")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
The differential effects of inspiratory, expiratory, and combined resistive breathing on healthy lung
Authors Loverdos K, Toumpanakis D, Litsiou E, Karavana V , Glynos C, Magkou C, Theocharis S, Vassilakopoulos T
Received 13 February 2016
Accepted for publication 11 April 2016
Published 19 July 2016 Volume 2016:11(1) Pages 1623—1638
DOI https://doi.org/10.2147/COPD.S106337
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Konstantinos Loverdos,1 Dimitrios Toumpanakis,1 Eleni Litsiou,1 Vassiliki Karavana,1 Constantinos Glynos,1 Christina Magkou,2 Stamatios Theocharis,3 Theodoros Vassilakopoulos1
1Department of Critical Care, Pulmonary Unit and Marianthi Simou Applied Biomedical Research and Training Center, Evangelismos General Hospital, University of Athens Medical School, 2Department of Pathology, Evangelismos General Hospital, 31st Department of Pathology, University of Athens Medical School, Athens, Greece
Abstract: Combined resistive breathing (CRB) is the hallmark of obstructive airway disease pathophysiology. We have previously shown that severe inspiratory resistive breathing (IRB) induces acute lung injury in healthy rats. The role of expiratory resistance is unknown. The possibility of a load-dependent type of resistive breathing-induced lung injury also remains elusive. Our aim was to investigate the differential effects of IRB, expiratory resistive breathing (ERB), and CRB on healthy rat lung and establish the lowest loads required to induce injury. Anesthetized tracheostomized rats breathed through a two-way valve. Varying resistances were connected to the inspiratory, expiratory, or both ports, so that the peak inspiratory pressure (IRB) was 20%–40% or peak expiratory (ERB) was 40%–70% of maximum. CRB was assessed in inspiratory/expiratory pressures of 30%/50%, 40%/50%, and 40%/60% of maximum. Quietly breathing animals served as controls. At 6 hours, respiratory system mechanics were measured, and bronchoalveolar lavage was performed for measurement of cell and protein concentration. Lung tissue interleukin-6 and interleukin-1β levels were estimated, and a lung injury histological score was determined. ERB produced significant, load-independent neutrophilia, without mechanical or permeability derangements. IRB 30% was the lowest inspiratory load that provoked lung injury. CRB increased tissue elasticity, bronchoalveolar lavage total cell, macrophage and neutrophil counts, protein and cytokine levels, and lung injury score in a dose-dependent manner. In conclusion, CRB load dependently deranges mechanics, increases permeability, and induces inflammation in healthy rats. ERB is a putative inflammatory stimulus for the lung.
Keywords: resistive breathing, lung injury, inflammation
Introduction
Resistive breathing is the hallmark of the pathophysiology of diseases of airway obstruction, such as asthma and COPD, especially during exacerbations.1 During resistive breathing, strenuous contractions of the inspiratory muscles produce large negative swings in intrathoracic pressure that can be injurious for the lung. Indeed, our group has previously shown that inspiratory resistive breathing (IRB) may exert an injurious and inflammatory effect with the characteristics of acute lung injury (ALI) on previously healthy lungs in an experimental rat model.2 In this model, we used inspiratory resistances resulting in peak inspiratory pressure amounting to 50% of maximum, which corresponds to the levels of inspiratory pressure observed during severe COPD exacerbations and asthma attacks.2 Whether lower inspiratory resistances resulting in lower inspiratory pressures can induce lung injury and inflammation has never been studied. The first aim of our study was to identify the lowest level of inspiratory pressure generated by inspiratory resistive loading that can induce lung injury and inflammation.
To our knowledge, the effects of pure expiratory resistance on the healthy lung have also never been studied before. Expiratory resistance results in dynamic hyperinflation and intrinsic positive end-expiratory pressure (PEEPi) induction; a potential proinflammatory role for dynamic hyperinflation per se has been proposed, mainly attributable to cellular stretch.3 The second aim of our study was to test the hypothesis that pure expiratory resistive breathing (ERB) can induce injury and inflammation and determine whether a load dependency exists.
In asthma and COPD, both inspiratory and expiratory airway resistances are elevated. Importantly, expiration is more seriously affected (ie, expiratory resistance is higher than inspiratory), as measured in patients with varying severity of spirometric airflow obstruction using body plethysmography and impulse oscillometry.4,5 This is due to the different intrathoracic pressure environments, with positive pressures during expiration that compress the airways and increase the airway resistance, in contrast to inspiration, where the negative intrathoracic pressures dilate the airways, thus reducing their resistance. Consequently, in order to simulate the consequences of an increased airway resistance per se (resistance being a pure mechanical stressor for the lung) observed in asthma and COPD, an experimental model of combined resistive breathing (CRB) with unequally increased inspiratory and expiratory resistances (expiratory greater than inspiratory) should be applied.
The effect of CRB (inspiratory and expiratory resistances being pure mechanical stressors) on the healthy lung has never been addressed. PEEPi induction during expiration, as a result of expiratory resistance, could oppose the large negative intrathoracic pressures producing pulmonary edema during inspiration,6 thus preventing the occurrence of ALI observed after IRB alone. On the other hand, ERB-induced hyperinflation could amplify the inflammatory process following IRB through the excessive deformation of resident lung cells. Patients with asthma and COPD present quite varying levels of airway obstruction, resulting in variable intrathoracic pressures, depending on the stage of the disease and/or the presence of exacerbation. It has previously been shown that the effect of mechanical stress on the lung is magnitude dependent. For example, in ventilator-induced lung injury (VILI) models, lung permeability increments occurred only when a cutoff tidal volume was reached.7,8 However, whether the effect of resistive breathing on the lung is load dependent is unknown. Thus, the third aim of our study was to test the hypothesis that CRB (with the expiratory resistance exceeding the inspiratory) causes ALI in previously healthy lungs and that the effects of resistive breathing on the lung are load dependent.
In order to address the above research questions, we expanded our previously described IRB model in rats to study IRB or ERB alone or in combination. All models of resistive breathing were examined for load dependence, so as to establish the lowest inspiratory, expiratory, and combined loads able to induce meaningful alterations.
Methods
Subjects
Adult female Wistar rats (8–12 weeks old, 200–260 g) were used in this study. Animals were purchased from the Hellenic Pasteur Institute and housed at the Experimental Surgery Department of Evangelismos General Hospital at 22°C±2°C temperature, 55%–60% humidity, 12 hours day–night cycle, and ad libitum food and water provision. All procedures were in accordance with the European Union Directive for care and use of laboratory animals and were approved by the Greek Veterinary Administration and the Ethical Committee of Evangelismos Hospital.
Experimental model
Three different models of resistive breathing were developed in healthy rats, according to our previously described model of IRB.1,2 Specifically, rats were anesthetized with a mixture of ketamine (75 mg/kg) and xylazine (5 mg/kg) injected intraperitoneally (ip) and tracheostomized with a tracheal cannula (14 G). After a short stabilization period (~10 minutes), a two-way nonrebreathing valve (Hans Rudolph Inc., Shawnee, KS, USA) was connected to the cannula. A pressure transducer (DirecWin; Raytech Instruments Inc., Vancouver, BC, Canada) connected to the cannula was used to monitor tracheal pressure, and maximal inspiratory or expiratory pressure (Pi,max and Pe,max) was measured during spontaneous breathing efforts through a totally occluded inspiratory or expiratory port, respectively, for ~10 seconds. In order to determine resistive loads, either the inspiratory or the expiratory port or both were then connected to a smaller tube (resistance), the diameter and length of which was selected so as to provide a peak tidal inspiratory (Pi) and/or expiratory pressure (Pe) at a predetermined percentage of Pi,max and/or Pe,max (IRB, ERB, or combined IRB and ERB).
Animals were randomly assigned to the following groups and subgroups:
- IRB group, consisting of animals expiring freely but inspiring through one of the three inspiratory loads of different magnitudes (Pi/Pi,max =20% [IRB 20%], Pi/Pi,max =30% [IRB 30%], and Pi/Pi,max =40% [IRB 40%]);
- ERB group, consisting of animals inspiring against no load but expiring through one of the four different loads (Pe/Pe,max =40% [ERB 40%], Pe/Pe,max =50% [ERB 50%], Pe/Pe,max =60% [ERB 60%], and Pe/Pe,max =70% [ERB 70%]); and
- CRB group, consisting of animals inspiring and expiring against different resistive loads (Pi/Pi,max =30% and Pe/Pe,max =50% [CRB 30%/50%], Pi/Pi,max =40% and Pe/Pe,max =50% [CRB 40%/50%], and Pi/Pi,max =40% and Pe/Pe,max =60% [CRB 40%/60%]).
The duration of resistive breathing was 6 hours. Animals breathing spontaneously against no load for equal time served as controls (ctrs). A total of 100% oxygen supply was delivered to all subjects throughout the experiment to prevent hypoxemia. Supplemental doses of ketamine (30 mg/kg ip), without xylazine, were administered during the procedure, in order to maintain adequately deep anesthesia, as estimated by pedal reflex testing.
Respiratory system mechanics
The mechanical properties of the respiratory system were determined with the use of a small animal ventilator (Scireq; FlexiVent, Montreal, QC, Canada). Following the completion of resistive breathing, the resistance(s) was removed, and animals were further ip anesthetized with a mixture of ketamine (75 mg/kg) and xylazine (10 mg/kg) and attached to the ventilator. They received a volume ctr ventilation mode with the ventilatory parameters set to 10 mL/kg tidal volume, 90 breaths/min respiratory rate and 3 cmH2O end-expiratory pressure. After 3 minutes, an ip injection of succinylcholine (8 mg/kg) was delivered to suspend spontaneous breathing, and 1 minute later, three consecutive forced oscillation perturbations with a 30-second interval were performed, followed by the construction of a static pressure – volume curve 30 seconds after the last forced oscillation. Prior to measurements (30 seconds), lung volume history was once standardized by occluding the expiratory line of the ventilator until the airway opening pressure reached 30 cmH2O. During ventilation, heart rate was monitored to ensure adequate depth of anesthesia. Repeated measures of mechanical parameters of each subject were averaged.
Forced oscillation technique
The forced oscillation perturbation consists of a pseudorandom waveform of low frequencies (0.5–19.75 Hz) applied for 8 seconds with a peak-to-peak volume of 3 mL/kg. Pressure and volume data are recorded, and the impedance of the respiratory system is calculated using the fast Fourier transformation. Impedance (Z) is then fitted to constant phase model: Zrs(f) = Rn + i2πfI + (G - iH)/(2πf)α, where Rn is the Newtonian resistance of the airways, i is the imaginary unit, f is the frequency, I is the inertance of the gas in the airways, G represents tissue viscance (viscous dissipitation of energy), H represents tissue elasticity, and α can be calculated through the equation α = (2/π)arctan(H/G).9 Data were accepted only when the coefficient of determination (fit of the model) was >0.9.
Static pressure–volume curves
Static pressure–volume (P–V) curves of the respiratory system were performed by gradually inflating and deflating the lungs with a total volume of 4.8 mL at seven steps each. Analysis of the static P–V curve was performed by identifying the lower and upper inflection points, and the static compliance (Cst) of the respiratory system was estimated by the slope of the mid linear part in the expiratory limb of the P–V curve, as previously described.2 Hysteresis was automatically calculated by the FlexiVent software.
Bronchoalveolar lavage
Following lung mechanics measurement, animals were detached from the ventilator and sacrificed by exsanguination (inferior vena cava and descending aorta dissection). The left main bronchus was temporarily ligated, and the right lung was lavaged with three aliquots of 2.5 mL of normal saline. Bronchoalveolar lavage (BAL) fluid withdrawn was immediately centrifuged at 300× g for 10 minutes at 4°C. The supernatant was collected and stored at −80°C, while the cell pellet was resuspended in 1 mL of normal saline.
Total and differential cell counts
Total cell counts in BAL fluid (BALF) were determined following Trypan blue dye staining on a Neubauer chamber. Aliquots (4×104 cells) were cytocentrifuged (450 rpm for 6 minutes), and the cytospins were stained with May-Grunwald/Giemsa. Percentages of monocytes/macrophages, neutrophils, and lymphocytes were determined after counting at least 300 cells per slide. Eosinophil and basophil counts were negligible and omitted from the differential cell analysis.
Total protein concentration in BALF
Total protein concentration in BALF was measured using a colorimetric protein assay according to the manufacturer’s instructions (Bio-Rad Laboratories Inc., Hercules, CA, USA). Bovine serum albumin was used to create standard curves.
Lung histology
Following BAL, the right main bronchus was also ligated, and the right lung was excised, immediately immersed in liquid nitrogen, and stored at −80°C. Left main bronchus ligation was then removed, and the left lung was fixed with 4% formaldehyde under a constant pressure of 20 cmH2O before excision and storage in formaldehyde. After 24 hours, the lung tissue was embedded in paraffin using conventional techniques, and paraffin blocks were cut into serial 5 μm sections, which were then stained with hematoxylin and eosin. A modified previously used lung injury score2 was determined based on the following histological features: 1) focal alveolar membrane thickening, 2) capillary congestion, 3) intraalveolar hemorrhage, 4) intraalveolar neutrophil infiltration, and 5) interstitial neutrophil infiltration. Each feature was ranked 0 to 3 depending on its absence (0) or presence to a mild (1), moderate (2), or severe (3) extent.
Lung myeloperoxidase assay
Myeloperoxidase (MPO) activity was measured in frozen lung tissue sections, as previously described.2 Briefly, lung sections were weighted and homogenized in a buffer containing 50 mM potassium phosphate (pH 6.0), 5 mM EDTA, and 0.5% hexadecyltrimethylammonium bromide. After incubation at 60°C for 2 hours, the homogenates were centrifuged at 12,000× g for 20 minutes at 4°C. The supernatant was collected and mixed (1:80) with buffer containing 0.0005% H2O2, 0.167 mg/mL o-dianisidine hydrochloride, and 100 mM potassium phosphate. Absorbance was measured at 460 nm every 20 seconds for 3 minutes, and results were expressed as difference in optical density in the first minute per gram of lung tissue (ΔOD/min/g).
Cytokine levels in lung tissue
Frozen lung tissue sections were homogenized with buffer containing 50 mM hepes (pH 7.5), 150 nM NaCl, 10% glycerol, 1% Triton X-100, 1 mM EDTA, 1.5 mM MgCl2, and a cocktail of protease and phosphatase inhibitors at a 1:1,000 concentration. The samples were then centrifuged at 10,000× g for 10 minutes. The supernatant was collected, and total protein concentration was estimated using a colorimetric protein assay according to the manufacturer’s instructions. Protein levels of interleukin (IL)-1β and IL-6 were determined in lung tissue homogenates using ELISA, according to the manufacturer’s protocol (DuoSet ELISA; R&D Systems, Inc., Mineapolis, MN, USA) and normalized to total protein content of lung homogenates.
Statistical analysis
Results are presented as mean ± standard error of the mean (SEM). Five to ten animals were studied in each group. Statistical analysis was performed by one-way analysis of variance (ANOVA) and Newman–Keuls post hoc test (Statistica software; StatSoft, Tulsa, OK, USA). Data for histology were ordinal and were analyzed with the nonparametric Kruskal–Wallis ANOVA. Mann–Whitney U-test was used for post hoc analysis. A P-value of <0.05 was chosen as statistically significant.
Results
Inspiratory resistive breathing
Respiratory system mechanics
In accordance with previous reports of our group,2 IRB was found to derange respiratory system mechanics. Both IRB 30% and IRB 40% produced significant increases in lung tissue elasticity (H), measured by the forced oscillation technique, compared to ctr (P<0.05 and P<0.001, respectively) (Figure 1A) and decreases in Cst as calculated using the static P–V curves of the respiratory system (P<0.01 and P<0.001 to ctr, respectively) (Figure 1B), the latter being shifted rightward and downward indicating the presence of lung injury (Figure 1C). Interestingly, the changes in tissue elasticity and Cst were load dependent.
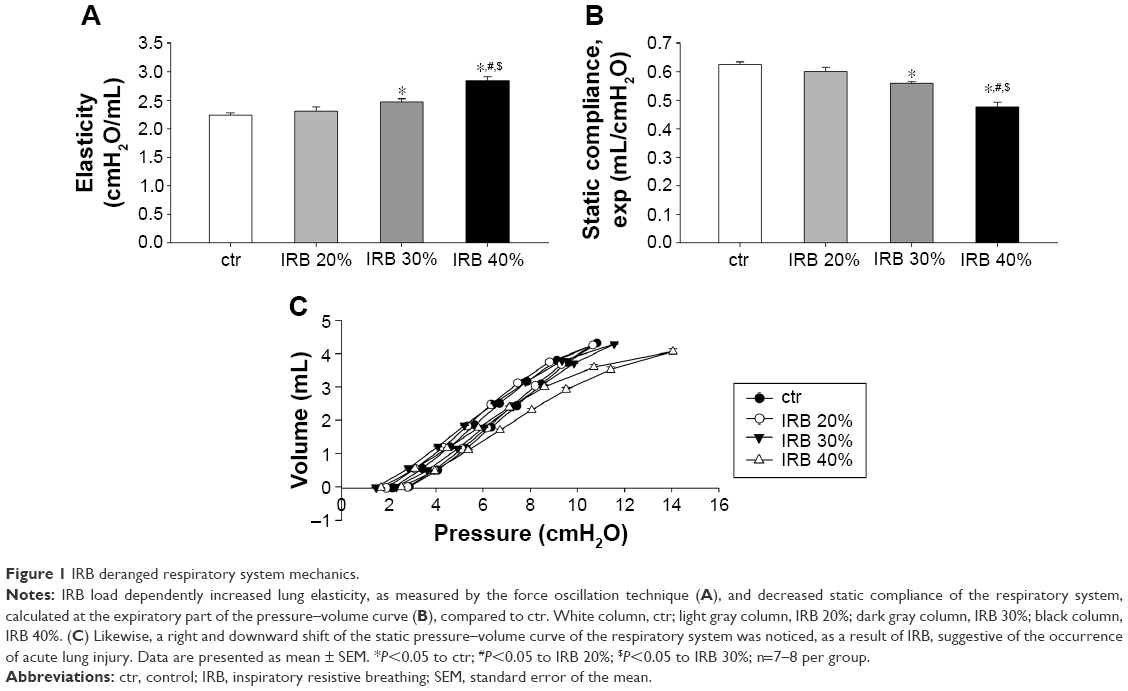 | Figure 1 IRB deranged respiratory system mechanics. |
IRB did not alter tissue viscance (G) (ANOVA, F=0.46, P=0.71; n=7–8 per group). As a result, hysteresivity (ie, the G-to-H ratio) was reduced, and the reduction reached statistical significance only in the IRB 40% group (ctr 0.22±0.01, IRB 30% 0.19±0.01, P=0.13 to ctr; IRB 40% 0.18±0.01, P<0.05 to ctr; n=7–8 per group). IRB did not significantly affect airway resistance (F=1.48, P=0.24; n=7–8 per group) or hysteresis of the P–V loop (F=1.54, P=0.23; n=6–8 per group). It should be emphasized that the lower inspiratory load of 20% did not induce significant mechanical derangements.
Inflammatory cells in BALF
IRB increased total BALF cellularity in a load-dependent manner (IRB 30%, P<0.01 to ctr; IRB 40%, P<0.001 to ctr; IRB 20%, P<0.05 to IRB 30%) (Figure 2A). Both macrophages and neutrophils were increased after IRB 40% compared to the ctr group. IRB 30% also resulted in BALF neutrophilia. The lower load of IRB 20% did not induce significant inflammatory changes in BALF cellularity.
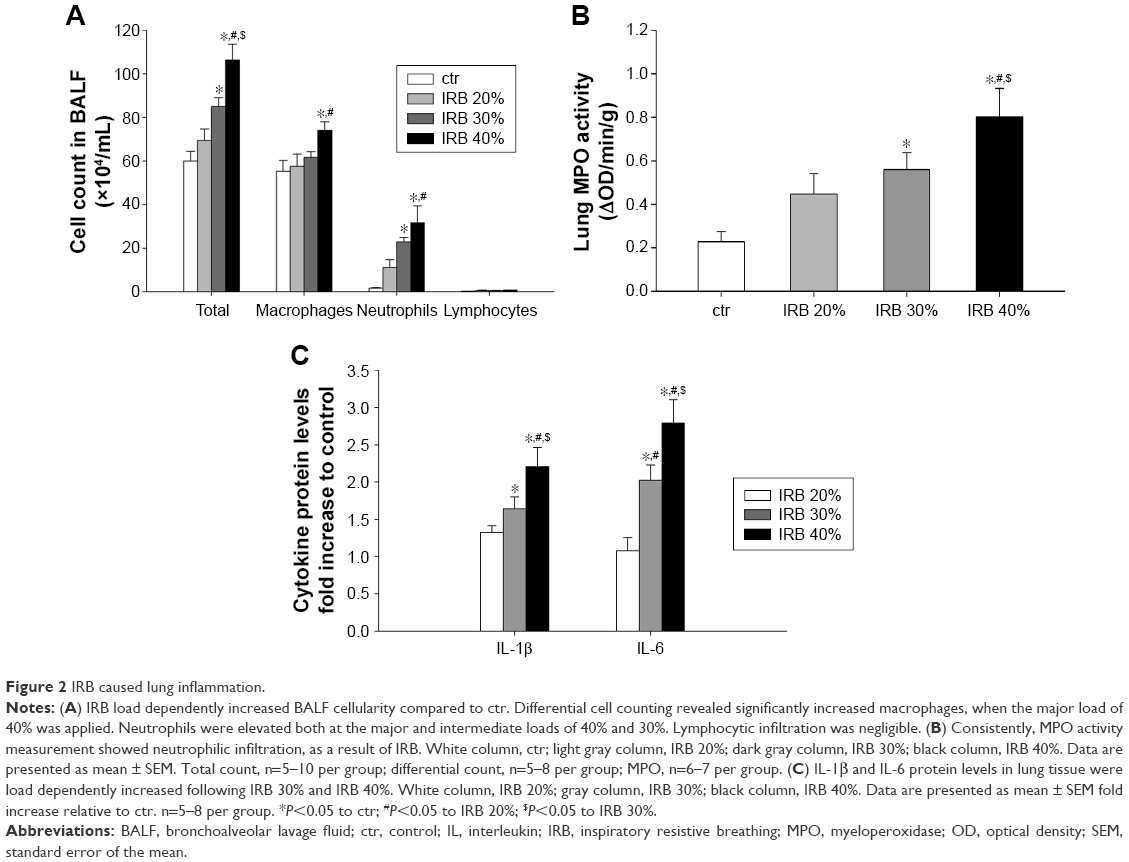 | Figure 2 IRB caused lung inflammation. |
Lung MPO activity
MPO activity in lung homogenates, an indicator of neutrophilic infiltration, was in-line with the results of BALF analysis. MPO activity increased in both IRB 30% and IRB 40% but not in IRB 20% compared to ctrs (IRB 30%, P<0.05 to ctr; IRB 40%, P<0.01 to ctr and P<0.05 to IRB 20%) (Figure 2B).
Lung tissue cytokine levels
Lung tissue IL-1β and IL-6 levels were load dependently elevated after both IRB 30% and IRB 40% (Figure 2C). The lower inspiratory load of 20% did not produce statistically significant increases in cytokines compared to the ctr group.
Total protein concentration in BALF
Total protein levels in BALF, an index of alveolar capillary barrier permeability, increased after IRB 30% and IRB 40% but not after IRB 20% (IRB 20%, P = ns to ctr; IRB 30%, P<0.01 to ctr and P<0.05 to IRB 20%; IRB 40%, P<0.001 to ctr and IRB 20% and P<0.05 to IRB 30%) (Figure 3A).
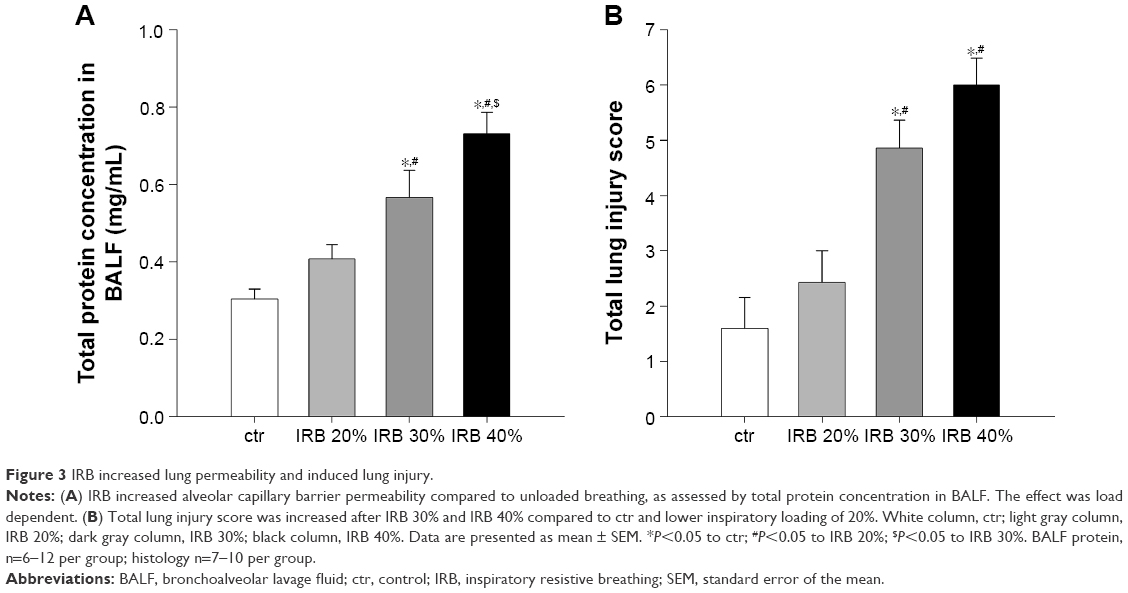 | Figure 3 IRB increased lung permeability and induced lung injury. |
Lung histology
Microscopic examination of hematoxylin–eosin-stained lung sections revealed the presence of ALI in IRB. More precisely, both IRB 30% and IRB 40% led to a significant total lung injury score increase compared to ctr and IRB 20% groups (Figures 3B and 10). This was mainly due to the result of neutrophil infiltration, both interstitial and intraalveolar (Table 1). Significant capillary congestion and focal thickening of the alveolar–capillary barrier compared to ctrs were also observed in the IRB 30% and IRB 40% groups, respectively (Table 1). Histological alterations were insignificant compared to unloaded animals in the IRB 20% group.
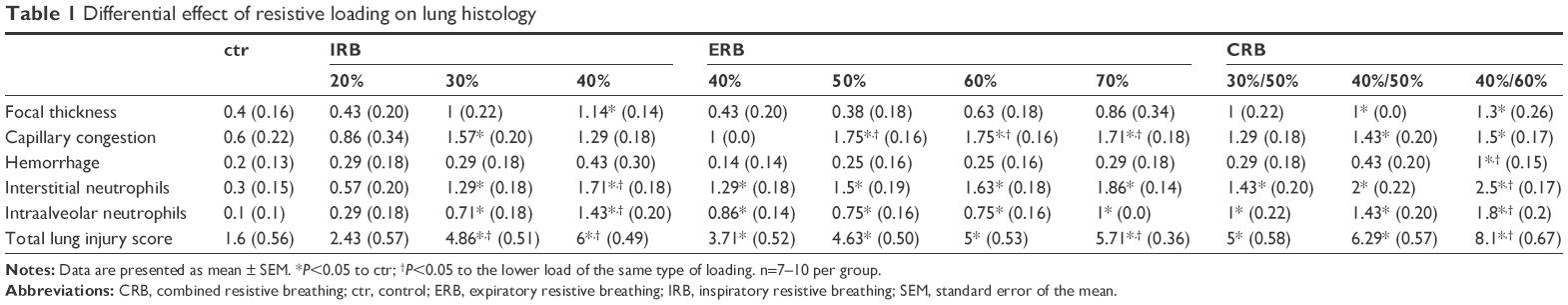 | Table 1 Differential effect of resistive loading on lung histology |
Expiratory resistive breathing
Respiratory system mechanics
ERB alone did not provoke any significant changes in the mechanical properties of the respiratory system (F=1.96, P=0.11 in H; F=0.48, P=0.79 in Cst), at the magnitude and duration applied (Figure 4).
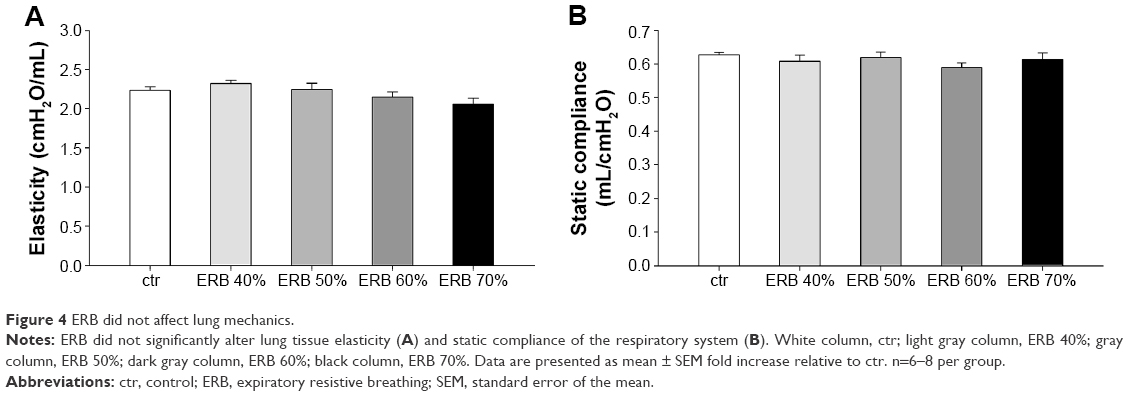 | Figure 4 ERB did not affect lung mechanics. |
Inflammatory cells in BALF
While total cell count was not significantly increased in any of the ERB groups (F=0.85, P=0.52), all of them were characterized by pronounced BALF neutrophilia (P<0.01 to ctr for all studied groups) (Figure 5A). The effect was not load dependent. Interestingly, neutrophilia was accompanied by a significant decrease in macrophage count in all ERB groups.
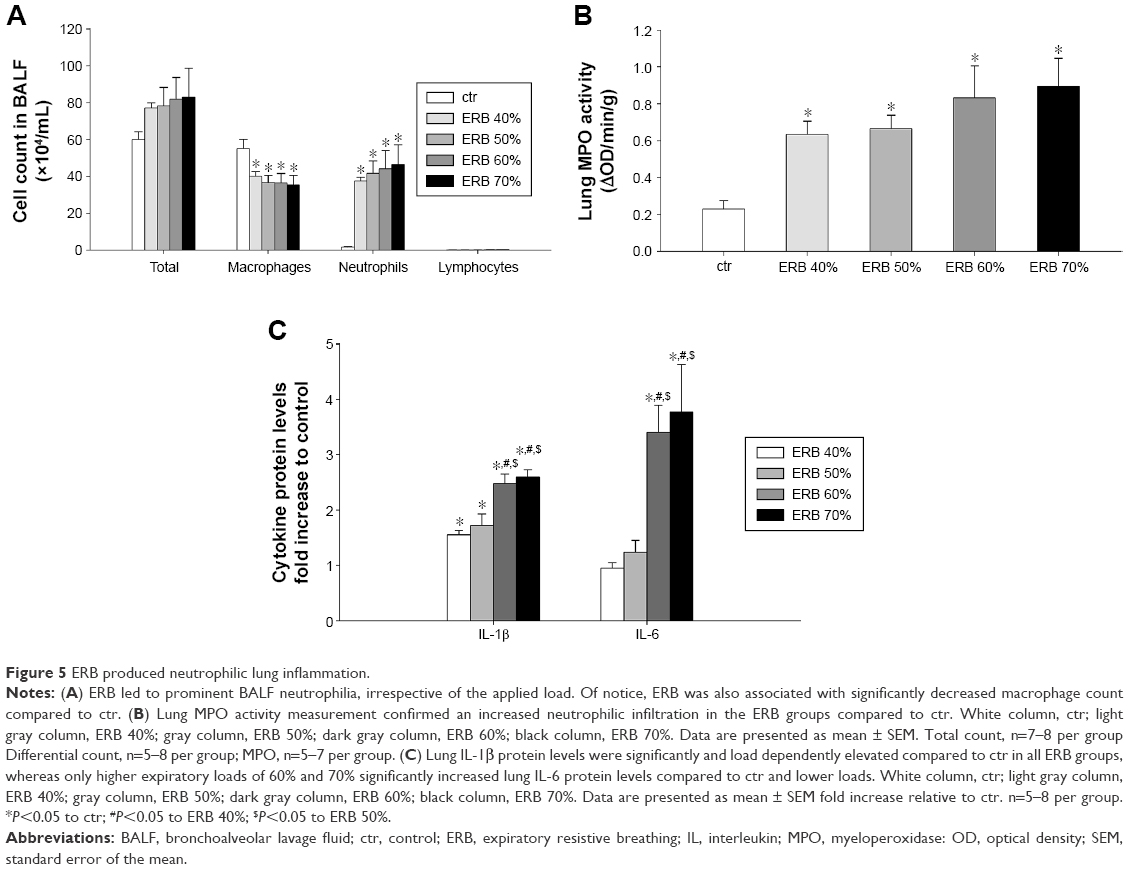 | Figure 5 ERB produced neutrophilic lung inflammation. |
Lung MPO activity
In consistence with the measurements in BALF, MPO activity in all ERB groups significantly exceeded that of ctrs (Figure 5B). Again, the result was not load dependent.
Lung tissue cytokine levels
The lower expiratory resistances of 40% and 50% used in our study significantly increased IL-1β protein levels but not IL-6 levels in lung tissue compared to ctrs (IL-1β, P<0.05 to ctr for both groups) (Figure 5C). Instead, both IL-1β and IL-6 were markedly upregulated (compared to ctr as well as to lower resistances), when higher expiratory loads of 60% and 70% were used (P<0.001 to ctr for both cytokines and loads).
Total protein concentration in BALF
ERB failed to increase BALF total protein content, with the exception of the higher resistance of 70% (Figure 6A).
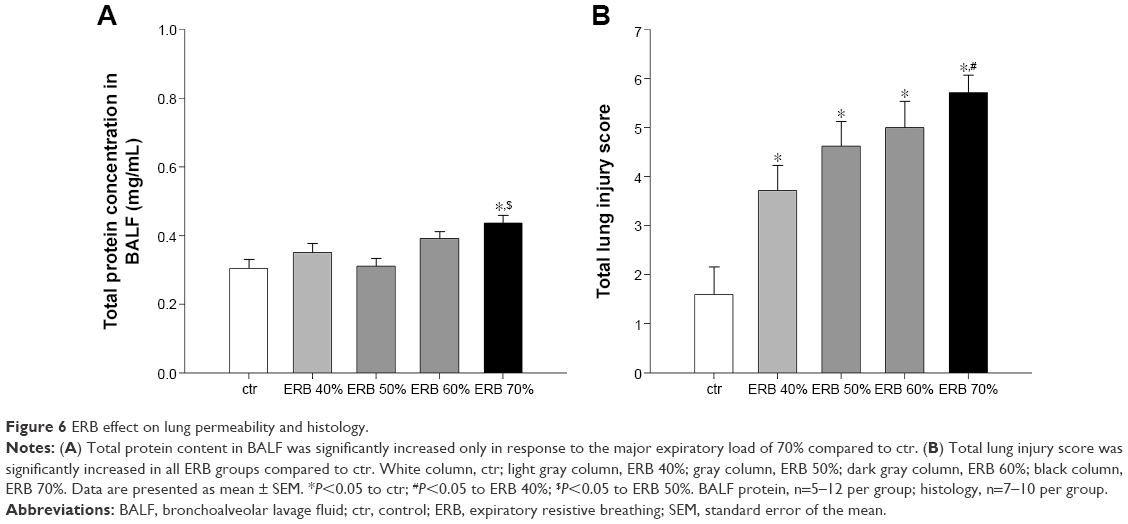 | Figure 6 ERB effect on lung permeability and histology. |
Lung histology
Total lung injury score was significantly elevated in all ERB groups compared to ctrs (Figures 6B and 10). More specifically, expiratory loading resulted in histologically prominent interstitial and intraalveolar neutrophilia, further supporting data from BAL and MPO activity analyses (Table 1). Furthermore, significant capillary congestion was observed in intermediate and higher expiratory loads compared to unloaded breathing and contributed to total score elevation in these groups. Focal alveolar membrane thickening and intraalveolar hemorrhage did not differ significantly compared to ctrs.
Combined inspiratory and expiratory resistive breathing
Respiratory system mechanics
CRB caused mechanical alterations similar to those observed in the IRB groups. Once again, the effect was load dependent. While CRB 30%/50% did not result in significant changes in elasticity and Cst, CRB 40%/50% and CRB 40%/60% increased tissue elasticity (CRB 40%/50%, P<0.05 to ctr; CRB 40%/60% P<0.001 to ctr and P<0.01 to CRB 30%/50%) (Figure 7A), decreased static Cst (CRB 40%/50%, P<0.05 to ctr; CRB 40%/60% P<0.001 to ctr, CRB 30%/50%, and CRB 40%/50%) (Figure 7B) and produced a downward shift to the P–V loop (Figure 7C). G was significantly elevated only in the CRB 40%/50% group (P<0.01 to ctr), and hysteresis increased only after CRB 40%/60% (P<0.01 to ctr and CRB 30%/50%). No statistical significant alterations in hysteresivity (F=2.21, P=0.11) or airway resistance (F=0.86, P=0.48) were noticed after CRB.
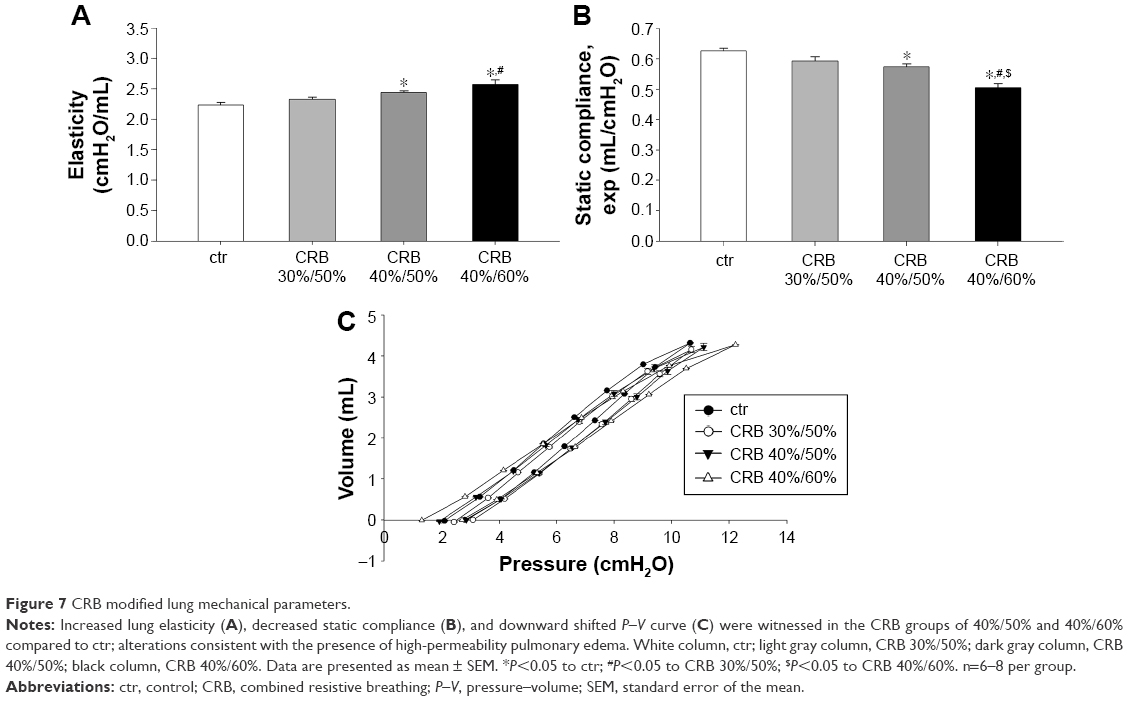 | Figure 7 CRB modified lung mechanical parameters. |
Inflammatory cells in BALF
CRB exhibited a marked load-dependent augmentative effect on BALF cellularity. CRB 40%/60% produced an ~2.3-fold increase in total cell count relative to the ctr value (P<0.001), by raising macrophage (P<0.05) and mostly neutrophil count compared to ctr (P<0.001) (Figure 8A). A less pronounced, though significant, total cell and neutrophil count increase was manifested, when CRB 40%/50% was used (P<0.01 and P<0.001 to ctr, respectively). CRB 30%/50% was incapable of significantly increasing total cellularity. It did, however, result in significant BALF neutrophilia compared to ctr.
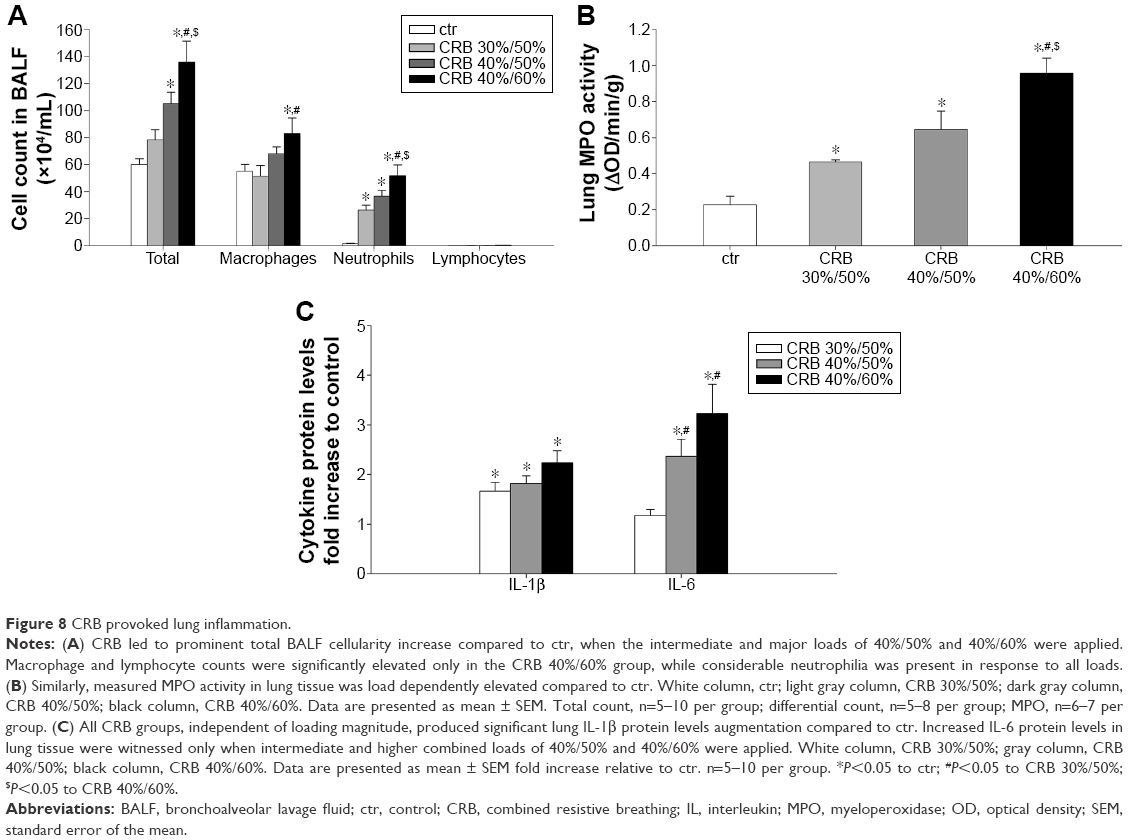 | Figure 8 CRB provoked lung inflammation. |
Lung MPO activity
CRB load dependently increased MPO activity in lung tissue compared to the ctr group (CRB 30%/50%, P<0.05 to ctr; CRB 40%/50%, P<0.01 to ctr; CRB 40%/60%, P<0.001 to ctr and CRB 30%/50% and P<0.01 to CRB 40%/50%) (Figure 8B), a finding in accordance with the results of BALF analysis.
Lung tissue cytokine levels
CRB resulted in the elevation of load-independent lung IL-1β protein levels compared to the ctr group (Figure 8C). In contrast, IL-6 protein levels in lung tissue were increased only in the intermediate and higher combined loading groups of 40%/50% (P<0.05 to ctr) and 40%/60% (P<0.001 to ctr).
Total protein concentration in BALF
Both CRB 40%/60% and CRB 40%/50% raised total protein concentration in BALF compared to ctrs (P<0.05 and P<0.001 to ctr, respectively) (Figure 9A), consistent with the presence of an increased lung permeability. The effect was load dependent (CRB 40%/60%, P<0.001 to CRB 30%/50% and P<0.05 to CRB 40%/50%) and not observed when CRB 30%/50% was used.
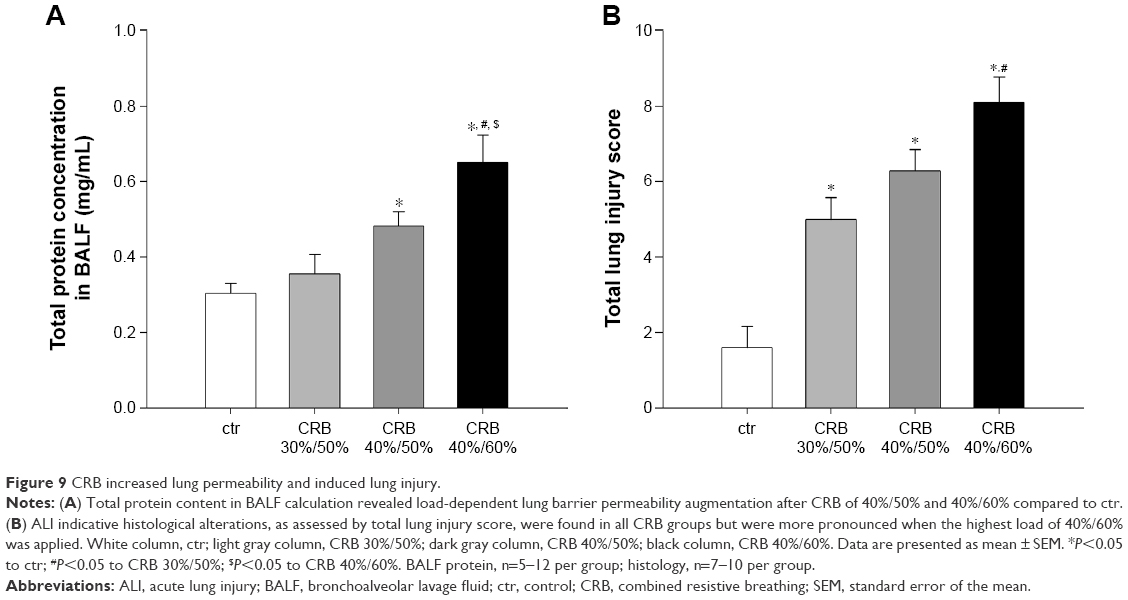 | Figure 9 CRB increased lung permeability and induced lung injury. |
Lung histology
All CRB groups caused parenchymal derangement, as assessed by total lung injury score (Figures 9B and 10). In CRB 30%/50%, this was mainly the result of inflammatory infiltration (Table 1). In CRB 40%/50% and CRB 40%/60%, most histological indices of ALI were prominent, including intraalveolar hemorrhage in the CRB 40%/60% group.
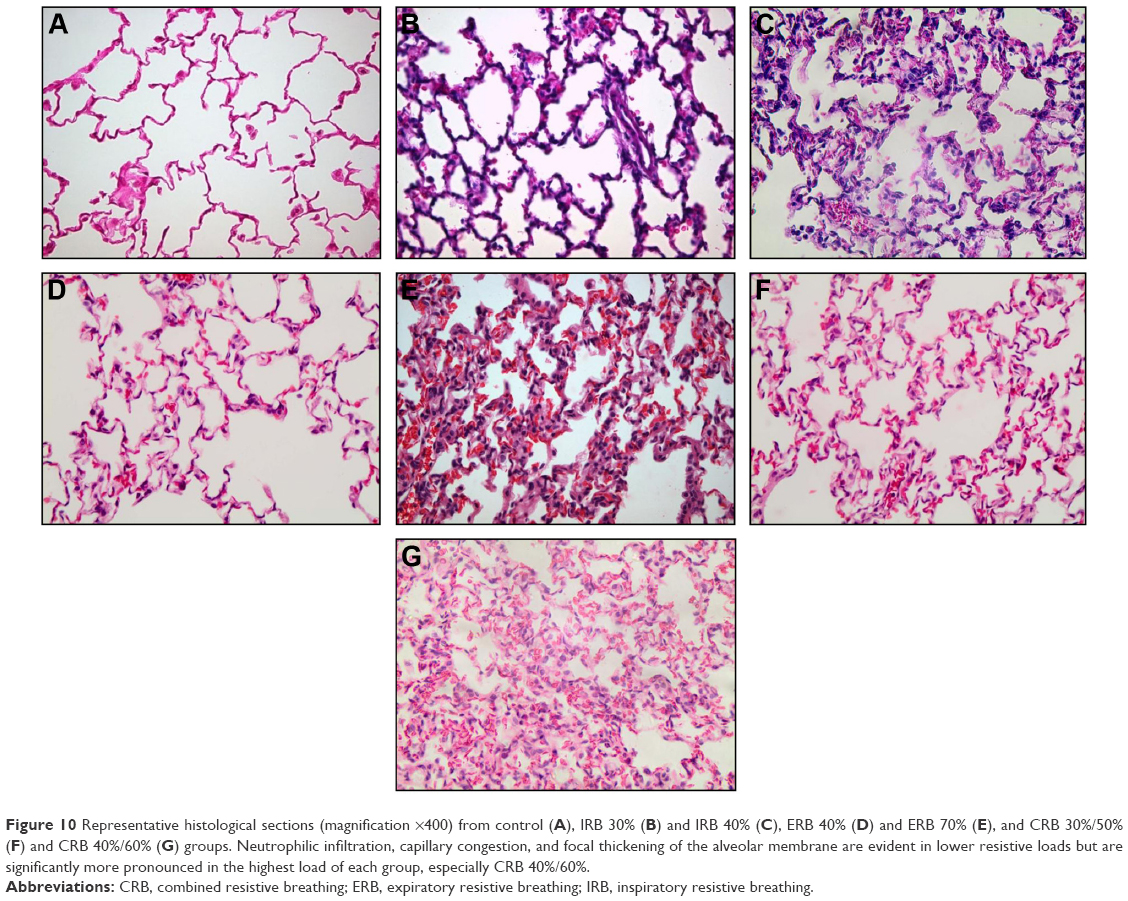 | Figure 10 Representative histological sections (magnification ×400) from control (A), IRB 30% (B) and IRB 40% (C), ERB 40% (D) and ERB 70% (E), and CRB 30%/50% (F) and CRB 40%/60% (G) groups. Neutrophilic infiltration, capillary congestion, and focal thickening of the alveolar membrane are evident in lower resistive loads but are significantly more pronounced in the highest load of each group, especially CRB 40%/60%. |
Discussion
The main findings of our study are as follows: 1) CRB increases lung elasticity and alveolar capillary barrier permeability and provokes lung inflammation and injury; 2) ERB alone is a putative inflammatory stimulus per se; and 3) the injurious effect of resistive breathing on the lung is load dependent.
In line with our previous report wherein we had used an inspiratory resistance of 50% of maximum,2 we found that lower inspiratory resistances of 30% and 40% but not 20% also induce mechanical, inflammatory, and permeability alterations consistent with the presence of ALI. IRB increased lung elasticity and produced (mainly) a neutrophilic lung inflammatory process, accompanied by IL-1β and IL-6 upregulations. BALF protein content increased and histological analysis revealed, apart from neutrophilic infliltration, capillary congestion, and focal thickening of the alveolar wall, which are well-known features of ALI.10
In IRB, airway obstruction leads to strenuous diaphragmatic contractions producing high negative pleural pressure swings during tidal breathing. Transmission of an increased negative pleural pressure causes significant alveolar and interstitial pressure reduction.11 Simultaneously, high negative intrathoracic pressure augments both venous return to the right ventricle and left ventricle afterload,12 thus increasing the pulmonary capillary pressure.13 As a result, increased capillary transmural pressure (ie, the difference between interstitial and capillary pressures) develops. The latter constitutes one of the principal mechanical forces acting on the blood–gas barrier and, when excessively elevated, may interfere with its integrity leading to stress failure with subsequent pulmonary edema and extravasation of inflammatory cells.14
Lung resident cells are able to sense mechanical forces and produce inflammatory responses mediated by intracellular signaling cascades activation, termed mechanotransduction.15–17 Indeed, mechanical stretch has been shown to induce proinflammatory cytokine release by both alveolar epithelial cells18 and alveolar macrophages19 in vitro. Similar cytokine and chemokine upregulations as well as nuclear factor-κB activation were observed when isolated perfused mice lungs were ventilated with an increased inspiratory pressure.20,21 Thus, the large negative intrathoracic pressures during IRB may account for the injurious effect of IRB, and the relative contribution of mechanotransduction to the observed inflammation could be larger in the case of lower inspiratory resistances that do not cause significant stress failure.
All IRB-induced alterations were load dependent, being less pronounced than those previously reported by our group with the use of a greater inspiratory resistance of 50% max (IRB 50%)2 than the one we used in the current study (IRB 40%). IRB 30% was identified as the lowest inspiratory resistance capable of producing significant derangements within the time frame (6 hours) used.
Load dependence has been described in previous studies examining the effects of mechanical stretch. In vitro magnitude-dependent cyclic stretch-induced cytokine and chemokine upregulations22,23 and oxidative stress generation24 were reported in bronchial and alveolar epithelial cells and macrophages. In vivo in a model of VILI in previously healthy animals, lung permeability increments occurred only when a cutoff tidal volume was reached.7,8
As noticed in IRB, CRB also provoked ALI and inflammation in the healthy rat lung. Our results are in agreement with those reported by our group for another animal model of CRB, via tracheal banding,25 in which mice were forced to breathe against a tracheal constriction-stenosis at half the initial surface area, accomplished by suturing a nylon band around their trachea. Although applied in a different species and for a different time period (24 hours in the mouse model vs 6 hours in the rat model), tracheal banding also produced mechanical, inflammatory, and histological indices of ALI, supporting the results of this study.
Interestingly, the addition of expiratory resistance differentially and load dependently influenced the diverse injurious alterations encountered during IRB alone. More specifically, all studied CRB groups load dependently increased BALF neutrophils and MPO activity similarly to the corresponding IRB groups. Remarkably, CRB 40%/60% led to markedly increased total BALF cellularity and prominent neutrophilia, unequivocally exceeding those caused by IRB 40% alone. Further supportive evidence for this observation was provided by histological analysis. The CRB 40%/60% group exhibited significantly more severe injury than the IRB 40% group, mainly attributable to interstitial neutrophil infiltration.
The addition of an expiratory resistance attenuated the elasticity increase produced by IRB alone. In the CRB 30%/50% group, elasticity and static Cst were practically unchangeable compared to ctrs. CRB 40%/50% and 40%/60% resulted in mechanical derangements, but to a lesser extent than the same inspiratory resistance alone (IRB 40%). Interestingly, the larger the expiratory resistance applied for the same inspiratory resistance, the worse the mechanical impairment was, as clearly witnessed by static Cst as well as hysteresis measurements. Identical protein content alterations were noted in BALF extracted by the CRB groups. Once again CRB 30%/50% did not significantly affect permeability, whereas BALF protein was found elevated in CRB 40%/50% and, even more, in CRB 40%/60%, approximating the levels encountered in IRB 40%.
Although not specifically measured in our experiments, it should be expected that raising expiratory resistance increased expiratory time constants (ie, the product of Cst and resistance) leading to incomplete lung emptying and, therefore, end-expiratory lung volume elevation and PEEPi generation, in other words dynamic hyperinflation.26,27 The effects of PEEP on lung injury are ambiguous. The protective effect of PEEP on injury encountered in animal models of mechanical stress-induced lung injury, such as VILI, is well documented28–32 and is attributed to the prevention of edema formation and shear stress, induced by cyclic recruitment and de-recruitment of lung units.33–36 In our model of CRB, PEEPi generation as a result of expiratory resistance addition could offset the effects of large negative intrathoracic pressures created as a result of inspiratory resistance. In this context, pulmonary edema resulting from upper airway obstruction occasionally ensues only after the removal of the obstruction, and this is supposed to occur due to the disruption of the preexisting balance between the opposing effects of simultaneous inspiratory and expiratory occlusions.37
On the other hand, it has also been shown that excessive PEEP and hyperinflation could have deleterious influence on lung structure and inflammatory status in the context of VILI, conceivably as a result of lung unit overdistention and concomitant alveolar wall stress failure and/or mechanotransduction stimulation. Dreyfuss and Saumon38 found increased lung permeability, despite normal tidal volume ventilation, when a large amount of PEEP was applied to rats. More recently, in mice subjected to noninjurious ventilation after hydrochloric acid inspiration, Thammanomai et al39 reported increased epithelial cell injury, as inferred by the appearance of soluble fragments of E-cadherin in BALF, when PEEP was doubled. A hypothetical causal relationship between dynamic hyperinflation and pulmonary inflammation has been proposed,3 mainly attributable to the proinflammatory potential of cell stretch resulting from hyperinflation. Indeed, in vitro excessive cyclic stretch of pulmonary endothelial cells has been shown to induce alterations promoting increased endothelial barrier permeability.40 Collectively, these findings could partially explain the ambiguous effects of our CRB model, attenuating lung permeability and elasticity increments compared to IRB alone, on one hand, and aggravating neutrophilic inflammation, on the other hand.
The possible proinflammatory action of hyperinflation caused by increased expiratory resistance may also account for our observation that ERB caused significant BALF neutrophilia, verified by concomitant MPO activity augmentation in lung tissue, IL-1β and Il-6 upregulations, and significant neutrophil infiltration in histological analysis. Interestingly, neutrophil count elevation in ERB BALF was accompanied by macrophage reduction in a load-independent manner. This is in-line with previous observations of intraalveolar macrophage depletion in in vivo and ex vivo models of VILI.41–43 It has been postulated that relocation of alveolar macrophages into the interstitium is the most likely explanation.43 Although reduced in count, macrophages have been shown to contribute to VILI pathogenesis.41 The observed lack of load dependency and the inability to determine a lower cutoff expiratory load for inflammation induction could partly be attributed to the relatively high loads used (≥40%).
In contrast to the devastating effects of IRB and CRB, ERB did not provoke alterations in lung mechanics. Increased BALF protein content was noticed in ERB only when the highest resistance of 70% was applied, indicating a smaller effect of ERB on lung permeability. This may reflect a potential protective effect of PEEPi on the formation of pulmonary edema and lung injury. Accordingly, Fu et al44 proved that rabbit lung inflation with a high lung volume results in blood–gas barrier disruption, imaged by electron microscopy as multiple epithelial, endothelial, and basement membrane breaks. In a subsequent experiment, the same group noticed that when increased PEEP (9 cmH2O) was applied to their ex vivo model, only mild interstitial thickening occurred without significant disruption, despite the expected lung volume increase induced by PEEP application.45
A potential limitation of our study is the administration of 100% oxygen to prevent hypoxemia during resistive breathing. Hyperoxia is a well-established model of ALI in rats,46,47 when applied for at least 40–50 hours.10 However, the significantly shorter time course (6 hours) used in our experiments renders a putative contribution of hyperoxia to the resistive breathing-induced lung injury rather unlikely. Moreover, the effect of hyperoxia was controlled by oxygen administration to unloaded animals as well.
Clinical implications
The present study expands the results of our previous report that IRB induces lung injury and inflammation.2 It should be acknowledged that pure IRB is never encountered in clinical practice. Breathing against an inspiratory resistance higher than expiratory resistance is observed in cases of upper airway obstruction, such as laryngospasm, upper airway tumors, obstructive sleep apnea and endotracheal tube obstruction in adults or epiglottitis, croup, and foreign body aspiration in children. On the other hand, CRB (with expiratory resistance higher than inspiratory resistance) is the hallmark of intrathoracic airway obstruction encountered in diseases, such as COPD and asthma. Our results imply that CRB causes injury and inflammation to previously healthy lungs in a load-dependent manner.
Resistances used in our study were of a magnitude observed in the clinical arena. In patients with COPD, with a wide range of airway obstruction (FEV1% predicted 21%–89%), Hayot et al48 reported the Pi/Pi,max values of 0.21±0.14 (SD). In stable hypercapnic COPD patients with severe obstruction (FEV1 0.79±0.35 [SD] L), Pi/Pi,max was 0.27±0.15 (SD).49 In another study measuring pleural pressures by an esophageal balloon, Ppleural/Ppleural,max was estimated at ~0.35 in stable COPD patients with equally severe obstruction (FEV1 0.57±0.06 [SEM] L), but less pronounced hypercapnia.50 Conceivably, significantly higher resistances and, thus, loading of the inspiratory muscles should be expected during COPD exacerbations, especially when severe enough to require mechanical ventilation or in COPD patients failing to wean. Indeed, in patients with COPD exacerbation who were intubated and invasively ventilated, Pi/Pi,max was measured 0.42±0.11,51 while a somewhat higher value of 0.49±0.09 was found in COPD patients failing to wean from mechanical ventilation.52 In patients with asthma, on the other hand, Misuri et al53 showed a Ppleural/Ppleural,max value of ~0.26, when bronchoconstriction was provoked by histamine, producing a FEV1% predicted drop of 42.7%±10.3% compared to baseline. Similarly, Lougheed et al54 induced bronchoconstriction by metacholine in asthmatic patients causing a FEV1% predicted fall to 44%. Ppleural/Ppleural,max under those circumstances was reported to be 0.33. Significantly higher Pi values, corresponding to more severe inspiratory loads, were measured in children with acute asthma in an earlier study.55 Based on these data, inspiratory and combined loads used in our study were chosen in order to reflect not only severe COPD and asthma exacerbations but also stable severely obstructed COPD patients.
Our results raise the intriguing, though speculative, possibility that in COPD and asthma patients some degree of lung parenchymal inflammation is the result of the increased airway resistance per se irrespective of the underlying airway inflammatory process involved in disease and/or exacerbation pathogenesis. The load dependency found in our study implies that this phenomenon may be present from earlier stages of the diseases, thus contributing to disease progression, although, during COPD exacerbations and possibly more severe asthma attacks, this inflammatory response is significantly more pronounced and accompanied by increased lung permeability. Interestingly, patients with frequent exacerbations of COPD (where severe resistive breathing is noted) exhibit more rapid decline in lung function.56,57
Furthermore, by allowing separate examination of inspiratory and expiratory resistances, our model gave us the opportunity to differentiate the effects of IRB from ERB on the lung. Reasonably assuming that ERB results in dynamic hyperinflation and PEEPi generation, our findings raise the interesting possibility that the latter may constitute an inflammatory stimulus for the lung per se. Our observations are in-line with those of Gatta et al,58 reporting a significant increase in C-reactive protein, a marker of systemic inflammation, in COPD patients with more pronounced dynamic hyperinflation, as assessed by inspiratory capacity. If hyperinflation resulting from ERB alone or in combination with inspiratory constitutes a potentially deleterious inflammatory stimulus per se, pharmacologic and other interventions known to reduce hyperinflation could exert anti-inflammatory effects in the context of COPD.59 Bronchodilators possess well-established deflating properties.27,60,61 Our findings lend support to the possibility of an additional anti-inflammatory action of bronchodilators, originating from their ability to diminish hyperinflation.
Conclusion
Combined IRB and ERB load dependently increases alveolar capillary barrier permeability and exerts inflammatory and injurious effects on healthy rat lung, thus providing new insights into the pathogenesis of obstructive airway diseases and their exacerbations.
Acknowledgments
The authors wish to express their gratitude to Zoi Kolia for technical assistance with experimental procedures. This research has been cofinanced by the European Union (European Social Fund) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework – Research Funding Program: Aristeia, 2106.
Disclosure
The authors report no conflicts of interest in this work.
References
Vassilakopoulos T, Divangahi M, Rallis G, et al. Differential cytokine gene expression in the diaphragm in response to strenuous resistive breathing. Am J Respir Crit Care Med. 2004;170(2):154–161. | ||
Toumpanakis D, Kastis GA, Zacharatos P, et al. Inspiratory resistive breathing induces acute lung injury. Am J Respir Crit Care Med. 2010;182(9):1129–1136. | ||
Agusti A, Soriano JB. Dynamic hyperinflation and pulmonary inflammation: a potentially relevant relationship? Eur Respir Rev. 2006;15(100):68–71. | ||
Paredi P, Goldman M, Alamen A, et al. Comparison of inspiratory and expiratory resistance and reactance in patients with asthma and chronic obstructive pulmonary disease. Thorax. 2010;65(3):263–267. | ||
Jarenback L, Ankerst J, Bjermer L, Tufvesson E. Flow-volume parameters in COPD related to extended measurements of lung volume, diffusion, and resistance. Pulm Med. 2013;2013:10. [Article ID 782052]. | ||
Willms D, Shure D. Pulmonary edema due to upper airway obstruction in adults. Chest. 1988;94(5):1090–1092. | ||
de Prost N, Dreyfuss D, Saumon G. Evaluation of two-way protein fluxes across the alveolo-capillary membrane by scintigraphy in rats: effect of lung inflation. J Appl Physiol. 2007;102(2):794–802. | ||
Carlton DP, Cummings JJ, Scheerer RG, Poulain FR, Bland RD. Lung overexpansion increases pulmonary microvascular protein permeability in young lambs. J Appl Physiol. 1990;69(2):577–583. | ||
Hantos Z, Adamicza A, Govaerts E, Daroczy B. Mechanical impedances of lungs and chest wall in the cat. J Appl Physiol. 1992;73(2):427–433. | ||
Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295(3):L379–L399. | ||
Lang SA, Duncan PG, Shephard DA, Ha HC. Pulmonary oedema associated with airway obstruction. Can J Anaesth. 1990;37(2):210–218. | ||
Wise RA. Effect of alterations of pleural pressure on cardiac output. South Med J. 1985;78(4):423–428. | ||
Fremont RD, Kallet RH, Matthay MA, Ware LB. Postobstructive pulmonary edema: a case for hydrostatic mechanisms. Chest. 2007;131(6):1742–1746. | ||
West JB. Invited review: pulmonary capillary stress failure. J Appl Physiol (1985). 2000;89(6):2483–2489. | ||
Dos Santos CC, Slutsky AS. Invited review: mechanisms of ventilator-induced lung injury: a perspective. J Appl Physiol (1985). 2000;89(4):1645–1655. | ||
Uhlig S. Ventilation-induced lung injury and mechanotransduction: stretching it too far? Am J Physiol Lung Cell Mol Physiol. 2002;282(5):L892–L896. | ||
Vlahakis NE, Hubmayr RD. Cellular stress failure in ventilator-injured lungs. Am J Respir Crit Care Med. 2005;171(12):1328–1342. | ||
Vlahakis NE, Schroeder MA, Limper AH, Hubmayr RD. Stretch induces cytokine release by alveolar epithelial cells in vitro. Am J Physiol. 1999;277(1 pt 1):L167–L173. | ||
Pugin J, Dunn I, Jolliet P, et al. Activation of human macrophages by mechanical ventilation in vitro. Am J Physiol. 1998;275(6 pt 1):L1040–L1050. | ||
Held HD, Boettcher S, Hamann L, Uhlig S. Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-kappaB and is blocked by steroids. Am J Respir Crit Care Med. 2001;163(3 pt 1):711–716. | ||
von Bethmann AN, Brasch F, Nusing R, et al. Hyperventilation induces release of cytokines from perfused mouse lung. Am J Respir Crit Care Med. 1998;157(1):263–272. | ||
Thomas RA, Norman JC, Huynh TT, Williams B, Bolton SJ, Wardlaw AJ. Mechanical stretch has contrasting effects on mediator release from bronchial epithelial cells, with a rho-kinase-dependent component to the mechanotransduction pathway. Respir Med. 2006;100(9):1588–1597. | ||
Wu J, Yan Z, Schwartz DE, Yu J, Malik AB, Hu G. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J Immunol. 2013;190(7):3590–3599. | ||
Davidovich N, DiPaolo BC, Lawrence GG, Chhour P, Yehya N, Margulies SS. Cyclic stretch-induced oxidative stress increases pulmonary alveolar epithelial permeability. Am J Respir Cell Mol Biol. 2013;49(1):156–164. | ||
Glynos C, Toumpanakis D, Loverdos K, et al. Guanylyl cyclase activation reverses resistive breathing-induced lung injury and inflammation. Am J Respir Cell Mol Biol. 2015;52(6):762–771. | ||
Demedts M. Mechanisms and consequences of hyperinflation. Eur Respir J. 1990;3(6):617–618. | ||
O’Donnell DE, Laveneziana P. Physiology and consequences of lung hyperinflation in COPD. Eur Respir Rev. 2006;15(100):61–67. | ||
Allen G, Lundblad LK, Parsons P, Bates JH. Transient mechanical benefits of a deep inflation in the injured mouse lung. J Appl Physiol (1985). 2002;93(5):1709–1715. | ||
Dreyfuss D, Soler P, Basset G, Saumon G. High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis. 1988;137(5):1159–1164. | ||
Kostic P, Zannin E, Andersson OM, et al. Positive end-expiratory pressure optimization with forced oscillation technique reduces ventilator induced lung injury: a controlled experimental study in pigs with saline lavage lung injury. Crit Care. 2011;15(3):R126. | ||
Martynowicz MA, Walters BJ, Hubmayr RD. Mechanisms of recruitment in oleic acid-injured lungs. J Appl Physiol (1985). 2001;90(5):1744–1753. | ||
Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis. 1974;110(5):556–565. | ||
Bilek AM, Dee KC, Gaver DP 3rd. Mechanisms of surface-tension-induced epithelial cell damage in a model of pulmonary airway reopening. J Appl Physiol (1985). 2003;94(2):770–783. | ||
de Prost N, Ricard JD, Saumon G, Dreyfuss D. Ventilator-induced lung injury: historical perspectives and clinical implications. Ann Intensive Care. 2011;1(1):28. | ||
Terragni PP, Rosboch GL, Lisi A, Viale AG, Ranieri VM. How respiratory system mechanics may help in minimising ventilator-induced lung injury in ARDS patients. Eur Respir J Suppl. 2003;42:15s–21s. | ||
Whitehead T, Slutsky AS. The pulmonary physician in critical care * 7: ventilator induced lung injury. Thorax. 2002;57(7):635–642. | ||
Ringold S, Klein EJ, Del Beccaro MA. Postobstructive pulmonary edema in children. Pediatr Emerg Care. 2004;20(6):391–395. | ||
Dreyfuss D, Saumon G. Role of tidal volume, FRC, and end-inspiratory volume in the development of pulmonary edema following mechanical ventilation. Am Rev Respir Dis. 1993;148(5):1194–1203. | ||
Thammanomai A, Hamakawa H, Bartolak-Suki E, Suki B. Combined effects of ventilation mode and positive end-expiratory pressure on mechanics, gas exchange and the epithelium in mice with acute lung injury. PLoS One. 2013;8(1):e53934. | ||
Birukov KG, Jacobson JR, Flores AA, et al. Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am J Physiol Lung Cell Mol Physiol. 2003;285(4):L785–L797. | ||
Frank JA, Wray CM, McAuley DF, Schwendener R, Matthay MA. Alveolar macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;291(6):L1191–L1198. | ||
Imanaka H, Shimaoka M, Matsuura N, Nishimura M, Ohta N, Kiyono H. Ventilator-induced lung injury is associated with neutrophil infiltration, macrophage activation, and TGF-beta 1 mRNA upregulation in rat lungs. Anesth Analg. 2001;92(2):428–436. | ||
Whitehead TC, Zhang H, Mullen B, Slutsky AS. Effect of mechanical ventilation on cytokine response to intratracheal lipopolysaccharide. Anesthesiology. 2004;101(1):52–58. | ||
Fu Z, Costello ML, Tsukimoto K, et al. High lung volume increases stress failure in pulmonary capillaries. J Appl Physiol (1985). 1992;73(1):123–133. | ||
Berg JT, Fu Z, Breen EC, Tran HC, Mathieu-Costello O, West JB. High lung inflation increases mRNA levels of ECM components and growth factors in lung parenchyma. J Appl Physiol (1985). 1997;83(1):120–128. | ||
Barry BE, Crapo JD. Patterns of accumulation of platelets and neutrophils in rat lungs during exposure to 100% and 85% oxygen. Am Rev Respir Dis. 1985;132(3):548–555. | ||
Naureckas ET, Factor P, Benjaminov O, Hoffer E, Sriram V, Sznajder JI. Pentoxifylline does not protect against hyperoxic lung injury in rats. Eur Respir J. 1994;7(8):1397–1402. | ||
Hayot M, Perrigault PF, Gautier-Dechaud V, et al. Tension-time index of inspiratory muscles in COPD patients: role of airway obstruction. Respir Med. 1998;92(6):828–835. | ||
Begin P, Grassino A. Inspiratory muscle dysfunction and chronic hypercapnia in chronic obstructive pulmonary disease. Am Rev Respir Dis. 1991;143(5 pt 1):905–912. | ||
Duranti R, Misuri G, Gorini M, Goti P, Gigliotti F, Scano G. Mechanical loading and control of breathing in patients with severe chronic obstructive pulmonary disease. Thorax. 1995;50(2):127–133. | ||
Zakynthinos SG, Vassilakopoulos T, Roussos C. The load of inspiratory muscles in patients needing mechanical ventilation. Am J Respir Crit Care Med. 1995;152(4 pt 1):1248–1255. | ||
Vassilakopoulos T, Zakynthinos S, Roussos C. The tension-time index and the frequency/tidal volume ratio are the major pathophysiologic determinants of weaning failure and success. Am J Respir Crit Care Med. 1998;158(2):378–385. | ||
Misuri G, Mancini M, Iandelli I, et al. Respiratory muscle overloading and dyspnoea during bronchoconstriction in asthma: protective effects of fenoterol. Pulm Pharmacol Ther. 1997;10(5–6):299–304. | ||
Lougheed DM, Webb KA, O’Donnell DE. Breathlessness during induced lung hyperinflation in asthma: the role of the inspiratory threshold load. Am J Respir Crit Care Med. 1995;152(3):911–920. | ||
Stalcup SA, Mellins RB. Mechanical forces producing pulmonary edema in acute asthma. N Engl J Med. 1977;297(11):592–596. | ||
Celli BR, Thomas NE, Anderson JA, et al. Effect of pharmacotherapy on rate of decline of lung function in chronic obstructive pulmonary disease: results from the TORCH study. Am J Respir Crit Care Med. 2008;178(4):332–338. | ||
Makris D, Moschandreas J, Damianaki A, et al. Exacerbations and lung function decline in COPD: new insights in current and ex-smokers. Respir Med. 2007;101(6):1305–1312. | ||
Gatta D, Aliprandi G, Pini L, Zanardini A, Fredi M, Tantucci C. Dynamic pulmonary hyperinflation and low grade systemic inflammation in stable COPD patients. Eur Rev Med Pharmacol Sci. 2011;15(9):1068–1073. | ||
Casaburi R, Porszasz J. Reduction of hyperinflation by pharmacologic and other interventions. Proc Am Thorac Soc. 2006;3(2):185–189. | ||
O’Donnell DE, Fluge T, Gerken F, et al. Effects of tiotropium on lung hyperinflation, dyspnoea and exercise tolerance in COPD. Eur Respir J. 2004;23(6):832–840. | ||
Rossi A, Centanni S, Cerveri I, et al. Acute effects of indacaterol on lung hyperinflation in moderate COPD: a comparison with tiotropium. Respir Med. 2012;106(1):84–90. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.