")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 12
The effect of A1 adenosine receptor in diabetic megalin loss with caspase-1/IL18 signaling
Authors Tian D, Shi X, Zhao Y , Peng X, Zou L, Xu L, Ma Y, Wen Y, Faulhaber-Walter R , Chen L
Received 13 May 2019
Accepted for publication 16 July 2019
Published 28 August 2019 Volume 2019:12 Pages 1583—1596
DOI https://doi.org/10.2147/DMSO.S215531
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ming-Hui Zou
Dongli Tian, 1 Xiaoxiao Shi, 1 Yumo Zhao, 1 Xiaoyan Peng, 1 Linfeng Zou, 1 Lubin Xu, 1 Ying Ma, 1 Yubin Wen, 1 Robert Faulhaber-Walter, 2 Limeng Chen 1
1Department of Nephrology, Peking Union Medical College Hospital, Peking Union Medical College & Chinese Academy of Medical Sciences, Beijing 100730, People’s Republic of China; 2Department Facharztzentrum Aarberg, Nephrology, Waldshut-Tiegen, Germany
Correspondence: Limeng Chen
Department of Nephrology, Peking Union Medical College Hospital, Peking Union Medical College, Chinese Academy of Medical Sciences, Road 1 Shuaifuyuan, Wangfujing Street, Beijing 100730, People’s Republic of China
Tel +86 106 915 5351
Fax +86 106 915 5058
Email [email protected]
Purpose: In our previous study, exacerbation of albuminuria was observed in A1 adenosine receptor knockout (A1AR−/-,) mice with diabetic nephropathy (DN), but the mechanism was unclear. Here, we investigated the relationship of megalin loss and albuminuria, to identify the protective effect of A1AR in megalin loss associated albuminuria by inhibiting pyroptosis-related caspase-1/IL-18 signaling of DN.
Methods: We successfully collected DN patients’ samples and built diabetes mice models induced by streptozotocin. Megalin, cubilin, and A1AR expression were detected in kidney tissue samples from DN patients and mice through immunohistochemical and immunofluorescent staining. A1AR, caspase-1, interleukin-18 (IL-18) expression were analyzed using Western blotting in wild-type and A1AR−/-, mice. Human renal proximal tubular epithelial cells (PTC) were cultured with high glucose to observe the effect of A1AR agonist and antagonist on caspase-1/IL-18 and megalin injury.
Results: The loss of megalin, co-localized with A1AR at PTC, was associated with the level of albuminuria in diabetic patients and mice. The injury of megalin-cubilin was accompanied with the A1AR upregulation (1.30± 0.1 vs 0.98± 0.2, P= 0.042), the caspase-1 (1.33± 0.1 vs 1.0± 0.2, P= 0.036), and IL-18 (1.26± 0.2 vs 0.96± 0.2, P= 0.026) signaling activation in mice with DN. More severe pathological injury, 24 hrs urine albumin excretion (170.8± 4.1 μg/d vs 132.0± 2.9 μg/d vs 17.9± 2.8 μg/d, P< 0.001) and megalin-cubilin loss were observed in A1AR−/-, DN mice with more pronounced caspase-1 (1.52± 0.03 vs 1.20± 0.01, P= 0.017) and IL-18 (1.42± 0.02 vs 1.21± 0.02, P= 0.018) secretion. High glucose could stimulate the secretion of caspase-1 (1.72 times, P≤ 0.01) and IL-18 (1.64 times, P≤ 0.01), which was abolished by A1AR agonist and aggravated by A1AR antagonist.
Conclusion: A1AR played a protective role in proximal tubular megalin loss associated albuminuria by inhibiting the pyroptosis-related caspase-1/IL-18 signaling in DN.
Keywords: A1 adenosine receptor, diabetic nephropathy, caspase-1, megalin
Corrigendum for this paper has been published
Introduction
Hyperglycemia-induced inflammation plays a very important role in progression of diabetic nephropathy (DN),1 which is one of the most common and most severe microvascular complications of diabetes.2 Albuminuria can indicate incipient DN and independently predict end stage renal disease.3 But the detailed mechanism of diabetic albuminuria is incompletely understood. Both glomerular filtration and proximal tubular reabsorption participate in this process,4 while most previous studies focused only on glomerular lesions.5 Megalin and cubilin, located at the brush border of proximal renal tubular cells (PTCs),6 are responsible for reabsorption of 97% of the albumin.7 Megalin loss is associated with albuminuria in a previous study,8 while the specific mechanism is not well known.
Pyroptosis, also called necroinflammation, is a programmed cell necrosis. Caspase-1, as the best-known sensor molecule subset of pyroptosis, is a crucial intracellular factor involved in the innate immunity that converts proIL-18 and proIL-1β to mature forms and triggers the inflammatory cascade reaction.9 It involves in pyroptosis, triggering necroptosis of tubular cells in cisplatin-induced acute kidney injury models.10,11 The same process causes tubular necrosis in contrast media–related renal dysfunction, which is entirely reversible by its inhibitor necrostatin-1.12 Whether the megalin loss associated albuminuria is induced by the pyroptosis signaling caspase-1 and IL-18 in DN has not been studied before.
A1 adenosine receptor (A1AR), widely distributed in renal PTCs and glomerular afferent arterioles,13 was known as an important regulator for renal tubule-glomerular feedback (TGF).14 In our previous study, A1AR deletion aggravated the histologic evidence of glomerular injury and albuminuria with marked elevation of GFR in diabetic mice without TGF response. It indicates that TGF is not required to cause the hyperfiltration associated albuminuria.15 It is not clear that whether the A1AR contribute to the proximal tubular megalin loss associated albuminuria. Recently, the protect effect of A1AR was observed in renal ischemia-reperfusion injury model by inhibiting the TNF-α and IL-1β,16 which was downstream molecule of caspase-1. However, the role of the A1AR in megalin loss associated albuminuria by inhibiting the caspase-1/IL18 signaling activation has not been investigated.
Thus in this study, we firstly to confirm the relationship between megalin loss and albuminuria in both diabetes patients and STZ-induced diabetes mice. Then, we established diabetes model in A1AR-deficient mice and cultured HK2 treated with A1AR agonist and antagonist to figure out the anti-inflammatory effect of A1AR on megalin loss. In the end to observe whether the effect was mediated by caspase-1and IL-18 inflammation process.
Materials and methods
All reagents and antibodies used in this study were listed in Table S1
Patients
Patients with biopsy-confirmed DN admitted to Peking Union Medical College Hospital (PUMCH) were enrolled from January 2015 to December 2017. The control group comprised the kidney slides from patients (n=14) with isolated microscopic hematuria diagnosed as glomerular minor lesion (GML) without the podocyte injury. The baseline clinical characteristics of these patients are shown in Table S2. The detailed pathologies of light microscopy and electron microscopy about these patients were described in our previous study.17,18 The study was approved by the Institutional Ethics Committee at PUMCH (ID: 2014–2-18), and the experiments were performed with the written informed consent from all subjects.
Animals
Healthy male C57BL/6 mice reared under specific pathogen-free conditions (age: 6–7 weeks, weight: 18~22 g) were obtained from Beijing Vital River Laboratory Animal Technology Company; A1AR−/-mice were provided by Professor Jurgen Schnermann in NIDDK of NIH (USA) and bred in Animal Experimental Center of Peking University Health Science Center. All mice underwent one week of adaption (ambient temperature 20–24°C, relative humidity 50–55%, light cycle 12–12 hrs, free drinking, and food) prior to the experiment.
Mice were genotyped as the protocol described by Sun D.19 Tail DNA was isolated and tested with A1AR and (Neo-R) specific primers generating PCR products of 444 bp from the wild-type (WT) allele and 457 bp from the mutant allele. Primer sequences were sense A1AR, 5ʹ-GTACATCTCGGCCTTCC AGG-3ʹ; antisense A1AR, 5ʹ-GAGAATACCTGGC TGACTAG-3ʹ; sense neo-R, 5ʹ-ACAA-CAGACAATCGG CTGCTC TGATG-3ʹ; and antisense neo-R, 5ʹ-TGCGCGCCTTGAGCCTGGCGAAC-3ʹ. The animal experimental protocol was approved by the PUMCH Institutional Ethics Committee of Animal Care and Use (ID: XHDW-2014–0024).
Treatment
Male WT and A1AR −/- mice with matched age, weight, and blood glucose were randomly divided into four groups (n=6 per group), including wild-type control group (WT-Control), wild-type diabetes group (WT-DM), A1AR knock out control (A1AR−/–Control), and diabetes group (A1AR−/–DM). For induction of diabetes, mice (n=6 per group) were injected with STZ (Sigma, USA) (120 mg/kg, i.p.) dissolved in sodium citrate buffer (pH=4.5) for two consecutive days.20 Controls mice were treated with sodium citrate buffer.
Sample collection
Mice weight was recorded weekly. Blood glucose was measured using tail venipuncture with One Touch Ultra Test Strips (Johnson & Johnson, USA). Four weeks after the successful establishment of the diabetes model, 24-hr urine collections were made in MMC100 metabolic cage (Hatteras, USA) at ambient room temperature with unrestricted access to tap water and standard rodent diet. After centrifugation (3500 rpm*10 mins), urine albumin was determined by automatic biochemical analysis (AU2700, Beckman Coulter Inc, USA).15,21 Then, the mice were anesthetized and sacrificed. After perfusing with 0.9% pre-cooled saline from the heart and aorta, kidneys were rapidly dissected and weighed to calculate the kidney index (kidney weight/body weight). Then, renal cortex and medulla were separated for the following study.
Histology
Standard staining techniques were used as previously described.22 Sections of 3 μm were cut from paraffin-embedded kidney tissues and stained with periodic acid-schiff, hematoxylin and eosin (HE) for light microscopy (Olympus, Japan) or stained for immunohistochemistry. Transmission electron microscope was used to observe the injury of proximal tubular microvilli and basement membrane (JEM-1400plus, Japan).
Immunohistochemistry and immunofluorescence
Immunohistochemical (IHC) staining was performed on serial sections using standard methods.22 Sections of 3 μm cut from paraffin-embedded tissue were deparaffinized, rehydrated, and heated with 0.01 mol/L citrate buffer (pH 6.0) to expose antigen, then incubated with the primary antibody overnight at 4°C. Secondary antibodies were HRP-conjugated goat- anti-rabbit (Immuno Reagents, USA). All section images were captured by the microscope (Eclipse 80i; Nikon, Japan) equipped with a digital photograph camera (DS-U1; Nikon, Japan). The analytical measurement of IHC staining was evaluated by calculating the percentage of positive area using Image Pro Plus 6.0. Scoring was performed by a “blinded” investigator on coded slides. At least ten fields per specimen were randomly selected to cover most of the cortex for photo-documentation. The micrographs of immunofluorescent stains were captured by confocal laser microscopy (Leica, Germany).
Immunoblotting
Renal cortical and medullary tissues were dissected and homogenized in ice-cooled isolation buffer. Total protein was extracted from cortex and HK2 as previously described and utilized for immunoblotting analysis with the primary antibodies for A1AR, megalin, caspase-1, and IL-18.23 The β-actin was used as an internal protein loading control. Secondary antibody was HRP-conjugated goat anti-rabbit, followed by the detection of immunoblotting signals with an enhanced chemiluminescence detection system (Tanon 5200, China). Results were analyzed by Image J Microsoft (NIH, USA).
Human renal proximal tubular epithelial cell lines
Human renal PTC lines (HK2) were purchased from Cell Resource Center of Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences. They were cultured in DMEM-F12 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and 1% penicillin-streptomycin solution, and were subcultured using 0.25% trypsin-EDTA digestion after confluence. The cellular morphological characteristics were identified by phase contrast microscopy, and positive cytokeratin18 and megalin staining were detected by immunofluorescence to be confirmed as HKC. DMEM Medium with low glucose (5 mmol/L), high glucose (25 mmol/L), or high mannitol (25 mmol/L) were cocultured with HK2 cells for 24 and 72 hrs. Appropriate stimulus concentration of A1AR agonist (CCPA, 0.1 μmol/L, sigma, USA) and antagonist (DPCPX, 1 μmol/L, sigma, USA) was selected by CCK8 and LDH assay to detect the effect of them on HKC in high glucose environment stimulation for 6, 12, and 24 hrs. The cells were lysed in RIPA buffer, and proteins were collected to detect A1AR, Caspase-1, IL18 and megalin expression by immunofluorescent staining and Western blotting analysis.
Statistical analysis
Unpaired t test (two tailed) was used for comparison between two groups. One-way ANOVA with Dunnett’s multiple comparison test was performed for comparison between multiple groups. P<0.05 was considered statistically significant. Values were presented as mean ± SEM.
Results
Association between albuminuria and megalin loss in diabetes patients and mice
After 72 hrs of STZ treatment, diabetes was confirmed with the random blood glucose higher than 16.7 mmol/L, along with polydipsia, polyphagia, polyuria, and emaciation in both wild-type and A1AR−/- mice. At week 4 after STZ treatment (Figure 1A), a higher blood glucose level (P<0.001, mean 28.8±4.5 mmol/L vs 7.1±0.7 mmol/L; 23.4±5.2 mmol/L vs 6.6±0.4 mmol/L, Figure 1B), and a lower body weight (P<0.001, mean 20.9±2.1 g vs 28.3±0.8 g; 16.1±2.3 g vs 26.2±0.8 g, Figure 1C) in A1AR−/- mice and WT mice treated by STZ were observed than A1AR−/- and WT control mice. Urine volume (P<0.001, mean 12.3±0.7 mL vs 2.1±0.3 mL; 10.3±1.6 mL vs 1.8±0.2 mL, Figure 1D), kidney index (P<0.05, mean 0.12±0.02 vs 0.09±0.01; 0.11±0.02 vs 0.07±0.01, Figure 1E) and 24 hrs urine albumin excretion (P<0.001, mean 170.8±4.1 μg/d vs 12.3±1.5 μg/d; 132.0±2.9 μg/d vs 17.9±2.8 μg/d, Figure 1F) were all significantly higher in diabetic mice than control mice.
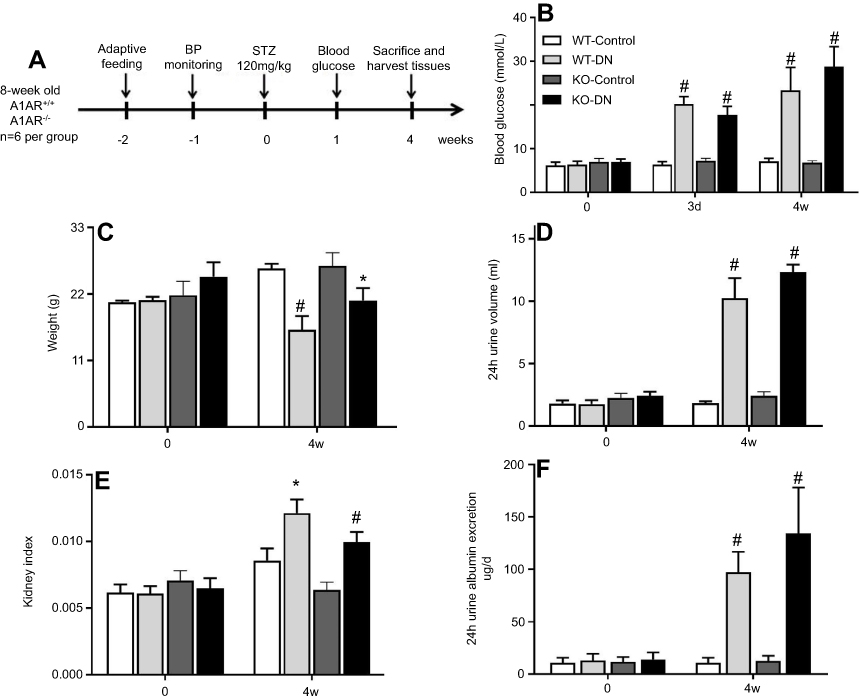 |
Figure 1 Successful establishment of type 1 diabetic nephropathy mice model induced by streptozotocin (STZ) for 4 weeks. (A) Flow chart of the mice model in this study. Blood glucose (B), body weight (C), 24 hrs urine volume (D), kidney weight index (E), and 24 hrs urine albumin excretion (F) were measured at day 0 and day 28 in three groups of wild-type non-diabetic mice (WT-control), wild-type mice with STZ-induced diabetes (WT-DN), A1AR-knockout mice with STZ-induced diabetes (KO-DN)). Blood glucose ≥16.7 mmol/L at 72 hrs after STZ-induction was defined to diagnose diabetes. Compared to WT-control mice, WT-DN and KO-DN mice showed significantly increased blood glucose, urine volume, kidney weight index and 24 hrs urine albumin excretion with decreased bodyweight, indicating successful type 1 diabetic nephropathy induction at fourth week. (N=6 per group,*P<0.05, #P<0.01). Abbreviation: Kidney weight index, kidney weight/body weight. |
The renal pathology of WT-control and A1AR−/–control mice had no significant difference. While compared to control mice, mild glomerular and proximal tubular hypertrophy was observed in WT-DN group mice(P=0.023), A1AR−/–DN mice presented with more severe glomerulomegaly, renal tubular hypertrophy, and vacuolization by light microscope (P=0.003, Figure 2A, 2B). Electron microscopy showed the typical changes in the early stage of DN that is thickening of the renal tubular basement membrane. The thickness in control groups was 110–130 nm, and that in groups with DN was 220–280 nm (Figure 2C).
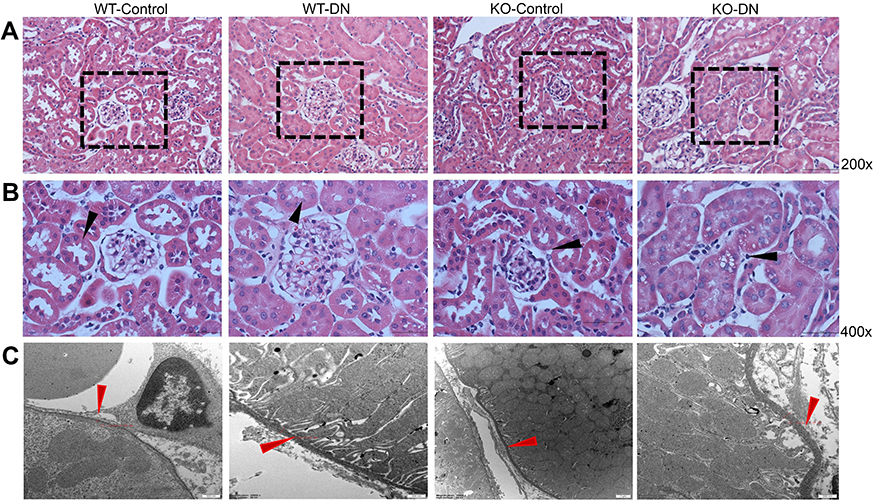 |
Figure 2 Representative images of renal histopathological change and tubular basement membrane. (A and B) Histopathological change in three groups (WT-control, WT-DN and KO-DN) at four weeks visualized by HE staining (A): Mild glomerulomegaly and brush border damage in proximal tubular epithelial cells, as typically for diabetic nephropathy, were shown in the WT-DN group while not in the WT-control group (P=0.023). Compared to the WT-DN mice group, KO-DN developed more severe glomerulomegaly (P=0.003), renal tubular hypertrophy and vacuolization (Original magnification =200×, 400× magnification reveals glomerulus and proximal renal tubule in black boxed area in 200×. Bar width =200 μm). The black arrows in panel B indicates P=0.023 (WT-DN VS WT-Control), P=0.003 (KO-DN VS KO-Control), P=0.005(KO-DN VS WT-DN). (C). Electron microscopy showed the typical changes in the early stage of DN that is thickening of the renal tubular basement membrane. The thickness in control groups was 110–130 nm, and that in groups with DN was 220–280 nm (Original magnification =25000×. Bar width =500 nm). The red arrows showed the tubular basement membrane. Abbreviation: HE, hematoxylin and eosin. |
Semiquantitative analysis showed that the percentage of positive area of megalin and cubilin staining in DN patients significantly decreased compared to GML patients (Figure 4D, 4E). And the positive area of megalin staining was negatively as sociated with 24 hrs urine protein in DN patients (R2=0.743, P=0.002, Figure 4F) and diabetic mice (R2=0.859, P<0.001, Figure 3C, 3E), and cubilin either (Figure 3D and F). It was accompanied with the shorter microvilli of proximal tubular in DN mice by immune electron microscopy (Figure 3A and B).
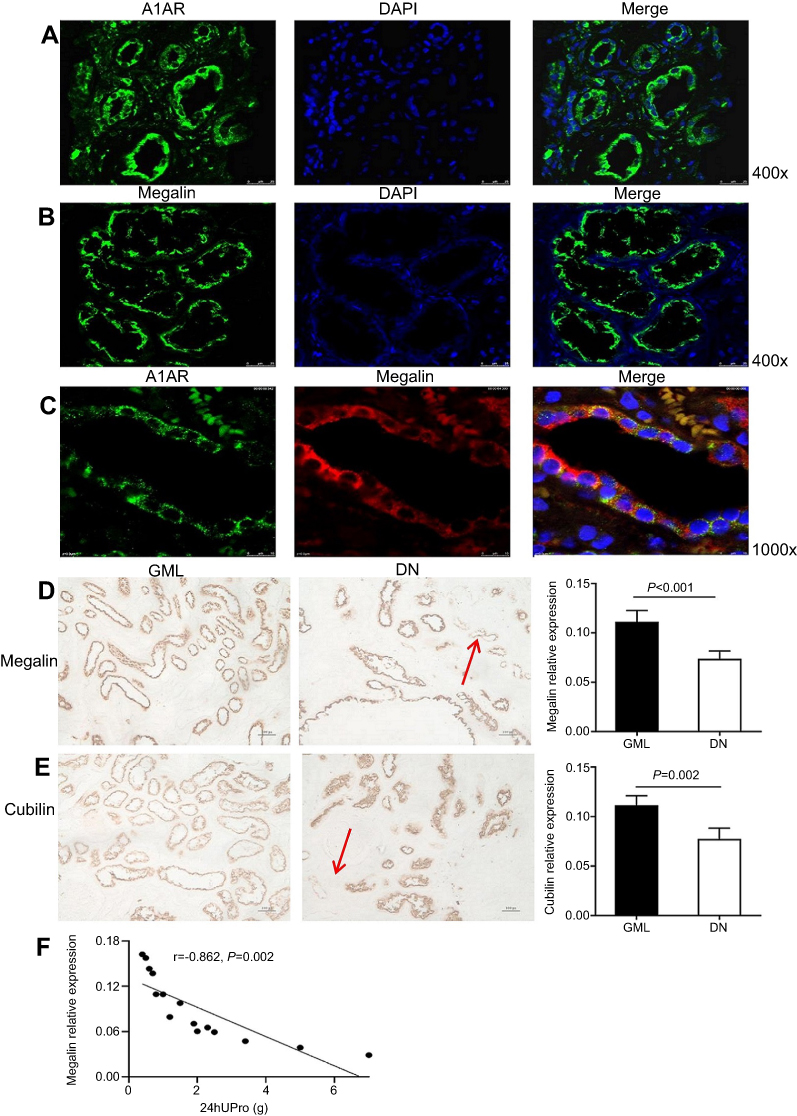 |
Figure 4 Expression of megalin, cubilin, and A1 adenosine receptor (A1AR) in kidneys of DN patients. (A) A1AR was located at the membrane of PTC by immunofluorescent staining. Green: A1AR; Blue: DAPI; Merge is A1AR with DAPI. (B) Megalin was visualized at the brush border of proximal tubular cells (PTC) in patients with diabetic nephropathy (DN) by immunofluorescent staining. Green: megalin; Blue: DAPI, Merge is megalin with DAPI. (C) Showed the coexpression of A1AR and megalin at PTC. Green: A1AR; Red: megalin; Blue: DAPI; Merge is A1AR with megalin. (D–E) Semiquantitative analysis of immunohistochemical staining showed the positive area of both megalin and cubilin expression decreased significantly in DN patients compared to that in GML patients. (F) Negative association of megalin positive expression area and 24 hrs urine protein in DN patients. Data are presented as the Mean ± SEM (n=12 per group). Bar width: 25 μm. The red arrows in panels D and E were megalin loss area.Abbreviation: DAPI, 4ʹ,6-diamidino-2-phenylindole. |
A1AR deletion aggravated caspase1/IL18 activation and megalin injury
Megalin was expressed at the brush border of PTC (Figure 4A), A1AR was expressed at the cellular membrane of PTC (Figure 4B). They were both located on PTC (Figure 4C).
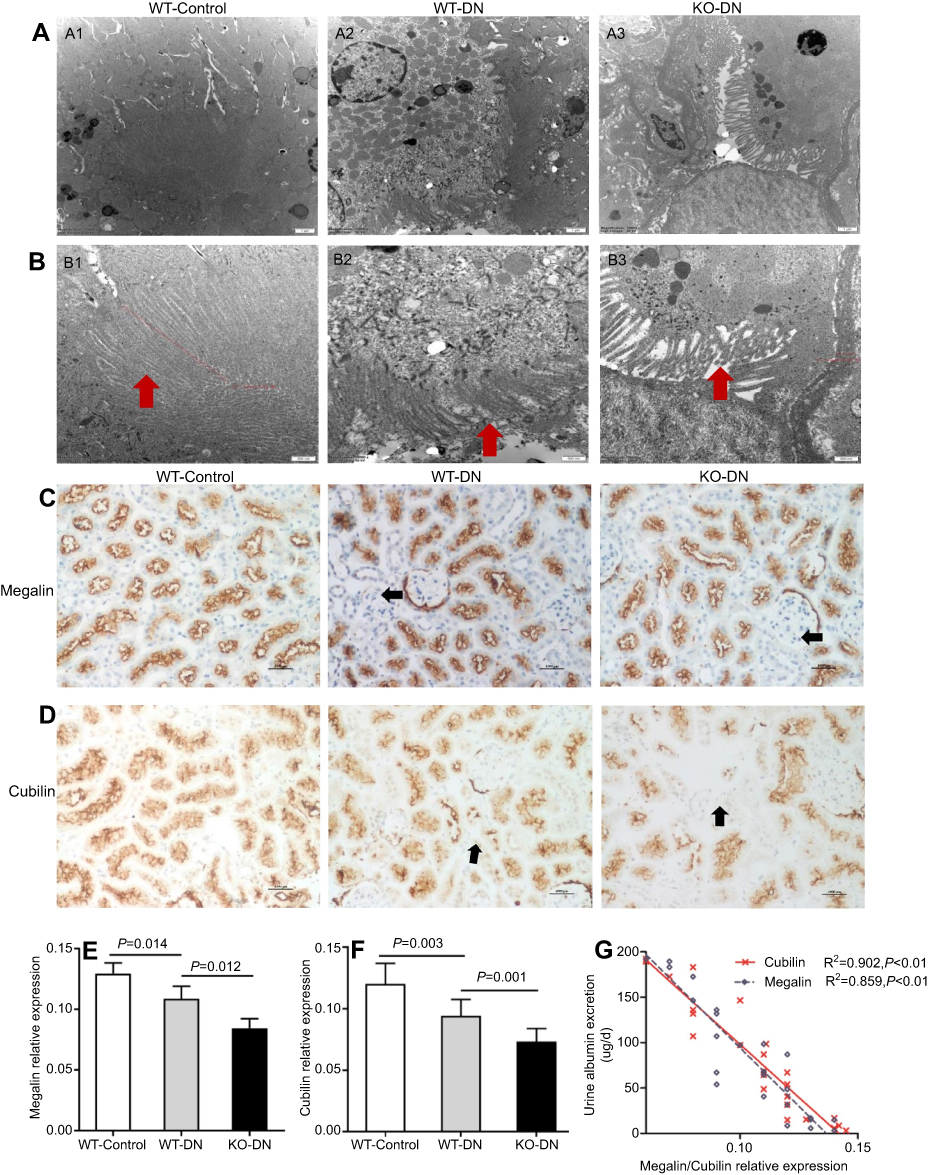 |
Figure 3 A1 adenosine receptor (A1AR) deletion aggravated renal tubular microvilli, megalin-cubillin loss , and albuminuria in DN mice. (A and B) Transmission electron microscope showed shortened length and reduced density tubular microvilli in DN mice compared to control mice, further shorter and more sparse density were observed in KO-DN mice than WT-DN mice. (A: Original magnification =10,000×. Bar width =1 μm; B: Original magnification =25,000×. Bar width =500 nm). (C and D). Immunohistochemical staining showed both megalin (C) and cubilin (D) expression decreased sequentially in WT-control, WT-DN, and A1AR knock out (KO)-DN mice at 4 weeks (100× magnification). (E) Semiquantitative analysis confirmed that down-regulation of megalin and cubilin expression was significant. (F) Compared to WT-DN, A1AR deletion aggravated urine albumin excretion (UAE) in KO-DN. (G). Level of UAE was negatively correlated with megalin (black dotted line) and cubilin (red solid line) relative expression. Red arrows in panels B showed microvilli of proximal tubular brush border. Black arrows in panels C and D showed megalin loss area of diabetes mice. Data were analyzed by the positive area proportion of megalin and cubilin at proximal tubules, and data were presented as mean ± SEM (N=6 per group). Bar width =100px. |
The upregulation of A1AR expression was detected by Western blotting (Figure 5A and E), while the pyroptosis signaling related protein expression of caspase-1 (P=0.036, Figure 5B and C) and the downstream inflammatory cytokine IL-18 (P=0.026, Figure 5B and D) also increased significantly in WT-DN mice compared to that in WT-control mice.
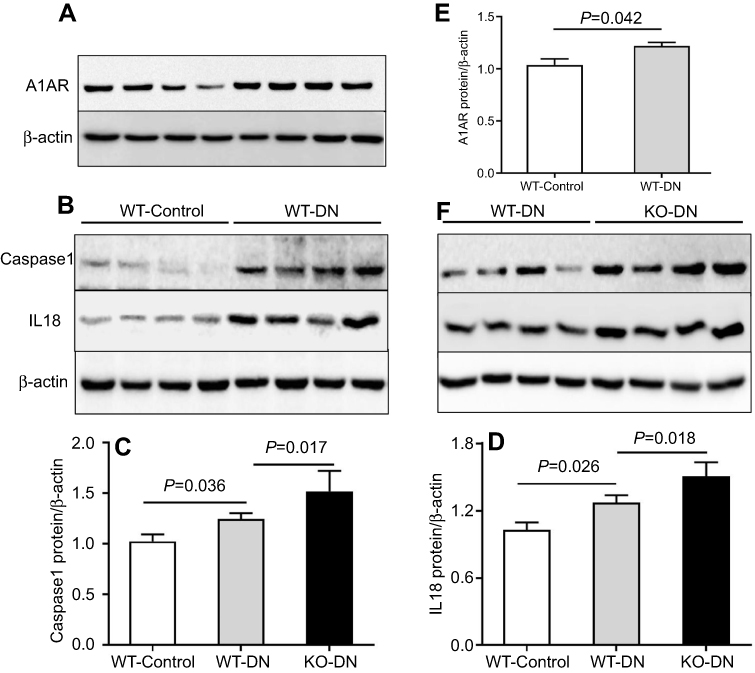 |
Figure 5 A1 adenosine receptor (A1AR) deletion aggravated diabetes-induced renal inflammation by activation caspase-1 and interleukin (IL)-18. (A). A1AR activation was observed in WT-DN mice, compared to WT-control mice by Western blotting. (C–D). Showed caspase-1/IL-18 protein expression by Western blotting and their quantification. Compared to WT-control, analyzed protein expression increased in WT-DN mice, while further more increasing in A1AR knockout (KO)-DN mice than in WT-DN mice. Data are shown as Mean ± SEM; n=6 per group. |
While in A1AR−/–DN mice, caspase-1 (P=0.017, Figure 5F and C) and IL-18 (P=0.018, Figure 5F and D) protein expression was further more increasing, accompanied by much shorter length of tubular microvilli (Figure 3A and B) and more decreasing expression of megalin and cubilin (Figure 3C and D) than in WT-DN mice.
The protective role of A1AR in megalin loss via inhibiting caspase-1/il-18 in vitro
In cultured HK2 cells with high glucose, the positive staining of CD68 (Figure S1A), caspase-1 (Figure S1B), and IL-18 (Figure S1C) was detected by immunofluorescence. Meanwhile, the upregulation of caspase-1 (1.76 times, P<0.001, Figure 6A and C)/IL-18 (1.85 times, P<0.001, Figure 6A and 6D) expression and downregulation of megalin expression (0.52 times, P<0.001, Figure 6A and E) were observed by Western blotting.
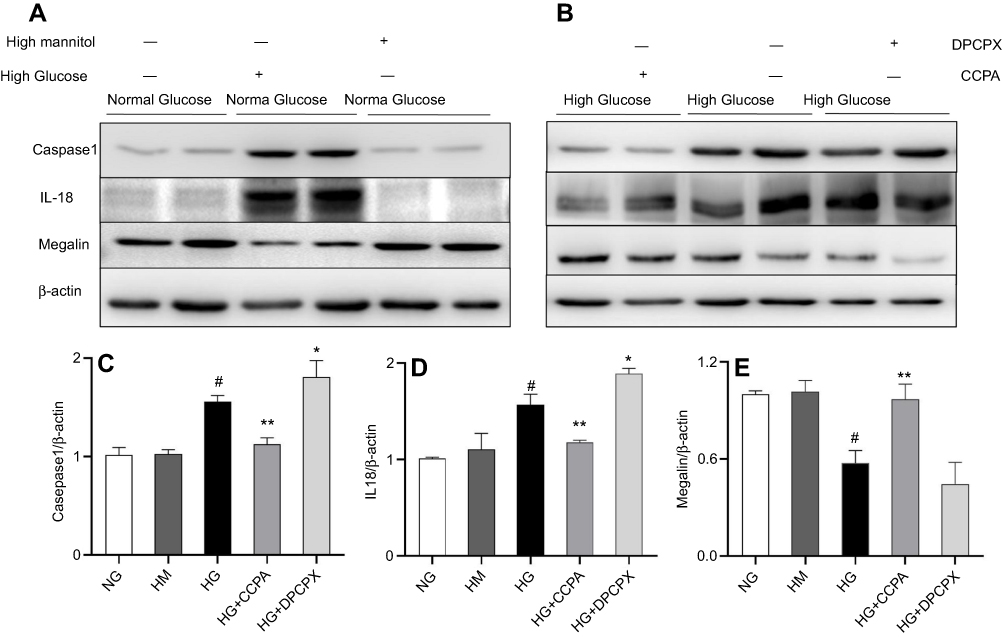 |
Figure 6 The role of A1AR antagonist DPCPX and agonist CCPA in caspase-1/IL18 activation and megalin loss in HK2 cell cocultured with high glucose. (A–B). Protein expression of caspase-1, IL-18, and megalin in high glucose, high glucose with DPCPX and CCPA by Western blotting. Quantitative analysis of caspase-1 (C), IL-18 (D), and megalin (E). Showed the upregulation of caspase-1/IL-18 and downregulation of megalin expression in high glucose. DPCPX further increased caspase-1/IL-18 expression and decreased megalin expression. However, CCPA inhibited caspase-1/IL-18 expression and improved megalin loss obviously. Data are shown as Mean ± SEM; *P<0.05 HG+DPCPX vs HG, #P<0.01 HG vs NG, **P<0.05 HG+CCPA vs HG. n=4 per group. |
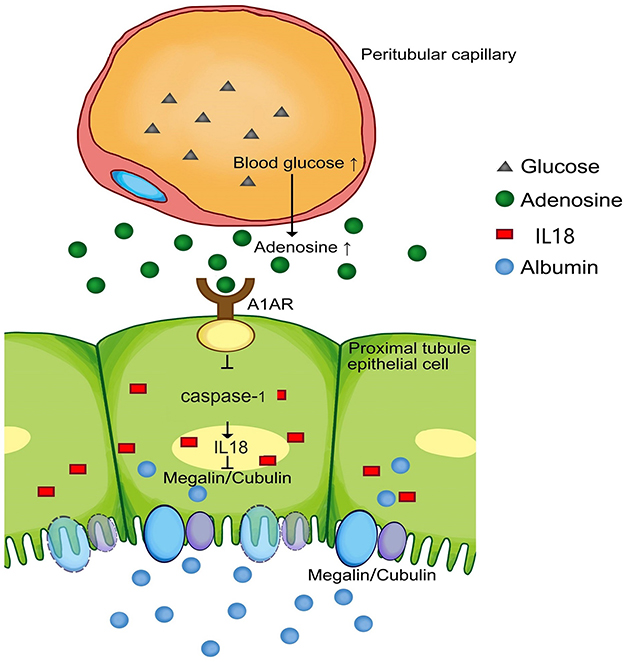 |
Figure 7 Model of the possible pathway in this study: Hyperglycemia could stimulate the increased production of adenosine, accompanied with high expression of its A1 receptor (A1AR), which widely distributes in renal proximal tubular epithelial cells. A1AR plays an anti-inflammatory role in megalin-cubilin loss by inhibiting pyroptosis related caspase-1/IL-18 signaling. |
In HK2 cells cocultured with high glucose medium and A1AR antagonist DPCPX, DPCPX further increased caspase-1 (1.96 times, P<0.05, Figure 6B and C)/IL-18 (2.0 times, P<0.05, Figure 6B and D) protein expression and decreased megalin expression (0.38 times, P<0.05, Figure 6B and E).
However, in HK2 cells cocultured with high glucose medium and A1AR agonist CCPA. CCPA inhibited caspase-1 (P<0.001, Figure 6B and C)/IL-18 (P<0.001, Figure 6B and D) expression and improved megalin (P<0.001, Figure 6B and E) expression obviously.
Discussion
This study confirms that albuminuria was associated with megalin-cubilin loss in DN patients and animal models. A1AR deletion aggravated megalin-cubilin loss associated albuminuria and renal pathological lesions in DN mice, through activating pyroptosis-related caspase-1/IL-18 signaling. While A1AR agonist could improve megalin loss via inhibiting caspase-1/IL-18 signaling in HK2 cells. To our knowledge, this is the first direct evidence of the protective effects of A1AR on megalin-cubilin loss associated albuminuria in DN. We summarized the role of A1AR in this process as Figure 7.
Microalbuminuria indicates incipient DN and independently predicts ESRD.24,25 But the mechanism of proteinuria is yet incompletely understood. Recently, decreasing protein reabsorption at PTC was found to contribute to diabetic proteinuria.6 Megalin-cubilin are multi-ligand protein receptors expressed at the brush border membrane of PTC involved in endocytosis.26 Megalin/LRP2 is known to bind and endocytosis more than 40 ligands and including some of the important signaling members in homeostasis of kidney, diabetes, neural stem cell niche in brain and eye.27–29 Low-molecular proteinuria due to failed endocytosis was observed in megalin-knockout rat and cubilin-deficient dogs.30 Thrailkill K. M. et al, proposed an association of megalin-cubilin with diabetic albuminuria, since urine megalin-cubilin ligands were significantly increased in type 1 diabetes patients with urine microalbuminuria compared to samples from non-albuminuria diabetes patients and healthy controls.8 In this study, we directly observed a significant loss of megalin-cubilin of PTC in patients and mice models with DN. Our finding was consistent with the reduction of albumin reabsorption of PTC as reported before.7
But the mechanism of megalin loss in diabetes is still not clear enough. In vitro studies, several mechanisms had been investigated that megalin could combine with advanced glycation end products (AGE). The overload or accumulation of AGE during the PTC endocytosis and activation of the renin-angiotensin system could induce the megalin injury.31,32 In the renal interstitium of patients with Fanconi syndrome, we observed the association between proinflammatory cytokine IL-1, IL-17, and megalin injury.18 A. Takeyama et al, also reported that megalin was downregulated via LPS-TNF-alpha-ERK1/2 signaling pathway in PTC.33 These data indicate that proinflammatory cytokines might play an important role in megalin loss of DN.
In this study, we firstly observed caspase-1/IL-18 signaling activation was associated with megalin loss in mice with DN. Caspase-1, which could detect endogenous signals, resulting in activation of IL-1β, IL-18, and other cytokines, is a well-characterized sensor molecule of the pyroptosis.34 It had reported that hyperglycemia could activate the pyroptosis-related proinflammatory cytokine IL1 family, which then mediated the occurrence and development of proteinuria through lysosomal damage and active oxygen cluster.35,36 In vitro PTC study, NLRP3 also could activate caspase-1 to release cytokines of the IL-1β, promote endoplasmic reticulum stress and inflammation, then lead megalin loss. IL-18, is also a member of the IL-1 family of inflammatory cytokines, and involved in the development and progression of DN.37 It is reasonable that the pyroptosis-related caspase1/IL18 signaling is associated with PTC injury and megalin loss in DN. Furthermore, besides the large number of clinical studies, recently, based on the inverse-variance weighted method, high IL-18 plasma levels were proved significantly increase the risk of T2DM in a Mendelian randomization study.38 Since IL-18 was recognized as the pathogenic mediators in diabetic megalin lost and proteinuria, it might be a new potential therapeutic targets.
A1AR, a member of the adenosine receptor family, has diverse function from role in diabetes and obesity to sleep deprivation in the human brain.39,40 In our previous study, exacerbation of albuminuria was observed in A1AR knockout (A1AR-/-) mice in the young (10–20w) Akita diabetic mice, without obvious pathological injury.15 In early stages of diabetes nephropathy, the upregulation of SLGT2 in PTC could increase the Na+/glucose reabsorption, and decrease the Na+ delivery to macula densa, impaired TGF and then elevate GFR. In the A1AR-/- diabetes mice, the abolished TGF, induced the more severer glomerular relaxation and hyperfiltration. But it is not clear, whether A1AR play a role in the proximal tubular albumin reabsorption, mediated by megalin loss.15
In this study, A1AR -deficiency was associated with more increasing activation effect of the caspase-1/IL-18 signaling, and augmented megalin-cubilin loss in DN mice. In WT mice with DN, the caspase1/IL-18 signaling could be activated by several well-known reasons, such as oxidative stress, hypoxia, and hyperglycemia41-43 The upregulation of A1AR might play an important role in the anti-inflammatory process, which had been demonstrated in acute lung injury and renal ischemia-reperfusion injury model.16,44 In a renal ischemia-reperfusion injury model, inflammatory factors TNF-α and IL-1β were suppressed by A1AR.16 The A1AR agonist was also reported to inhibit the IL-18 releases by alleviating macrophage infiltration.45 Thus, A1AR could protect against megalin injury in mice with DN through inhibiting the caspase-1/IL-18 signaling activation. Since megalin loss is obviously related to albuminuria and inflammation, megalin KO mouse will be helpful to confirm this conclusion in future experiments.
Conclusion
A1AR played a protective role in renal proximal tubular megalin loss associated albuminuria via inhibition of caspase-1/IL-18 signaling in DN.
Ethics statement
This study of patients was approved by the Institutional Ethics Committee of Peking Union Medical College Hospital (2014-2-18), and the experiments were performed with the written informed consent from all subjects in accordance with the Declaration of Helsinki. The mice were maintained in the Animal Care of the Peking Union Medical College Hospital, Beijing, China. All procedures involving animal treatment were approved by the Peking Union Medical College Hospital institutional committee of animal care and use (XHDW-2014-0024).
Acknowledgments
We thank Professor Schnermann from National Institutes of Health, USA for providing us with A1AR gene deficient mice complimentarily. Parts of this work were previously presented at the Annual Meeting of the 53rd European Renal Association-European Dialysis and Transplant Association (ERA-EDTA) in 2016, and the International Society of Nephrology-World Congress of Nephrology (ISN-WCN) in 2017.
Author contributions
TD, SX, and CL contributed in the conception and design of the study; TD and PX organized the database; TD and MY performed the statistical analysis; TD wrote the first draft of the manuscript and ZY, XL, and ZL wrote sections of the manuscript. WY performed diagnosis and analysis of pathological sections. WRF and CL revised the manuscript critically for important intellectual content. All authors contributed to data analysis, drafting or revising the articles, gave final approval of the vision to be published, and agree to be accountable for all aspects of the work.
Disclosure
Dr Dongli Tian reports grants from the National Natural Scientific Foundation, China (81470937, 81641024 to CL), Key Research and Development Program of Ningxia Hui Autonomous Region (2018BFG02010 to CL), National Key-point Research Program Precision Medicine Grant (2016YFC0901500 to CL), Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (CIFMS 2016-12M-2-004 to CL), and Xiehe Scholar Grant (to CL), during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Garcia-Garcia PM, Getino-Melian MA, Dominguez-Pimentel V, Navarro-Gonzalez JF. Inflammation in diabetic kidney disease. World J Diabetes. 2014;5(4):431–443. doi:10.4239/wjd.v5.i4.431
2. Gallagher H, Suckling RJ. Diabetic nephropathy: where are we on the journey from pathophysiology to treatment? Diabetes Obes Metab. 2016;18(7):641–647. doi:10.1111/dom.12630
3. Balakumar P, Arora MK, Reddy J, Anand-Srivastava MB. Pathophysiology of diabetic nephropathy: involvement of multifaceted signalling mechanism. J Cardiovasc Pharmacol. 2009;54(2):129–138. doi:10.1097/FJC.0b013e3181ad2190
4. Tramonti G, Kanwar YS. Review and discussion of tubular biomarkers in the diagnosis and management of diabetic nephropathy. Endocrine. 2013;43(3):494–503. doi:10.1007/s12020-012-9820-y
5. Mori KP, Yokoi H, Kasahara M, et al. Increase of total nephron albumin filtration and reabsorption in diabetic nephropathy. J Am Soc Nephrol. 2017;28(1):278–289. doi:10.1681/ASN.2015101168
6. Dickson LE, Wagner MC, Sandoval RM, Molitoris BA. The proximal tubule and albuminuria: really! J Am Soc Nephrol. 2014;25(3):443–453. doi:10.1681/ASN.2013090950
7. Nielsen R, Christensen EI, Birn H. Megalin and cubilin in proximal tubule protein reabsorption: from experimental models to human disease. Kidney Int. 2016;89(1):58–67. doi:10.1016/j.kint.2015.11.007
8. Thrailkill KM, Nimmo T, Bunn RC, et al. Microalbuminuria in type 1 diabetes is associated with enhanced excretion of the endocytic multiligand receptors megalin and cubilin. Diabetes Care. 2009;32(7):1266–1268. doi:10.2337/dc09-0112
9. Mulay SR, Linkermann A, Anders HJ. Necroinflammation in kidney disease. J Am Soc Nephrol. 2016;27(1):27–39. doi:10.1681/ASN.2015040405
10. Zhu Y, Cui H, Xia Y, Gan H. RIPK3-mediated necroptosis and apoptosis contributes to renal tubular cell progressive loss and chronic kidney disease progression in rats. PLoS One. 2016;11(6):e0156729. doi:10.1371/journal.pone.0156729
11. Xu Y, Ma H, Shao J, et al. A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol. 2015;26(11):2647–2658. doi:10.1681/ASN.2014080741
12. Xiao X, Du C, Yan Z, Shi Y, Duan H, Ren Y. Inhibition of necroptosis attenuates kidney inflammation and interstitial fibrosis induced by unilateral ureteral obstruction. Am J Nephrol. 2017;46(2):131–138. doi:10.1159/000478746
13. Vallon V, Osswald H. Adenosine receptors and the kidney. Handb Exp Pharmacol. 2009;(193):443–470. doi:10.1007/978-3-540-89615-9_15
14. Osswald H, Muhlbauer B, Schenk F. Adenosine mediates tubuloglomerular feedback response: an element of metabolic control of kidney function. Kidney Int Suppl. 1991;32:S128–S131.
15. Faulhaber-Walter R, Chen L, Oppermann M, et al. Lack of A1 adenosine receptors augments diabetic hyperfiltration and glomerular injury. J Am Soc Nephrol. 2008;19(4):722–730. doi:10.1681/ASN.2007060721
16. Lee HT, Gallos G, Nasr SH, Emala CW. A1 adenosine receptor activation inhibits inflammation, necrosis, and apoptosis after renal ischemia-reperfusion injury in mice. J Am Soc Nephrol. 2004;15(1):102–111. doi:10.1097/01.asn.0000102474.68613.ae
17. Jiang L, Chen C, Yuan T, et al. Clinical severity of Gitelman syndrome determined by serum magnesium. Am J Nephrol. 2014;39(4):357–366. doi:10.1159/000360773
18. Wang J, Wen Y, Zhou M, et al. Ectopic germinal center and megalin defect in primary Sjogren syndrome with renal Fanconi syndrome. Arthritis Res Ther. 2017;19(1):120. doi:10.1186/s13075-017-1317-x
19. Sun D, Samuelson LC, Yang T, et al. Mediation of tubuloglomerular feedback by adenosine: evidence from mice lacking adenosine 1 receptors. Proc Natl Acad Sci U S A. 2001;98(17):9983–9988. doi:10.1073/pnas.171317998
20. Tesch GH, Allen TJ. Rodent models of streptozotocin-induced diabetic nephropathy. Nephrology. 2007;12(3):261–266. doi:10.1111/j.1440-1797.2007.00796.x
21. Breyer MD, Bottinger E, Brosius FC
22. Chen L, Faulhaber-Walter R, Wen Y, et al. Renal failure in mice with Gsalpha deletion in juxtaglomerular cells. Am J Nephrol. 2010;32(1):83–94. doi:10.1159/000314635
23. Chen L, Kim SM, Oppermann M, et al. Regulation of renin in mice with Cre recombinase-mediated deletion of G protein Gsalpha in juxtaglomerular cells. Am J Physiol Renal Physiol. 2007;292(1):F27–F37. doi:10.1152/ajprenal.00193.2006
24. Schena FP, Gesualdo L. Pathogenetic mechanisms of diabetic nephropathy. J Am Soc Nephrol. 2005;16 Suppl 1:S30–S33. doi:10.1681/asn.2004110970
25. Perkins BA, Ficociello LH, Roshan B, Warram JH, Krolewski AS. In patients with type 1 diabetes and new-onset microalbuminuria the development of advanced chronic kidney disease may not require progression to proteinuria. Kidney Int. 2010;77(1):57–64. doi:10.1038/ki.2009.399
26. Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol. 2002;3(4):256–266. doi:10.1038/nrm778
27. Gajera CR, Emich H, Lioubinski O, et al. LRP2 in ependymal cells regulates BMP signaling in the adult neurogenic niche. J Cell Sci. 2010;123(Pt 11):1922–1930. doi:10.1242/jcs.065912
28. Kur E, Christa A, Veth KN, et al. Loss of Lrp2 in zebrafish disrupts pronephric tubular clearance but not forebrain development. Dev Dyn. 2011;240(6):1567–1577. doi:10.1002/dvdy.22624
29. De S, Kuwahara S, Hosojima M, et al. Exocytosis-mediated urinary full-length megalin excretion is linked with the pathogenesis of diabetic nephropathy. Diabetes. 2017;66(5):1391–1404. doi:10.2337/db16-1031
30. Sirac C, Bridoux F, Essig M, Devuyst O, Touchard G, Cogne M. Toward understanding renal Fanconi syndrome: step by step advances through experimental models. Contrib Nephrol. 2011;169:247–261. doi:10.1159/000313962
31. Saito A, Nagai R, Tanuma A, et al. Role of megalin in endocytosis of advanced glycation end products: implications for a novel protein binding to both megalin and advanced glycation end products. J Am Soc Nephrol. 2003;14(5):1123–1131. doi:10.1097/01.asn.0000062962.51879.f8
32. Li XC, Zhuo JL. Mechanisms of AT1a receptor-mediated uptake of angiotensin II by proximal tubule cells: a novel role of the multiligand endocytic receptor megalin. Am J Physiol Renal Physiol. 2014;307(2):F222–F233. doi:10.1152/ajprenal.00693.2013
33. Takeyama A, Sato H, Soma-Nagae T, et al. Megalin is downregulated via LPS-TNF-alpha-ERK1/2 signaling pathway in proximal tubule cells. Biochem Biophys Res Commun. 2011;407(1):108–112. doi:10.1016/j.bbrc.2011.02.118
34. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311–320. doi:10.1038/nature14191
35. Ralston JC, Lyons CL, Kennedy EB, Kirwan AM, Roche HM. Fatty acids and NLRP3 inflammasome-mediated inflammation in metabolic tissues. Annu Rev Nutr. 2017;37:77–102. doi:10.1146/annurev-nutr-071816-064836
36. Qiu YY, Tang LQ. Roles of the NLRP3 inflammasome in the pathogenesis of diabetic nephropathy. Pharmacol Res. 2016;114:251–264. doi:10.1016/j.phrs.2016.11.004
37. Elsherbiny NM, Al-Gayyar MM. The role of IL-18 in type 1 diabetic nephropathy: the problem and future treatment. Cytokine. 2016;81:15–22. doi:10.1016/j.cyto.2016.01.014
38. Zhuang H, Han J, Cheng L, Liu SL. A positive causal influence of IL-18 levels on the risk of T2DM: a mendelian randomization study. Front Genet. 2019;10:295. doi:10.3389/fgene.2019.00295
39. Dhalla AK, Chisholm JW, Reaven GM, Belardinelli L. A1 adenosine receptor: role in diabetes and obesity. In: Wilson C, Mustafa S, editors. Adenosine Receptors in Health and Disease. Handbook of Experimental Pharmacology. Heidelberg: Springer; 2009:271–295.
40. Elmenhorst D, Meyer PT, Winz OH, et al. Sleep deprivation increases A1 adenosine receptor binding in the human brain: a positron emission tomography study. J Neurosci. 2007;27(9):2410–2415. doi:10.1523/JNEUROSCI.5066-06.2007
41. Chen C, Ma X, Yang C, et al. Hypoxia potentiates LPS-induced inflammatory response and increases cell death by promoting NLRP3 inflammasome activation in pancreatic beta cells. Biochem Biophys Res Commun. 2018;495(4):2512–2518. doi:10.1016/j.bbrc.2017.12.134
42. Li X, Zeng L, Cao C, et al. Long noncoding RNA MALAT1 regulates renal tubular epithelial pyroptosis by modulated miR-23c targeting of ELAVL1 in diabetic nephropathy. Exp Cell Res. 2017;350(2):327–335. doi:10.1016/j.yexcr.2016.12.006
43. Chen Y, Wang L, Pitzer AL, Li X, Li PL, Zhang Y. Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. J Mol Med (Berl). 2016;94(12):1335–1347. doi:10.1007/s00109-016-1481-5
44. Ngamsri KC, Wagner R, Vollmer I, Stark S, Reutershan J. Adenosine receptor A1 regulates polymorphonuclear cell trafficking and microvascular permeability in lipopolysaccharide-induced lung injury. J Immunol. 2010;185(7):4374–4384. doi:10.4049/jimmunol.1000433
45. DeOliveira CC, Paiva Caria CR, Ferreira Gotardo EM, Ribeiro ML, Gambero A. Role of A1 and A2A adenosine receptor agonists in adipose tissue inflammation induced by obesity in mice. Eur J Pharmacol. 2017;799:154–159. doi:10.1016/j.ejphar.2017.02.017
Supplementary materials
 |
Table S1 Reagents used in this study |
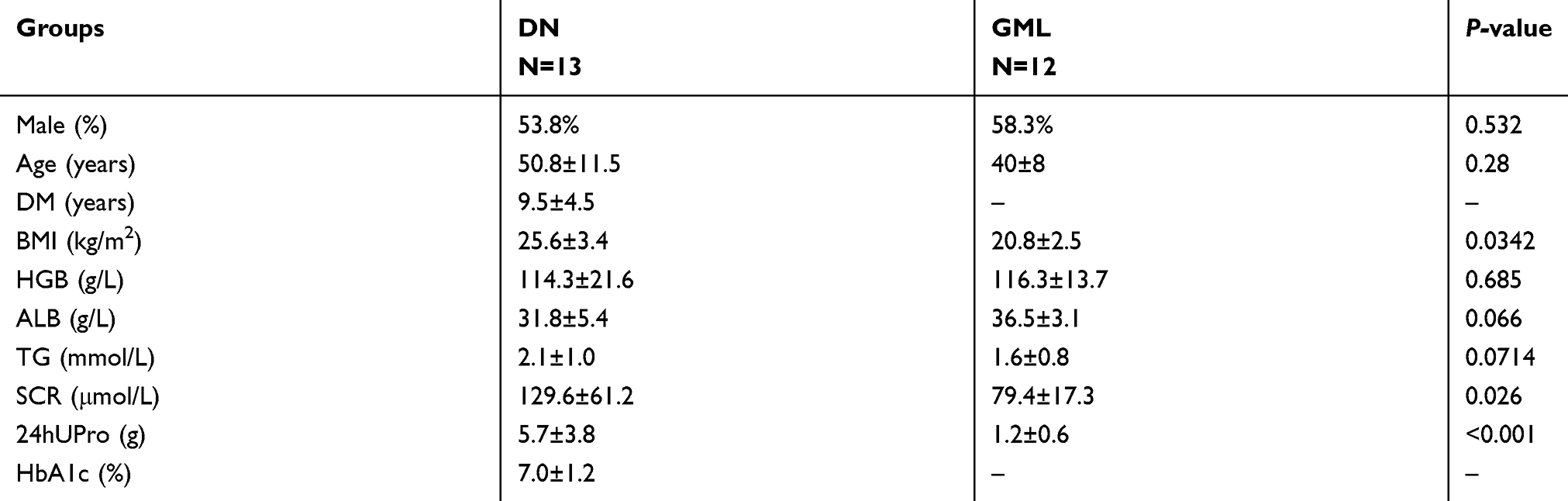 |
Table S2 Baseline characteristics of DN patients in biopsy |
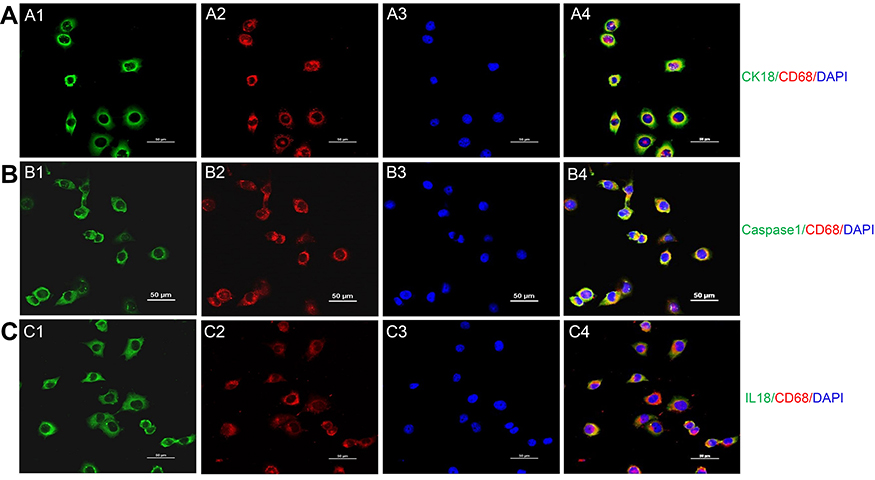 |
Figure S1 Transdifferentiation of proximal tubular epithelial cell to macrophage in a high glucose environment. (A) Costaining of PTC immunolabeled by cytokeratin (CK)-18 and macrophage immunolabelled by CD68. Green: cytokeratin (CK)-18; Red: CD68; Blue: DAPI; Merge is CK18 with CD68. (B) Costaining of pyroptosis signaling immunolabeled by caspase-1 and macrophage immunolabeled by CD68. Green: Caspase-1; Red: CD68; Blue: DAPI; Merge is caspase-1 with CD68. (C) Costaining of pyroptosis signaling immunolabeled by IL18 and macrophage immunolabeled by CD68Green: IL-18; Red: CD68; Blue: DAPI; Merge is IL-18 with CD68. Original magnification =400x. Bar width =50 μm. Abbreviation: DAPI, 4ʹ,6-diamidino-2-phenylindole. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.