")
Back to Journals » International Journal of General Medicine » Volume 14
The Diabetes Syndrome – A Collection of Conditions with Common, Interrelated Pathophysiologic Mechanisms
Authors Rachfal AW , Grant SFA, Schwartz SS
Received 3 February 2021
Accepted for publication 8 March 2021
Published 18 March 2021 Volume 2021:14 Pages 923—936
DOI https://doi.org/10.2147/IJGM.S305156
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Amy W Rachfal,1 Struan FA Grant,2– 4 Stanley S Schwartz5,6
1Stage Gate Partners, LLC, Ardmore, PA, USA; 2Center for Spatial and Functional Genomics, Division of Human Genetics, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; 3Department of Pediatrics, University of Pennsylvania, Perlman School of Medicine, Philadelphia, PA, USA; 4Department of Genetics, University of Pennsylvania, Perlman School of Medicine, Philadelphia, PA, USA; 5Stanley Schwartz MD, LLC, Main Line Health System, Wynnewood, PA, USA; 6University of Pennsylvania, Perlman School of Medicine, Philadelphia, PA, USA
Correspondence: Stanley S Schwartz
Stanley Schwartz, MD, LLC, 233 E Lancaster Aver, Suite 305, Ardmore, PA, USA
Tel +1 610 642 6800
Fax +1 610 642 6850
Email [email protected]
Abstract: The four basic pathophysiologic mechanisms which damage the β-cell within diabetes (ie, genetic and epigenetic changes, inflammation, an abnormal environment, and insulin resistance [IR]) also contribute to cell and tissue damage and elevate the risk of developing all typical diabetes-related complications. Genetic susceptibility to damage from abnormal external and internal environmental factors has been described including inflammation and IR. All these mechanisms can promote epigenetic changes, and in total, these pathophysiologic mechanisms interact and react with each other to cause damage to cells and tissues ultimately leading to disease. Importantly, these pathophysiologic mechanisms also serve to link other common conditions including cancer, dementia, psoriasis, atherosclerotic cardiovascular disease (ASCVD), nonalcoholic fatty liver disease (NAFLD), and nonalcoholic steatohepatitis (NASH). The “Diabetes Syndrome”, an overarching group of interrelated conditions linked by these overlapping mechanisms, can be viewed as a conceptual framework that can facilitate understanding of the inter-relationships of superficially disparate conditions. Recognizing the association of the conditions within the Diabetes Syndrome due to common pathophysiologies has the potential to provide both benefit to the patient (eg, prevention, early detection, precision medicine) and to the advancement of medicine (eg, driving education, research, and dynamic decision-based medical practice).
Keywords: cancer, psoriasis, dementia, atherosclerotic cardiovascular disease, nonalcoholic steatohepatitis, nonalcoholic fatty liver disease
Introduction
Although many pathways lead to hyperglycemia in diabetes – the so-called “Egregious Eleven” (Listed in Table 1) - β-cell dysfunction is the core defect.1,2 Four basic pathophysiologic mechanisms damage the β-cell, namely, genes and epigenetic changes, inflammation, an abnormal environment [especially fuel excess], and insulin resistance (IR).1,2 Further, the same factors that damage the β-cell may also lead to cell and tissue damage and elevate the risk of developing typical diabetes-related complications, such as retinopathy, neuropathy, and nephropathy.1–3 The interplay between these pathophysiologic mechanisms influences the specific risk of development and progression of complications in an individual patient. Importantly, we these mechanisms can also predispose patients to other common conditions including diabetes mellitus (DM), cancer, dementia, psoriasis, atherosclerotic cardiovascular disease (ASCVD), and nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH).4–15
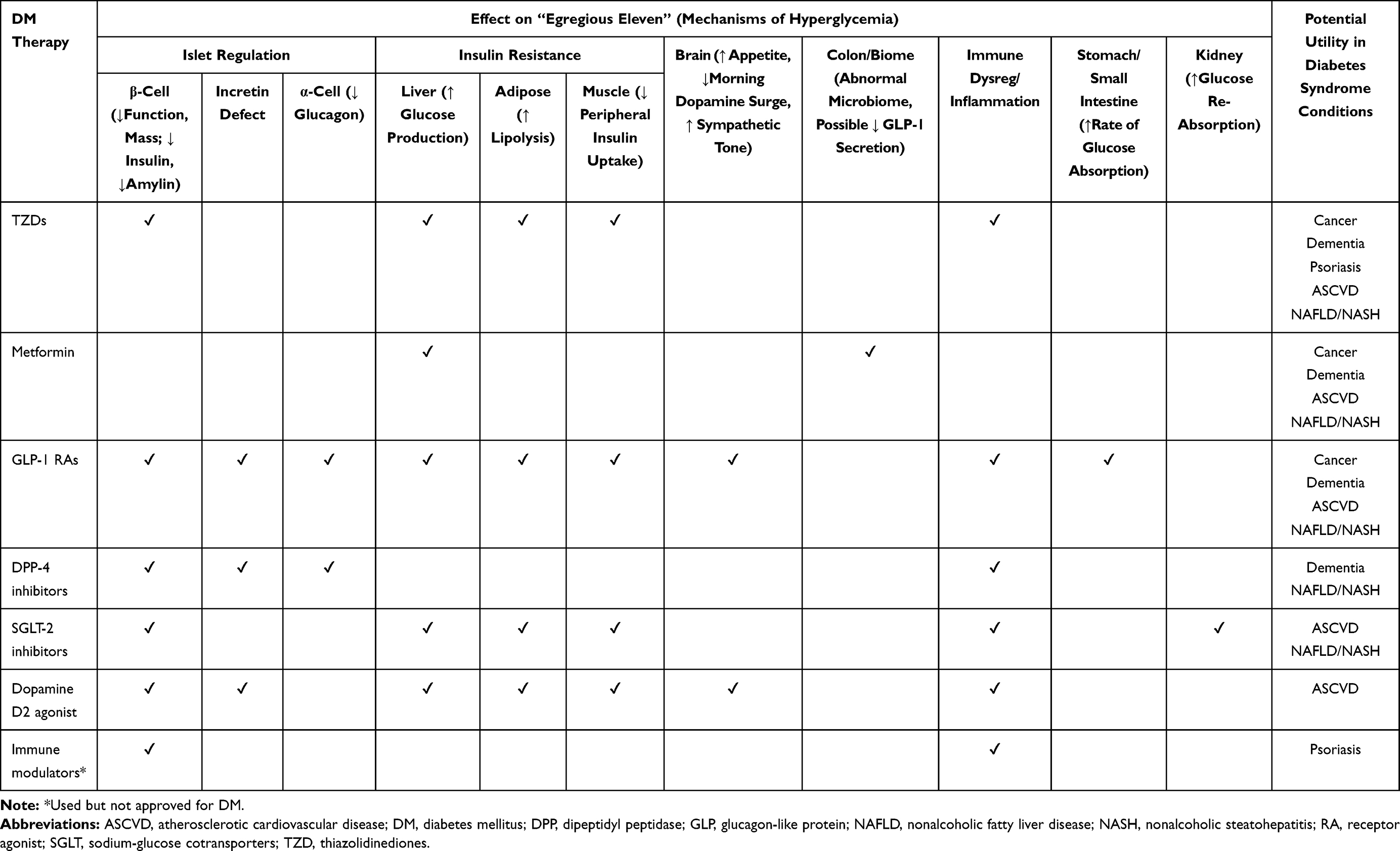 |
Table 1 Targets and Potential Utility of Diabetes Therapies in Conditions of the Diabetes Syndrome |
In clinical practice we often encounter these common diseases, frequently within one individual patient and they are treated as independent conditions. However, we believe their epidemiologic associations is, in part, due to the same underlying pathophysiologies driving β-cell damage and diabetic complications. That is, the same pathophysiologic mechanisms that damage the β-cell and promote diabetes-specific complications also have key roles in the pathogenesis of these diseases. The overlapping epidemiology of these conditions highlights this connection and the significant associations that have been found between DM and all these other phenotypes.7–15 However, we propose these connections go beyond mere epidemiologic links due to overlapping pathophysiology. In fact, these conditions occur together in enough frequency and have common overlapping pathophysiologic drivers that we have created a conceptual framework called “The Diabetes Syndrome”. The name is inspired by the Greek meaning of syndromē (sun- [together] + dramein [to run]) as the conditions, indeed, run together (Figure 1).
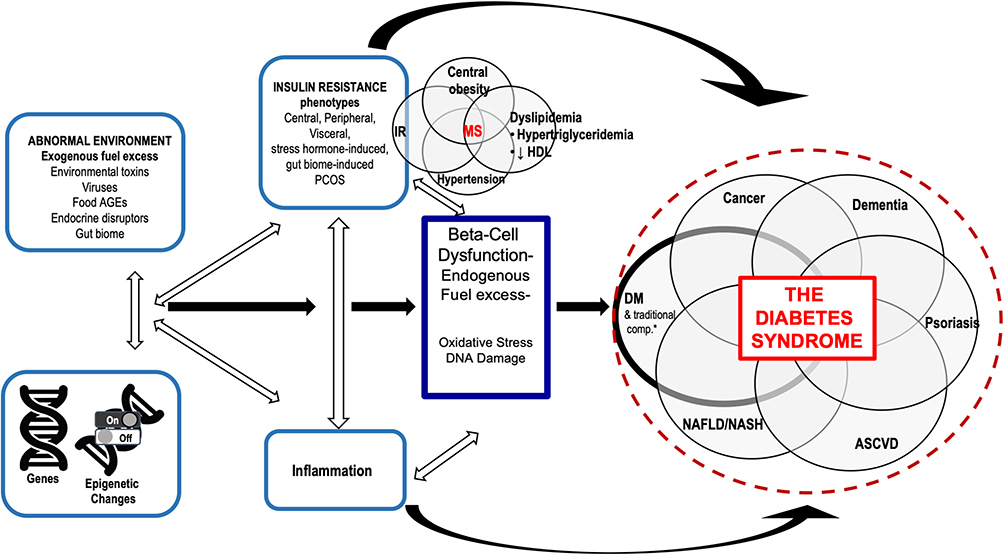 |
Figure 1 Overlapping pathophysiologic mechanisms that lead to the diabetes syndrome. Abbreviations: AGE, advanced glycosylation end products; ASCVD, atherosclerotic cardiovascular disease; DM, diabetes mellitus; DNA, deoxynucleic acid; HDL, high density lipoprotein; IR, insulin resistance; MS, metabolic syndrome; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; PCOS, polycystic ovary disease. Notes: Insulin resistance, its complications, and other conditions arise from common pathophysiologies. Metabolic syndrome, obesity, and PCOS, etc, can be viewed as phenotypes related to genes and epigenetic changes associated with IR. Insulin resistance along with inflammation increase the risk of β-cell dysfunction as well as the Diabetes Syndrome. *Traditional DM complications include retinopathy, nephropathy, neuropathy-tissues that do not require insulin to get glucose into cells. |
This article will describe the shared pathophysiologic and etiologic factors across these prevalent and related diseases within the Diabetes Syndrome conceptual framework discussed within the context of the 4 basic pathophysiologic mechanisms – genes and epigenetic changes, abnormal environment, inflammation, and IR – with a focus on commonalities between these diseases and DM. In brief, genetics can mediate susceptibility to damage from abnormal external and internal environmental factors, including inflammation and IR. All these mechanisms can promote epigenetic changes. In total, these 4 pathophysiologic mechanisms cross-react with each other to cause damage to cells and tissues ultimately leading to disease.2 If we as physicians begin to recognize and understand the inter-relatedness of these conditions, it may result in better screening, therapy, and cross-purposing of therapies rather than treating these diseases as independent conditions. Furthermore, as more is learned through continued research and clinical experience, this framework of Diabetes Syndrome may grow to include other related conditions.
The Diabetes Syndrome as a Conceptual Framework
The common, frequently concomitant chronic conditions of DM, cancer, dementia, psoriasis, ASCVD, NAFLD, and NASH are related beyond shared epidemiologic associations. We propose these conditions are linked by shared pathophysiologies namely, genes and epigenetic changes, abnormal environment, inflammation, and IR. Because these conditions occur together in reasonable regularity and have common overlapping pathophysiologic drivers we have created a conceptual framework called “The Diabetes Syndrome”. Of note, this new term is not proposed to be used for “diagnosis” of the Diabetes Syndrome, rather to heighten clinicians’ awareness of the inter-relationships between these conditions to enhance to quality of care to our patients. This paper will discuss the common links based on current understanding, but it is expected as new findings and data are revealed and viewed within this conceptual framework, that the conditions and diseases of the Diabetes Syndrome framework may grow and evolve.
Epidemiology
The conditions of the Diabetes Syndrome are common and often occur within the same patient. Even just considering DM alone, nearly 1 in 10 people worldwide are living with diabetes, translating to a prevalence of 463 million adults.16 Diabetes and the conditions within the Diabetes Syndrome are often considered co-morbidities and/or risk factors of each other.
Cancer is a broad and diverse disease, however, links have been found between certain types of cancer and DM, with the strongest associations found with pancreatic and hepatic cancer.14 Moreover, multiple studies demonstrate a higher incidence of cancer in patients with DM than without.12 The hypothesized connection between DM and carcinogenesis include hyperglycemia, chronic inflammation, and hyperinsulinemia.12,14
Dementia is estimated to affect 5–7% of elderly patients worldwide, with Alzheimer’s disease (AD) representing a majority of cases.17 There is a growing body of research that reveals a link between DM and cognitive decline/dysfunction, and dementia (including AD).9,10,18 Indeed, a phenotypic consequence of IR or deficiency can result in AD, sometimes referred to as “type 3” diabetes or diabetes of the brain.19 Patients with DM are at increased risk for dementia and both diseases have similar risk factors and biologic mechanisms.9,10,18,19 Indeed, AD is twice as frequent in diabetic patients compared to patients without DM and shared pathophysiologic features include IR, amyloid aggregations, inflammatory stress, and cognitive disturbances.10,20
Approximately 8 million people in the US are diagnosed with psoriasis, a chronic, systemic inflammatory condition of the skin condition.21,22 It is characterized by underlying inflammatory processes including regulatory immune cells, cytokines, and adipokines.21 Psoriasis is often associated with other chronic conditions including cardiometabolic disease, cancer, NAFLD, NASH, psoriatic arthritis, inflammatory bowel disease, chronic kidney disease, cancer, and mood disorders.22–24
Cardiovascular disease is the leading cause of premature mortality and disability worldwide with the vast majority due to underlying atherosclerotic pathogenesis including coronary artery disease, cerebrovascular disease, venous thromboembolism, and peripheral vascular disease.25 These conditions can lead to myocardial infarction, cardiac arrhythmias, and stroke.25 Common risk factors for both DM and ASCVD include hyperlipidemia, IR, inflammation, hypertension, obesity, smoking, and lack of physical activity.4,25 Indeed, CVD is the leading cause of death in patients with Type 2 DM (T2DM), with both impaired micro and macrovascular circulation leading to CVD in patients with DM.13
Nonalcoholic fatty liver disease, the most common form of chronic liver disease in the world, is associated with potentially severe sequela.26,27 It encompasses a spectrum of disorders ranging from simple steatosis to the more aggressive necro-inflammatory form, NASH, which can result in cirrhosis, hepatocellular carcinoma, and end-stage liver disease.26,27 Type 2 DM is considered a risk factor for NAFLD and, conversely, DM appears to accelerate progression of disease in NAFLD.15 The “multiple-hit” pathogenesis of NAFLD is thought to include nutritional factors, gut microbiota, genetic and epigenetic factors, IR, and hormones secreted from the adipose tissue.6
Clearly, there are many shared comorbidities and associations between the conditions within the proposed Diabetes Syndrome construct. However, it is our contention that this is due to shared overlapping pathophysiologies that link them. This paper will discuss the common links based on current understanding; however, it is anticipated that as new research comes to light and is viewed within this conceptual framework that the conditions and diseases conscribed within the Diabetes Syndrome Framework may be refined.
Linking Conditions of the Diabetes Syndrome Through Genes and Epigenetics
Perhaps the most powerful and compelling connection of the conditions within the Diabetes Syndrome conceptual framework is genetic. Genes and genetic variation can mediate susceptibility to damage from abnormal external and internal environmental factors, including inflammation and IR. All these mechanisms can promote epigenetic changes.2,28,29 Epigenetics, physical genomic alterations that lead to changes in gene expression in response to environmental triggers are believed to have a generally strong influence on patient phenotype, likely more influential than specific base changes in the genes themselves.2,29 In fact, nutritional state and exposure to stress are hypothesized to have phenotypic consequences extending to children and even grandchildren.29 Furthermore, intriguing research using genetic variation in a Mendelian Randomization (MR) framework is being conducted to assess causality for disease, including epigenetic state.30–33
Although observational epidemiological studies have a valuable role in identifying risk factors or comorbid conditions associated with disease, MR studies harness genetic data in population and observational studies to offer new insights regarding causality.30,33 Mendelian randomization studies rely on the random assortment of genetic variants in a population at birth to avoid confounding, similar to randomization to an experimental drug or controlling in randomized controlled trials.30,33 In addition, MR analyses avoid reverse causality as genetic randomization precedes the disease onset and are not modified by progression of a disease.30,33
Many of the connections we propose within the Diabetes Syndrome are testable by leveraging current genetic knowledge from efforts that include powerful MR approaches which, in turn, can inform implementation of precision medicine.30,31 Further, in the absence of MR studies, genetic and epigenetic links can also be elucidated by genetic susceptibility loci, microRNA (short, single-stranded noncoding RNAs that influence and regulate gene expression), chromatin marks, 3D genomic architecture, etc.
Although not all MR studies have identified causative associations between DM and cancer in general, there have been reports of important connections to specific cancer types. In a recent MR study, genetic predisposition to T2DM was associated with higher odds of pancreatic, kidney, uterine, and cervical cancer, lower odds of melanoma and esophageal cancer, and no association with 16 other types of specific cancer types.34 Although there is limited evidence of causal association between fasting glucose and cancer, genetically predicted insulin levels have been reported to be positively associated with uterine, kidney, pancreatic, and lung cancer in a two-sample, MR study.34 This causal association between common DM features and pancreatic cancer has also been shown in another MR study, where genetically predicted increasing BMI and fasting insulin levels were causally associated with increased risk of pancreatic cancer.35 However, interestingly, no evidence of such a causal relationship between T2DM or dyslipidemia and pancreatic cancer has been reported to date.
Furthermore, a variety of converging genetic pathways between DM and cancer have been identified.12,36 For example, various genetic features at the TCF7L2 locus increase risk for colorectal cancer and T2DM pathogenesis, while the T2D THADA, JAZF1, and TCF2 loci have yielded associations for both prostate cancer and T2DM.12 Epigenetic changes and modifications may occur due to sustained excess fuel/nutrition, leading in turn to stress and the formation of reactive oxygen species (ROS) which may activate oncogenes or deactivate tumor suppressor genes.12 Hundreds of epigenetically regulated genes involved in glucose metabolism and adaptive survival have been identified and it is likely that shared pathways between energy metabolism and tumorigenesis exist.12 Indeed, a number of noncoding RNAs have been identified that regulate the insulin growth factor-1 receptor (IGF-1R), a master regulator in DM and cancer.37
Although observational trials have demonstrated that DM is associated with major subtypes of dementia, in particular dementias with obvious vascular pathologies (eg, vascular dementia and unspecified dementia), MR has not shown T2DM to be a direct cause of AD in a recent two-sample study.38 The MR approach has not been viable to date to test the causal effect between T2DM and vascular and unspecified dementia due to the lack of publicly available genetic data.38
Recent genetic association studies have identified 86 loci common to dementia, DM, and metabolic syndrome and 159 common between dementia and DM.36,39 Gene set enrichment analyses have revealed common co-localized genes involved in energy metabolism, metabolic pathways, and immune responses.39 Multiple environmental factors, abnormal metabolic environment, and exogenous fuel excess can engender epigenetic changes. For example, increased expression of histone deacetylases associated with altered expression of synaptic proteins was found in the brains of diabetic patients compared with controls.40 Furthermore, it has been hypothesized that altered expression of DM genes in AD brains may be exacerbated by peripheral IR or DM.20
Multiple genetic susceptibility loci have been identified that are shared between DM and psoriasis.41,42 Examination of DM susceptibility loci in thousands of psoriasis and control patients identified 3 loci significant in psoriasis and DM related to immune signaling regulation, glucose metabolism, and T cell activation.42 Further, shared epigenetic changes have been described. For example, several microRNAs, have been found that are implicated in both DM and psoriasis, including an microRNA that is upregulated in T helper cells in psoriatic skin lesions as well as activated as a promotor of genes in Type 1 DM (T1DM).43
Two MR studies have included T2DM as the exposure in a putative causal relationship with coronary heart disease (CHD) as the outcome.33,44 Using publicly available data from consortia of risk loci, these MR studies demonstrated a strong causal relationship between higher T2DM risk and higher CHD risk.33,44
Shared genetic predisposition for DM and coronary artery disease includes signals on a chromosome 9p21 involving a risk allele for coronary artery disease and variation within the adiponectin gene.45 Moreover, epigenetic changes and signatures have been identified that link DM and CV dysfunction.36,46 For example, histone modifications impacting the expression of NF-kB dependent genes are thought to contribute to vascular dysfunction in patients with DM.46,47 Further, elevated renin and growth differentiation factor 15 and lower adiponectin were shared both T2DM and coronary artery disease in a proteomic study.48
Nonalcoholic fatty liver disease, T2DM and obesity are epidemiologically correlated with each other, but until recently their causal relationship had not been fully elucidated. An MR study demonstrated that NAFLD causally promotes T2DM with a late-onset type 1-like diabetic sub-phenotype and central obesity.49 Conversely, T2DM, obesity, and central obesity all causally increase the risk of NAFLD.49 The authors suggested that these results indicate that closely related diseases should be stratified into subtypes in order to aid prevention, diagnosis, and treatment of these conditions.49
Common genetic susceptibility to NAFLD and NASH are also found in DM36,50 Moreover, genetic commonality between NAFLD and T2DM has been suggested based on gene/protein co-occurrences.50 From an epigenetics perspective, differential DNA methylation in the liver of patients with NASH has been associated with expression of genes linked with insulin metabolism.51
Linking Conditions of the Diabetes Syndrome Through Abnormal Environment
Abnormal metabolic environment and fuel excess are hallmarks of DM. A wide-angled MR study intended to produce an atlas of T2DM risk factors found 8 factors associated with T2DM after adjustment for BMI.52 Systolic blood pressure, smoking, insomnia, and alanine aminotransferase levels were positively associated with T2DM, while testosterone, sex hormone binding globulin, high density lipoprotein and total cholesterol levels were inversely associated with T2DM.52 Further, another MR study demonstrated a causal association between childhood adiposity and T1DM risk.53
Additionally, an abnormal metabolic environment and exogenous fuel excess have been associated with increased risk of cancer or increased cancer morality.12 For example, higher BMI, high nutrient intake, hyperglycemia, hyperinsulinemia as well as IR and inflammation have been independently associated with increased cancer risk and decreased cancer survival.12 Increasing BMI (as predicted by genes) was reported as causally associated with increased risk of pancreatic cancer using an MR approach.35 Similarly, in another MR study, genetically predicted post-prandial glucose and BMI were positively associated with breast cancer.54 In addition, dietary advanced glycation end products (AGEs) trigger various downstream changes to gene expression and have been hypothesized to be potential endocrine disruptors.2,55 Manufacture of processed food, increasingly more prevalent, involves extreme temperatures that result in the formation of AGEs which interfere with endogenous hormone production and action.55 With regard to tumorigenesis, dietary AGEs have been found to be pro-tumorigenic on estrogen breast cancer cells, human pancreatic ductal adenocarcinoma cell lines, and a mouse pancreatic cancer model.55
Diabetes and cognitive dysfunction share many shared environmental risk factors including smoking, physical inactivity, pollution/pesticides, etc.10 These environmental toxicants are thought to induce pathogenesis through interaction with genetic factors leading to downstream oxidative stress, mitochondrial dysfunction, inflammation, and IR, all common with pathogenesis of DM.10 Food AGEs, discussed above, accumulate with long-lived structural protein, contribute to complications in both DM and AD, and are thought to have a role in neurodegenerative diseases.55 Furthermore, an altered metabolic environment is thought to drive both conditions with common links of altered energy metabolism, cholesterol modifications, and dysfunctional protein O-GlcNAcylation formation.20 Finally, it has been found that high plasma glucose (as predicted by genes) is causally related to unspecified dementia, but not AD or vascular dementia risk in a recent MR study.56
Environmental risk factors believed to play a key role in psoriasis including UV exposure, medications, smoking, diet and obesity, alcohol intake, infections, and stress, are also common environmental risk factors for DM.2,57 Many of these are associated with downstream dysregulation of the immune system, likely though epigenetic modifications.2,57 The gastrointestinal tract has a large role in mediating glucose homeostasis though stimulation of incretin hormones (eg, glucagon-like protein-1 [GLP-1]) which prompt postprandial insulin release.58 Interestingly, level and function of incretin hormones appear to be reduced in patients with DM as well as psoriasis patients compared with controls.2,58,59 Mendelian Randomization has leveraged such genetic markers to determine causality between obesity/BMI, a risk factor for DM, and psoriasis.60,61
Abnormal metabolic environment is a common pathophysiologic mechanism associated with ASCVD and DM, with elevated lipids and glucose both important drivers.2,4 Intracellular fuel excess leads to oxidative stress and production of ROS that result in inflammation and induction of transcription factors that subsequently lead to changes in gene expression that finally results in cell dysfunction – not only the β-cell but also other cells including cardiomyocytes and vascular smooth muscle cells.2 Indeed, in a 2-sample MR study, genetic predisposition to childhood obesity was causally associated with an increased risk of both T2DM and coronary artery disease in adults.62 As with other conditions within the Diabetes Syndrome, a relationship between the gut microbiome and ischemic heart disease has also been suggested. For example, an MR approach demonstrated a beneficial association of the gut bacteria Bifidobacterium with ischemic heart disease, adiposity, high-density lipid cholesterol (HDL-C), and insulin resistance.63 Environmental risk factors including life-style choices such as smoking, in addition to air pollutants, pesticides, as well as food AGEs impact DM and CV mortality.45,55 Indeed, restriction of food AGEs in human studies has been associated with beneficial effects on IR and CV disease.55
Nutrition, fuel excess, and abnormal metabolic environment are associated with conditions across the Diabetes Syndrome including DM and NAFLD/NASH, leading to cell and tissue damage and dysfunction.2,6 Further, aberrant gut biome is implicated in the pathogenesis of both DM and NAFLD/NASH.2,6 Changes in the microbiome are thought to cause inflammation, alter intestinal permeability, and modulate metabolism of fatty acids resulting in.6,64 Decreased GLP-1 secretion (incretin effect) from the gut flora has also been associated with DM.2,58 Endocrine disruptors such as food AGEs are implicated in IR, DM, and NAFLD.55,65 Furthermore, a common link of endocrine disruptors to various conditions including DM, NAFLD, PCOS, and obesity may be due to their impact on IR through modification of transcription of various IR-related genes.36,65
Linking Conditions of the Diabetes Syndrome Through Inflammation
Inflammation negatively impacts a wide swath of other organ systems outside of those involved in DM specifically.2 Hyperinsulinemia which can occur as a result of IR or exogenous insulin therapy is associated with inflammation, a common biological process of both DM and cancer.1,37 Moreover, exposure to hyperglycemia and glucolipotoxicity leads to ROS which activate pathways (eg, polyol flux, AGE formation, protein kinase C activation, hexosamine flux) which lead to inflammation and, in turn, can lead to altered gene expression and epigenetic changes.2,3,12 Inflammation within adipose tissue can lead to production of cytokines that interfere with insulin signaling. Overproduction of inflammation cytokines (such as TNF-α and IL-6) has been found to be associated with IR and the development and progression of DM and cancer.37
Inflammatory responses are associated with peripheral and central IR.66 Elevated cytokines have been detected in the cerebral spinal fluid of patients with AD and mouse models have revealed inflammation may interact with both processing and deposit of Aβ.20 Chronic inflammation is frequent in both DM and AD and it is commonly known that neuroinflammation occurs in AD.20,66,67
Systemic inflammation plays an important role in both psoriasis and DM pathogenesis.68 Of note, inflammatory mediators involved in the development of IR including TNF-α, IL-6, leptin, and adiponectin have been found to be altered in patients with psoriasis.69 Leptin activates induction of proinflammatory cytokines associated with IR and keratinocyte proliferation.69 Adiponectin, an inducer of anti-inflammatory cytokines and enhanced insulin sensitivity, is reduced in patients with DM and psoriasis.69,70
Inflammation and IR are intimately intertwined in all conditions of the Diabetes Syndrome, not the least of which with ASCVD. Insulin resistance and compensatory hyperinsulinemia promote inflammation, vascular smooth muscle growth and proliferation, and atherogenesis.71 Further, excess fat in adipocytes induces inflammation and secretion of IR-provoking and proinflammatory cytokines while inhibiting adiponectin, an insulin sensitizer.71 Formation of atherosclerotic lesions is hypothesized to be driven by local inflammation in the vascular wall triggered by dyslipidemia.72
Increased lipotoxicity in patients with NAFLD leads to mitochondrial dysfunction and oxidative stress.6 Hepatocyte fat content and local liver inflammation lead to a state of chronic inflammation which can lead to fibrosis and NASH.6 In addition, inflammation and NF-kβ activation can also promote carcinogenesis which may play a role in hepatocellular cancer development, and potential downstream outcome of NASH.6
Finally, although auto-immunity links T1DM with other autoimmune diseases, much remains unknown. To wit, although there is a high degree of overlap in involved variants in autoimmune diseases with similar pathophysiology, in disease with differing pathophysiology (eg, DM and IBD), the same variants are often implicated in opposite roles.73 Further, even in diseases with differing pathophysiology and many non-overlapping variants and oppositely implicated shared variants, variants which are overlapping or shared still exist.73 This is to say, for now, the addition of autoimmune diseases to the Diabetes Syndrome framework is not warranted until we understand more.
Linking Conditions of the Diabetes Syndrome Through Insulin Resistance
Inflammation and IR are intimately connected and play robust roles in the etiology and progression of diseases within the Diabetes Syndrome.2,74 In our view, IR can be expressed in a variety of different “phenotypes” that result from genetic and epigenetic variation in response to an abnormal environment, exogenous fuel excess, and/or inflammation including metabolic syndrome, obesity, PCOS, etc. Indeed, IR may have a role in the development of prediabetes phenotypes as well.28 Importantly, IR has been suggested as a critical role in both traditionally classified T1 and T2DM in the accelerator hypothesis.75 The postulation is that DM should be viewed as a continuum and the interaction between IR and genetic responses determines that age at which critical B-cell loss occurs.75 In this hypothesis, autoimmunity retains its role, though not necessarily as the primary causative factor in T1DM.75
Insulin resistance and resultant hyperinsulinemia drive increased ROS, overexpression or overactivation of insulin-related receptors, Ras/MEK activation leading to tumorigenesis, and PTEN and mTOR activation leading to cell growth and survival.12 It has been hypothesized that cell growth regulation is “reprogrammed” due to altered circulating hormones (including insulin), substrate availability, and adipose cell dysfunction.76 Moreover, hyperinsulinemia and increased availability of cellular substrate appear to allow cancer cells to bypass cell growth checkpoints.12 Genetically predicted increasing fasting insulin levels have been causally associated with increased risk of pancreatic cancer estrogen receptor positive breast cancer in MR studies.45,54
Insulin resistance is a common pathophysiologic mechanism between both DM and AD and is a common feature of AD patients even without concomitant DM.20,66 It has been proposed that AD should be considered a degenerative metabolic disease caused by brain IR and insulin deficiency.66 Indeed, it is thought that IR may also occur in the brain, possibly earlier than the development of peripheral IR, potentially triggered by Aβ oligomers and cognitive decline.20 Mendelian randomization analyses suggest that, in fact, insulin sensitivity can impact AD risk more so than overall T2DM.77
Insulin resistance is an important pathophysiologic mechanism implicated in both DM and psoriasis. Patients with psoriasis have IR and the degree of IR is correlated with psoriasis area and severity.68,69,78 Further, hyperinsulinemia increases insulin binding to IGF receptors which lead to keratinocyte and fibroblast proliferation.70
Insulin resistance has been linked to ASCVD via multiple studies.71 Insulin resistance contributes to ASCVD by reduction of nitric oxide production (a vasodilator and antiatherogenic agent) leading to endothelial dysfunction, and, as a result of compensatory hyperinsulinemia, excessive stimulation of the MAPK pathway resulting in inflammation, vascular smooth muscle cell proliferation, and atherogenesis.48 Furthermore, IR is associated with a variety of phenotypes (see Figure 1) and risk factors, all independently associated with ASCVD.71 The fact that some anti-diabetic agents are associated with CV benefits despite modest impact on hyperglycemia highlights that other mechanisms are in place.71 (see also Thiazolidinediones Section below)
Insulin resistance is a key factor in the progression of NAFLD to NASH.6 Increased hepatic lipogenesis and impaired inhibition of adipose tissue lipolysis as a result of IR lead to increased fatty acids in the liver.6 In addition, IR-stimulated adipose tissue dysfunction leads to altered production and secretion of adipokines and inflammatory cytokines.6 Further, cluster analysis of diabetes phenotypes related to age, obesity, glycemia, and insulin deficiency or resistance has linked patients with T2DM with severe IR-related diabetes to increase incidence of hepatic fibrosis.79 A bi-directional MR study demonstrated genetic predisposition to higher fasting insulin, but not T2DM, was related to increased circulating ALT, markers of NAFLD.80 Genetically predicted higher circulating ALT and AST were related to increased risk of T2DM.80 These outcomes provide support for the potential of IR resulting in NAFLD which, in turn, increases T2DM risk.80
Anti-Diabetic Therapies Impact on Other Related Conditions – What Can We Learn?
In earlier work, we have espoused the logic of combination therapy with the least number of safe and efficacious agents to treat the highest number of overlapping pathophysiologies contributing to DM.1,2,81 This same concept can be further applied to the Diabetes Syndrome to allow us to expand our treatment armamentarium. Indeed, there is promise for many therapies traditionally associated with DM management to provide benefit in the Diabetes Syndrome (Table 1). The potential impact on patient management of using existing and new medicines and classes with this in mind is considerable. Continued investigations and research are warranted.
Thiazolidinediones
Thiazolidinediones (TZDs) are PPARλ agonists (eg, pioglitazone) extensively used for DM treatment as insulin sensitizers. Potential benefits of TZD use in cancer, dementia, psoriasis, ASCVD, and NASH have also been reported. In general, TZD use is associated with reduced cancer risk including liver, stomach, and colorectal (although increased bladder cancer risk has been reported).82 Furthermore, risk of dementia in pioglitazone users is lower than in non-users.83–86 Moreover, recent studies have suggested that TZDs may be antipsoriatic due to PPARs’ ability to promote keratinocyte differentiation, inhibit epidermal growth, and reduce inflammatory responses.87 A meta-analysis of randomized controlled trials concluded pioglitazone was efficacious for psoriasis treatment.87 In addition, pioglitazone reduced CV events and retarded the atherosclerotic process in high-risk DM patients in multiple large outcome trials.71 Finally, pioglitazone was associated with a trend toward improved histology and a significant reduction in liver enzymes (ALT, AST), hepatic steatosis, and lobular inflammation in patients with NASH compared with placebo.88
Metformin
Metformin, often used as first-line treatment in DM and PCOS based on its ability to decrease the rate of hepatic gluconeogenesis and potentially decrease IR, is also being evaluated as an agent to treat other conditions including cancer, AD, ASCVD, metabolic syndrome, etc.74 Reduced risk for pancreatic, breast, lung, prostate, colorectal, and liver cancer has been associated with metformin use.82 Further, meta-analysis results prompted the recommendation that metformin should be used as first-line therapy for DM at risk for developing dementia or AD.76,83 Trials in patients without DM, although smaller, show positive results with regard to effect of metformin on dementia symptoms.82 Further, the potential protective effect of metformin in youth and adults with T1DM and in adults with pre- and T2DM has been suggested based on surrogate measures of ASCVD, though based on mixed study results whether metformin improves CV outcomes remains uncertain.71,89 Finally, although increasing AMPK activity (which is reduced by inflammation, obesity, and DM) through metformin use has considered as a potential treatment for NAFLD and NASH, to date results have been mixed.90,91
Glucagon-Like Peptide-1 Receptor Agonists
GLP-1 RAs are approved for the treatment of DM and to reduce risk of MACE events in DM patients and promote weight loss.92 GLP-1 has multiple metabolic functions including delayed gastric emptying, appetite suppression, enhanced liver glucose update, peripheral insulin sensitivity as well as glucose-dependent insulin secretion and inhibition of glucagon release from α cells.58 Although there have been reports of potential for cancer with GLP-1 RA use, a recent meta-analysis of GLP-1 RA did not reveal increased cancer risk in T2DM patients and showed a decreased risk with albiglutide.93 Studies using neurodegenerative disease animal models support potential for GLP-1 receptor stimulation to improve cognitive impairment (regardless of DM status).94 Further, reduction of MACE events has been recently added to GLP-1 RA labels on the basis of large CV outcomes trials.71 Multiple RCTs have been conducted in NAFLD/NASH that demonstrated significant reduction in liver steatosis, fibrosis, and fat content.88,95,96
Dipeptidyl Peptidase-4 Inhibitors
Dipeptidyl peptidase-4 inhibitors inhibit the degradation of GLP-1 and are approved for DM. A DPP-4 inhibitor (sitagliptin) has been associated with improvement in cognitive function in elderly patients with and without AD.97 To date, results of DDP-4 inhibitors have been mixed with regard to liver enzymes, fat content, and fibrosis in patients with NAFLD/NASH.88
Sodium Glucose Transport Protein 2 Inhibitors
SGLT-2 inhibitors block the reabsorption of glucose in the kidney and are approved for DM. Recent studies have shown CV protective effect of SGLT-2 inhibitors, including reduction in MACE, hospitalization for heart failure, and CV mortality in high-risk patients with DM.71 In addition to reduction of hyperglycemia, it is hypothesized that increased levels of ketone bodies, potent anti-inflammatory molecules, may contribute to this effect.98 Moreover, based on a recent systematic review, SGLT-2 inhibitors improved liver enzymes, decreased liver fat and fibrosis, and improved other metabolic parameters (obesity, IR, glycemia, lipid parameters) in T2DM patients with NAFLD.99
Dopamine D2 Agonists
Bromocriptine QR, approved for DM, acts centrally and while it is only associated with modest improvement of glycemic control, it confers a clinically meaningful CV benefit.100,101 Indeed, the reduction in CV disease in patients treated with bromocriptine compared with placebo over 12 months in patients with good glycemic control at baseline (HbA1c ≤7%), in addition to higher odds of remaining in good glycemic control suggests that in patients with T2DM there are important mechanisms beyond glycemic control (eg, reducing vascular sympathetic tone/reducing endothelial dysfunction) that may be contributing to the CV risk reduction observed with this dopamine D2 agonist.100
Immune Modulators
Abatacept, an immunomodulator that inhibits T cell activation is approved for rheumatoid arthritis, juvenile idiopathic arthritis, and adult psoriatic arthritis.102 It has been used for DM management due to reports of improved insulin sensitivity in patients with rheumatoid arthritis.103 It is currently under evaluation in the T1DM setting for deceleration of β-cell damage in newly diagnosed patients, prevention of DM in at risk patients, and prevention of destruction/rejection of transplanted pancreatic islets.103–105
Conclusion
The Diabetes Syndrome can be thought of as a supra-structure that can facilitate understanding the inter-relationships of superficially disparate conditions. It allows physicians and researchers to immediately place new ideas and articles into a coherent whole. The structure will allow new science to be added easily and facilitates the evaluation of therapeutics for the management of disease. Further, many of the connections we propose within the Diabetes Syndrome are testable by leveraging current genetic knowledge from efforts that include powerful MR approaches which, in turn, can inform implementation of precision medicine.30,31 The structure should be considered organic and will be refined and modified as we grow in our basic understanding of the interplay between genetics and epigenetics, inflammation, environment, and IR. Furthermore, recognizing the association of the conditions with the “Diabetes Syndrome” due to interconnection of common driving elements and the differential contribution of the various pathophysiologies for individual patients has the potential to provide both benefit to the patient (eg, prevention, early detection, precision medicine) and to the advancement of medicine (eg, driving education, research, and dynamic decision-based medical practice).
Disclosure
Dr. Rachfal is a clinical drug development consultant. None of her past or current clients has provided compensation related to this manuscript. Dr. Rachfal has been compensated by Dr. Schwartz for assistance in medical editing and preparation of the manuscript. Dr. Rachfal is self-employed at Stage Gate Partners, LLC with no compensation from Stage Gate Partners related to the manuscript. Dr. Grant has active NIH grants R01 DK122586 and UM1 DK126194, which cover the topic of functional mechanisms of Type 1 diabetes and Type 2 diabetes risk variants, respectively, and their target genes using 3D epigenomics and single cell approaches. Dr. Schwartz is on advisory boards for Salix Pharmaceuticals and Arkay Therapeutics and on the Speaker’s Bureau for Salix Pharmaceuticals, Johnson and Johnson, Arkay, Novo, Janssen Pharmaceuticals, Boehringer Ingelheim, Eli Lilly, and Merck. Dr. Schwartz is employed and receive financial compensation from his clinical practice, Stanley Schwartz, MD, LLC (affiliated with Main Line Health [with no monetary connection]) with no compensation related to the manuscript. He is also an emeritus professor for University of Pennsylvania, Perlman School of Medicine, Philadelphia, PA, USA. With the exception of the listed potential conflict of interests herein, the authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Schwartz SS, Epstein S, Corkey BE, et al. The time is right for a new classification system for diabetes: rationale and implications of the β-cell-centric classification schema. Diabetes Care. 2016;39(2):179–186. doi:10.2337/dc15-1585
2. Schwartz SS, Epstein S, Corkey BE, et al. A unified pathophysiological construct of diabetes and its complications. Trends Endocrinol Metab. 2017;28(9):645–655. doi:10.1016/j.tem.2017.05.005
3. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. doi:10.2337/diabetes.54.6.1615
4. American Diabetes Association. Cardiometabolic risk, type 2 diabetes and cardiovascular disease; 2015. Available from: https://professional.diabetes.org/sites/professional.diabetes.org/files/media/cardiometabolicrisk2015.pdf.
5. Aksentijevich M, Lateef SS, Anzenberg P, et al. Chronic inflammation, cardiometabolic diseases and effects of treatment: psoriasis as a human model. Trends Cardiovasc Med. 2019;30(8):
6. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65(8):1038–1048. doi:10.1016/j.metabol.2015.12.012
7. Holm JG, Thomsen SF. Type 2 diabetes and psoriasis: links and risks. Psoriasis (Auckl). 2019;9:1–6. doi:10.2147/PTT.S159163
8. Mamizadeh M, Tardeh Z, Azami M. The association between psoriasis and diabetes mellitus: a systematic review and meta-analysis. Diabetes Metab Syndr. 2019;13(2):1405–1412. doi:10.1016/j.dsx.2019.01.009
9. Ninomiya T. Epidemiological evidence of the relationship between diabetes and dementia. Adv Exp Med Biol. 2019;1128:13–25.
10. Paul KC, Jerrett M, Ritz B. Type 2 diabetes mellitus and Alzheimer’s disease: overlapping biologic mechanisms and environmental risk factors. Curr Environ Health Rep. 2018;5(1):44–58. doi:10.1007/s40572-018-0176-1
11. Perumpail BJ, Khan MA, Yoo ER, et al. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J Gastroenterol. 2017;23(47):8263–8276. doi:10.3748/wjg.v23.i47.8263
12. Schwartz SS, Grant SFA, Herman ME. Intersections and clinical translations of diabetes mellitus with cancer promotion, progression and prognosis. Postgrad Med. 2019;131(8):597–606. doi:10.1080/00325481.2019.1657358
13. Strain WD, Paldánius PM. Diabetes, cardiovascular disease and the microcirculation. Cardiovasc Diabetol. 2018;17(1):57. doi:10.1186/s12933-018-0703-2
14. Wojciechowska J, Krajewski W, Bolanowski M, et al. Diabetes and cancer: a review of current knowledge. Exp Clin Endocrinol Diabetes. 2016;124(5):263–275. doi:10.1055/s-0042-100910
15. Younossi ZM, Golabi P, de Avila L, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta-analysis. J Hepatol. 2019;71(4):793–801. doi:10.1016/j.jhep.2019.06.021
16. International Diabetes Federation. IDF Diabetes Atlas.
17. Campbell JM, Stephenson MD, de Courten B, et al. Metformin use associated with reduced risk of dementia in patients with diabetes: systematic review and meta-analysis. J Alzheimers Dis. 2018;65(4):1225–1236. doi:10.3233/JAD-180263
18. Biessels GJ, Despa F. Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat Rev Endocrinol. 2018;14(10):591–604. doi:10.1038/s41574-018-0048-7
19. Nguyen TT, Ta QTH, Nguyen TKO, et al. Type 3 diabetes and its role implications in Alzheimer’s disease. Int J Mol Sci. 2020;21(9):E3165. doi:10.3390/ijms21093165
20. Blázquez E, Velázquez E, Hurtado-Carneiro V, et al. Insulin in the brain: its pathophysiological implications for States related with central IR, type 2 diabetes and Alzheimer’s disease. Front Endocrinol (Lausanne). 2014;9(5):161.
21. National Psoriasis Foundation. Statistics; 2019. Available from: https://www.psoriasis.org/content/statistics.
22. Takeshita J, Grewal S, Langan SM, et al. Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol. 2017;76(3):377–390. doi:10.1016/j.jaad.2016.07.064
23. Armstrong AW, Harskamp CT, Armstrong EJ. Psoriasis and the risk of diabetes mellitus: a systematic review and meta-analysis. JAMA Dermatol. 2013;149(1):84–91. doi:10.1001/2013.jamadermatol.406
24. Wan MT, Shin DB, Hubbard RA, et al. Psoriasis and the risk of diabetes: a prospective population-based cohort study. J Am Acad Dermatol. 2018;78(2):315–322.e1. doi:10.1016/j.jaad.2017.10.050
25. Flora GD, Nayak MK, Brief A. Review of cardiovascular diseases, associated risk factors and current treatment regimes. Curr Pharm Des. 2019;25(38):4063–4084.
26. Bertot LC, Adams LA. The natural course of non-alcoholic fatty liver disease. Int J Mol Sci. 2016;17(5):E774. doi:10.3390/ijms17050774
27. Manne V, Handa P, Kowdley KV. Pathophysiology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Clin Liver Dis. 2018;22(1):23–37. doi:10.1016/j.cld.2017.08.007
28. Häring HU. Novel phenotypes of prediabetes? Diabetologia. 2016;59(9):1806–1818. doi:10.1007/s00125-016-4015-3
29. Goyal D, Limesand SW, Goyal R. Epigenetic responses and the developmental origins of health and disease. J Endocrinol. 2019;242(1):T105–T119. doi:10.1530/JOE-19-0009
30. Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. JAMA. 2017;318(19):1925–1926. doi:10.1001/jama.2017.17219
31. Hemani G, Zheng J, Elsworth B, et al. The MR-base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408. doi:10.7554/eLife.34408
32. Juvinao-Quintero DL, Hivert MF, Sharp GC, et al. DNA methylation and type 2 diabetes: the use of mendelian randomization to assess causality. Curr Genet Med Rep. 2019;7(4):191–207. doi:10.1007/s40142-019-00176-5
33. Swerdlow DI. Mendelian randomization and type 2 diabetes. Cardiovasc Drugs Ther. 2016;30(1):51–57. doi:10.1007/s10557-016-6638-5
34. Yuan S, Kar S, Carter P, et al. Is type 2 diabetes causally associated with cancer risk? evidence from a two-sample mendelian randomization study. Diabetes. 2020;69(7):1588–1596. doi:10.2337/db20-0084
35. Carreras-Torres R, Johansson M, Gaborieau V, et al. The role of obesity, type 2 diabetes, and metabolic factors in pancreatic cancer: a mendelian randomization study. J Natl Cancer Inst. 2017;109(9):djx012. doi:10.1093/jnci/djx012
36. Piñero J, Ramírez-Anguita JM, Saüch-Pitarch J, et al. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020;48(D1):D845–D855. doi:10.1093/nar/gkz1021
37. Chen B, Li J, Chi D, et al. Non-coding RNAs in IGF-1R signaling regulation: the underlying pathophysiological link between diabetes and cancer. Cells. 2019;8(12):E1638. doi:10.3390/cells8121638
38. Thomassen JQ, Tolstrup JS, Benn M, et al. Type-2 diabetes and risk of dementia: observational and Mendelian randomisation studies in 1 million individuals. Epidemiol Psychiatr Sci. 2020;29:e118. doi:10.1017/S2045796020000347
39. Zhang W, Xin L, Lu Y. Integrative analysis to identify common genetic markers of metabolic syndrome, dementia, and diabetes. Med Sci Monit. 2017;23:5885–5891. doi:10.12659/MSM.905521
40. Wang J, Gong B, Zhao W, et al. Epigenetic mechanisms linking diabetes and synaptic impairments. Diabetes. 2014;63(2):645–654. doi:10.2337/db13-1063
41. David T, Ling SF, Barton A. Genetics of immune-mediated inflammatory diseases. Clin Exp Immunol. 2018;193(1):3–12. doi:10.1111/cei.13101
42. Wang H, Wang Z, Rani PL, et al. Identification of PTPN22, ST6GAL1 and JAZF1 as psoriasis risk genes demonstrates shared pathogenesis between psoriasis and diabetes. Exp Dermatol. 2017;26(11):1112–1117. doi:10.1111/exd.13393
43. Granata M, Skarmoutsou E, Trovato C, et al. Obesity, type 1 diabetes, and psoriasis: an autoimmune triple flip. Pathobiology. 2017;84(2):71–79. doi:10.1159/000447777
44. Ahmad OS, Morris JA, Mujammami M, et al. A Mendelian randomization study of the effect of type-2 diabetes on coronary heart disease. Nat Commun. 2015;6(1):7060. doi:10.1038/ncomms8060
45. Murea M, Ma L, Freedman BI. Genetic and environmental factors associated with type 2 diabetes and diabetic vascular complications. Rev Diabet Stud. 2012;9(1):6–22. doi:10.1900/RDS.2012.9.6
46. Costantino S, Mohammed SA, Ambrosini S, et al. Epigenetic processing in cardiometabolic disease. Atherosclerosis. 2019;281:150–158. doi:10.1016/j.atherosclerosis.2018.09.029
47. Paneni F, Costantino S, Battista R, et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ Cardiovasc Genet. 2015;8(1):150–158. doi:10.1161/CIRCGENETICS.114.000671
48. Ferrannini G, Manca ML, Magnoni M, et al. Coronary artery disease and type 2 diabetes: a Proteomic Study. Diabetes Care. 2020;43(4):843–851. doi:10.2337/dc19-1902
49. Liu Z, Zhang Y, Graham S, et al. Causal relationships between NAFLD, T2D and obesity have implications for disease subphenotyping. J Hepatol. 2020;73(2):263–276. doi:10.1016/j.jhep.2020.03.006
50. Sookoian S, Pirola CJ. Genetics of nonalcoholic fatty liver disease: from pathogenesis to therapeutics. Semin Liver Dis. 2019;39(2):124–140. doi:10.1055/s-0039-1679920
51. de Mello VD, Matte A, Perfilyev A, et al. Human liver epigenetic alterations in non-alcoholic steatohepatitis are related to insulin action. Epigenetics. 2017;12(4):287–295. doi:10.1080/15592294.2017.1294305
52. Yuan S, Larsson SC. An atlas on risk factors for type 2 diabetes: a wide-angled Mendelian randomisation study [published online ahead of print, 2020 Sep 8]. Diabetologia. 2020;
53. Censin JC, Nowak C, Cooper N, et al. Childhood adiposity and risk of type 1 diabetes: a Mendelian randomization study. PLoS Med. 2017;14(8):e1002362. doi:10.1371/journal.pmed.1002362
54. Shu X, Wu L, Khankari NK, et al. Associations of obesity and circulating insulin and glucose with breast cancer risk: a Mendelian randomization analysis. Int J Epidemiol. 2019;48(3):795–806. doi:10.1093/ije/dyy201
55. Ravichandran G, Lakshmanan DK, Raju K, et al. Food advanced glycation end products as potential endocrine disruptors: an emerging threat to contemporary and future generation. Environ Int. 2019;123:486–500. doi:10.1016/j.envint.2018.12.032
56. Benn M, Nordestgaard BG, Tybjærg-Hansen A, et al. Impact of glucose on risk of dementia: Mendelian randomisation studies in 115,875 individuals. Diabetologia. 2020;63(6):1151–1161. doi:10.1007/s00125-020-05124-5
57. Zeng J, Luo S, Huang Y, et al. Critical role of environmental factors in the pathogenesis of psoriasis. J Dermatol. 2017;44(8):863–872. doi:10.1111/1346-8138.13806
58. Nauck MA, Meier JJ. Incretin hormones: their role in health and disease. Diabetes Obes Metab. 2018;20(Suppl 1):5–21. doi:10.1111/dom.13129
59. Gyldenløve M, Vilsbøll T, Zachariae C, et al. Impaired incretin effect is an early sign of glucose dysmetabolism in nondiabetic patients with psoriasis. J Int Med. 2015;278(6):660–670. doi:10.1111/joim.12388
60. Budu-Aggrey A, Brumpton B, Tyrrell J, et al. Evidence of a causal relationship between body mass index and psoriasis: a mendelian randomization study. PLoS Med. 2019;16(1):e1002739. doi:10.1371/journal.pmed.1002739
61. Ogawa K, Stuart PE, Tsoi LC, et al. A transethnic mendelian randomization study identifies causality of obesity on risk of psoriasis. J Invest Dermatol. 2019;139(6):1397–1400. doi:10.1016/j.jid.2018.11.023
62. Fang X, Zuo J, Zhou J, et al. Childhood obesity leads to adult type 2 diabetes and coronary artery diseases: a 2-sample mendelian randomization study. Medicine (Baltimore). 2019;98(32):e16825. doi:10.1097/MD.0000000000016825
63. Yang Q, Lin SL, Kwok MK, et al. The roles of 27 genera of human gut microbiota in ischemic heart disease, type 2 diabetes mellitus, and their risk factors: a mendelian randomization study. Am J Epidemiol. 2018;187(9):1916–1922. doi:10.1093/aje/kwy096
64. Sircana A, Framarin L, Leone N, et al. Altered gut microbiota in type 2 diabetes: just a coincidence? Curr Diab Rep. 2018;18(10):98. doi:10.1007/s11892-018-1057-6
65. Polyzos SA, Kountouras J, Deretzi G, et al. The emerging role of endocrine disruptors in pathogenesis of insulin resistance: a concept implicating nonalcoholic fatty liver disease. Curr Mol Med. 2012;12(1):68–82. doi:10.2174/156652412798376161
66. de la Monte SM. Insulin resistance and neurodegeneration: progress towards the development of new therapeutics for Alzheimer’s disease. Drugs. 2017;77(1):47–65.
67. Newcombe EA, Camats-Perna J, Silva ML, Valmas N, Huat TJ, Medeiros R. Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J Neuroinflammation. 2018;15(1):276.
68. Chiu HY, Hung CJ, Muo CH, et al. The bidirectional association between type 2 diabetes and psoriasis: two retrospective cohort studies. Indian J Dermatol Venereol Leprol. 2020;86(4):366–374. doi:10.4103/ijdvl.IJDVL_428_18
69. Polic MV, Miskulin M, Smolic M, et al. Psoriasis severity-a risk factor of insulin resistance independent of metabolic syndrome. Int J Environ Res Public Health. 2018;15(7):1486. doi:10.3390/ijerph15071486
70. Hu Y, Zhu Y, Lian N, et al. Metabolic syndrome and skin diseases. Front Endocrinol. 2019;10.
71. Di Pino A, DeFronzo RA. Insulin resistance and atherosclerosis: implications for insulin-sensitizing agents. Endocr Rev. 2019;40(6):1447–1467.
72. Katakami N. Mechanism of development of atherosclerosis and cardiovascular disease in diabetes mellitus. J Atheroscler Thromb. 2018;25(1):27–39. doi:10.5551/jat.RV17014
73. Roizen JD, Bradfield JP, Hakonarson H. Progress in understanding type 1 diabetes through its genetic overlap with other autoimmune diseases. Curr Diab Rep. 2015;15(11):102. doi:10.1007/s11892-015-0668-4
74. Bost F, Rena G, Viollet B. Editorial: metformin: beyond diabetes. Front Endocrinol (Lausanne). 2019;10(10):851. doi:10.3389/fendo.2019.00851
75. Wilkin TJ. The accelerator hypothesis: a review of the evidence for insulin resistance as the basis for type I as well as type II diabetes. Int J Obes (Lond). 2009;33(7):716–726. doi:10.1038/ijo.2009.97
76. Martin SD, McGee SL. Metabolic reprogramming in type 2 diabetes and the development of breast cancer. J Endocrinol. 2018;237(2):R35–R46. doi:10.1530/JOE-18-0037
77. Walter S, Marden JR, Kubzansky LD, et al. Diabetic phenotypes and late-life dementia risk: a Mechanism-Specific Mendelian Randomization Study. Alzheimer Dis Assoc Disord. 2016;30(1):15–20. doi:10.1097/WAD.0000000000000128
78. Gyldenløve M, Storgaard H, Holst JJ, et al. Patients with psoriasis are insulin resistant. J Am Acad Dermatol. 2015;72(4):599–605. doi:10.1016/j.jaad.2015.01.004
79. Zaharia OP, Strassburger K, Strom A, et al.; German Diabetes Study Group. Risk of diabetes-associated diseases in subgroups of patients with recent-onset diabetes: a 5-year follow-up study. Lancet Diabetes Endocrinol. 2019;7(9):684–694. doi:10.1016/S2213-8587(19)30187-1.
80. De silva NMG, Borges MC, Hingorani AD, et al. Liver function and risk of type 2 diabetes: bidirectional Mendelian randomization study. Diabetes. 2019;68(8):1681–1691. doi:10.2337/db18-1048
81. Scheiner G, Schwarz SS, Herman ME. Managing diabetes by maintaining healthier beta cells: a fresh perspective for diabetes educators. AADE Pract. 2018;6(4):12–18. doi:10.1177/2325160318781529
82. Pareek KK, Mathur G, Ramchandani GD. Anti-diabetic agent and cancer. J Assoc Physicians India. 2019;67(10):66–69.
83. Bendlin BB. Antidiabetic therapies and Alzheimer disease. Dialogues Clin Neurosci. 2019;21(1):83–91.
84. Chou PS, Ho BL, Yang YH. Effects of pioglitazone on the incidence of dementia in patients with diabetes. J Diabetes Complications. 2017;31(6):1053–1057. doi:10.1016/j.jdiacomp.2017.01.006
85. Lu CH, Yang CY, Li CY, et al. Lower risk of dementia with pioglitazone, compared with other second-line treatments, in metformin-based dual therapy: a population-based longitudinal study. Diabetologia. 2018;61(3):562–573. doi:10.1007/s00125-017-4499-5
86. Tseng CH. Pioglitazone reduces dementia risk in patients with type 2 diabetes mellitus: a retrospective cohort analysis. J Clin Med. 2018;7(10):E306. doi:10.3390/jcm7100306
87. Chang G, Wang J, Song J, et al. Efficacy and safety of pioglitazone for treatment of plaque psoriasis: a systematic review and meta-analysis of randomized controlled trials. J Dermatolog Treat. 2019;1–7.
88. Oseini AM, Sanyal AJ. Therapies in non-alcoholic steatohepatitis (NASH). Liver Int. 2017;37(Suppl 1):97–103. doi:10.1111/liv.13302
89. Jenkins AJ, Welsh P, Petrie JR. Metformin, lipids and atherosclerosis prevention. Curr Opin Lipidol. 2018;29(4):346–353. doi:10.1097/MOL.0000000000000532
90. Iogna Prat L, Tsochatzis EA. The effect of antidiabetic medications on non-alcoholic fatty liver disease (NAFLD). Hormones (Athens). 2018;17(2):219–229. doi:10.1007/s42000-018-0021-9
91. Smith BK, Marcinko K, Desjardins EM, et al. Treatment of nonalcoholic fatty liver disease: role of AMPK. Am J Physiol Endocrinol Metab. 2016;311(4):E730–E740. doi:10.1152/ajpendo.00225.2016
92. Aroda VR. A review of GLP-1 receptor agonists: evolution and advancement, through the lens of randomised controlled trials. Diabetes Obes Metab. 2018;20(Suppl 1):22–33. doi:10.1111/dom.13162
93. Cao C, Yang S, Zhou Z. GLP-1 receptor agonists and risk of cancer in type 2 diabetes: an updated meta-analysis of randomized controlled trials. Endocrine. 2019;66(2):157–165. doi:10.1007/s12020-019-02055-z
94. Grieco M, Giorgi A, Gentile MC, et al. Glucagon-like peptide-1: a focus on neurodegenerative diseases. Front Neurosci. 2019;18(13):1112. doi:10.3389/fnins.2019.01112
95. Petit JM, Vergès B. GLP-1 receptor agonists in NAFLD. Diabetes Metab. 2017;43(Suppl 1):2S28–2S33. doi:10.1016/S1262-3636(17)30070-8
96. Seghieri M, Christensen AS, Andersen A, et al. Future perspectives on GLP-1 receptor agonists and GLP-1/glucagon receptor co-agonists in the treatment of NAFLD. Front Endocrinol (Lausanne). 2018;9:649. doi:10.3389/fendo.2018.00649
97. Isik AT, Soysal P, Yay A, et al. The effects of sitagliptin, a DPP-4 inhibitor, on cognitive functions in elderly diabetic patients with or without Alzheimer’s disease. Diabetes Res Clin Pract. 2017;123:192–198. doi:10.1016/j.diabres.2016.12.010
98. Prattichizzo F, De Nigris V, Micheloni S, et al. Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: is low-grade inflammation the neglected component? Diabetes Obes Metab. 2018;20(11):2515–2522. doi:10.1111/dom.13488
99. Raj H, Durgia H, Palui R, et al. SGLT-2 inhibitors in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: a systematic review. World J Diabetes. 2019;10(2):114–132. doi:10.4239/wjd.v10.i2.114
100. Chamarthi B, Gaziano JM, Blonde L, et al. Timed bromocriptine-QR therapy reduces progression of cardiovascular disease and dysglycemia in subjects with well-controlled type 2 diabetes mellitus. J Diabetes Res. 2015;2015:157698. doi:10.1155/2015/157698
101. Gaziano JM, Cincotta AH, Vinik A, et al. Effect of bromocriptine-QR (a quick-release formulation of bromocriptine mesylate) on major adverse cardiovascular events in type 2 diabetes subjects. J Am Heart Assoc. 2012;1(5):e002279. Erratum in: J Am Heart Assoc. 2015; 4(10). doi:10.1161/JAHA.112.002279
102. Ursini F, Russo E, De Giorgio R, et al. Current treatment options for psoriatic arthritis: spotlight on abatacept. Ther Clin Risk Manag. 2018;14:1053–1059. doi:10.2147/TCRM.S148586
103. Ursini F, Russo E, Letizia Hribal M, et al. Abatacept improves whole-body insulin sensitivity in rheumatoid arthritis: an observational study. Medicine (Baltimore). 2015;94(21):e888. doi:10.1097/MD.0000000000000888
104. Rachid O, Osman A, Abdi R, et al. CTLA4-Ig (abatacept): a promising investigational drug for use in type 1 diabetes. Expert Opin Investig Drugs. 2020;29(3):221–236. doi:10.1080/13543784.2020.1727885
105. Simmons K, Michels AW. Lessons from type 1 diabetes for understanding natural history and prevention of autoimmune disease. Rheum Dis Clin North Am. 2014;40(4):797–811. doi:10.1016/j.rdc.2014.07.008
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.