")
Back to Journals » ImmunoTargets and Therapy » Volume 6
The current status of immunobased therapies for metastatic renal-cell carcinoma
Authors Sathianathen NJ , Krishna S, Anderson JK, Weight CJ, Gupta S, Konety BR, Griffith TS
Received 9 September 2017
Accepted for publication 27 October 2017
Published 5 December 2017 Volume 2017:6 Pages 83—93
DOI https://doi.org/10.2147/ITT.S134850
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Niranjan J Sathianathen,1 Suprita Krishna,1 J Kyle Anderson,1,2 Christopher J Weight,1,2 Shilpa Gupta,2,3 Badrinath R Konety,1,2 Thomas S Griffith1,2,4,5
1Department of Urology, 2Masonic Cancer Center, 3Division of Hematology, Oncology, and Transplantation, 4Center for Immunology, 5Microbiology, Immunology, and Cancer Biology Graduate Program, University of Minnesota, Minneapolis, MN, USA
Abstract: The management of metastatic renal-cell carcinoma (mRCC) represents an important clinical challenge. Since being approved in the early 1990s, aspecific immunotherapy has been a mainstay of treatment for mRCC and the only therapy that has demonstrated long-term cures for mRCC. However, in recent times there have been landmark advances made in the field of specific immunotherapy for a number of malignancies, including kidney cancer. This review outlines the range of immunobased agents currently available for the treatment of mRCC.
Keywords: immunotherapy, cryosurgery, kidney neoplasm, vaccines, targeted therapy, immunocheckpoints
Introduction
Renal-cell carcinoma (RCC) accounts for 2%–3% of all adult malignancies, with rising worldwide incidence. While the majority of patients present with localized cancer that is effectively managed with radical nephrectomy, 25%–30% of first presentations are patients with disseminated disease. In addition, nearly a third of patients will experience recurrence or progression following primary radical treatment. Metastatic RCC (mRCC) is generally incurable, with median survival of only 18 months.1 Several risk factors have been identified for RCC, including smoking, hypertension, and obesity.2 One of the classic hallmarks of cancer is immunoevasion,3,4 and RCC has emerged as a candidate for immunotherapy, because RCC tumors have a high somatic mutation frequency,5 display a high number of tumor-infiltrating lymphocytes, and an abysmal 5-year survival rate, despite the development of seven novel targeted therapies currently used in the clinic.6
While the use of classical cytokine-based interventions (eg, IFN and IL2) has been approved since the 1990s, they only induce durable responses in a small fraction (7%–8%) of mRCC patients.7 The treatment options for RCC have undergone a substantial transformation in recent years, from using cytokines to stimulate the immune system to developing agents targeting small molecules and monoclonal antibodies (mAbs) specific for checkpoint inhibitors. The emphasis on “reprogramming” the immune system for cancer has centered on the development and use of treatments designed to engage the adaptive immune system and primarily induce T-cell responses.8 Indeed, Ab-based therapeutics targeting immunocheckpoint regulators, such as CTLA4 and PD1, have now generated durable complete responses in selected patients who were otherwise resistant to all forms of conventional cytotoxic and targeted therapy.9,10 Recent clinical data have shown that RCC responds to checkpoint immunotherapy, with long-term survival data being compiled, but showing the potential to induce durable remissions.11,12 This review provides an overview of the role of immunotherapy in the treatment of RCC, and details the various therapies used over the years that engage the immune system, discusses the basic biology underpinning these modalities, and provides an insight into the future direction of treatments for this cancer.
Antitumor immunoresponse
The potential to utilize the immune system against cancer was first demonstrated by William Coley, when he observed the regression of sarcomas following intratumoral injection of erysipelas.13 Our understanding of the immune system and its role in tumor biology has advanced substantially since the days of Coley, and the neoplastic process is now recognized as being based on a combination of mutations that lead to a loss of cellular regulatory processes.14 Moreover, a series of events must occur to mount an antitumor immunoresponse (Figure 1). It is within these processes that immunotherapeutic agents can be employed to facilitate and/or potentiate the generation of antitumor immunity.
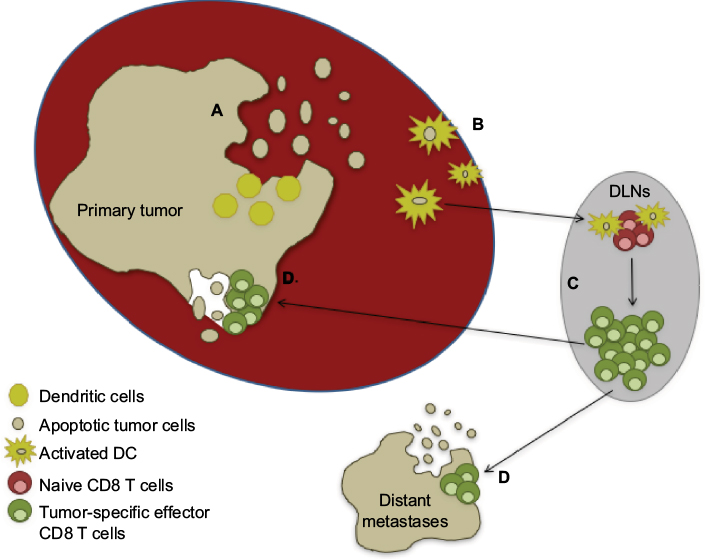 | Figure 1 Basic steps needed for the generation of an antitumor immunoresponse and clearance of tumors. Notes: (A) Normal cellular turnover or application of a death-inducing treatment leads to the release of tumor-cell fragments, which are phagocytosed by local dendritic cells. (B) The dendritic cells become activated by local cytokines and/or other inflammatory stimuli and migrate to the draining lymph node, where they (C) cross-present tumor antigens to naïve CD8 T cells. This leads to the activation and expansion of tumor-specific CD8 T-cell effectors (D), which then migrate to the primary tumor and distant metastases and mediate further tumor cell killing and systemic antitumor immunity. Abbreviations: DC, dendritic cells; DLNs, draining lymph nodes. |
The initiating step in an adaptive cellular immunoresponse against a tumor is the acquisition of tumor-derived antigens by immature dendritic cells (DCs) as a result of the phagocytosis of dying/dead tumor cells generated during normal cellular turnover or after exposure to some death-inducing agent (eg, radiation, chemotherapy, thermal ablation, or cytotoxic cytokines). These DCs then undergo a maturation process via cytokines and/or Toll-like receptor agonists that is critical to avoid tolerance and produce an effector T-cell response.15 The DCs upregulate major histocompatibility complex (MHC) class I and II surface molecules, costimulatory molecules (eg, CD80 and CD86), and the ability to secrete proinflammatory cytokines (eg, IL12 and IFNγ) during this maturation process.16 Migration of the DCs to lymph nodes is aided by the acquisition of CCR7.17 Fujii et al suggested CD40 ligation acts a critical signal required for DC maturation,18 which gives them the capability to launch an antitumor T-cell response. Dysfunctional DCs have been characterized in patients with RCC (as well as other tumor types), and have been implicated as a mechanism contributing to immunoevasion by tumors.19
Cytokines have also been demonstrated to have an important function at this stage of the cancer-immunity cycle. It is hypothesized the cumulative effects of a range of cytokines, including TNFα, IL6, IL8, IL10, IL18, macrophage migration-inhibitory factor, and TGFβ, result in an environment of simultaneous immunostimulation and immunosuppression.20 The immunosuppressive tumor microenvironment promotes the dysfunction of immunoeffector cells, and thus compromises both the innate and adaptive immune systems. Consequently, there is reduced detection and subsequent eradication of the tumor by the defense system, a process that facilitates tumor survival.
The type of T-cell response induced by the tumor antigen-laden DCs is dictated by several factors, including the type of immunogenic maturation signal initially received, the availability of prosurvival cytokines, and the relationship between costimulatory molecules on DCs and corresponding surface receptors on T cells.21 Higher frequencies of TH1 CD4 T cells in RCC patients protect against recurrence following surgery, and are important in the process of tumor regression.22 TH2 CD4 T-cell responses have the direct opposite effect by blunting the TH1 response and facilitating a tumor microenvironment conducive to disease progression.23 The importance of costimulatory molecules in immunoresponse was first suggested in the early 1990s and especially when the role of CD28 was highlighted by its antagonism, which prevented pancreatic islet-xenograft rejection and induced tolerance.24,25 Salomon and Bluestone found that CD28 signaling via CD80 and CD86 had an important role in T-cell expansion, IL2 production, and suppressing apoptotic mechanisms.26 In recent years, additional costimulatory molecules, such as OX40, 41BB, and CD27, have been shown to play important roles in CD4 and CD8 T-cell clonal expansion, differentiation, and the production of memory cells.27 Conversely, CTLA4 and PD1 transduce signals that negatively regulate lymphocyte activation.28 CTLA4 functions by inhibiting CD28 binding to both CD80 and CD86, and thus impairs T-cell proliferation and IL2 secretion.29 PD1 expression is increased on activated T cells, but ligation with PDL1 or PDL2 generates an inhibitory signaling pathway within the T cell that decreases effector function and survival.30 PD1 expression has been correlated with settings where T cells become repeatedly stimulated with antigens, as in states of chronic infection and cancer.31 These molecules and pathways have been the focus of a number of new therapeutic options in the treatment of cancer, and will be discussed in later sections of this review.
Following successful priming and activation, T cells need to migrate to the tumor site (which can be either the primary tumor or a metastatic lesion) to exert their function. Inflammatory chemokines, such as CCL3, CCL5, CCL20, and CXCL10, are involved in the recruitment of T cells, which occurs in a selective manner to employ the optimal combination of “cell fighters” against the tumor.32–35 On arrival, the immune cells infiltrate the tumor using molecular pairings (such as CD103 with E-cadherin or LFA1 with ICAM1) to mediate the important interaction between T cells and target cells.36 Once the T cells have recognized the tumor cells, a number of effector mechanisms can be used to kill the tumor cells to perpetuate and amplify this cycle. However, the tumor has its own set of defenses to circumvent the action of the immune system. Many tumors express PDL1, which engages with T-cell surface receptors and causes anergy.37 Hypoxia in the tumor microenvironment aids multiple anti-immune mechanisms. For example, the production of CCL28 recruits regulatory T (Treg) cells, which promote tumor tolerance and angiogenesis.38 Furthermore, Munn et al have outlined the potent immunosuppressive qualities of the IDO enzyme within the tumor microenvironment.39
Myeloid-derived suppressor cells (MDSCs), which are now recognized as formidable immunosuppressive cells capable of blunting an antitumor immunoresponse, are also expanded within the tumor-bearing host.40 Six primary populations of MDSCs have been characterized in RCC, of which the granulocytic subsets (GMDSCs) are the most prevalent.41 These GMDSCs suppress T-cell activity by potentiating the synthesis of arginase, leading to the depletion of arginine, which is required to maintain cell proliferation.42 The predominance of GMDSCs is at least partly driven by the increased expression of VEGF in RCC due to von Hippel–Lindau silencing.43 Youn et al have also hypothesized that transcriptional silencing of the retinoblastoma gene facilitates differentiation of MDSCs to GMDSCs.44 The important role MDSCs play in tumor progression is underlined in the demonstrated association between blood MDSC levels in kidney cancer patients and tumor progression.41
Much like T cells, macrophages adopt a specific phenotype dependent on the signals present in the environment within which it resides and which determine its function in the tumor microenvironment.45 These phenotypes are commonly described as being on a spectrum between the “classical” M1 and the “alternative” M2 forms.46 The M1 phenotype in general enhances the antitumor immunoresponse by potentiating the production of proinflammatory cytokines and the ability to phagocytose foreign antigens. On the other hand, M2 macrophages, also referred to as tumor-associated macrophages, have an immunosuppressive effect. One mechanism of immunosuppression is the increased the secretion of IL10 driven by the lipoxygenase pathway.47 The same study demonstrated that tumor-associated macrophages upregulate CTLA4- and FOXP3-expression T cells. The latter molecule has been implicated in increasing the numbers of Treg cells, which is associated with poor overall and disease-free survival outcomes.48–50 Ning et al found that not only are Treg-cell numbers increased in individuals with RCC compared to controls but there was also a correlation between the number of Treg cells and both pathological stage and nuclear grade.51 Lastly, there was also a positive association with VEGF expression and microvascular density score, suggesting a role for VEGF in angiogenesis.
These data demonstrate the complexity of immunoresponse to cancer, where a number of cellular and molecular processes interact with and against one another to either lead to tumor regression or survival, depending on the overall balance. While this cycle and components of each step are simplified to an extent, it is now clear that these (and many other) cells and molecules are viable targets of immunotherapeutic agents designed to tip the balance of processes toward antitumor immunity and tumor regression.
Use of vaccines in the treatment of RCC
The importance of the immune system for overall health is dramatically illustrated by individuals with immune-system defects being highly susceptible to serious and often life-threatening infections. Stimulating immunity against microbes through vaccination is the most effective way to protect individuals against infection, and the development of vaccines to infectious pathogens is one of the great successes of immunology. Vaccination exploits the property of immunological memory to provide long-lasting protection against infection. With data generated over decades investigating the mechanisms behind the power of vaccination for pathogens, it seems logical to extend this approach into the treatment of cancer, including RCC.
Single- and multiple-peptide vaccination schemes
One of the first therapeutic vaccines developed for mRCC was IMA901, consisting of multiple tumor-associated peptides (TumAPs). The Phase I trial recruited 28 (HLAA)*02+ patients to receive GM-CSF, followed by a maximum of eight doses of IMA901. In this cohort, 20 demonstrated a response to at least one TUMAP, and eight patients showed a T-cell response to multiple TUMAPs. It was found that a T-cell response to multiple TUMAPs was significantly associated with better disease control. The results from this study led to a Phase II trial where cyclophosphamide was used as an immunomodulator in one arm. While progression-free survival was comparable in both groups of this study, patients that received cyclophosphamide had an increase in overall survival, although this did not reach statistical significance (23.5 months vs 14.8 months, P=0.09). The study also demonstrated a 20% reduction in Treg cells in the cyclophosphamide group, the absence of which was associated with better overall response in both the Phase I and Phase II trials.52 Following these trials, the Phase III, open-label IMPRINT trial recruited 339 patients from 124 different sites. Patients were randomized 3:2 to receive IMA901 with sunitinib or sunitinib alone. Patients in the combination group received GM-CSF and cyclophosphamide, along with the IMA901 vaccine. The results from the trial showed no significant survival advantage when sunitinib was added to IMA901. Around 57% of the combination group experienced grade ≥3 adverse events, and four patients died. The adverse risk profile deterred further studies on the vaccine.53 An HSP–Gp96 peptide complex-based therapeutic vaccine was evaluated as an adjuvant treatment following nephrectomy, but did not decrease recurrence rates compared to no adjuvant treatment (HR 0.92, 95% CI 0.73–1.17). Subgroup analysis on patients with early-stage disease suggested that adjuvant therapy with the autologous vaccine reduced disease recurrence (HR 0.58, 95% CI 0.32–1.02, P=0.056).
Dendritic cells
The primary function of DCs is to acquire, process, and present peptides in the context of MHCI and -II to stimulate CD8 and CD4 T-cell-mediated immunity. Because of their vital role in this context, DC-based therapies to treat cancer have been pursued for over 20 years. Interest in DC-based cancer therapies peaked in 2010 with the approval of Provenge for prostate cancer, leading to the testing of similar approaches in other cancers, including RCC. AGS003 is one such immunotherapy approach using mature monocyte-derived DCs, patient tumor RNA, and synthetic CD154 RNA to induce a potent patient-specific antitumor T-cell response. A Phase II study was designed to evaluate the efficacy of AGS003 in combination with sunitinib in patients who were postnephrectomy for intermediate- or poor-risk RCC. Sunitinib, considered to be first-line standard of care for mRCC, decreases tumor-cell survival, migration, and proliferation and vascular changes associated with angiogenesis by inhibiting the signaling of PDGF receptors and VEGF receptors, as well as suppressing the function of Treg cells and MDSCs.54,55 Patients received a median of six doses of AGS003 and at least one 6-week cycle of sunitinib. Of the 21 patients enrolled, nine experienced a partial response, with a median overall survival of 61.9 months and 9.1 months for intermediate-risk and poor-risk patients, respectively.56 It should be noted that more than half the cohort experienced a survival benefit >30 months. The multi-institutional Phase III ADAPT trial (NCT01582672) was evaluating the survival benefit conferred by sunitinib with AGS003 compared to sunitinib alone. However, preliminary analysis of the trial data suggested there was unlikely to be a survival advantage in the combination arm, and it was subsequently recommended to close the trial early.
Oncofetal antigens
The 5T4 oncofetal antigen is a cell-surface protein found on human placental cells. While it is not normally present in adult tissue, it is overexpressed in RCC.57 The restricted expression of 5T4 to the tumor suggested that it could act as a target for immunobased therapy and led to the development of the TroVax vaccine. TroVax (MVA5T4) is an attenuated, modified vaccinia Ankara (MVA) engineered to deliver the oncofetal antigen 5T4. The TRIST trial (NCT00397345), a Phase III randomized, double-blind, placebo-controlled study for mRCC patients postnephrectomy, evaluated the efficacy of this vaccine. In this study, patients who initially received either IL2, IFNα, or sunitinib based on local practice were randomized to receive either 13 doses of MVA5T4 or placebo. It was found that the addition of MVA5T4 compared to placebo did not prolong the overall survival of patients with mRCC, but the subgroup of patients who received MVA5T4 plus IL2 showed a significant survival benefit.58
Cryoablation
While the concept of tumor vaccines is logical, clinical trials using synthetic peptide vaccines derived from tumor antigens have had limited success.21,59 The suboptimal efficacy of peptide-based vaccines may come from MHC restriction, diminishing its utility in diverse populations,60 as well as emergence of tumor cells no longer expressing the protein/antigen of interest and immunological changes limiting the generation of antitumor immunity. Autologous whole tumor cells contain all the antigens a person’s immune system could potentially respond to, representing a more exhaustive repertoire of tumor antigens. However, generation of autologous whole tumor-cell vaccines currently requires tumor-tissue collection and careful and costly ex vivo processing.61 In situ “vaccination” therapies, in contrast, represent an alternative approach where local treatments are designed to induce tumor-cell death within the body, leading to the release of a range of tumor antigens, including neoantigens.62,63 Methods of inducing tumor-cell death in situ also circumvent the technical/financial constraints of other tumor vaccines by letting the host immune system respond to the most relevant tumor antigen, but it is now accepted that the type of death induced can have a profound influence on how the tumor-cell debris is processed/presented and the type of ensuing immunoresponse generated. The induction of immunogenic-cell death (ICD) is linked to the generation of an adaptive immunoresponse against dead-cell-associated antigens,64 and a number of stimuli can trigger ICD. Cells release or express molecules, such as calreticulin, HSP90, and HMGB1, during ICD that act as damage-associated molecular patterns to stimulate the immune system.65 Among the known therapies capable of causing ICD, thermal ablative therapies – especially cryotherapy – have become an approved and effective means of treating RCC.
Gases like oxygen, nitrogen, and argon exhibit the Joule–Thomson effect when they rapidly expand at room temperature. This principle is used in cryoablation by targeted freezing of tissue and thawing, leading to cell death.66 Cellular death is caused by both direct and indirect methods (Figure 2). The intracellular component has higher osmolarity and freezes more slowly than the extracellular component; as a result, fluid shifts out of the cells into the extracellular space. In the thawing phase, there is a reversal in the osmotic gradient, and fluid rushes back into the intracellular space, causing the cells to swell and rupture. In contrast to the coagulative necrosis seen in the central zone, the periphery demonstrates cell death via delayed apoptosis, due to the sublethal nature of injury by the cryoprobe. The balance between necrosis and apoptosis influences the immunomodulatory effects of cryoablation.66,67 When cells are frozen, the cellular structure is maintained, which then stimulates DCs to mount a T-cell response (ie, ICD).
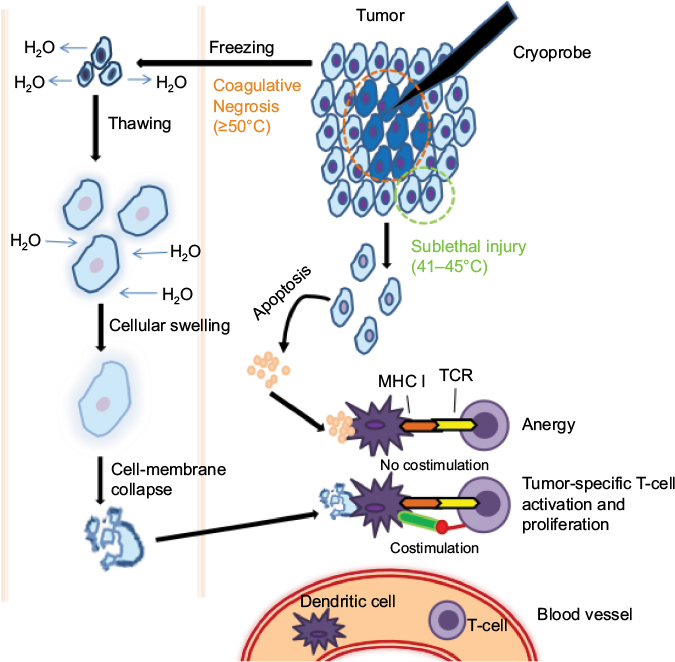 | Figure 2 Mechanism of cryoablation. Notes: Cryoablation involves both direct and indirect methods of cellular injury. The central zone, where the cryoprobe is in direct contact with the tissue, undergoes coagulative necrosis, while the cells in the peripheral zone undergo apoptosis. The cells in the central zone freeze first, causing higher extracellular osmolality compared to intracellular osmolality. Water from the intracellular space rushes out and causes cellular shrinkage. During the thawing phase, the extracellular ice melts first, causing an osmotic imbalance that results in fluid shift intracellularly, cellular swelling, and rupture. Dendritic cells phagocytose the dying/dead tumor cells, process tumor-derived protein, and present the tumor-derived peptides to T cells. It is classically thought that apoptotic cells do not release any danger signals (ie, damage-associated molecular patterns), resulting in the generation of a tolerogenic signal to T cells that can lead to anergy and/or clonal deletion. Apoptosis may also induce immunosuppression through stimulation of IL10 and TGFβ. Conversely, necrotic cell death has long been considered to be more immunogenic in nature, due to the rapid and violent release of cellular components, such as DNA, RNA, HSP70, and uric acid, which can stimulate and activate dendritic cells to express costimulatory molecules and proinflammatory cytokines. This balance between increased and reduced antitumor activity is the basis of cryoablation. Abbreviations: MHC, major histocompatibility complex; TCR, T cell receptors. |
Most studies using cryoablation for RCC have focused on laparoscopic approaches, largely due to its minimally invasive nature, for the treatment of localized RCC, especially in older individuals who are not good surgical candidates.68 The percutaneous approach has even been found to be an option for local tumor control, particularly for posteriorly located tumors. In addition, cryoablation can offer palliation in mRCC, with an estimated 5-year survival rate of 27%.69 Animal models using orthotopically implanted RCC have shown an increase in neutrophils, macrophages, CD4 T cells, and CD8 T cells into the treatment site following cryoablation.70 Interestingly, there are limited data to suggest cryotherapy alone is sufficient to induce a systemic immunoresponse, but the combination of cryotherapy and immunomodulators has resulted in the formation of systemic immunity. For example, Thakur et al examined the immune status of mRCC patients who received cryoablation treatment of lung metastases in combination with local administration of aerosolized GM-CSF.71 Both humoral and cellular responses were detected in the majority of patients receiving this combination therapy, and the magnitude of the antitumor response correlated with the clinical response. Therefore, these data suggest that the generation of a large amount of tumor antigens resulting from the cryoablative reduction in tumor burden could initiate a systemic immunoresponse when the immune system is given additional cues (GM-CSF, in this specific case) to boost the immunogenic nature of the tumor antigens.
Cytokine therapy
With the natural function of effector cells of the immune system being heavily influenced by various cytokines, the use of immunostimulatory cytokines was one of the first immunobased treatments approved for the treatment of RCC. The most widely studied cytokines used to manage RCC, especially mRCC, have been IFNα and IL2. IFNs exhibit their antitumor effects by prolonging the neoplastic cell cycle72 and upregulating MHCI expression, both of which result in a more potent immunoreaction,73,74 and by inhibiting angiogenesis.75 The first trial to establish the therapeutic benefit of IFNα in mRCC was conducted by the Medical Research Council, where patients were randomized into two groups receiving either 10 MU IFNα three times a week for 12 weeks or medroxyprogesterone acetate (300 mg/day) for 12 weeks. IFNα offered a 1-year survival benefit of 43% compared to 31% in the medroxyprogesterone acetate group. The toxicity profile, though similar, was severe in both groups.76 In contrast, Gore et al94 reported combination therapy with IFNα, IL2, and fluorouracil did not improve overall compared to IFNα alone (HR 1.05, 95% CI 0.9–1.21; P=0.55) in an open-label randomized trial of 1,006 patients with mRCC. There was no difference in progression-free survival between the arms either. However, the best overall response rate was higher in the combination group compared to the control group (23% vs 16%, P=0.0045), suggesting there still could be a role for combination-cytokine therapy and highlighting the importance of developing methods to identify patients who are most likely to respond to treatment. As an alternative to IFNα, polyethylene glycolated (PEGylated) IFN was developed by covalent attachment of a 40 kDa branched methoxy-PEG molecule to IFNα. The benefit of PEG-IFN includes slower and sustained rate of absorption, thereby avoiding the frequent dosing required by IFNα. A Phase II study evaluating the efficacy of PEG-IFN demonstrated a major response in five of 40 patients (12.5%) and a 63% 1-year survival benefit, but the side-effect profile was found to be comparable to IFNα.77
IL2 has been used as a single agent and in combination therapy since the early 1990s. The role of IL2 in cancer immunotherapy includes differentiation of naïve CD8+ T cells, maintaining Treg homeostasis, and natural-killer-cell activation. The first Phase II study evaluating the effect of IL2 in RCC had 255 patients who received high-dose bolus IL2 600,000 IU/kg for 14 doses or 720,000 IU/kg for 12 doses every 8 hours. The median overall survival was found to be 15.8 months.78 Following this, a 20-year follow-up study was conducted by the National Cancer Institute, where 9% of the patients showed complete response and only four developed disease recurrence.79 The adverse effects of high-dose IL2 therapy demanded further research to be conducted exploring alternative dosing regimens. The Cytokine Working Group randomized patients to receive either a bolus of IL2 or a continuous infusion. The overall response rate in the bolus arm was 20% versus 15% in the infusion arm, but there were no significant differences in adverse effects.80 The discovery of other immunomodulatory treatment modalities led to studies combining IL2 with other agents, such as IFNα, sorafenib, and vaccines.
Checkpoint inhibitors
CTLA4 and PD1 expression by activated T cells act as negative immunoregulators to suppress T-cell activity. Therefore, it was thought that inhibition of checkpoint receptors should reenable the body to mount an effective T-cell response. This theoretical basis drove the development and testing of mAbs that block the interaction of these checkpoint receptors with their cognate ligands, with the anti-CTLA4 mAb ipilimumab (Yervoy; Bristol-Myers Squibb, New York, NY, USA) being the first to receive approval for the treatment of unresectable or metastatic melanoma.81 Since this breakthrough, a number of other agents targeting checkpoint receptors have entered the clinic and shown significant potential to improve overall survival in a range of tumors.82 In the setting of mRCC, CTLA4 and PD1 inhibitors have been the most studied therapies, and the exciting results obtained in a number of clinical trials have led to US Food and Drug Administration (FDA) approval of these agents in the treatment of RCC.
CTLA4 inhibitors
Following the early success of ipilimumab in melanoma, Yang et al conducted a Phase II trial to assess its efficacy in treating mRCC.83 A total of 61 patients were enrolled in the study, of which two-thirds were administered a high dose of 3 mg/kg every 3 weeks for up to 1 year and the remaining third were administered a loading dose of 3 mg/kg, but then received only 1 mg/kg for their subsequent doses. Unfortunately, no patients displayed a complete response as per Response Evaluation Criteria in Solid Tumors, but 12.5% (95% CI 4%–27%) of the patients receiving the higher dose of the drug for the duration of the study had a partial response of 7–21 months. This was at the expense of 43% of this group experiencing clinically significant immunomediated toxicities that included enteritis, hypophysitis, adrenal insufficiency, and even aseptic meningitis. Interestingly, the patients who experienced autoimmune toxicity were significantly more likely to respond to ipilimumab than those who did not. A similar relationship was also experienced in the original melanoma study. Given the lack of complete regression observed in this study and the relative success of other immunotherapeutic agents in the mRCC setting, CTLA4-inhibitor monotherapy is not generally used currently. A Phase I study examined another CTLA4 inhibitor – tremelimumab – in combination with sunitinib, but reported multiple incidences of rapid-onset renal failure, despite the 43% partial-response rate.84
PD1–PDL1 pathway inhibitors
The anti-PD1 mAb nivolumab followed the trend of other immunotherapies that first showed potential in melanoma before receiving wider approval for other cancers. CheckMate 025 was an open-label, Phase III, randomized trial that compared nivolumab with everolimus, an mTOR inhibitor that at the time was recommended in advanced RCC following failed sorafenib or sunitinib treatment.12 This international trial was prematurely halted, as the primary end point was met early on, with the nivolumab group demonstrating superior median overall survival of 25 months (95% CI 21.8 months to not estimable) compared to 19.6 months (95% CI 17.6–23.1 months) in the everolimus group. The patients who received nivolumab were also statistically less likely to die from any cause (HR 0.73, 98.5% CI 0.57–0.93; P=0.002). Furthermore, those receiving nivolumab were nearly six times more likely to experience some degree of response (OR 5.98, 95% CI 3.68–9.72; P<0.001), with median duration of response and progression-free survival of 12 and 4.6 months, respectively. Additionally, nivolumab was also relatively well tolerated, with only 19% of patients experiencing a grade 3 or 4 complication. The promising results of this trial led to FDA approval of nivolumab for advanced RCC in late 2015.
Recently, further analysis was conducted on the CheckMate 025 trial to characterize those patients who were most likely to benefit from nivolumab.85 The superiority of this treatment was underlined by the improved overall survival across all patient subgroups regardless of Memorial Sloan Kettering Cancer Center and International Metastatic Renal Cell Carcinoma Database Consortium risk groups, age, site of metastasis, and/or prior therapy. This study found that those with poor Memorial Sloan Kettering Cancer Center risk status (defined as having at least two of low Karnofsky performance status [<80%], low serum hemoglobin, or high corrected calcium) had the most to benefit from nivolumab treatment (HR 0.48, 95% CI 0.32–0.7). Adverse effects of treatment were also less prevalent in this group.
Anti-PDL1 mAbs are closely related to PD1 inhibitors in their blocking function, but are specific to the PD1–PDL1 signaling pathway (PD1–PDL2 interaction can still occur). Compared to CTLA4 and PD1 inhibitors, research on this class of agent is still in its infancy. Brahmer et al published data from a Phase I trial using the anti-PDL1 mAb BMS936559 across a range of cancers, of which 17 subjects had RCC.86 Among the patients with RCC, 12% experienced an objective response lasting 4-17 months and a further 41% had stable disease lasting at least 24 weeks. This level of responsiveness was comparable to efficacy across all cancer patients enrolled in the study, which reported an objective response of 6%–17% and stabilization rate of 12%–41%. Furthermore, the incidence of grade 3 or 4 toxicities was only 9%. More recently, McDermott et al studied atezolizumab in patients with both clear-cell and non-clear-cell RCC.87 Patients with clear-cell disease displayed median overall survival and progression-free survival of 28.9 and 5.6 months, respectively, and the objective response rate was 15% (95% CI 7%–26%).
Checkpoint-inhibitor combination therapy
It has been hypothesized that a combination of checkpoint inhibitors is likely to be more efficacious than monotherapy, since CTLA4 and PD1 inhibitors target different inhibitory pathways on lymphocytes.28 The positive results seen among untreated melanoma patients further support this theory.88 While a number of trials are currently examining the efficacy of checkpoint-inhibitor combination therapy in mRCC, no results have been published to date. Preliminary results of the CheckMate 214 trial were presented recently at the 2017 European Society for Medical Oncology meeting.89 This Phase III randomized study evaluated the combination of nivolumab and ipilimumab in the treatment of advanced RCC or mRCC compared to sunitinib. At 17.5 months of follow-up, the objective response rate was greater in the combination group compared to sunitinib alone among intermediate/poor-risk patients (41.6% vs 26.5%, P<0.001). There was also an improvement in median progression-free survival with combination therapy (HR 0.82, P=0.03). Another important finding in this study was that efficacy outcomes varied according to PDL1 expression, with those who had a baseline PDL1 expression of at least 1% demonstrating superior outcomes. Moreover, the level of PDL1 expression differed between risk groups, and may at least partially explain the superiority of sunitinib in favorable-risk patients. Furthermore, one arm of the Phase I CheckMate 016 study included the combination of nivolumab and ipilimumab at two different doses: one agent at 3 mg/kg and the other at 1 mg/kg, and vice versa.90 The group of patients receiving the higher dose of ipilimumab reported slightly greater objective response rate of 48% compared to 43% in the high-dose nivolumab group. However, all patients in the former group experienced some degree of treatment side effects, of which 60.9% were grade 3 or 4, but only just over a quarter required treatment to be ceased. The subgroup who received the higher dose of nivolumab tolerated treatment better, with only 9.5% halting therapy as a result of adverse effects. Although it is hard to draw conclusions from these preliminary results, owing to the limitations in the study, the data obtained do suggest the high potential of combination checkpoint-inhibitor therapy for mRCC and the need for future trials to provide clarity on its efficacy and safety.
Clinical studies have also explored combining checkpoint inhibitors with other types of agents, with some early success. Amin et al combined nivolumab with either sunitinib or pazopanib in a Phase I trial.91 Preliminary results suggest these combinations are safe and have the potential to change the course of disease, with a response rate of 52% and 45% in the sunitinib and pazopanib arms, respectively. Similarly, initial reports suggest the combination of PDL1 inhibitors with bevacizumab have a synergistic effect as well.92 An ongoing Phase III randomized study (KEYNOTE 426; NCT02853331) is evaluating the efficacy and safety of pembrolizumab plus axitinib versus sunitinib alone in the treatment of naïve mRCC. Several other studies evaluating a variety of combination treatments, such as avelumab plus axitinib, bevacizumab plus atezolizumab, cabozantinib plus nivolumab, and lenvatinib plus pembrolizumab, are under way, and will be important in providing answers in this landscape.
Future direction
The aforementioned therapies highlight the considerable potential of immunobased treatment in altering the disease course of mRCC. However, it is important to recognize the efficacy of these treatments are not identical across all patients and they all potentially have serious adverse effects. Therefore, much effort is currently going into identifying markers to identify those patients most and least likely to respond to immunotherapies. Utilizing biomarkers as a tool to predict responsiveness to immunotherapy has yielded promising results in other malignancies,93 with the majority of research focused on PDL1 expression in non-small-cell lung cancer, melanoma, or bladder cancer. It is thus imperative that biomarkers predicting responsiveness to PD1 and CTLA4 inhibitors – which have demonstrated the greatest potential in mRCC – are developed to minimize cost and toxicity.
The early results from immunobased treatments highlight their potential to transform the clinical paradigm for mRCC, and this is further supported by the recent preliminary report of the CheckMate 214 trial. FDA approval of the anti-PD1 mAb nivolumab represented a shift in this landscape from aspecific to targeted immunotherapies. While checkpoint inhibitors are currently garnering the attention, the efficacy of cytokine therapy (especially IL2), immunobased vaccines, and cryoablation cannot be overlooked. However, many of these agents lack Phase III data supporting their efficacy, which will be required before they become available in routine clinical practice.
Disclosure
The authors report no conflicts of interest in this work.
References
Patil S, Ishill N, Deluca J, Motzer RJ. Stage migration and increasing proportion of favorable-prognosis metastatic renal cell carcinoma patients: implications for clinical trial design and interpretation. Cancer. 2010;116(2):347–354. | ||
Kabaria R, Klaassen Z, Terris MK. Renal cell carcinoma: links and risks. Int J Nephrol Renovasc Dis. 2016;9:45–52. | ||
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100(1):57–70. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218. | ||
Bhatt JR, Finelli A. Landmarks in the diagnosis and treatment of renal cell carcinoma. Nat Rev Urol. 2014;11(9):517–525. | ||
Gabrilovich DI, Bronte V, Chen SH, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67(1):425. | ||
Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–581. | ||
Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33(17):1889–1894. | ||
McDermott DF, Drake CG, Sznol M, et al. Survival, durable response, and long-term safety in patients with previously treated advanced renal cell carcinoma receiving nivolumab. J Clin Oncol. 2015;33(18):2013–2020. | ||
Nakano O, Sato M, Naito Y, et al. Proliferative activity of intratumoral CD8+ T-lymphocytes as a prognostic factor in human renal cell carcinoma: clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001;61(13):5132–5136. | ||
Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. | ||
Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. Am J Med Sci. 1893;105:487–511. | ||
Tian T, Olson S, Whitacre JM, Harding A. The origins of cancer robustness and evolvability. Integr Biol. 2011;3(1):17–30. | ||
Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106(3):255–258. | ||
Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. | ||
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–277. | ||
Fujii S, Liu K, Smith C, Bonito AJ, Steinman RM. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med. 2004;199(12):1607–1618. | ||
Gigante M, Blasi A, Loverre A, et al. Dysfunctional DC subsets in RCC patients: ex vivo correction to yield an effective anti-cancer vaccine. Mol Immunol. 2009;46(5):893–901. | ||
Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. Lancet Oncol. 2013;14(6):e218–e228. | ||
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. | ||
Kondo T, Nakazawa H, Ito F, et al. Favorable prognosis of renal cell carcinoma with increased expression of chemokines associated with a Th1-type immune response. Cancer Sci. 2006;97(8):780–786. | ||
Tatsumi T, Kierstead LS, Ranieri E, et al. Disease-associated bias in T helper type 1 (Th1)/Th2 CD4+ T cell responses against MAGE-6 in HLA-DRB10401+ patients with renal cell carcinoma or melanoma. J Exp Med. 2002;196(5):619–628. | ||
Linsley PS, Wallace PM, Johnson J, et al. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science. 1992;257(5071):792–795. | ||
Lenschow DJ, Zeng Y, Thistlethwaite JR, et al. Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4lg. Science. 1992;257(5071):789–792. | ||
Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. 2001;19:225–252. | ||
Redmond WL, Ruby CE, Weinberg AD. The role of OX40-mediated co-stimulation in T cell activation and survival. Crit Rev Immunol. 2009;29(3):187–201. | ||
Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543–9553. | ||
Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182(2):459–465. | ||
Riella LV, Paterson AM, Sharpe AH, Chandraker A. Role of the PD-1 pathway in the immune response. Am J Transplant. 2012;12(10):2575–2587. | ||
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–499. | ||
Fushimi T, Kojima A, Moore MA, Crystal RG. Macrophage inflammatory protein 3α transgene attracts dendritic cells to established murine tumors and suppresses tumor growth. J Clin Invest. 2000;105(10):1383–1393. | ||
Luster AD, Leder P. IP-10, a -C-X-C- chemokine, elicits a potent thymus-dependent antitumor response in vivo. J Exp Med. 1993; 178(3):1057–1065. | ||
Maric M, Liu Y. Strong cytotoxic T lymphocyte responses to a macrophage inflammatory protein 1α-expressing tumor: linkage between inflammation and specific immunity. Cancer Res. 1999;59(21):5549–5553. | ||
Mule JJ, Custer M, Averbook B, et al. RANTES secretion by gene-modified tumor cells results in loss of tumorigenicity in vivo: role of immune cell subpopulations. Hum Gene Ther. 1996;7(13):1545–1553. | ||
Franciszkiewicz K, Le Floc’h A, Boutet M, Vergnon I, Schmitt A, Mami-Chouaib F. CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Res. 2013;73(2):617–628. | ||
Kooi S, Zhang HZ, Patenia R, Edwards CL, Platsoucas CD, Freedman RS. HLA class I expression on human ovarian carcinoma cells correlates with T-cell infiltration in vivo and T-cell expansion in vitro in low concentrations of recombinant interleukin-2. Cell Immunol. 1996;174(2):116–128. | ||
Facciabene A, Peng X, Hagemann IS, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and Treg cells. Nature. 2011;475(7355):226–230. | ||
Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37(3):193–207. | ||
Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–179. | ||
Walter S, Weinschenk T, Stenzl A, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012;18(8):1254–1261. | ||
Zea AH, Rodriguez PC, Atkins MB, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65(8):3044–3048. | ||
Kaelin WG Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8(11):865–873. | ||
Youn JI, Kumar V, Collazo M, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol. 2013;14(3):211–220. | ||
De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23(3):277–286. | ||
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. | ||
Daurkin I, Eruslanov E, Stoffs T, et al. Tumor-associated macrophages mediate immunosuppression in the renal cancer microenvironment by activating the 15-lipoxygenase-2 pathway. Cancer Res. 2011;71(20):6400–6409. | ||
Griffiths RW, Elkord E, Gilham DE, et al. Frequency of regulatory T cells in renal cell carcinoma patients and investigation of correlation with survival. Cancer Immunol Immunother. 2007;56(11):1743–1753. | ||
Li JF, Chu YW, Wang GM, et al. The prognostic value of peritumoral regulatory T cells and its correlation with intratumoral cyclooxygenase-2 expression in clear cell renal cell carcinoma. BJU Int. 2009;103(3):399–405. | ||
Sell K, Barth PJ, Moll R, et al. Localization of FOXP3-positive cells in renal cell carcinoma. Tumour Biol. 2012;33(2):507–513. | ||
Ning H, Shao QQ, Ding KJ, et al. Tumor-infiltrating regulatory T cells are positively correlated with angiogenic status in renal cell carcinoma. Chin Med J (Engl). 2012;125(12):2120–2125. | ||
Walter S, Weinschenk T, Stenzl A, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012;18(8):1–12. | ||
Rini BI, Stenzl A, Zdrojowy R, et al. IMA901, a multipeptide cancer vaccine, plus sunitinib versus sunitinib alone, as first-line therapy for advanced or metastatic renal cell carcinoma (IMPRINT): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2016;17(11):1599–1611. | ||
Finke JH, Rini BI, Ireland J, et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14(20):6674–6682. | ||
Ko JS, Zea AH, Rini BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–2157. | ||
Amin A, Dudek AZ, Logan TF, et al. Survival with AGS-003, an autologous dendritic cell-based immunotherapy, in combination with sunitinib in unfavorable risk patients with advanced renal cell carcinoma (RCC): phase 2 study results. J Immunother Cancer. 2015;3:14. | ||
Griffiths RW, Gilham DE, Dangoor A, et al. Expression of the 5T4 oncofoetal antigen in renal cell carcinoma: a potential target for T-cell-based immunotherapy. Br J Cancer. 2005;93(6):670–677. | ||
Amato RJ, Hawkins RE, Kaufman HL, et al. Vaccination of metastatic renal cancer patients with MVA-5T4: A randomized, double-blind, placebo-controlled phase III study. Clin Cancer Res. 2010;16(22):5539–5547. | ||
Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. | ||
Rohn TA, Reitz A, Paschen A, et al. A novel strategy for the discovery of MHC class II-restricted tumor antigens: identification of a melanotransferrin helper T-cell epitope. Cancer Res. 2005;65(21):10068–10078. | ||
Chiang CL, Benencia F, Coukos G. Whole tumor antigen vaccines. Semin Immunol. 2010;22(3):132–143. | ||
Brody JD, Ai WZ, Czerwinski DK, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol. 2010;28(28):4324–4332. | ||
Salazar AM, Erlich RB, Mark A, Bhardwaj N, Herberman RB. Therapeutic in situ autovaccination against solid cancers with intratumoral poly-ICLC: case report, hypothesis, and clinical trial. Cancer Immunol Res. 2014;2(8):720–724. | ||
Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12(12):860–875. | ||
Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000;191(3):423–433. | ||
Erinjeri JP, Clark TWI. Cryoablation: mechanism of action and devices. J Vasc Interv Radiol. 2010;21(8):S187–S191. | ||
Gage AA, Baust J. Mechanisms of tissue injury in cryosurgery. Cryobiology. 1998;37(3):171–186. | ||
American Urological Association. Guideline for management of the clinical stage 1 renal mass. 2009. Available from: https://www.auanet.org/documents/education/clinical-guidance/Renal-Mass.pdf. Accessed October 30, 2017. | ||
Wen B, Lampe JN, Roberts AG, Atkins WM, Rodrigues AD, Nelson SD. Cysteine 98 in CYP3A4 contributes to conformational integrity required for P450 interaction with CYP reductase. Arch Biochem Biophys. 2007;454(1):42–54. | ||
Matin SF, Sharma P, Gill IS, et al. Immunological response to renal cryoablation in an in vivo orthotopic renal cell carcinoma murine model. J Urol. 2010;183(1):333–338. | ||
Thakur A, Littrup P, Paul EN, Adam B, Heilbrun LK, Lum LG. Induction of specific cellular and humoral responses against renal cell carcinoma after combination therapy with cryoablation and granulocyte-macrophage colony stimulating factor: a pilot study. J Immunother. 2011;34(5):457–467. | ||
Hanley JP, Haydon GH. The biology of interferon-α and the clinical significance of anti-interferon antibodies. Leuk Lymphoma. 1998;29(3–4):257–268. | ||
Baron S, Tyring SK, Fleischmann WR, et al. The interferons: mechanisms of action and clinical applications. JAMA. 1991;266(10):1375–1383. | ||
Giacomini P, Aguzzi A, Pestka S, Fisher PB, Ferrone S. Modulation by recombinant DNA leukocyte (α) and fibroblast (β) interferons of the expression and shedding of HLA- and tumor-associated antigens by human melanoma cells. J Immunol. 1984;133(3):1649–1655. | ||
Folkman J. Antiangiogenic therapy. In: Devita VT, Hellman S, Rosenberg SA, editors. Cancer: Principles and Practice of Oncology. 5th ed. Philadelphia: Lippincott Williams and Wilkins; 1997:3075–3085. | ||
Santhanam S, Decatris M, O’Byrne K. Potential of interferon-α in solid tumours: part 2. BioDrugs. 2002;16(5):349–372. | ||
Motzer RJ, Bacik J, Murphy BA, Russo P, Mazumdar M. Interferon-α as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol. 2002;20(1):289–296. | ||
Rogers E, Bredin H, Butler M, et al. Combined subcutaneous recombinant α-interferon and interleukin-2 in metastatic renal cell cancer: results of the Multicentre All Ireland Immunotherapy Study Group. Eur Urol. 2000;37(3):261–266. | ||
Klapper JA, Downey SG, Smith FO, et al. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma: a retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer. 2008;113(2):293–301. | ||
Weiss GR, Margolin KA, Aronson FR, et al. A randomized phase II trial of continuous infusion interleukin-2 or bolus injection interleukin-2 plus lymphokine-activated killer cells for advanced renal cell carcinoma. J Clin Oncol. 1992;10(2):275–281. | ||
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | ||
Alexander W. The checkpoint immunotherapy revolution: what started as a trickle has become a flood, despite some daunting adverse effects; new drugs, indications, and combinations continue to emerge. P T. 2016;41(3):185–191. | ||
Yang JC, Hughes ME, Kammula US, et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother. 2007;30(8):825–830. | ||
Rini BI, Stein M, Shannon P, et al. Phase 1 dose-escalation trial of tremelimumab plus sunitinib in patients with metastatic renal cell carcinoma. Cancer. 2011;117(4):758–767. | ||
Escudier B, Sharma P, McDermott DF, et al. CheckMate 025 randomized phase 3 study: outcomes by key baseline factors and prior therapy for nivolumab versus everolimus in advanced renal cell carcinoma. Eur Urol. Epub 2017 Mar 3. | ||
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. | ||
McDermott DF, Sosman JA, Sznol M, et al. Atezolizumab, an anti-programmed death-ligand 1 antibody, in metastatic renal cell carcinoma: long-term safety, clinical activity, and immune correlates from a phase IA study. J Clin Oncol. 2016;34(8):833–842. | ||
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. | ||
Escudier B, Tannir N, McDermott DF, et al. CheckMate 214: efficacy and safety of nivolumab + ipilimumab (N+I) v sunitinib (S) for treatment-naïve advanced or metastatic renal cell carcinoma (mRCC), including IMDC risk and PD-L1 expression subgroups. Ann Oncol. 2017;28 Suppl 5:v605–v649. | ||
Hammers H, Plimack ER, Infante JR, et al. Phase I study of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma (MRCC). J Clin Oncol. 2014;32(15 Suppl):4504. | ||
Amin A, Plimack ER, Infante JR, et al. Nivolumab (anti-PD-1; BMS-936558, ONO-4538) in combination with sunitinib or pazopanib in patients (pts) with metastatic renal cell carcinoma (mRCC). J Clin Oncol. 2014;32(15 Suppl):5010. | ||
Lieu C, Bendell J, Powderly JD, et al. Safety and efficacy of MPDL3280A (anti-PDL1) in combination with bevacizumab (Bev) and/or chemotherapy (chemo) in patients (pts) with locally advanced or metastatic solid tumors. Ann Oncol. 2014;25 Suppl 4:iv361. | ||
Yuan J, Hegde PS, Clynes R, et al. Novel technologies and emerging biomarkers for personalized cancer immunotherapy. J Immunother Cancer. 2016;4:3. | ||
Gore ME, Griffin CL, Hancock B. Interferon alfa-2a versus combination therapy with interferon alfa-2a, interleukin-2, and fluorouracil in patients with untreated metastatic renal cell carcinoma (MRC RE04/EORTC GU 30012): an open-label randomised trial. Lancet. 2010;375(9715):641–648. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.