")
Back to Journals » OncoTargets and Therapy » Volume 13
The Ectopic Expression of SurvivinT34A and FilC Can Enhance the Oncolytic Effects of Vaccinia Virus in Murine Gastric Cancer
Authors Wang M, Luo Y, Sun T, Mao C, Jiang Y, Yu X, Li Z, Xie T, Wu F, Yan H , Teng L
Received 13 September 2019
Accepted for publication 16 January 2020
Published 3 February 2020 Volume 2020:13 Pages 1011—1025
DOI https://doi.org/10.2147/OTT.S230902
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Minglong Wang,1,* Yanxi Luo,2,* Ting Sun,2 Chenyu Mao,1 Yili Jiang,2 Xiongfei Yu,1 Zhongqi Li,1 Tian Xie,3 Fusheng Wu,1 Hui Yan,2,3 Lisong Teng1
1Department of Surgical Oncology, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, People’s Republic of China; 2Institute of Materia Medica, Zhejiang Academy of Medical Sciences, Hangzhou, People’s Republic of China; 3Holistic Integrative Pharmacy Institutes, Hangzhou Normal University, Hangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lisong Teng
Department of Surgical Oncology, The First Affiliated Hospital, School of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou 310003, People’s Republic of China
Email [email protected]
Hui Yan
Institute of Materia Medica, Zhejiang Academy of Medical Sciences, Hangzhou 310013, People’s Republic of China
Email [email protected]
Background/Aims: Anti-tumor vaccines have been shown to be effective in cancer therapeutics ever since the anti-HPV vaccine was developed. Compared to conventional chemotherapy, anti-tumor vaccines can specifically target cancer cells and they have lower side effects. We developed a recombinant vaccinia virus (VACV) (Western Reserve) WR strain, and we tested its anti-tumor effects in an animal model.
Methods: A recombinant VACV WR strain expressing mutant survivin T34A (SurT34A) and FilC was constructed and validated. Its oncolytic effect was tested in vitro using a CCK-8 assay, and its tolerance and anti-tumor effects were tested in a murine gastric cancer model. The proportion of lymphocytes in the spleen and tumor was determined after antibody-mediated immuno-depletion.
Results: The recombinant VACV showed a stronger replication ability in tumor cells, and it was safe in vivo, even at high doses. The combination of vv-SurT34A and vv-FilC resulted in a stronger anti-tumor effect compared to either construct alone. However, the inhibitory effect of vv-SurT34A was stronger than the combination. The recombinant VACV activated the host immune response, as indicated by lymphocyte infiltration in the spleen and tumor tissues.
Conclusion: The recombinant VACV WR strain expressing SurT34A and FilC is a safe and effective anti-tumor vaccine.
Keywords: vaccinia virus WR strain, survivin T34A, FilC, anti-tumor effect
Introduction
Gastric cancer is the second most common malignancy in China, with 42.7 million new cases and about 30.1 million deaths recorded in 2013, making it the third most common cause of cancer-related mortality in China.1 The major therapies of gastric carcinoma are surgery, radiotherapy, chemotherapy, and molecular targeted therapy. Despite new drugs in clinical practice, the results have been far from satisfactory. In China, the 5-year overall survival rate is only 46%.2 Biological therapies, including anti-cancer vaccines, have gained considerable attention in recent years, and the vaccinia virus (VACV) is considered to be a suitable vector for anti-tumor vaccine therapy.3
Survivin or Birc-5 (Sur) is a member of the apoptotic inhibitory protein (IAP) family, and it is widely expressed in almost all tumor cells, but usually not in normal cells.4 Recent studies have shown that survivin can stimulate anti-tumor cytotoxic T-cell (CTL) responses.5,6 Krieg et al reported that the high levels of survivin in gastric cancer cells was correlated with a high mortality rate, recurrence, metastasis, and other adverse outcomes.7 Our previous study showed that the survivin T34A mutant (Sur-T34A), when fused with the TAT cell-penetrating peptide and transduced into cancer cells, can induce apoptosis in melanoma cells in vitro and in vivo.8 Ogura et al further demonstrated that Sur-T34A induced apoptosis in cancer cells by activating mitochondrial pathways,9 while other researchers showed little toxicity of Sur-T34A in normal cells both in vitro and in vivo.10,11 Therefore, SurT34A is a potential antigen for developing therapeutic anti-tumor vaccines.
Bacterial flagellin (FilC) is a pathogen-associated molecular pattern (PAMP) ligand that binds to the Toll-like receptor 5 (TLR-5) on the surface of dendritic cells (DC), triggers cytokine release, and activates innate and adaptive immune responses.12,13 Previous research also showed that FilC is widely used as a key protein and peptides encoded by both DNA vaccines or viral vectors, which showed a favorable adjuvant effect, are also key.14,15 Lee et al reported that FilC acted as a potent adjuvant by activating the TLR5 signaling pathway in a cervical cancer mouse model.16 Thus, we examined whether FilC could be used as a molecular adjuvant with the viccinia virus Western Reverse (WR) strain as a vector.
A number of viruses, such as adenoviruses (Ads), HSV, alphaviruses, rhabdoviruses, and Newcastle disease virus (NDV), among others, have been used as oncolytic viruses in a number of animal models and clinical trials.17–21 VACV has been used as a vaccine for decades, and the WR strain is a particularly attractive oncolytic viral vector due to its high lytic ability and natural selectivity against tumor cells.22 Despite the therapeutic potential of VACV in gastric cancer, the combination of Sur-T34A and FilC encoded by the VACV WR strain has never been reported previously.
In this study, we developed a recombinant VACV-based anti-tumor vaccine expressing SurT34A and FilC. We expected to observe both therapeutic and prophylactic effects from SurT34A, and enhanced immunogenicity from FilC, eventually leading to synergically enhanced anti-tumor effects.
Materials and Methods
Cell Lines
Vero (African green monkey kidney epithelial), B-16 (murine melanoma), NIH3T3 (murine embryo fibroblast), and hepa1-6 (murine hepatoma) cell lines were cultured in DMEM supplemented with 10% fetal calf serum (FCS).
AGS, SGC-7901 (human gastric carcinoma), and MFC (murine forestomach carcinoma) cell lines were cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS).
The 4T1 (murine breast carcinoma) cell lines were cultured in RPMI 1640 medium with 10% FCS. All of the cells lines were cultured at 37°C and 80% humidity under 5% CO2.
Plasmids
The sequence of the mouse survivin gene was obtained from Genbank, NCBI (NM_009689.2), and the pUC57-Survivin T34A plasmid was generated by GenScript Co. Ltd(Nanjing, Chnia). after T34A mutagenesis and codon optimization for the vaccinia virus system. The pCZ vector was constructed by incorporating the mcherry, pgt, and Zeocin genes into the pCB plasmid, and it was used as the empty vector control for the Thymidine Kinase(TK) gene deletion virus. The pCZ-SurT34A-ch vector was constructed by digesting pUC57-Survivin T34A and pCZ wtih Bgl-II and SwaI (New England Biolabs), and cloning SurT34A into pCZ using T4 DNA ligase (NEB). To construct the pCZ-fliC-ch vector, the genomic DNA of Salmonella ZJ111 was extracted and a FilC gene fragment was first amplified using the primers P1 and P2 (see below) and digested with Swal and BanHI. The pCZ vector was then digested with Bgl-II and Swal, and the FilC fragment was inserted into the pCZ Vector with T4 DNA ligase. All of the plasmids were extracted with a SanPrep Column Plasmid Mini-Prep Kit (Shanghai Sangon Biotech Co. Ltd.), and sequenced by Sangon Biotech (Shanghai Sangon Biotech Co. Ltd.). The primer sequences were as follows:
P1: 5ʹgcgcatttaaatgcggccgcattaacgcagtaaagagag 3ʹ,
P2: 5ʹ gaaggatccatcgatgaattcactagtgccaccatggcacaagtcattaatacaaac 3ʹ.
Construction of the TK Gene Deleted Recombinant VAVC
All of the viruses used in this study are based on the WR strain. The wild-type (WT) virus was kindly provided by the NIH-AIDS Research & Reference Reagent Program. Vero cells were transfected with the plasmids pCZ-SurT34A-ch, pCZ-FilC-ch, and pCB-MCZ using Lipofection (NEB). A mixture of 4 µg plasmid, 10 µL Lipofection, and 500 µL serum-free DMEM was added onto Vero monolayers and incubated for 4 hrs. The cells were then infected with the parental virus (WT VACV) at the multiplicity of infection (MOI) of 0.1 PFU/cell. After 48–72 hrs, the recombinant vaccinia viruses were harvested, freeze/thawed thrice, and then used to serially infect new Vero monolayers. The infected cells were selected by xanthine-guanine phosphoribosyltransferase (XGPRT) due to the presence of the gpt gene in the recombinant virus. After several plaque purification passages, the TK-deleted VACV infected cells were selected and isolated with the help of the mCherry reporter gene. Following several rounds of selection, the virus was plaque tested to confirm purity. The pure VACVs were then amplified in Vero cells, extracted by sucrose gradient centrifugation, and quantified by a plaque assay in terms of the number of plaque-forming units per milliliter (PFU/mL).
CCK-8 Assay
Cells were seeded in a 96-well plate at the density of 104 cells/well in 100 μL medium, and cultured in a CO2 incubator at 37°C for 24 hrs. After adding 10 μL of recombinant viruses at different MOIs (0.01 PFU/cell, 0.1 PFU/cell, 1 PFU/cell, 10 PFU/cell), the cells were incubated for another 48 hrs. Ten microliters of CCK-8 solution (Sangon Biotech, China) was added to each well, and the plate was incubated for 1 hr, after which the absorbance was measured at 450 nm using a microplate reader (Thermo, USA).
Western Blotting
The virus-infected cells were incubated at 37°C for 48 hrs, and the total protein and recombinant viruses were extracted. After separating the proteins using 13% SDS-PAGE, the bands were transferred onto a polyvinylidene fluoride (PVDF) membrane. The latter was blocked with 5% milk for 2 hrs at 37°C, and incubated overnight with anti-survivin antibodies (1:1000, Abcam ab182132) and anti-flagellin antibodies (1:1000, Abcam ab93713) at 4°C. After washing with TBST, the membrane was incubated with HRP-conjugated goat anti-Rabbit IgG (1:5000) for 1 hr at room temperature. The protein bands were detected using the SuperLumia ECL Plus Western blotting detection reagents (Abbkine) and bands were quantified with NIH ImageJ.
Flow Cytometry Analysis
Mice inoculated with viruses or PBS were sacrificed, and their spleens and tumors were removed and collected in ice-cold PBS containing 1% FBS and 2 mM EDTA. After homogenizing the spleen tissues, the resulting splenocytes were incubated with Fc Block (mouse anti-CD16/CD32; ab25235), followed by anti-CD3 (ab16669), anti-CD4 (ab25475), anti-CD8 (ab22378), anti-CD11b (ab8878), anti-CD19 (ab31947), anti-CD11c (ab11029), anti-MHC II (ab23990), and anti-foxp3 (ab20034) antibodies. The stained cells were acquired on BD FACSCalibur, and data were analyzed using FlowJo software (v.10.0.7; Tree Star, Ashland, OR, USA). The CD4+ T lymphocytes were characterized as CD3+/CD4+, CD8+ T lymphocytes as CD3+/CD8+, B lymphocytes as CD11b+/CD19+, DCs as CD11c+/MH II +, and T-reg cells as CD4+/foxp3+.
Virus Replication and Cytotoxicity Assay in vitro
To verify the infection ability of the TK gene-deleted recombinant VACV in different cancer cells, 5 × 106 cells of each line were infected at the MOI of 0.1 PFU/cell. The viral titers were measured at 12, 24, 48, and 72 hrs post infection (hpi). All of the cell lines were then infected at different MOIs, and cell viability was determined with a CCK-8 assay at 72 hpi.
Viral Toxicity Assay in vivo
The mice were intraperitoneally (i.p.) injected with recombinant VACV, WT VACV (1×107 PFU/mouse, 100 μL), or PBS. The tumor weights and survival duration of the mice were recorded daily for 15 days. In addition, some mice were sacrificed at 24, 72, 120, and 168 hpi, and their parenchymal organs were removed. Viral titers in the homogenates of the brain, liver, and ovaries and peritoneal effusion were then measured (schemes are shown in Figure 1A).
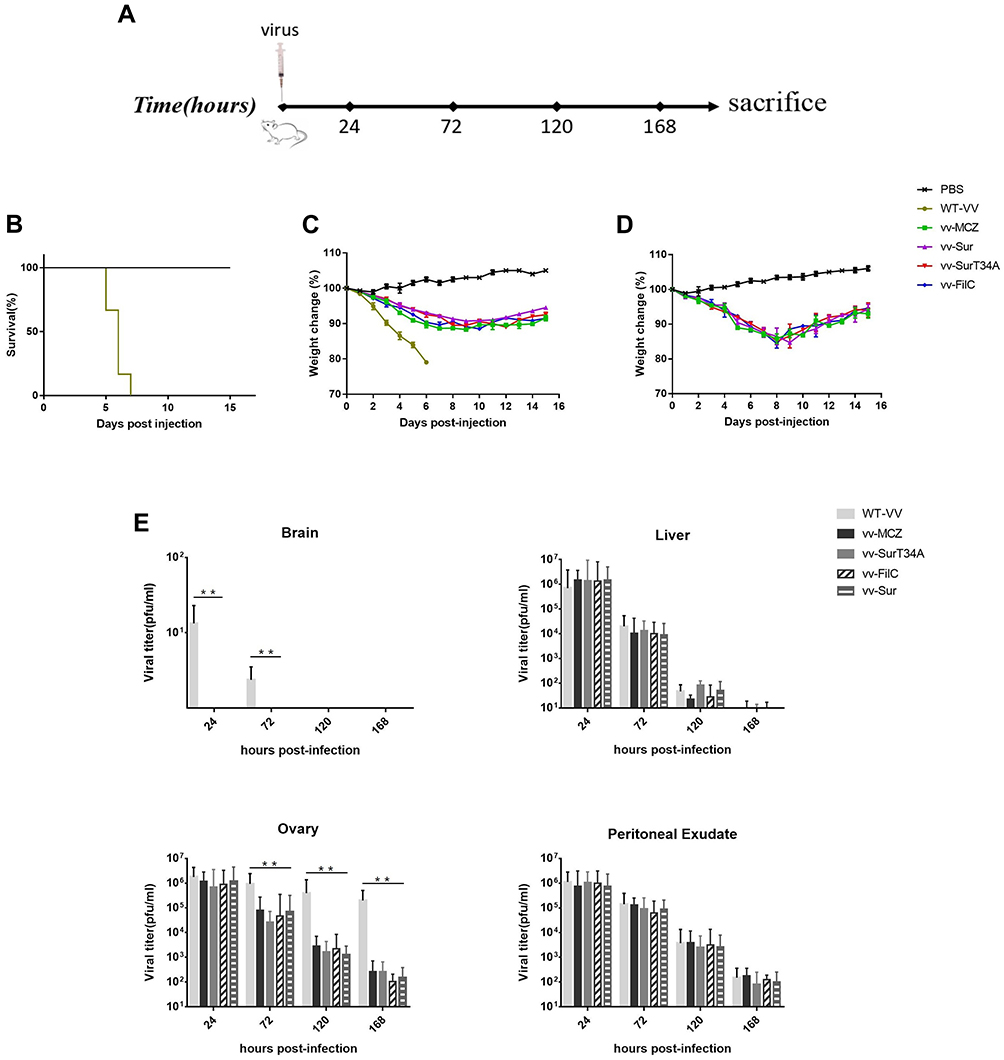 |
Figure 1 Viral toxicity and replication in vivo. Female BALB/c mice (4 mice each group) were inoculated (i.p.) with 100 µL of different viruses (WT-VV, vv-MCZ, vv-SurT34A, vv-Sur and vv-FilC at 1×107 or 1×108 PFU/mouse), and their weight and survival time were recorded daily until day 15. (A) Scheme of virus injection; (B) Survival time of mice inoculated with 1 × 107 PFU/mouse, mice were sacrificed when body weight reduction was > 20%; (C) Inoculation of 1 × 107 PFU/mouse, weights of the mice were recorded and evaluated for percent weight change; (D) Inoculation of 1 × 108 PFU/mouse, weights of the mice were recorded and evaluated for percent weight change; (E) Inoculation of 1 × 107 PFU/mouse, viral titer in the brain, liver, ovary, and peritoneal effusion at 24 hpi, 72 phi, 120 hpi, and 168 phi. Data are shown as the mean ± SD. **p < 0.01. |
Tumor Modelling and Vaccination Protocol
Female BALB/c mice (6–8 weeks old, weighing 18 ± 2 g) were purchased from the Experimental Animal Center, Zhejiang Academy of Medical Sciences. The animals were housed under controlled environmental conditions (temperature and humidity), and they were given access to food and water ad libitum.
To establish the xenograft tumor model, the mice were injected subcutaneously with 5 × 105 MFC cells under their right flanks. Once the tumor volume reached ~50 mm3, the recombinant VACV (1 × 108 PFU) or PBS was injected i.p. Some mice (3 mice each group) were sacrificed on days 3, 6, 9, and 12 post injection, and their tumors and spleens were removed. The percentage of CD4+, CD8+, and CD19+ lymphocytes were analyzed as described, and the viral titers of the tumor homogenates were determined with a plaque assay. In another cohort, mice were injected i.p. with recombinant VACV (1 × 108 PFU/mouse, 100 μL) or 100 μL PBS 4 times. The size of the tumor was measured every 3 days and survival duration was recorded. A part of the cohort (n = 6) was sacrificed on day 30 and their tumors were weighed, and the tumor volume was calculated as (length × width 2) × 2.23
To vaccinate the mice, the mice were injected i.p. with recombinant VACV (1 × 108 pfu/mouse, 100 μL) or 100 µL PBS on days 7, 14, 21, and 28 after acclimatization. The MFCs (5 × 105/100 μL/mouse) were then transplanted at 7 days after inoculation. Tumor growth and survival durations were recorded every 3 days, and the volume of the tumors was calculated as above.
Antibody-Mediated Immuno-Depletion in vivo
To deplete specific immune cell subsets prior to and during recombinant VACV therapy, the mice were injected with purified monoclonal antibodies against CD4+ (GK 1.5) and CD8+ (53–6.7) T-cells on days −4 and −2, relative to day 0 when MFC cells (1 × 106/100 μL/mouse) were inoculated. The recombinant VACV (1 × 108 pfu/mouse, 100 μL) was injected on days 5, 9, and 13. Immuno-depletion was repeated before and after every vaccination on days 4, 7, 11, and 15. T-cell depletion was confirmed by flow cytometry on days 0, 5, 9, and 13, and it was consistently > 95%. In addition to the isotype control, unvaccinated (injected with PBS instead of VACV and not immuno-depleted), therapeutic (injected only with recombinant VACV), and non-depleted controls were also included.
Statistical Analysis
All of the data were obtained from at least three independent experiments, and the data are presented as the mean ± SD. Statistical analyses were carried out using SPSS v22.0 (IBM, USA). Significance for comparison between groups was determined using a one-way ANOVA test with Bonferroni correction for multiple comparisons. Differences in tumor growth were analyzed using a two-tailed Student’s t-test. Survival between groups was determined with a Log rank test and survival Kaplan-Meier survival curve. Differences were considered to be statistically significant when *p < 0.05 or **p < 0.01.
Results
Construction and Identity of Recombinant Virus
The recombinant VACV expressing Sur-T34A and FilC was constructed using GPT, Zeocin, and mCherry as the selecting genes. The cloning scheme is shown in Figure 2A–D, respectively, show fluorescent virus-infected cells, FilC expression in vv-FilC, and expression of survivin protein in vv-SurT34A-infected cell lines.
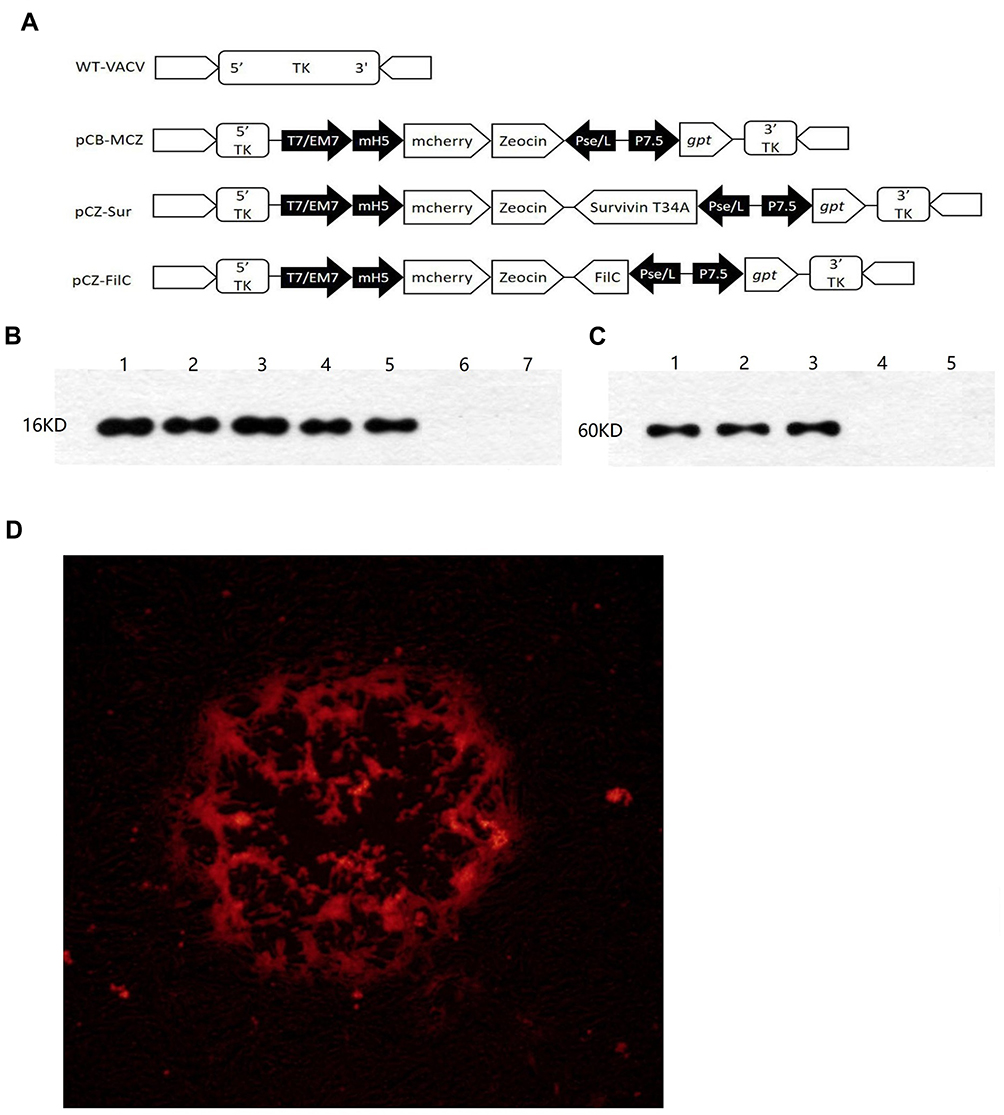 |
Figure 2 Characterization of vv-SurT4A and vv-FilC. (A) Cloning scheme; (B) Survivin expression in virus and cells: Lane 1: vv-Sur (1 × 107 PFU), Lane 2: vv-SurT34A (1 × 107 PFU), Lane 3: B-16 cells, lane 4: MFC cells, Lane 5: Hepa1-6, Lane 6: VERO cells and Lane 7: wild-type VACV; (C) FilC expression in virus and cells: Lane 1: vv-FilC (1 × 107 PFU), Lane 2: Escherichia coli, BL21, Lane 3: Salmonella ZJ-111, Lane 4: VERO cells and Lane 5: wild-type VACV; (D) Red fluorescent of VERO infected with recombinant VACV. |
Viral Replication in vitro
To verify the replication ability of recombinant VACV, different cells were infected at the same MOI (0.1 PFU/cell), and after harvesting the viruses at different times (12 h, 24 h, 48 h, and 72 h), their titers were determined using the Vero cells. As shown in Figure 3, the titers were stable in all of the cell lines after 48 hrs of infection, as no significant difference was seen between the 48 hpi and 72 hpi viral titers. The replication ability of the recombinant VACV was not weaker than that of the parental strain, except in the NIH3T3 cells, where lower titers were obtained. In the cancer cell lines, the recombinant virus had a stronger replication ability than the WT VACV, while both recombinant and WT VACV were less replicative in 4T1 cells.
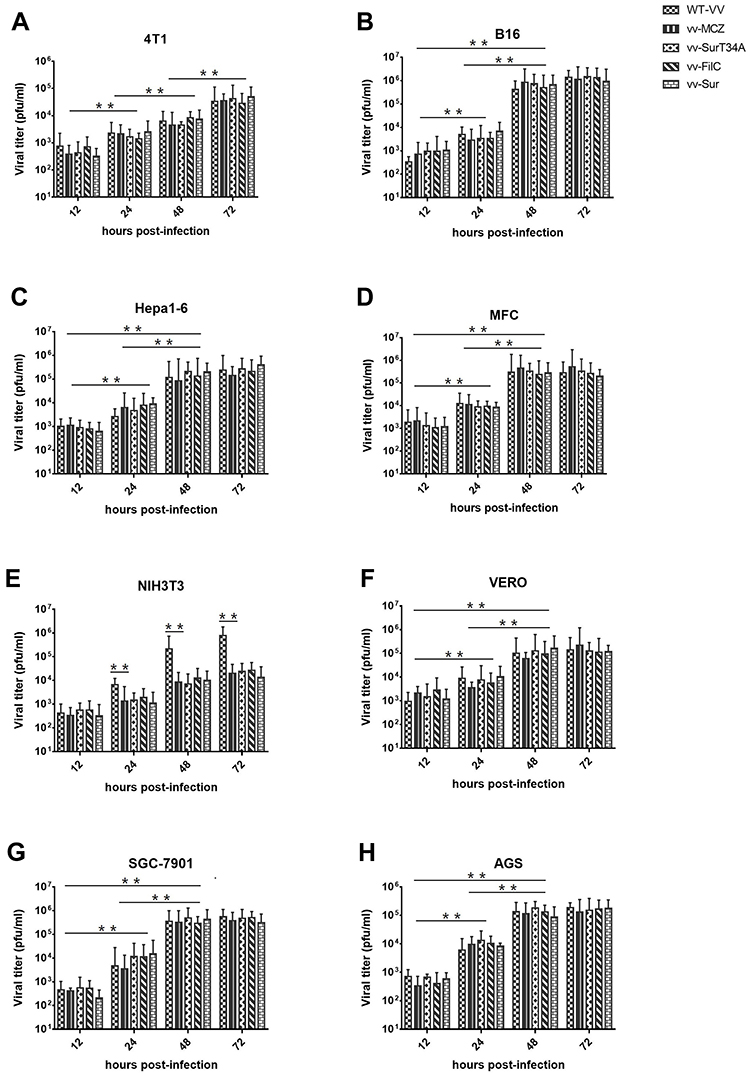 |
Figure 3 Viral replication in vitro. Murine tumor cell lines (A–D, B16, Hepa1-6, MFC and 4T1), the normal cell line (E, NIH3T3), tool cells (F, VERO), and human gastric carcinoma cell lines (G, H, SGC-7901, and AGS) were infected with wild type VACV (WT-VV), vv-MCZ, vv-SurT34A, vv-FilC, and vv-Sur at 0.1 MOI and samples were collected at indicated times. Viral titers were determined in Vero cells. Each bar represents the mean ± SD of three separate experiments (n = 4). **p < 0.01. |
Viral Cytotoxicity in vitro
To evaluate the oncolytic ability of the recombinant virus, different cell lines were infected with different MOIs of the WT and recombinant VACV, and cell viability was measured with a CCK-8 assay at 72 hpi. Both viruses had poor oncolytic potency in the 4T1, while the WT VACV had a stronger oncolytic potency than the recombinant VACV in NIH3T3 cells. In the three tumor cell lines and Vero cells, the oncolytic effect of both viruses were similar, and the cell viability was less than 20% at the MOI of 10 PFU/cell at 72 hpi (Figure 4).
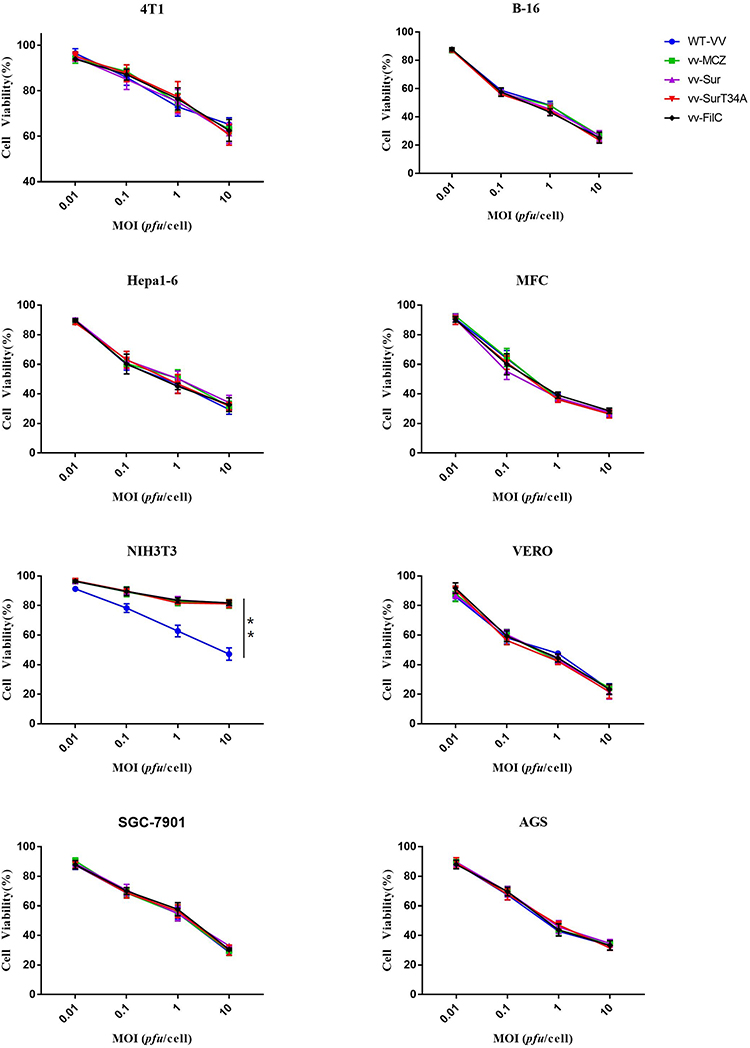 |
Figure 4 Viral cytotoxicity assay in vitro. B-16, Hepa1-6, MFC, 4T1, NIH3T3, SGC-7901, AGS, and Vero cells were infected with WT-VV, vv-MCZ, vv-FilC, vv-Sur, and vv-SurT34A at different MOIs (0.01 PFU/cell, 0.1 PFU/cell, 1 PFU/cell, and 10 PFU/cell). Cell viability was measured with a CCK-8 assay at 72 hrs hpi. Each bar represents the mean ± SD of three separate experiments (n = 4). **p < 0.01. |
Viral Toxicity and Replication in vivo
To evaluate the viral toxicity in mice, they were injected with 100 µL recombinant VACV (1 × 107PFU/1 × 108 PFU), WT VACV (1 × 107PFU/1 × 108PFU; control group), or PBS (mock-group). As shown in Figure 1B–D, the main adverse effect of VACV was weight loss; when inoculation of 1 × 107 PFU/mouse, the control group mice rapidly lost weight and died within 1 week. Mice injected with the recombinant VACV started regaining weight 7–10 days after injection; when inoculation was 1 × 108 PFU/mouse, mice injected with recombinant VACV started regaining weight 11–14 days after injection. Therefore, a high dose of WT VACV was not suitable in vivo due to its toxicity.
To evaluate the viral replication in mice, they were sacrificed at 24, 72, 120, and 168 h after inoculation of 1 × 107 PFU/mouse. As shown in Figure 1E, viral titers in the peritoneal exudates were initially significantly lower than the injected dose, and they gradually decreased over time. There was little virus in the brain due to the blood-brain barrier. Viral titers decreased rapidly in the liver and was eradicated in 168 h. Interestingly, the WT VACV showed more replication in the ovaries compared to the recombinant VACV.
In vivo Oncolytic Ability of Recombinant VACV
The anti-tumor effect of recombinant VACV in vivo was determined as described in the Methods section (Figure 5A). The tumor volume of the vv-MCZ and PBS groups was similar during the first 10 days post virus injection, and it decreased significantly in the vv-MCZ mice after 20 days. The tumor volume of the vv-SurT34A and vv-FilC groups were significantly smaller than that of the vv-MCZ group after the first injection. The combination of vv-Sur-T34A and vv-FilC showed a stronger anti-tumor effect than either virus alone (Figure 5B). As shown in Figure 5C, the unvaccinated mice had a poor prognosis, while those injected with recombinant VACV survived longer. The median survival duration of the PBS, vv-MCZ, vv-SurT34A, vv-FilC, and vv-SurT34A/vv-FilC groups were 19, 35, 44, 41, and 50 days, respectively. The weight of the tumors correlated significantly with tumor volume and mice survival (Figure 5D).
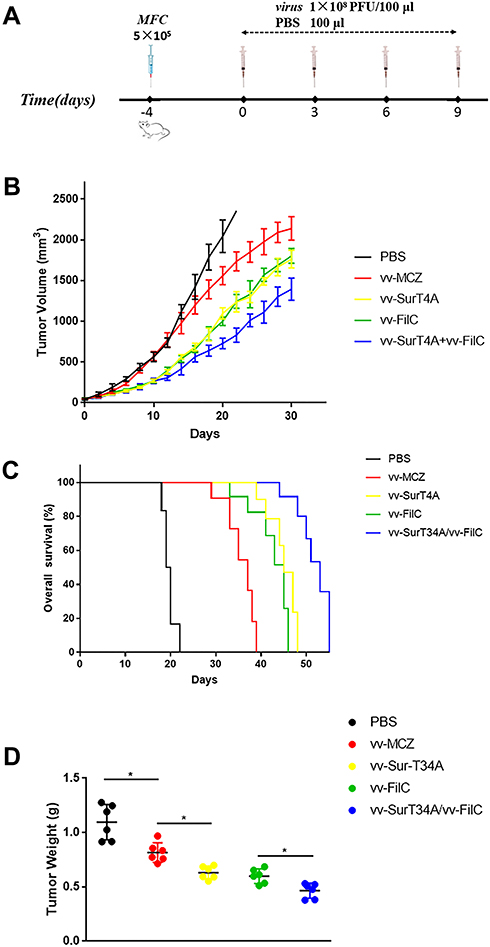 |
Figure 5 Tumor model and therapeutic protocol. MFC (5 × 105 cells·100 μL/mice, 6 mice each group) were injected subcutaneously under the right flank. When the tumor volume reached 50 mm3, the mice were injected (i.p.) with recombinant VACV (1 × 108PFU/100 μL) or 100 μL PBS at the interval of 3 days, with a total of 4 injections. Some of the mice were sacrificed at day 30. Combination groups were 5 × 107PFU/50 μL of vv-SurT34A and vv-FilC. (A) Scheme of viral infection; (B) The tumor volume of mice in each group; (C) Kaplan-Meier survival curves for tumor bearing mice; (D) Weights of tumors at day 30. * p < 0.05. |
Recombinant Virus Activates Mice Immunity
High viral titers were maintained in the tumor homogenates even 12 days after injection (schemes of virus infection are shown in Figure 6A), with no significant differences between the three recombinant viruses (Figure 6B). Since the spleen is the key organ in the adaptive immune response, we evaluated the splenic lymphocytes to assess the immune response elicited by recombinant VACV. As shown in Figure 6C–E, there was no significant difference in the proportion of the CD4+, CD8+, and CD19+ cells between the groups three days after virus inoculation. However, the recombinant VACV group had more CD4+ and CD8+ splenic T-cells compared to the uninfected controls, peaking on the ninth day post infection (Figure 6C and D). In addition, the proportion of CD4+ and CD8+ T-cells in the vv-SurT34A/vv-FilC group was higher than that of the vv-MCZ group (p<0.05). CD19+ B-cells were abundant in the vv-FilC and vv-SurT34A/vv-FilC groups from the 9th day onwards and lasted until the 12th day, but they were lower in the vv-SurT34A, vv-MCZ, and PBS groups (Figure 6E).
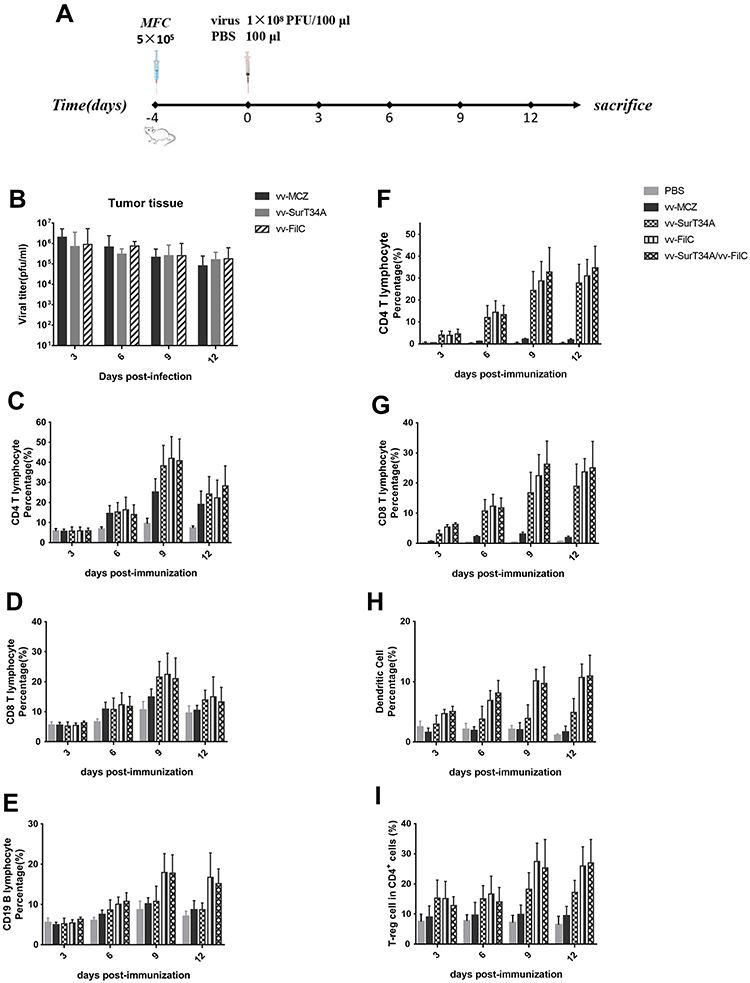 |
Figure 6 Recombinant VACV activates immunity of mice. MFC cells (5 × 105) were injected subcutaneously under the right flank. Recombinant VACV (1 × 108 PFU/100μL) or PBS was injected (i.p.) when the tumor volume reached 50 mm3. Mice were sacrificed at 3, 6, 9, and 12 days post injection, and the tumor and spleen were removed. The CD4+ T lymphocytes were characterized as CD3+/CD4+, CD8+ T lymphocytes as CD3+/CD8+, B lymphocytes as CD11b+/CD19+, DCs as CD11c+/MH II +, and T-reg cells as CD4+/foxp3+. (A) Scheme of virus infection; (B) Tumor viral titer in each group; (C) Percentage of CD4+ T lymphocytes in the spleen; (D) Percentage of CD8+ T lymphocytes in the spleen; (E) Percentage of CD19+ cells in the spleen; (F) Percentage of CD4+ T lymphocytes in the tumor tissue; (G) Percentage of CD8+ T lymphocytes in the tumor tissue; (H) Percentage of dendritic cells in the tumor tissue; (I) Percentage of T-reg cells in CD4+ cells; Data are shown as the mean ± SD. |
We also analyzed T-cell infiltration in the tumor tissues of different groups (Figure 6F–I), and we observed significantly increased recruitment of CD4+ and CD8+ T-cells in the recombinant VACV group, compared to the PBS and vv-MCZ groups, peaking on the 9th day and remaining sustained until the 12th day (Figure 6F and G). The proportion of DCs in the vv-FilC and combination groups was about 2-fold higher compared to that in the vv-SurT34A group on the 6th day, and 3-fold higher on the 9th and 12th days (Figure 6H). The proportion of T-reg cells in the vv-FilC and combination groups increased to 30% between days 6 and 12, while that in the vv-SurT34A group remained at 10% (Figure 6I). These results clearly indicated that vv-FilC acted as an adjuvant and enhanced the immunogenicity of the recombinant VACV vaccine.
Tumor Inhibition of Recombinant VACV
To evaluate the anti-tumor effect of recombinant VACV, the mice were immunized with four inoculations of the recombinant VACV or PBS, and then challenged with MFC cells subcutaneously (Figure 7A). The median oncogenesis time in the PBS and vv-FilC groups was 4 days, 5 days in the vv-MCZ group, and 7 days in the vv-SurT34A group. As shown in Figure 7B, the vv-FilC group had similar tumor growth rate as the PBS group, while vv-SurT34A had a stronger inhibitory effect on tumor growth than vv-MCZ, and significantly inhibited tumor growth from day 10 to 20 after inoculation. Surprisingly, in Figure 7C, the combination and vv-FilC groups did not show superior tumor inhibitory activity compared to the vv-SurT34A group. The median survival time of the PBS, vv-MCZ, vv-SurT34A, vv-FilC, and vv-SurT34A/vv-FilC groups were 18, 23, 39, 20, and 20 days, respectively. The rate of the tumor growth correlated with mice survival.
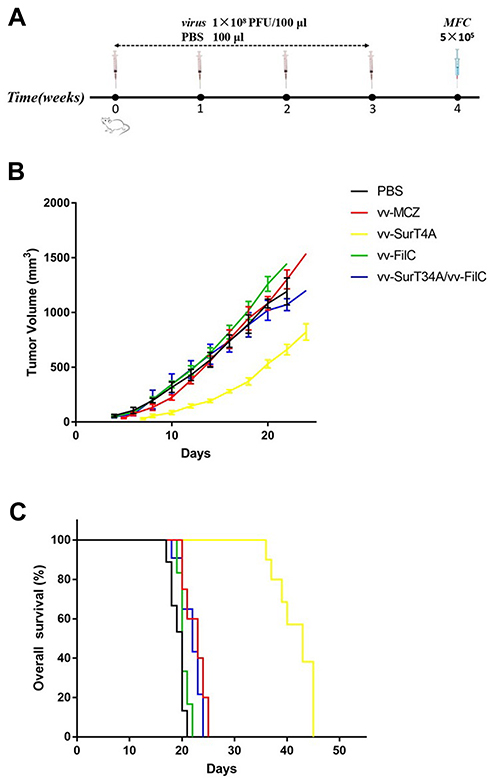 |
Figure 7 Tumor inhibition of recombinant VACV. Mice were inoculated with recombinant VACV (1 × 108PFU/100 μL) or 100 μL PBS 4 times, and the right flank was injected subcutaneously with MFC (5 × 105cells/100 μL). (A) Scheme of virus infection; (B) The tumor volume of mice in each group; (C) Kaplan-Meier survival curves for tumor-bearing mice. |
Antibody-Mediated Immuno-Depletion Diminished the Anti-Tumor Response
To evaluate the role of tumor-specific CD4+ and CD8+ T-cells in recombinant VACV-mediated anti-tumor responses, we depleted the above cells in the mice using specific antibodies (Figure 8A) before and after vaccination. As shown in Figure 8B and C, depletion of CD8+ T-cells significantly reduced the survival of the vaccinated mice compared to the non-depleted and isotype control groups (p<0.01), to that of the unvaccinated mice. Vaccinated mice depleted of both CD4+ and CD8+ T-cells showed an even worse prognosis compared to the CD8+-depleted and unvaccinated groups. Although depletion of CD4+ T-cells also reduced survival, the effect was less severe. The median survival time of anti-CD4+, anti-CD8+, anti-CD4+/CD8+, PBS, and non-depleted groups vaccinated with vv-SurT34A was 20, 17, 14, 18, and 31 days, respectively, and 17, 14, 13, 16, and 28 days, respectively, in the mice vaccinated with vv-FilC.
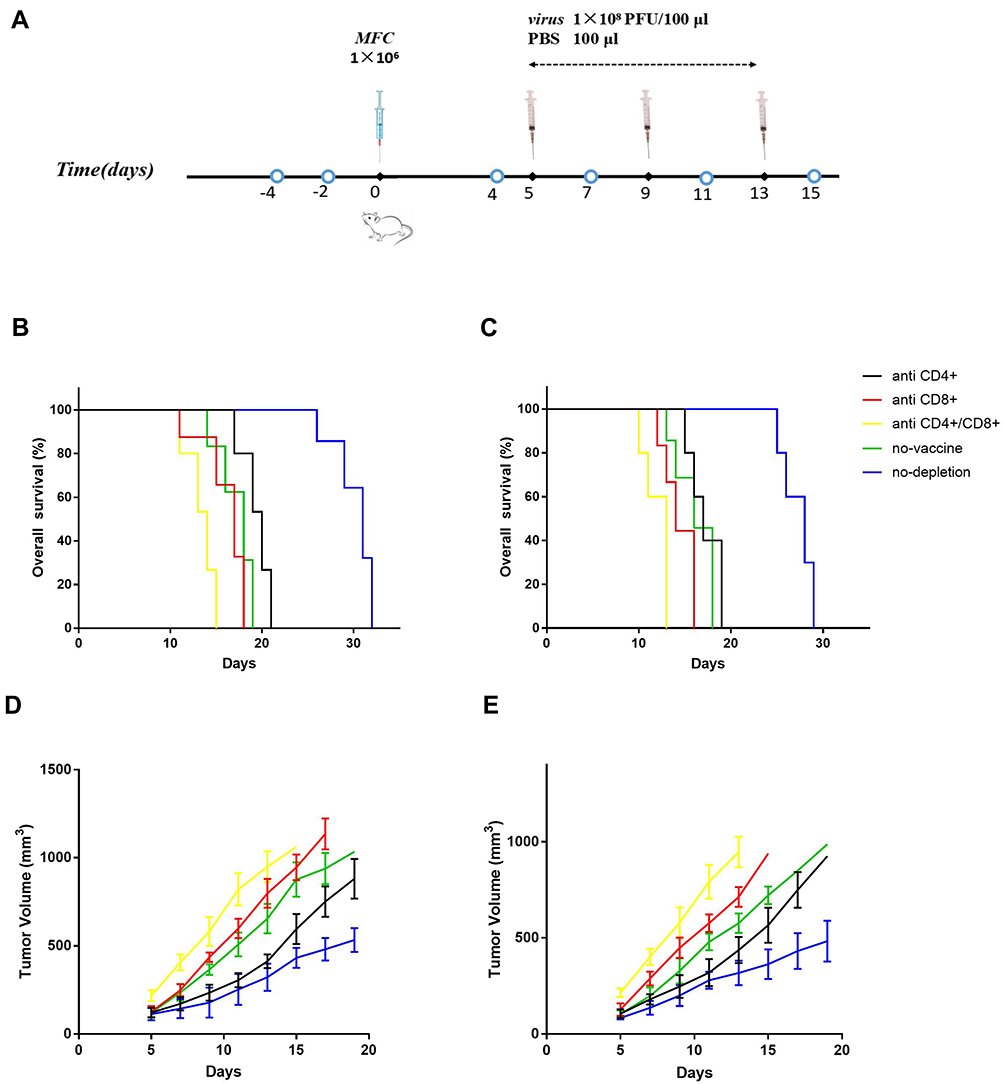 |
Figure 8 Antibody-mediated immuno-depletion diminished the anti-tumor response. The antibodies were injected on days −4 and −2, followed by MFC (1 × 106/100 μL/mouse) transplantation on day 0, and recombinant VACV (1 × 108 pfu/mouse, 100 μL) inoculation on days 5, 9, and 13. Further depletion was carried out before and after each vaccination on days 4, 7, 11, and 15. (A) Scheme of antibodies and virus injection; (B) Kaplan-Meier survival curves for immuno-depleted mice treated with vv-SurT34A; (C) Kaplan-Meier survival curves for immuno-depleted mice treated with vv-FilC; (D) The tumor volume in mice treated with vv-SurT34A; (E) The tumor volume in mice treated with vv-FilC. |
To assess the effect of specific T cells in tumor progression, we measured the tumor volume every 2 days after MFC implantation. As shown in Figure 8D and E, the mean tumor volumes of the anti-CD4+, anti-CD8+, anti-CD4+/CD8+, PBS, and non-depleted groups vaccinated with vv-SurT34A were 121.5±13.2 mm3, 126.8±16.2 mm3, 217.2±15.5 mm3, 121.5±14.4 mm3, and 113.4±17.1 mm3, respectively, and 105.8±11.2 mm3, 126.5±16.3 mm3, 214.5±11.2 mm3, 103.5±10.6 mm3, and 82.6±9.3mm3, respectively, in mice vaccinated with vv-FilC, at 5 days post-implantation. Mice depleted of both T-cell subtypes had the largest tumor volume and the fastest tumor growth, whereas CD8+ T-cell depletion also accelerated tumor growth. Until day 10 post-implantation, tumor growth in the anti-CD4+ group paralleled that in the non-depleted and IgG2 groups, and accelerated thereafter.
Discussion
Following the success of immune checkpoint inhibitors (PD-1, PDL-1, and CTLA-4), the next promising anti-tumor immunotherapeutic strategy was based on oncoltytic viruses.23 These viruses specifically infect and replicate within tumor cells, and lyse them to release their progeny, thereby continuing an infection-lysis cycle until all of the tumor cells are cleared. We chose VACV in this study since it is the only virus that replicates in the cytoplasm, and larger exogenous genes (about 25 kb) can be inserted into its genome without any risk of integrating into the host genome.24 The modified, highly attenuated Ankara strain of VACV (MVA) is used as a broad spectrum vaccine, but compared to the WR strain, it is poorly immunogenic in BALB/c mice; the WR strain elicited a 20-fold stronger CD8-T+ lymphocyte response compared to MVA.25
Thymidine kinase (TK) is the key enzyme in the pyrimidine synthesis pathway, and it is present in significantly higher levels in tumor cells compared to normal cells. The TK gene-deleted VACV therefore requires thymidine triphosphate (TTP) from the host cells to replicate, and the TK-deficient VACV selectively infect and replicate in the rapidly dividing tumor cells, which have higher levels of nucleotides compared to the healthy, less replicative cells.26,27 For its tumor cell selectivity and lower virulence in mice, TK-deficient VACV is often used as an oncolytic virus vector.28,29 The ability to infect different tumor cells is not diminished after TK gene deletion, and with significantly less replicative ability in normal cells, even high doses of recombinant VACV were found to be safe for the BALB/c mice, thereby providing the basis for high titer viral therapy. The New York City Board of Health vaccine (Wyeth) strain is TK gene deleted (VACV:JX-594), and it expresses granulocyte-macrophage colony-stimulating factor (GM-CSF).30 It has shown good host tolerance in a Phase I clinical trial, and longer survival in a Phase II trial, after both intra-tumoral and intravenous injections.31–33 However, this vaccine has a poorer anti-tumor effect in vitro compared to the wild-type (WT) strain.34 Recombinant VACV has shown therapeutic effects in several murine tumor models, including those of glioblastoma, pancreatic cancer, and melanoma.35–38 Interestingly, we found that both WT and recombinant VACV were less replicative in the murine triple-negative breast cancer (TNBC) cell line 4T1, which shows high levels of TTP,39 both in vitro and in vivo. Furthermore, TNBC cells from different species differ in terms of TTP expression, which affects the replication of recombinant VACV.
This is the first study to express the survivin T34A mutant in the VACV WR vector, as previous studies have only cloned wild-type survivin in the vaccinia Ankara vector or truncated survivin in other vectors.40,41 The wild-type survivin inhibits apoptosis in tumor cells and enables the cells to escape host immune surveillance, since it is usually not recognized by the immune system due to its low antigenicity. However, the SurT34A mutant reversed the immunosuppressive tumor microenvironment and induced apoptosis in tumor cells. However, we did not observe a stronger anti-tumor effect of vv-SurT34A compared to vv-MCZ, vv-FilC, or even wild-type VACV in vivo or in vitro, possibly because the high anti-tumor effect of VACV made the apoptosis induced by SurT34A redundant.
Tumor growth is frequently assisted by the evasion of host immune responses by the tumor cells.42 In the murine tumor microenvironment, DCs are involved in the innate immune response and the CD4+, CD8+, and T-reg cells mediate the specific immune response. Tumor cell lysis releases the tumor-associated antigens that attract more antigen-presenting cells (APCs), thereby intensifying the anti-tumor immune response. The VACVs elicited 1.4-fold higher CD4+ and CD8+ T-cells in the spleen, and 15-, 10-, and 2-fold higher CD4+, CD8+, and regulatory T-cells, respectively, in the tumors compared to vv-MCZ. In addition, vv-FilC acted as a strong adjuvant that augmented immune cell production. The depletion of CD4+ and CD8+ T-cells resulted in larger tumor volumes and shorter survival, thereby proving the vital role of the adaptive immune responses in tumor progression and regression, as well as the potent anti-tumor effects of reversing immunosuppression. Taken together, the combination of vv-SurT34A and vv-FilC had a significantly stronger anti-tumor effect than either construct alone.
Helper T cell (Th) function is critical for the adaptive immune response against tumors, since the APCs present the processed tumor antigen to CD4+ Th cells via MHC-II molecules. The activated CD4+ T-cells subsequently stimulate the antigen-specific CD8+ cytotoxic T lymphocytes (CTLs), which clear the antigen-harboring tumor cells. However, the survival and tumor growth in the VACV- or vv-FilC-inoculated mice were similar to that of the unvaccinated and vv-MCZ groups. This is consistent with previous reports that showed early accelerated tumor growth in the tumor-bearing BALB/c mice treated with flagellin compared to the saline-treated group, which can be explained by a flagellin-induced shift in the Th1/Th2 response, resulting in a decreased IFN-γ:IL-4 ratio and increased proportion of CD4+ and CD25+ T-cells.43–47 These lymphocyte are associated with increased immune tolerance that desensitize the anti-tumor immune effector mechanisms, thereby accelerating tumor growth.48 In this study, the proportion of CD4+ and CD25+ T-cells might have decreased before MFC transplantation, and, therefore, did not accelerate tumor growth.43
We incorporated only a single gene fragment into the VACV vector. To enhance its antigenicity and increase its tumor spectrum, other tumor-associated genes can be incorporated into the survivin T34A construct. Furthermore, other adjuvants should also be tested since vv-FilC was not particularly strong. Finally, other animal models should also be tested for recombinant VACV anti-tumor function and safety.
Ethics
All of the animal experiments were conducted in accordance with the Zhejiang University animal welfare guidelines, and they were approved by the Ethics Committee of Animal Experimentation of Zhejiang University. All of the cell lines were obtained from the National Infrastructure of Cell Line Resource (Beijing, China).
Acknowledgments
This research was supported by a grant from the Zhejiang Provincial Natural Science Foundation of China (Grant Numbers: LZ16H160001, LY15H160026, LY15H160012, and LGF19H160032); Key Projects of National Natural Science Foundation of China (Grant Number: 81730108); Zhejiang Science and Technology Project (Grant Number: 2017C37171); and National Health and Family Planning Commission Research Fund & Zhejiang Provincial Medical and Health Major Science and Technology Plan Project (No. 2017209495).
Disclosure
The authors declare that they have no conflicts of interest with the contents of this article.
References
1. Chen W, Zheng R, Zhang S, et al.Cancer incidence and mortality in China, 2013. Cancer Lett. 2017;401:63–71. doi:10.1016/j.canlet.2017.04.024.
2. Strong VE, Wu AW, Selby LV, et al. Differences in gastric cancer survival between the U.S. and China. J Surg Oncol. 2015;112:31–37. doi:10.1002/jso.23940
3. Kim M, Nitschké M, Sennino B, et al. Amplification of oncolytic vaccinia virus widespread tumor cell killing by sunitinib through mutiple mechanisms. Cancer Res. 2018;78:922–937. doi:10.1158/0008-5472.CAN-15-3308
4. Andersen MH, Svane IM, Becker JC, Straten PT. The universal character of the tumor-associated antigen survivin. Clin Cancer Res. 2007;13:5991–5994. doi:10.1158/1078-0432.CCR-07-0686
5. Rohayem J, Diestelkoetter P, Weigle B, et al. Antibody response to the tumor-associated inhibitor of apoptosis protein survivin in cancer patients. Cancer Res. 2000;60:1815–1817.
6. Schmidt SM, Schag K, Müller MR, et al. Survivin is a shared tumor-associated antigen expressed in a broad variety of malignancies and recognized by specific cytotoxic T cells. Blood. 2003;102:571–576. doi:10.1182/blood-2002-08-2554
7. Krieg A, Baseras B, Tomczak M, Verde PE, Stoecklein NH, Knoefel WT. Role of survivin as prognostic and clinicopathological marker in gastric cancer:a meta-analysis. Mol Biol Rep. 2013;40:5501–5511. doi:10.1007/s11033-013-2650-z
8. Yan H, Thomas J, Liu T, et al. Induction of melanoma cell apoptosis and inhibition of tumor growth using a cell-permeable survivin antagonist. Oncogene. 2006;25:6968–6974. doi:10.1038/sj.onc.1209676
9. Ogura A, Watanabe Y, Iizuka D, et al. Radiation-induced apoptosis of tumor cells is facilitated by inhibition of the interaction between survivin and Smac/DIABLO. Cancer Lett. 2008;259:71–81. doi:10.1016/j.canlet.2007.09.017
10. Wall NR, O’Connor DS, Plescia J, Pommier Y, Altieri DC. Suppression of survivin phosphorylation on Thr34 by flavopiridol enhances tumor cell apoptosis. Cancer Res. 2003;63(1):230–235.
11. Peng XC, Yang L, Yang LP, et al. Efficient inhibition of murine breast cancer growth and metastasis by gene transferred mouse survivin Thr34->Ala mutant. J Exp Clin Cancer Res. 2008;27:46–59. doi:10.1186/1756-9966-27-46
12. López-Yglesias AH, Zhao X, Quarles EK, et al. Flagellin induces antibody responses through a TLR5- and inflammasome-independent pathway. J Immunol. 2014;192:1587–1596. doi:10.4049/jimmunol.1301893
13. Kang X, Pan Z, Jiao X. Amino acids 89–96 of Salmonella flagellin: a key site for its adjuvant effect independent of the TLR5 signaling pathway. Cell Mol Immunol. 2017;14:1023–1025. doi:10.1038/cmi.2017.125
14. Ajamian L, Melnychuk L, Jean-Pierre P, Zaharatos GJ. DNA vaccine-encoded flagellin can be used as an adjuvant scaffold to augment HIV-1 gp41 membrane proximal external region immunogenicity. Viruses. 2018;10:100–122. doi:10.3390/v10030100
15. Sanos SL, Kassub R, Testori M, et al. NLRC4 inflammasome-driven immunogenicity of a recombinant MVA mucosal vaccine encoding flagellin. Front Immunol. 2018;8:1988. doi:10.3389/fimmu.2017.01988
16. Lee SE, Hong SH, Verma V, et al. Flagellin is a strong vaginal adjuvant of a therapeutic vaccine for genital cancer. Oncoimmunology. 2016;5:e1081328. doi:10.1080/2162402X.2015.1081328
17. Wang Y, Zhang J, Wu Y, et al. Mannan-modified adenovirus targeting TERT and VEGFR-2: a universal tumour vaccine. Sci Rep. 2015;5:11275. doi:10.1038/srep11275
18. Thomas ED, Meza-Perez S, Bevis KS, et al. IL-12 expressing oncolyticherpes simplex viruspromotes anti-tumor activity and immunologic control of metastatic ovariancancerin mice. J Ovarian Res. 2016;9:70. doi:10.1186/s13048-016-0282-3
19. Riabov V, Tretyakova I, Alexander RB, Pushko P, Klyushnenkova EN. Anti-tumor effect of thealphavirus-based virus-like particle vector expressing prostate-specific antigen in a HLA-DR transgenic mouse model of prostatecancer. Vaccine. 2015;33:5386–5395. doi:10.1016/j.vaccine.2015.08.062
20. Pol JG, Zhang L, Bridle BW, et al. Maraba virus as a potent oncolytic vaccine vector. Mol Ther. 2014;22:420–429. doi:10.1038/mt.2013.249
21. Cuadrado-Castano S, Ayllon J, Mansour M, et al. Enhancement of the proapoptotic properties ofnewcastle disease viruspromotes tumor remission in syngeneic murinecancermodels. Mol Cancer Ther. 2015;14:1247–1258. doi:10.1158/1535-7163.MCT-14-0913
22. Ehrig K, Kilinc MO, Chen NG, et al. Growth inhibition of different human colorectal cancer xenografts after a single intravenous injection of oncolytic vaccinia virus GLV-1h68. J Transl Med. 2013;11:79. doi:10.1186/1479-5876-11-79
23. Naito S, von Eschenbach AC, Giavazzi R, Fidler IJ. Growth and metastasis of tumor cells isolated from a human renal cell carcinoma implanted into different organs of nude mice. Cancer Res. 1986;46:4109–4115.
24. Fukuhara H, Ino Y, Todo T. Oncolyfic virus therapy:a new era of cancer treatment at dawn. Cancer Sci. 2016;107:1373–1379. doi:10.1111/cas.13027
25. Russell TA, Tscharke DC. Strikingly poor CD8+ T-cell immunogenicity of vaccinia virusstrainMVAin BALB/c mice. Immunol Cell Biol. 2014;92:466–469. doi:10.1038/icb.2014.10
26. Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9:64–71. doi:10.1038/nrc2545
27. Puhlmann M, Brown CK, Gnant M, et al. Vaccinia as a vector for tumor-directed gene therapy: biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther. 2000;7:66–73. doi:10.1038/sj.cgt.7700075
28. Buller RM, Smith GL, Cremer K, Notkins AL, Moss B. Decreased virulence of recombinant vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature. 1985;317:813–815. doi:10.1038/317813a0
29. Chan WM, McFadden G. Oncolytic poxviruses. Annu Rev Virol. 2014;1:119–141. doi:10.1146/annurev-virology-031413-085442
30. Parato KA, Breitbach CJ, Le Boeuf F, et al. The oncolytic poxvirusJX-594selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther. 2012;20:749–758. doi:10.1038/mt.2011.276
31. Park BH, Hwang T, Liu TC, et al. Use of a targeted oncolytic poxvirus,JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9:533–542. doi:10.1016/S1470-2045(08)70107-4
32. Park SH, Breitbach CJ, Lee J, et al. Phase 1b trial of biweekly intravenous pexa-vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus in colorectal cancer. Mol Ther. 2015;23:1532–1540. doi:10.1038/mt.2015.109
33. Breitbach CJ, Moon A, Burke J, Hwang TH, Kirn DH. A Phase 2, open-label, randomized study of pexa-Vec (JX-594) administered by intratumoral injection in patients with unresectable primary hepatocellular carcinoma. Methods Mol Biol. 2015;1317:343–357.
34. Thorne SH, Hwang TH, O’Gorman WE, et al. Rational strain selection and engineering creates a broad-spectrum,systemically effective oncolytic poxvirus, JX-963. J Clin Invest. 2007;117:3350–3358. doi:10.1172/JCI32727
35. Deng L, Fan J, Ding Y, et al. Oncolytic efficacy of thymidine kinase-deleted vaccinia virus strain Guang9. Oncotarget. 2017;8:40533–40543. doi:10.18632/oncotarget.17125
36. Wu Y, Mou X, Wang S, Liu XE, Sun X. ING4 expressing oncolytic vaccinia virus promotes anti-tumor efficiency and synergizes with gemcitabine in pancreatic cancer. Oncotarget. 2017;8:82728–82739. doi:10.18632/oncotarget.21095
37. Li G, Mei S, Cheng J, Wu T, Luo J. Haliotis discus discus sialic acid-binding lectin reduces the oncolytic vaccinia virus induced toxicity in a glioblastoma mouse model. Mar Drugs. 2018;16:141. doi:10.3390/md16050141
38. White M, Freistaedter A, Jones GJB, Zervos E, Roper R. Development of improved therapeutic mesothelin-based vaccines for pancreatic cancer. PLoS One. 2018;13:e0193131. doi:10.1371/journal.pone.0193131
39. Mejías-Pérez E, Carreño-Fuentes L, Esteban M. Development of a safe and effective vaccinia virus oncolytic vector WR-Δ4 with a set of gene deletions on several viral pathways. Mol Ther Oncolytics. 2017;8:27–40. doi:10.1016/j.omto.2017.12.002
40. Ishizaki H, Manuel ER, Song GY, et al. Modified vaccinia Ankara expressing survivin combined with gemcitabine generates specific antitumor effects in a murine pancreatic carcinoma model. Cancer Immunol Immunother. 2011;60:99–109. doi:10.1007/s00262-010-0923-0
41. Zhang H, Liu C, Zhang F, et al. MUC1 and survivin combination tumor gene vaccine generates specific immune responses and anti-tumor effects in a murine melanoma model. Vaccine. 2016;34:2648–2655. doi:10.1016/j.vaccine.2016.04.045
42. Umemura S, Shirane M, Takekoshi S, Tokuda Y, Mori K, Osamura RY. High expression of thymidine phosphorylase in basal-like breast cancers: stromal expression in EGFR- and/or CK5/6-positive breast cancers. Oncol Lett. 2010;1:261–266. doi:10.3892/ol_00000046
43. Sfondrini L, Rossini A, Besusso D, et al. Antitumor activity of the TLR-5 ligand flagellin in mouse models of cancer. J Immunol. 2006;176:6624–6630. doi:10.4049/jimmunol.176.11.6624
44. McSorley SJ, Ehst BD, Yu Y, Gewirtz AT. Bacterial flagellin is an effective adjuvant for CD4+ T cells in vivo. J Immunol. 2002;169:3914–3919. doi:10.4049/jimmunol.169.7.3914
45. Didierlaurent A, Ferrero I, Otten LA, et al. Flagellin promotes myeloid differentiation factor 88-dependent development of Th2-typeresponse. J Immunol. 2004;172:6922–6930. doi:10.4049/jimmunol.172.11.6922
46. Agrawal S, Agrawal A, Doughty B, et al. Cutting edge: different toll-like receptor agonists instruct dendritic cells to induce distinct thresponses via differential modulation of extracellular signal-regulated kinase-mitogen-activatedprotein kinase and c-Fos. J Immunol. 2003;171:4984–4989. doi:10.4049/jimmunol.171.10.4984
47. Cunningham AF, Khan M, Ball J, et al. Responses to the soluble flagellar protein FliC are Th2,while those to FliC on Salmonella are Th1. Eur J Immunol. 2004;34:2986–2995. doi:10.1002/eji.200425403
48. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integratingimmunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi:10.1126/science.1203486
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.