formulations was further quantified and the results are shown in Figure 6C. The quantitative distribution results of DOX in vivo were in accordance with the qualitative results obtained by fluorescence imaging, indicating that HA-VES/DOX micelles mainly accumulated in the tumor, followed by lung. Therefore, higher accumulation of HA-VES/DOX in tumor would be conducive to exert better antitumor effects in vivo.
Moreover, to observe the distribution of DOX in tumor tissues, the tumors were frozen, sectioned, stained with DAPI, and visualized under CLSM (Figure 6D). The red fluorescence of DOX was barely visualized in tumors treated with DOX-Sol. On the contrary, more red fluorescence was observed in tumors treated with HA-VES/DOX, especially in HA-VES7/DOX and HA-VES12/DOX treated groups, demonstrating that HA-VES/DOX micelles could enhance tumor permeability and accumulation of DOX. Enhanced accumulation of HA-VES/DOX in tumors could be attributed to the small particle size, EPR, and CD44-targeting effects, leading to the extension of circulation time in vivo, and better permeability in tumors.
Antitumor effects in vivo
The antitumor efficacy of DOX formulations was determined in mice bearing 4T1 tumor. Due to the fast growth of tumor, the body weight of saline-treated mice exhibited fast increase (shown in Figure 7A). The body weight of the mice treated with DOX-Sol was markedly decreased, demonstrating serious side effects of DOX-Sol. However, the body weights in the HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX groups did not significantly vary, which indicates that DOX encapsulated in micelles could remarkably reduce the systemic toxicity of DOX. As shown in Figure 7B and C, the tumor weight and tumor volume were significantly affected by DOX formulations compared with saline group. Excitingly, enhanced antitumor efficiency was achieved in mice treated with HA-VES/DOX micelles compared with DOX-Sol group, almost completely inhibiting the growth of tumor. HA-VES12/DOX exhibited superior tumor growth inhibition among the three groups of micelles with different DS, which was perhaps due to the increased cellular uptake, the synergistic effect of nanocarrier and DOX, improved cytotoxicity, and smaller particle size. At the end of the experiments, the tumors were excised and photographed, and HA-VES/DOX showed higher antitumor efficiency compared with DOX-Sol, once again proving that HA-VES/DOX could improve tumor accumulation and further increase antitumor activity (Figure 7D).
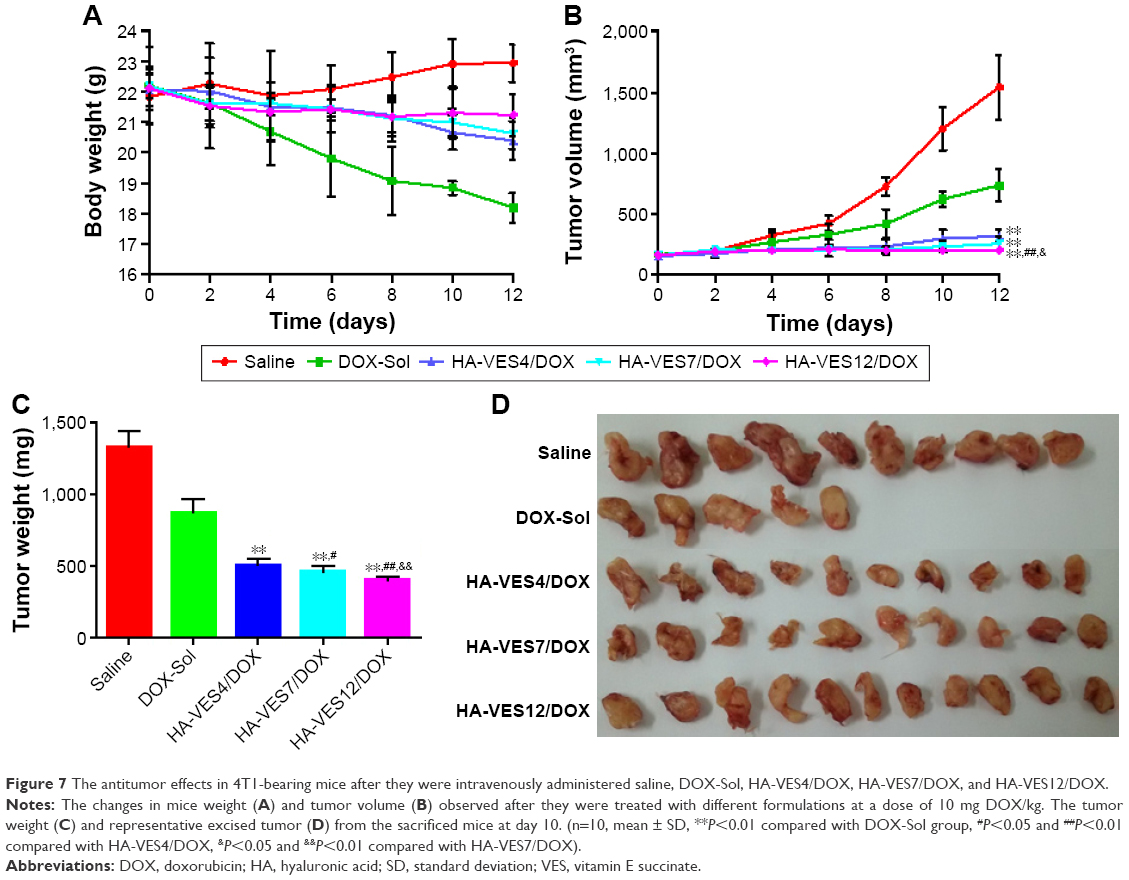 | Figure 7 The antitumor effects in 4T1-bearing mice after they were intravenously administered saline, DOX-Sol, HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX. |
To further investigate the toxicity of DOX formulation in vivo, the important hepatic and cardiac health indicators, that is, AST, LDH, CK, CKMB, were employed to evaluate the hepatic toxicity and cardiotoxicity. As shown in Supplementary materials, the values of CKMB, CK, LDH, and AST in the mice treated with DOX-Sol were significantly higher than in saline-treated group, indicating that DOX caused serious hepatic and cardiac toxicity in vivo. However, the four parameters reduced remarkably in mice treated with HA-VES/DOX compared with DOX-Sol treated group and they were the same as in saline-treated group, indicating that HA-VES/DOX could significantly alleviate the serious hepatic toxicity and cardiotoxicity of DOX and could even cause no damage to the liver and heart during treatment.
To further evaluate the therapeutic efficacy and toxicity of HA-VES/DOX to mice in vivo, the histological analysis of major organs including heart, liver, spleen, lung, kidney, and tumors was performed after 12 days of intravenous administration of different formulations and the results are shown in Figure 8. Results of hematoxylin and eosin staining shown in Figure 8A reveal obvious pathological damage in myocardial cells and spleen cells, such as membranolysis and nuclear lysis and fusion, of mice treated with DOX-Sol, while the mice treated with all HA-VES/DOX exhibited no histological and pathological changes. These results further confirmed that HA-VES/DOX micelles could reduce the systemic and organizational toxicity of DOX.
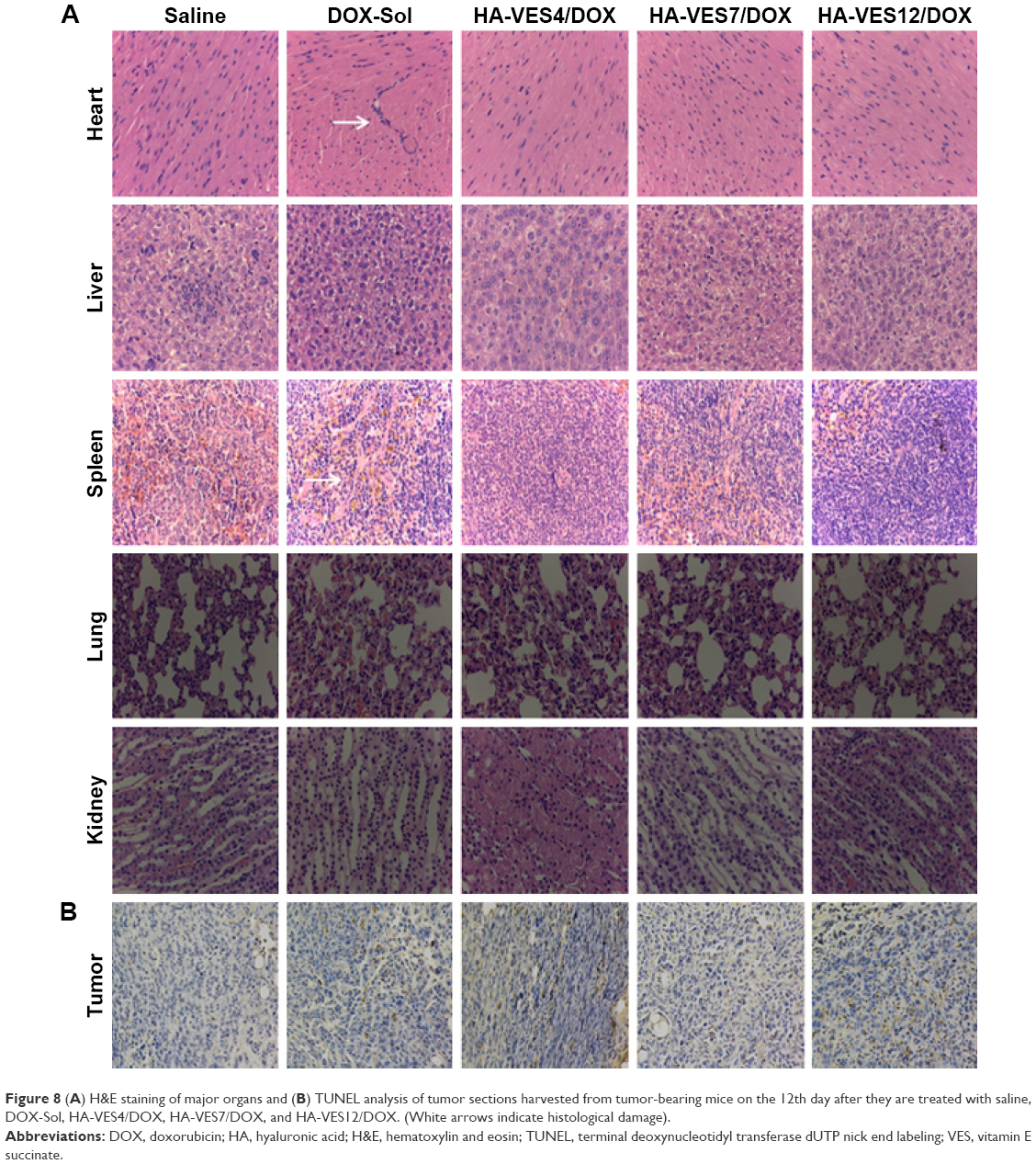 | Figure 8 (A) H&E staining of major organs and (B) TUNEL analysis of tumor sections harvested from tumor-bearing mice on the 12th day after they are treated with saline, DOX-Sol, HA-VES4/DOX, HA-VES7/DOX, and HA-VES12/DOX. (White arrows indicate histological damage). |
Moreover, the apoptosis tumor cells were stained dark brown by TUNEL assay (shown in Figure 8B). The apoptosis cells with dark brown staining were scarce in saline group, but some dark brown spots were found in DOX-Sol treated group, demonstrating that DOX-Sol exerted its effects on tumor through inducing tumor cell apoptosis. Obviously, enhanced dark brown spots were found in tumors treated with all HA-VES/DOX compared with DOX-Sol group, indicating that DOX loaded in HA-VES could significantly improve the ability of DOX to induce tumor cell apoptosis and exhibit better antitumor effects. It could be attributed to the nanosize and CD44 targeting of HA-VES/DOX micelles, by which the micelles could be absorbed through EPR effects from blood into the tumors sites, through CD44 receptor target on the tumor cell surface, and then are transported into the tumor cells by endocytosis bypassing the P-gp efflux (Figure 9). Above all, HA-VES/DOX could significantly improve the antitumor effects of DOX and reduce the systemic toxicity in vivo.
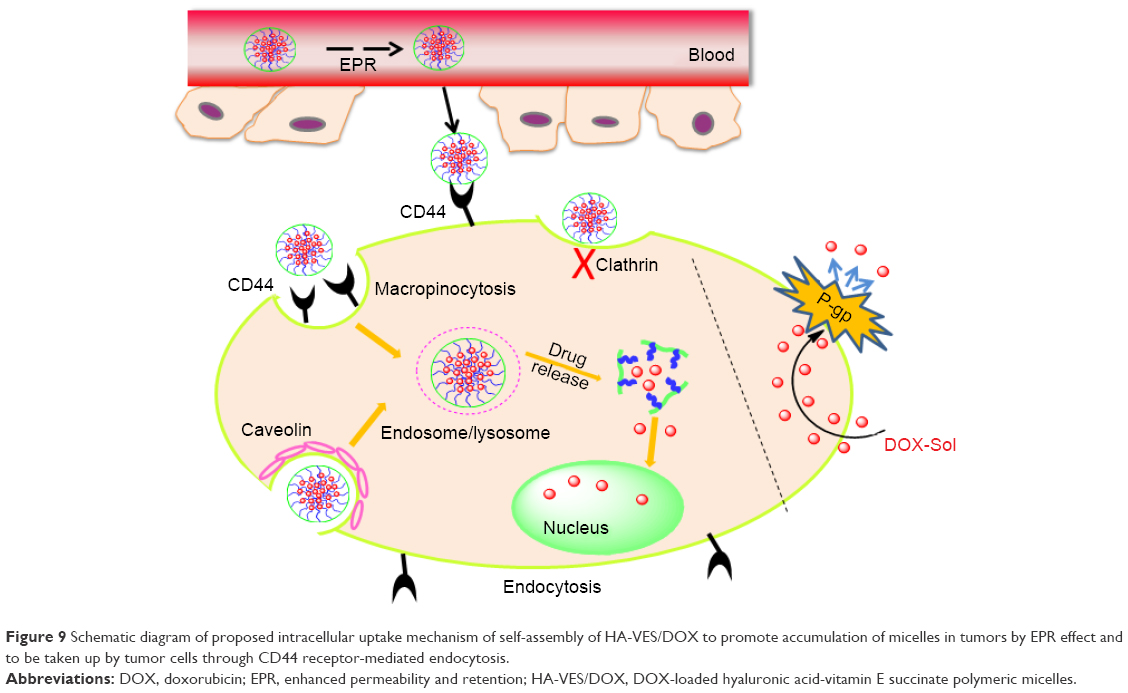 | Figure 9 Schematic diagram of proposed intracellular uptake mechanism of self-assembly of HA-VES/DOX to promote accumulation of micelles in tumors by EPR effect and to be taken up by tumor cells through CD44 receptor-mediated endocytosis. |
Conclusion
In summary, three different DS of HA-VES copolymers with synergistic antitumor effect and CD44 targeting were synthesized and used as nanocarriers to improve the targeted delivery and antitumor efficiency of DOX. In vitro, HA-VES/DOX, especially HA-VES12/DOX with high DS of 12%, exhibited enhanced cytotoxicity with synergistic antitumor effects of HA-VES copolymers, induced cell apoptosis, reversed MDR, and improved cellular uptake based on EPR effects and active recognition between HA and CD44 receptors on the tumor surface. Importantly, HA-VES/DOX showed excellent antitumor effects with low systemic toxicity in vivo. The dual-functional HA-VES/DOX, especially HA-VES12/DOX, could be a promising vehicle for effective and targeted tumor therapy.
Acknowledgments
Specialized Research Fund for the Doctoral Program of Higher Education (No 20130013120008) and National Natural Science Foundation of China (No 81503262) are gratefully acknowledged for financial support.
Disclosure
The authors report no conflicts of interest in this work.
References
Wu Q, Li G, Deng S, et al. Enhanced antitumor activity and mechanism of biodegradable polymeric micelles-encapsulated chetomin in both transgenic zebrafish and mouse models. Nanoscale. 2014;6(20):11940–11952. | ||
Zhao Y, Zhou Y, Wang D, et al. pH-responsive polymeric micelles based on poly(2-ethyl-2-oxazoline)-poly(D,L-lactide) for tumor-targeting and controlled delivery of doxorubicin and P-glycoprotein inhibitor. Acta Biomater. 2015;17:182–192. | ||
Xiong XB, Falamarzian A, Garg SM, Lavasanifar A. Engineering of amphiphilic block copolymers for polymeric micellar drug and gene delivery. J Control Release. 2011;155(2):248–261. | ||
Li PY, Lai PS, Hung WC, Syu WJ. Poly(L-lactide)-Vitamin E TPGS nanoparticles enhanced the cytotoxicity of doxorubicin in drug-resistant MCF-7 breast cancer cells. Biomacromolecules. 2010;11(10):2576–2582. | ||
Mohamed S, Parayath NN, Taurin S, Greish K. Polymeric nano-micelles versatile platform for targeted delivery in cancer. Ther Deliv. 2014;5(10):1101–1121. | ||
Pan J, Feng SS. Targeted delivery of paclitaxel using folate-decorated poly(lactide)-vitamin E TPGS nanoparticles. Biomaterials. 2008;29(17):2663–2672. | ||
Wang M, Thanou M. Targeting nanoparticles to cancer. Pharmacol Res. 2010;62(2):90–99. | ||
Lim EK, Kim HO, Jang E, et al. Hyaluronan-modified magnetic nanoclusters for detection of CD44-overexpressing breast cancer by MR imaging. Biomaterials. 2011;32(31):7941–7950. | ||
Chen H, Zhang T, Zhou Z, et al. Enhanced uptake and cytotoxity of folate-conjugated mitoxantrone-loaded micelles via receptor up-regulation by dexamethasone. Int J Pharm. 2013;448(1):142–149. | ||
Yang G, Wang J, Wang Y, Li L, Guo X, Zhou S. An implantable active-targeting micelle-in-nanofiber device for efficient and safe cancer therapy. ACS Nano. 2015;9(2):1161–1174. | ||
Sun B, Feng SS. Trastuzumab-functionalized nanoparticles of biodegradable copolymers for targeted delivery of docetaxel. Nanomedicine. 2009;4(4):431–445. | ||
Du W, Fan Y, Zheng N, et al. Transferrin receptor specific nanocarriers conjugated withfunctional 7peptide for oral drug delivery. Biomaterials. 2013;34(3):794–806. | ||
Xu W, Siddiqui IA, Nihal M, et al. Aptamer-conjugated and doxorubicin-loaded unimolecular micelles for targeted therapy of prostate cancer. Biomaterials. 2013;34(21):5244–5253. | ||
Han X, Li Z, Sun J, et al. Stealth CD44-targeted hyaluronic acid supramolecular nanoassemblies for doxorubicin delivery: probing the effect of uncovalent pegylation degree on cellular uptake and blood long circulation. J Control Release. 2015;197:29–40. | ||
Li J, He Y, Sun W, et al. Hyaluronic acid-modified hydrothermally synthesized iron oxide nanoparticles for targeted tumor MR imaging. Biomaterials. 2014;35(11):3666–3677. | ||
Li J, Yin T, Wang L, Yin L, Zhou J, Huo M. Biological evaluation of redox-sensitive micelles based on hyaluronic acid-deoxycholic acid conjugates for tumor-specific delivery of paclitaxel. Int J Pharm. 2015;483(1–2):38–48. | ||
Ge Z, Chen Q, Osada K, et al. Targeted gene delivery by polyplex micelles with crowded PEG palisade and cRGD moiety for systemic treatment of pancreatic tumors. Biomaterials. 2014;35(10):3416–3426. | ||
Choi KY, Yoon HY, Kim JH, et al. Smart nanocarrier based on PEGylated hyaluronic acid for cancer therapy. ACS Nano. 2011;5(11):8591–8599. | ||
Datir SR, Das M, Singh RP, Jain S. Hyaluronate tethered, “smart” multiwalled carbon nanotubes for tumor-targeted delivery of doxorubicin. Bioconjug Chem. 2012;23(11):2201–2213. | ||
Upadhyay KK, Bhatt AN, Mishra AK, et al. The intracellular drug delivery and anti tumor activity of doxorubicin loaded poly(gamma-benzyl L-glutamate)-b-hyaluronan polymersomes. Biomaterials. 2010;31(10):2882–2892. | ||
Yadav AK, Mishra P, Mishra AK, Jain S, Agrawal GP. Development and characterization of hyaluronic acid-anchored PLGA nanoparticulate carriers of doxorubicin. Nanomedicine. 2007;3(4):246–257. | ||
Li J, Huo M, Wang J, et al. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials. 2012;33(7):2310–2320. | ||
De Santis S, Diociaiuti M, Cametti C, Masci G. Hyaluronic acid and alginate covalent nanogels by template cross-linking in polyion complex micelle nanoreactors. Carbohydr Polym. 2014;101:96–103. | ||
Thomas RG, Moon M, Lee S, Jeong YY. Paclitaxel loaded hyaluronic acid nanoparticles for targeted cancer therapy: in vitro and in vivo analysis. Int J Biol Macromol. 2015;72:510–518. | ||
Wang J, Sun J, Chen Q, et al. Star-shape copolymer of lysine-linked di-tocopherol polyethylene glycol 2000 succinate for doxorubicin delivery with reversal of multidrug resistance. Biomaterials. 2012;33(28):6877–6888. | ||
Youk HJ, Lee E, Choi MK, et al. Enhanced anticancer efficacy of alpha-tocopheryl succinate by conjugation with polyethylene glycol. J Control Release. 2005;107(1):43–52. | ||
Ireland DJ, Kissick HT, Beilharz MW. Alpha-Tocopheryl succinate: toxicity and lack of anti-tumour activity in immuno-competent mice. Food Chem Toxicol. 2008;46(2):508–512. | ||
Kanai K, Kikuchi E, Mikami S, et al. Vitamin E succinate induced apoptosis and enhanced chemosensitivity to paclitaxel in human bladder cancer cells in vitro and in vivo. Cancer Sci. 2010;101(1):216–223. | ||
Liang D, Wang AT, Yang ZZ, Liu YJ, Qi XR. Enhance cancer cell recognition and overcome drug resistance using hyaluronic acid and alpha-tocopheryl succinate based multifunctional nanoparticles. Mol Pharm. 2015;12(6):2189–2202. | ||
Qiu L, Li Z, Qiao M, et al. Self-assembled pH-responsive hyaluronic acid-g-poly((L)-histidine) copolymer micelles for targeted intracellular delivery of doxorubicin. Acta Biomater. 2014;10(5):2024–2035. | ||
Liu Y, Sun J, Lian H, Cao W, Wang Y, He Z. Folate and CD44 receptors dual-targeting Hydrophobized hyaluronic acid paclitaxel-loaded polymeric micelles for overcoming multidrug resistance and improving tumor distribution. J Pharm Sci. 2014;103(5):1538–1547. | ||
Chen D, Sun J, Sun K, Liu W, Wu Z. In vivo evaluation of novel ketal-based oligosaccharides of hyaluronan micelles as multifunctional CD44 receptor-targeting and tumor pH-responsive carriers. Artif Cells Nanomed Biotechnol. 2016;44(3):898–902. | ||
Zhang P, Ling G, Pan X, et al. Novel nanostructured lipid-dextran sulfate hybrid carriers overcome tumor multidrug resistance of mitoxantrone hydrochloride. Nanomedicine. 2012;8(2):185–193. | ||
Zha X, Hu Z, He S, Wang F, Shen H, Zhang H. TSC1/TSC2 inactivation inhibits AKT through mTORC1-dependent up-regulation of STAT3-PTEN cascade. Cancer Lett. 2011;313(2):211–217. | ||
Zhao H, Ma B, Wang Y, et al. miR-34a inhibits the metastasis of osteosarcoma cells by repressing the expression of CD44. Oncol Rep. 2013;29(3):1027–1036. | ||
Wang T, Ma W, Sun Y, et al. Ultra-sensitive assay for paclitaxel in intracellular compartments of A549 cells using liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;912:93–97. | ||
Huang J, Zhang H, Yu Y, et al. Biodegradable self-assembled nanoparticles of poly(D,L-lactide-co-glycolide)/hyaluronic acid block copolymers for target delivery of docetaxel to breast cancer. Biomaterials. 2014;35(1):550–566. | ||
Dong H, Dube N, Shu JY, et al. Long-circulating 15 nm micelles based on amphiphilic 3-helix peptide–PEG conjugates. ACS Nano. 2012;6(6):5320–5329. | ||
Duan X, Xiao J, Yin Q, et al. Smart pH-sensitive and temporal-controlled polymeric micelles for effective combination therapy of doxorubicin and disulfiram. ACS Nano. 2013;7(7):5858–5869. | ||
Yan J, Du YZ, Chen FY, You J, Yuan H, Hu FQ. Effect of proteins with different isoelectric points on the gene transfection efficiency mediated by stearic acid grafted chitosan oligosaccharide micelles. Mol Pharm. 2013;10(7):2568–2577. | ||
Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145(3):182–195. | ||
Bareford LM, Swaan PW. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 2007;59(8):748–758. | ||
Mo R, Jin X, Li N, et al. The mechanism of enhancement on oral absorption of paclitaxel by N-octyl-O-sulfate chitosan micelles. Biomaterials. 2011;32(20):4609–4620. | ||
Mathot F, des Rieux A, Arien A, Schneider YJ, Brewster M, Preat V. Transport mechanisms of mmePEG750P(CL-co-TMC) polymeric micelles across the intestinal barrier. J Control Release. 2007;124(3):134–143. | ||
He B, Jia Z, Du W, et al. The transport pathways of polymer nanoparticles in MDCK epithelial cells. Biomaterials. 2013;34(17):4309–4326. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.