")
Back to Journals » Biologics: Targets and Therapy » Volume 15
The Current Status of Gene Therapy for the Treatment of Cancer
Authors Belete TM
Received 22 January 2021
Accepted for publication 4 March 2021
Published 18 March 2021 Volume 2021:15 Pages 67—77
DOI https://doi.org/10.2147/BTT.S302095
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shein-Chung Chow
Tafere Mulaw Belete
Department of Pharmacology, College of Medicine and Health Sciences, University of Gondar, Gondar, Amhara Region, Ethiopia
Correspondence: Tafere Mulaw Belete P.o.box 196 Tel +251 9 18 04 59 43
Email [email protected]
Abstract: Gene therapy is the administration of foreign genomic material into the host tissue to modify the expression of a gene product or to change the biological properties of cells for therapeutic use. Initially, the major objective of gene therapy was to manage genetic diseases, but now different disorders with several patterns of acquired and inherited disorders are targets of gene therapy. Over three decades, the advancement of Genome engineering technologies facilitated gene therapy for the prevention and management of intractable diseases. Researchers are advancing with cautious optimism that safe and effective treatment will give to patients with single-gene disorders and complex acquired disorders. To date, over 3000 genes associates with disease-causing mutations, and about 2600 gene therapy trials are undergoing for the management of various disorders. This review summarizes the principles of genome-editing approaches, such as zinc finger nucleases, transcription activator-like effector nucleases, meganucleases, and the CRISPR/Cas9 system with the underlying mechanisms. This review also explains the types of gene delivery systems as viral [adenoviral, adeno association, herpes simplex virus] and nonviral delivery systems (physical: DNA bombardment, electroporation) and (chemical: Cationic lipids, cationic polymers). Finally, this review summarizes gene therapy medicines approved to treat cancer in detail, including names, indications, vectors, and mode of gene therapy. Gene therapy becomes an alternative to an existing management for different diseases. Therefore, gene products with safe vectors and better biotechnologies play a significant role in the prophylaxis and management of various disorders in the future.
Keywords: gene therapy, gene product, gene delivery, cancer, clinical trial
Background
After DNA helical structure discovery, the world continuous staircase outburst of several advanced technologies, which are currently heading toward translation into clinical practice. Over the last decades, several molecular techniques developed that help to edit the DNA codes and modify mRNA by post-transcriptional modifications. Gene therapy is the delivery of specific genetic material to modify the encoding of a gene product or to change the biological properties of tissues for the management of various disorders.1 Gene therapy overcomes the limitations associated with the recombinant therapeutic use of peptides, such as low bioavailability, instability, severe toxicity, clearance rates, and high production cost.2 Gene therapies act by different mechanisms including, replacing malfunction genes with the therapeutic genes, gene knockdown, or deactivating problem genes, and insert a new gene to treat a disease.3 Gene therapy can be done in either somatic or germline cells. In somatic cells, gene therapy only the modified tissues will be affected, but in germline cell gene therapy, genetic changes transmit to the offspring. So, there is no clinical trial on human germline gene therapy.4 Currently, somatic gene therapy is safe for the management of several disorders in human beings. Gene therapy effectively treats several diseases due to increased understanding of disease pathogenesis and improved gene delivery technologies.5 Gene therapy uses genetic material (ie, RNA or DNA) via a vector that facilitates the delivery of foreign genetic material into the host organ. The genetic material is administered into the target organ (in vivo gene therapy) or used to modify cells taken from the host that are then re-administered (ex vivo gene therapy). Gene therapy aims to provide a functional gene copy of the damaged gene(s), increase the availability of disease-modifying genes or suppress the activity of a damaged gene.6,7 Gene therapy has a broad spectrum of applications, from gene replacement and knockdown for genetic disorders including cancer, hemophilia, hypercholesterolemia, and neurodegenerative diseases to vaccination, each with different requirements for gene administration.8 Gene delivery systems consist of three components: a gene that expresses essential therapeutic peptides, a plasmid-based gene encoding system that regulates the activity of a gene in the target organ, and a gene delivery system that regulates the administration of the encoding gene to host tissue.9
Gene Editing Tools
Conventional gene therapy mostly depends on viral-based delivery of genes that either randomly integrates into the host genome (eg retroviruses) or remains as extrachromosomal DNA copy (eg AAV]) and expresses a protein that is missing or mutated in human disorder. In contrast to traditional gene therapy, gene editing provides more versatile tools for gene therapy, for example, precisely correct point variants, place an extra, healthy gene at a safe genomic location or disrupt a gene. The Current gene-editing process depends on the introduction of endogenous double-strand DNA breaks (DSBs) and repair mechanisms. When DSBs occur by nucleases, cellular DNA repair mechanisms are activated. There are two main mechanisms for repairing double-strand breaks, non-homologous end joining (NHEJ) and homology-directed repair (HDR). Genome-editing nucleases can be modified to recognize and break the genome at specific DNA sequences, resulting in DSBs, which are efficiently repaired by either NHEJ or HDR.10,11
NHEJ repair damaged DNA without a homologous template. Due to this reason, NHEJ may lead to deletions or insertions of nucleotides in the damaged loci; thus, it is error-prone. HDR differs from NHEJ since it repairs DNA damages using a homologous template. Generally, having used a homologous sequence, this form of DNA repair has less chance to cause errors. From a clinical viewpoint, HDR is favorable for restoring mutations in genes or for integrating genes for therapeutic purposes.10–13
Currently, there are four different gene-editing nuclease enzymes available based on their structures: meganucleases, zinc-finger nucleases, transcription activator-like effector nucleases, and CRISPR-associated nucleases.
Meganucleases (MNs)
Are sequence-specific endonucleases that recognize unique large (14–40 bp) target sites. It has low cytotoxicity that makes it an attractive tool for genome editing. Existing engineering techniques include the creation of fusion protein from existing MN domains and engineering MN specificity via the direct alteration of protein residues in the DNA-binding domain. The complexity in re-engineering and low editing efficiency limits the uses of MNs.14
Zinc Finger Nucleases (ZFNs)
Artificially produced by fusing site-specific zinc finger protein with the non-specific cleavage domain of the FokI restriction endonuclease. The DNA-binding component has 3–6 zinc finger repeats, and each can identify between 9 and 18 base pairs. ZFN has three zinc fingers that each identifies three base pair DNA sequence to form a three-finger array that attaches to nine base pair target sites and the non-specific cleavage domain.14,15 ZFPs deliver a site-specific DSB to the genome and facilitate local homologous recombination that enhances targeted genome editing. The ZFN-encoding plasmid-based targeted administration of the required genes decreases the limitations of viral administration. If ZFNs are not specific at the target site, off-target break may occur. Such off-target breakage may cause DBS that causes cell death. An Off-target break may facilitate the random integration of donor DNA.15,16
Transcription Activator-Like Effector Nucleases (TALENs)
Are artificial DNA nucleases formed by fusing a DNA-binding domain with a nonspecific nuclease domain derived from Fok I endonuclease that specifically cut the required DNA sequence.15 TALE effectors DNA-binding domain has a repeating unit of 33–35 conserved amino acids. Each repeat is similar, except positions 12 and 13, which are variable and have a strong correlation with specific nucleotide recognition. DNA cleavage domain is nonspecific from FokI endonuclease. The FokI domain acts as a dimer that needs two constructs with unique DNA binding for sites in the target genome. Both the number of amino acids between the TALE DNA binding domain and the FokI cleavage domain are essential for better activity. TALEN uses to edit genomes by inducing DSB that cells respond to with repair mechanisms.17,18
CRISPR-Cas
CRISPR is a heritable, adaptive immune system of bacteria that provides them with the memory of previous virus infections and defends against re-infection. Contrary to the human adaptive immune system, CRISPR is passed on to the next generation of bacteria, rendering the colony immune to future virus infections. CRISPR immunity depends on the integration of the invader’s DNA (virus or plasmid) into the bacterial genome.19 CRISPR helps the bacterium to identify the viral sequences and break. CRISPR stands for Clustered Regularly Interspaced Short Palindromic Repeats, which are interrupted by “spacer” sequences. These “spacer” sequences are viral sequences integrated during past viral infections when transcribed into short RNA sequences, are capable of guiding the Cas endonuclease to complementary sequences of viral DNA. Upon target identification, Cas binds to the viral DNA and cleaves it, protecting the prokaryotic cell from infection.20,21 CRISPR immune system modified to create a gene-editing tool that can target changes to the DNA. The most common is CRISPR/Cas9, which posses the Cas9 endonuclease and a short noncoding guide RNA (gRNA) that contains two components: a target-specific CRISPR RNA (crRNA) and a helper trans-activating RNA (tracrRNA). The gRNA unit guides Cas9 to a specific genomic locus via base pairing between the crRNA sequence and the target sequence.22 CRISPR-Cas-mediated gene repair, disruption, insertion, or deletion are thus finding applications in several areas of biomedical research, medicine, agriculture, and biotechnology.22,23
Gene Delivery Technologies
Since the emergence of recombinant DNA technology that helps gene-therapy, how to effectively and safely administer gene products is the major challenge. Vector is a vehicle that uses to deliver the gene of interest. An ideal vector can administer a gene to a specific tissue, accommodate enough foreign gene size, achieve the level and duration of transgenic expression enough to correct the defect gene, non-immunogenic, and safe. Delivery of the gene products done by Viral Vectors, Bactofection, and none viral Vectors (chemical and physical) method as summarized in Figure 1.24 The most important step in achieving gene therapy is choosing the vectors.
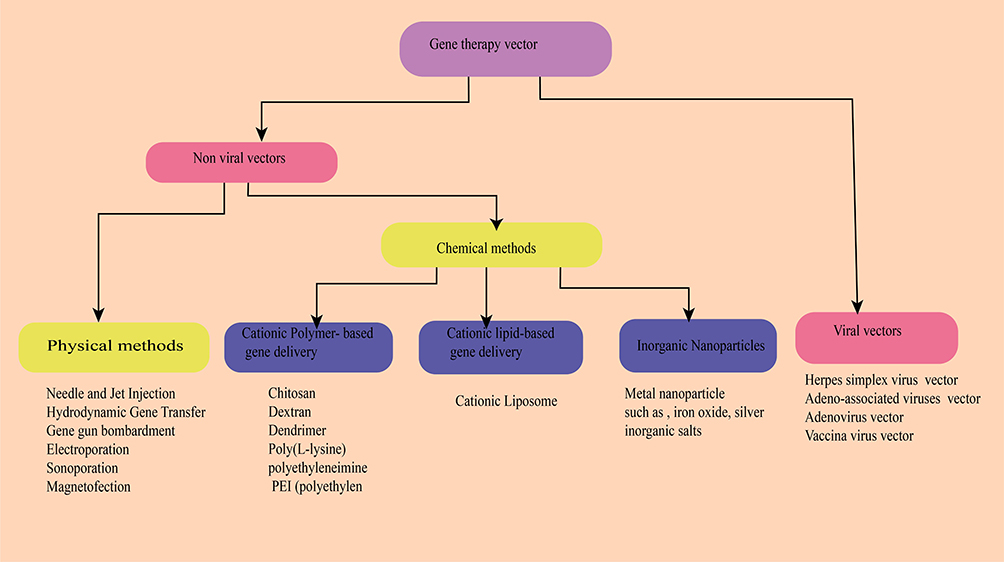 |
Figure 1 Overview of the delivery systems used in gene therapy. |
Viral Vectors Used for Gene Delivery
Viruses were the first and the most widely used vectors to administer genes into the target tissue. Viral vectors ensure that almost all cells can infect, without affecting cell viability. Viruses have distinctive features that make them suitable for gene delivery in clinical practice. Surface proteins on viruses interact with their host receptors, which activate endocytosis. Once entered, viruses release their genome into the nucleus for viral gene expression.25,26 Herpes simplex virus (HSV), adenovirus (Ad), adeno-associated virus (AAV), and lentivirus (LV) are the most important viral vectors.27,28
Bacterial Mediated Gene Transfer (Bactofection)
Some bacteria specifically target tumor cells leading to RNA interference (RNAi) and gene silencing by inhibiting RNA activity, such as protein synthesis. Several in vivo and in vitro studies revealed that intracellular bacteria such as Salmonella spp., Listeria monocytogenes, Shigella flexneri, Bifidobacterium longum, E. coli, and Yersinia enterocolitica use to deliver plasmids pro-drug converting enzymes and cytotoxic agents into the target cell.29 Phase I trial is undergoing by using Listeria, Bifidobacterium, Salmonella, Shigella, and Clostridium gene therapy against cancer. Another clinical trial is ongoing on the effects of Lactococcus synthesizing interleukin 10 against colitis in Phase II.30,31
Chemical-Based Nonviral Vectors
Viral-vectors-based gene transfer displays better and long-term gene encoding but has some limitations like immunogenicity, less specific to the target cell, carcinogenicity, high cost and cannot deliver large genome size. Non-viral methods display better advantages due to relatively safe, can deliver a large genome, and ease for production.32–35 Chemical vectors, also known as non-viral vectors grouped as organic and inorganic vectors. The organic vectors consist of cationic lipid-based vectors: synthetic cationic polymers-based vector and peptide-based vectors. These cationic organic vectors form complexes with negatively charged DNA via an electrostatic bond. The complexes protect the genomic material and enhance cell uptake and intracellular delivery. Generally, non-viral vectors help to deliver small DNA, large DNA (plasmid DNA), and RNA (Si RNA, m RNA) into the target tissue.36–38 Physical methods use different mechanical forces to facilitate the administration of gene material into the host tissues. It is an alternative to viral and chemical methods to decrease barriers that limit DNA delivery into the host tissues.39 It is feasible to deliver genes into target tissues by mechanical force. Indeed, there are several methods, and most have a similar mode of gene delivery, ie, physically formed transient pores in the cell membrane through which the genetic material enters into the host cell.40,41 Needle and jet injection, hydrodynamic gene transfer, electroporation, sonoporation, magnetofection, and gene gun bombardment are examples of physical DNA delivering methods.42–44
Gene Therapy for Cancer Treatment
Cancer occurs due to disrupting the normal cell proliferation and apoptosis process. Advances in cancer therapy need a novel therapeutic agent with novel mode of action, several mechanisms of cell death, and synergy with conventional management. Gene therapies possess all these profiles. Several gene therapy approaches were developed for the management of cancer, including anti-angiogenic gene therapy, suicide gene therapy, immunotherapy, siRNA therapy, pro-apoptotic gene therapy, oncolytic virotherapy, and gene directed-enzyme prodrug therapy.45 By November 2017, greater than 2597 clinical trials were conducted on gene therapy in the world. Among these trials, greater than 65% are associated with cancer, followed by monogenetic and cardiovascular diseases.8 The use of CAR T cell therapy showed promising results for the management of both myeloid and lymphoid leukemia. Until August 2019, only 22 gene products were approved for the treatment of different disorders. Most gene products used for the treatment variety types of cancers as shown in Table 1. Immuno-gene therapy is a potential treatment approach for the treatment of p53-deficient tumors (Imlygic, Gendicine, Yescarta, and Kymriah.47
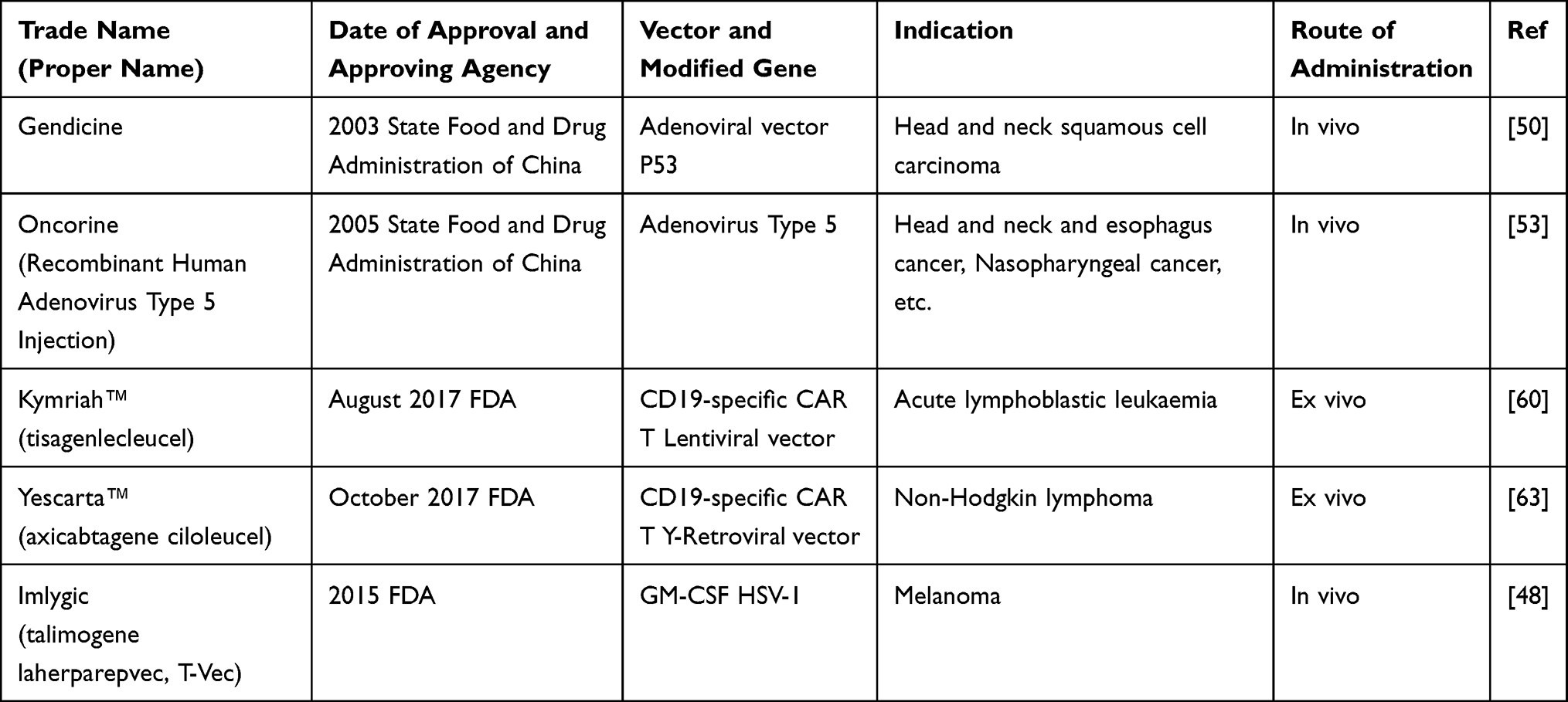 |
Table 1 Gene Therapies Products Approved for Therapeutic Use |
Oncolytic Virotherapy
Oncolytic virotherapy (OV) is the most promising approach for tumor immunotherapy. OV uses replication-competent viruses that can proliferate selectively at tumor cells. Oncolytic viruses grouped as naturally occurring or genetically modified viruses. Natural occurring viruses like parvoviruses, and Newcastle disease viruses that selectively replicate in tumor cell without genetic modification. The second virus category, such as vesicular stomatitis viruses, adenoviruses, measles viruses, HSV and vaccinia viruses, genetically modified to improve the safety, tumor-specificity, and decrease virus pathogenicity. The therapeutic use of oncolytic viruses for cancer treatment is an immune-related treatment alternative. Oncolytic viruses act by directly lyses tumor cells and by introducing wild-type tumor suppressor genes into cells that lack the tumor suppressor gene.48,49 Change in p53 gene function is present in half of all malignancies, and the induction of wild-type p53 gene re-establishes the normal p53 expression. Several recombinant OVs expressing p53 were developed with the aim of producing more potent OVs that act in combination with host immunity or with other treatments’ modality to destroy tumor cells.49,50
Gendicine (Recombinant Human P53 Adenovirus [Ad5RSV-P53])
Was the first approved gene product for the management of neck and head squamous cell carcinoma in 2003.50 Gendicine is a non-replicative an adenoviral vector, where the E1 gene is replaced with the tumor suppressor p53 cDNA gene. The expression of p53 in tumor cells triggers the antitumor effect by activating the apoptotic pathway, inhibit damaged DNA repair, and anti-apoptotic activity. P53 gene mutation is prevalent in several cancers. Therefore, Gendicine induces the expression of p53 restores its activity and destroys the tumor cells. Generally, Gendicine management showed 30–40% complete response and 50–60% partial response with a total response rate of 90%–96% in different therapeutic use. Up-to-date greater than 30,000 patients managed by Gendicine.50,51
Oncorine (rAd5-H101)
It is the first replicative, oncolytic recombinant ad5 (rAd5-H101) approved to treat refractory nasopharyngeal cancer. Loss of p53 gene linked with drug resistance and survival rate reduction in non-small cell cancer patients.50 Oncorine is an ad5 virus with a deletion in the E1B 55K gene. Host cell p53 gene inactivation is essential for wild-type to block the activation of apoptotic pathway. The removal of the E1B 55K gene inhibits viral proliferation in normal cells, allowing only proliferate in p53-deficient host cells. In tumor cells, viral proliferation causes oncolysis that is the mechanism to treat solid tumors. Following cancer cell lysis, adenoviruses release and infect another cell activating a serious of Oncorine-mediated cell death.52,53
Imlygic (Talimogene Laherparepvec)
It is a genetically modified oncolytic HSV-1 approved in Europe in 2015 for the management of non-resectable metastatic melanoma. Imlygic is the first oncolytic virus used for the management of advanced melanoma.48 The replacement of γ34.5 and α47 genes with the human granulocyte-macrophage colony-stimulating factor (GM-CSF) gene modifies the HSV-1 gene. The γ34.5 gene deletion causes tumor cell-selective replication and suppression of pathogenicity. The γ34.5 gene blocks protein synthesis of the host cell during viral infection. Thus, suppressing γ34.5 seizes the virus proliferation in normal cells. In tumor cells, the γ34.5 gene deleted HSV-1 can replicate. The α47gene inhibits the host cell transporter associated with antigen presentation. The depletion of α47gene reduces MHC class I expression that increases antitumor immune activity.53 Besides, two human GM-CSF genes inserted into the virus providing high levels of GM-CSF production, and stimulate immune responses. Administration of Imlygic causes apoptosis of tumor cell enhanced antigen presentation and increased antitumor response.49,54
Rexin-G (Mx-dnG1)
Is the first targeted injectable vector approved for the management of metastatic cancers. It is a replication-incompetent retroviral vector showing a SIG-binding peptide to bind to abnormal Signature (SIG) proteins in the tumor cell that increase vector concentration in tumor cells and express a dominant-negative human cyclin G1 inhibitor. After the entrance into the tumor cells, Rexin-G synthesizes cytocidal dnG1 proteins that inhibit the cell cycle in the G1 phase resulting in apoptosis of cancer cells.55,56
Chimeric Antigen Receptor (Car) T Cell Therapy
T cells destroy infected and tumor cells by detecting nonself antigens with the T cell receptor (TCR). CAR T is a T cell transduced with a chimeric antigen receptor specific to a tumor-associated antigen. CAR is “chimeric” because it contains the antigen-binding site of the B cell receptor and an intracellular TCR activation domain. CAR has three domains, an extracellular domain that has cancer-specific epitopes (scfv region) made from light (VL) and heavy (VH) chains of immunoglobin that target antigen (such as CD19), a transmembrane domain, and intracellular TCR derived stimulatory domains as showed in Figure 2. The scfv component binds to the target antigen in the MHC independent way leading to CAR clustering and stimulating T-cell via intracellular region that posses the TCR-derived CD3ζ chain, with or without co-stimulatory domains. Stimulated CAR T-cells give target-specific memory cells that inhibit tumor relapse.57 CD19‐targeted CAR T cells were the first CARs to be studied. CD19 is a promising target due to its expression limited to the B cell. First‐generation, CD19‐targeted CAR T cells were safe but ineffective. Second-generation CARs have a costimulatory domain with the CD3ζ activation domain show enhanced T cell activity. Two second‐generation, CD19‐targeted CARs are in clinical use contain a 41BB costimulatory domain (19‐BBz) and a CD28 costimulatory domain and those with more than one additional co-stimulatory molecule are known as “third-generation” CAR.57–59
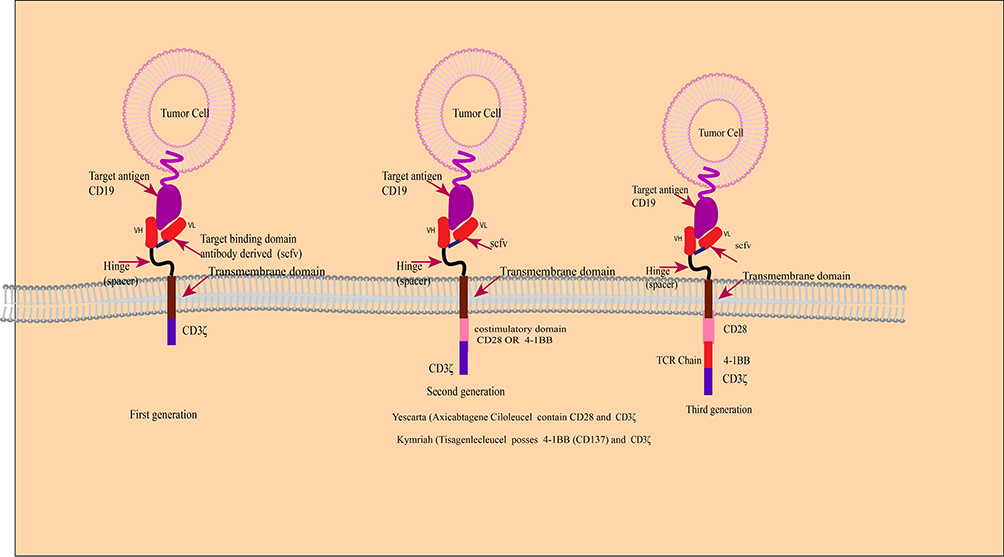 |
Figure 2 Schematic diagram of CAR-T-cell products. |
Kymriah (Tisagenlecleucel)
It is the first FDA approved CAR T-cell-based gene product to treat relapsed B-cell acute lymphoblastic leukemia. Kymriah has autologous T cells, modified with the lent virus to encode a CAR consist of a murine single-chain antibody fragment (scFv) selective for CD19, an intracellular domain 4–1BB (CD137), and CD3 zeta with CD8 transmembrane hinge. After binding to CD19 antigen-expressing cells, Kymriah initiates the antitumor effect via the CD3 domain. The intracellular 4–1BB co-stimulatory domains enhance the antitumor activity. The CD19 antigen is a 95-kD glycoprotein encoded as a surface antigen in diffuse large B-cell lymphoma (DLBCL) and other B-cell lymphomas.60,61 High response rates were recorded in patients with refractory DLBCL in Phase 2 clinical trials. The response rate was 50% at 3 months, 43% with a complete response at 6 months, and there were no patients with a complete response at 6 months who had a relapse by the median of 28.6 months.62
Yescarta (Axicabtagene Ciloleucel)
It is another CAR T-cell therapy used for the management of aggressive non-Hodgkin lymphoma. It is CD19 antigen-specific ex-vivo modified autologous T cells infected with a gamma-retroviral. It encodes a CAR comprising an extracellular murine anti-CD19 single-chain variable fragment fused to a cytoplasmic domain that possesses CD28 and CD3-zeta co-stimulatory domains.63,64
Zalmoxis (Allogenic T Cells Encoding LNGFR and HSV-TK)
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) uses for the management of several hematopoietic malignancies. But, acute graft-versus-host-disease (aGvHD) and Graft rejection are barriers to its success. The treatment strategy for haplo-HSCT depends on T-cell depletion or administration of lymphotoxin agents like cyclophosphamide after stem cell infusion to selectively deplete activated alloreactive lymphocytes but causes prolonged immunodeficiency post-transplantation. Thus, treatment to enhance immune reconstitution after transplantation is necessary.65 Zalmoxis is a genetically modified allogeneic T cell using a retroviral vector encoding a human low-affinity nerve growth factor receptor (ΔLNGFR) and HSV-TK Mut2 to transduce the allogeneic T immune cells. The ΔLNGFR expression uses as a marker of the transduced T cells, and the HSV-TK Mut2 expression provides the suicide gene induction during the administration of the prodrug ganciclovir (GCV). Administration of the genetically modified donor T cells to T cell-depleted transplant patients (HSCT) reconstitutes the immunity to defend from infections. But, donor cells may specifically act as the host cells leading to Graft Versus Host Disease (GVHD). In this case, induction of suicide gene by GCV administration may kill the donor T cells encoding HSV-TK and control GVHD. Zalmoxis is a potential curative agent for HSCT patients when the matched donor does not exist. Zalmoxis provides post-transplant GvHD control, Graft versus Leukemia (GvL) improvement, relapse decrease, and immune reconstitution causes reduced infection.52,66
Gene Silencing
Gene silencing therapy is RNA interference (RNAi)-mediated knockdown of specific genes in tumor cells. RNAi is single or double-stranded noncoding RNAs (21 ribonucleotides) that induce sequence-specific degradation of complementary mRNAs via the cells’ internal machinery.67 siRNA is vital because most genes do not have inhibitors due to a lack of ligand binding sites and amino acid sequence homology with other proteins that limit target selectivity. RNAi consists of microRNA (miRNA), Small Interfering RNA (siRNA) and short hairpin RNA (shRNA). Two decades later after the discovery of RNAi, ONPATTRO™ (patisiran) approved for the first time for the management of the polyneuropathy of hereditary transthyretin‐mediated (hATTR) amyloidosis.68 Tumor suppressor genes, oncogenes, genes involved in cancer progression, and drug-resistance are promising targets for gene silencing by RNAi-based cancer treatment due to selective gene silencing effect and relatively fewer adverse effects than conventional chemotherapy.69 The merits of RNAi in cancer treatment are targeting several genes of different cellular pathways involved in cancer progression and develop a drug for a specific patient.70 Several studies conducted on animals revealed that targeting vital proteins in the cell cycle, such as Protein kinase N3 (PKN3), kinesin spindle protein (KSP), and polo-like kinase 1 (PLK1) by siRNA displayed a potent antitumor effect. Several liposomal siRNA dose preparations are in Phase 1 trials, such as treatments for pancreatic cancer (PKN3 siRNA), liver cancer (CEBPA siRNA), and neuroendocrine tumors (PLK1 siRNA).71
Suicide Gene Therapy
Suicide gene therapy uses viral or bacterial genes into malignant cells that metabolize non-toxic prodrug into a toxic compound. Several suicide gene systems were identified including the HSV-thymidine kinase gene (HSV-TK) with ganciclovir (GCV) and the cytosine deaminase gene (CD) with 5-fluorocytosine (5-FC).72 Gene-mediated cytotoxic immunotherapy is one strategy where an adenoviral vector possessing the herpes virus thymidine kinase gene (AdV-TK) is administered locally into the tumor site that causes local expression of the HSV-TK gene to the synthesis of viral thymidine kinase that converts GCV to GCV monophosphate. The next step is the administration of GCV that is a substrate of HSV-TK and phosphorylated to produce GCV monophosphate. Then, cellular kinases metabolize GVC-monophosphate into GVC-triphosphate. GCV triphosphate is a deoxyguanosine triphosphate analog, incorporated into the DNA chain causing chain termination and tumor cell death.73
The anti-tumor effect of the TK/GCV system showed promising results in animal models. A study on hormone-refractory prostate cancer patients treated with HSV-TK delivered by adenovirus followed by GCV. The result showed response was at the surrogate marker level and safe. Several studies are in Phase III trials.74 The cytosine deaminase (CD) enzyme exists in fungi and bacteria but not in mammalian cells, metabolizes cytosine into uracil. CD metabolizes the non-toxic prodrug 5-FC into 5-FU, which is subsequently metabolized by cellular enzymes into 5-FdUMP, 5-FdUTP, and 5-FUTP. Inhibition of thymidylate synthase and production of (5-FU) DNA and RNA are the mode of cell death induced by the CD/5-FC suicide system. 5-FU uses for cancer treatment but requires a high dose. This suicide system results in tumor-targeted chemotherapy with few side effects. The CD/5-FC system improved by the inclusion uracil phosphoribosyltransferase (UPRT) gene that phosphorylates 5-FU to 5-fluorouridine mono-phosphate, the first step of its pathway to activation.75 The anti-tumor effect of the CD/5-FC combination showed a better efficacy in animal models. A study on refractory cancer patients that involved intratumoral administration of TAPET-CD attenuated Salmonella bacterium encoding the E. coli CD gene in three patients. The study showed a significant effect and lack of side effects. An oncolytic adenovirus possessing a CD/HSV-1 TK gene was used in a phase I study in patients with prostate cancer. The result showed that the transgene encoding persistence in the prostate for 3 weeks after administration.76
Anti-Tumor Angiogenesis
Tumor-driven angiogenesis several growth factors are involved, such as vascular endothelial growth factor (VEGF), fibroblast growth factor-2 (FGF-2), angiopoietins or IL-8, to secure oxygen and nutrients supply. Two major approaches are being pursued to block tumor angiogenesis. The first approach is down-regulation of pro-angiogenic factors expression, such as VEGF, and the second approach is up-regulation of expression of anti-angiogenic factors such as angiostatin, endostatin, and human soluble FMS-like tyrosine kinase receptor. Despite the successful therapeutic use of mAb like Bevacizumab for targeted therapy of cancer, the production and administration of therapeutic mAb are limited due to costly production. Therefore, gene-based studies were done to develop an angiogenesis-targeted cancer treatment.77,78
Conclusion
Gene therapy represents a novel alternative for the management of diseases that have no satisfactory cure. Gene therapy for cancer treatment has good progress in the last three decades, few drugs approved, while others are still in trials. Relatively gene therapy has better safety with tolerable adverse effects than chemotherapy for the treatment of cancer. In the future, tumor genomic analysis, assessment of host humoral and cellular immunity will facilitate a better selection of the most appropriate patient for gene therapy. Recent progress in developing safe and effective vectors for gene delivery, and understanding the activity of nucleases facilitate future genome editing as new treatment approaches for untreatable diseases like cancer.
The success of using autologous and allogenic chimeric antigen receptor integrated T-lymphocytes in mediating adoptive immunotherapy enhances the safety and effectiveness of gene therapy. Besides, the enhanced biological research, cheaper gene vectors will be available in the market, which increases gene therapy accessibility for most cancer patients. This will change the future of cancer treatment, from generalized cancer treatment strategies to individualized cancer treatment, based on the patient’s specific genome, immune status, and genetic profile of the tumor. Gene therapy is expected to be fast, effective, less toxic, and inexpensive, with higher cure rates. In November 2017, more than 2597 clinical trials are ongoing in several countries and a few of them are listed in Table 2. Until August 2019, 22 gene medicines had been approved by the drug regulatory agencies from various countries.79 Gene therapy gradually accepted by the government and the public since the 1980s and has become an important alternative to the existing treatments in the past few years. Therefore, gene therapy drugs, with safe vectors and advanced biotechnologies, would play a greater role in the prophylaxis and management of cancer in the future.
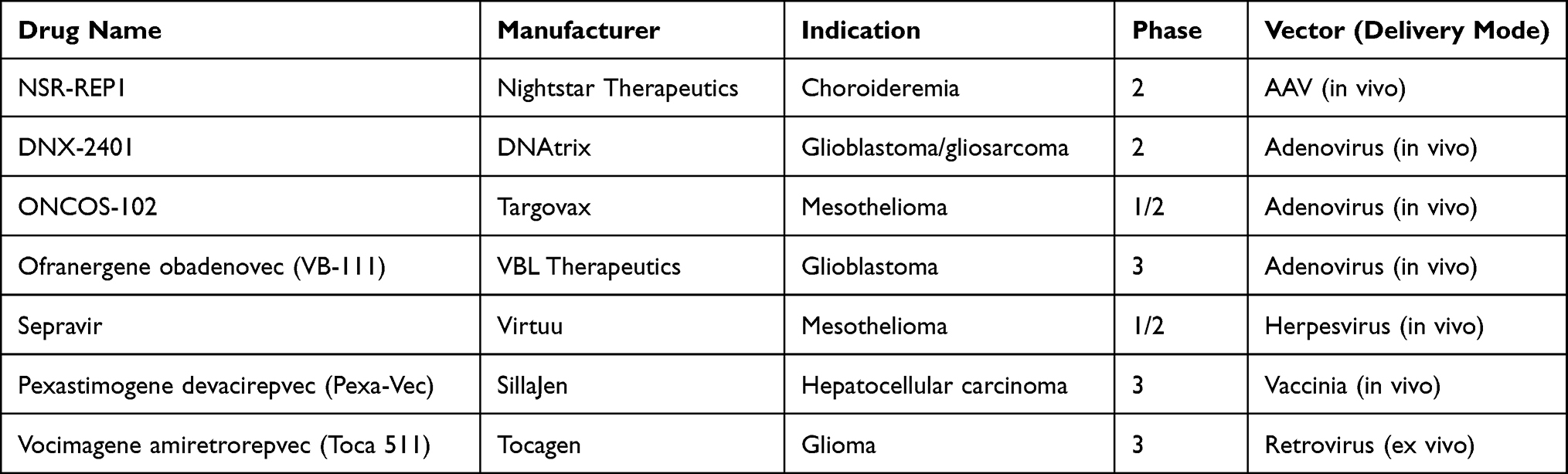 |
Table 2 Gene Therapies Products Candidates Under Clinical Trial |
Abbreviations
ADA, adenosine deaminase; Ad, adenovirus; AAV, adeno-associated virus; aGvHD, acute graft-versus-host-disease; allo-HSCT, allogeneic hematopoietic stem cell transplantation; CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats; CAR, chimeric antigen receptor; DSBs, double-strand breaks; ERT, enzyme replacement therapy; HDR, homology-directed repair; HSV, herpes simplex virus; IRDs, inherited retinal degenerations; LV, lentivirus; NHEJ, non-homologous end joining; NMDs, neuromuscular disorders; OV, oncolytic virotherapy; tracrRNA, trans-activating RNA; TCR, T cell receptor; MNs, meganucleases.
Data Sharing Statement
All data are provided in the manuscript or found from published papers as cited.
Acknowledgment
I would like to acknowledge Mrs Fasika Abu for editing the manuscript for English Style.
Disclosure
The authors declare no competing interests in this work.
References
1. Wirth T, Parker N, Ylä-Herttuala S. History of gene therapy. Gene. 2013;525(2):162–169. doi:10.1016/j.gene.2013.03.137
2. Sharma AR, Kundu SK, Nam JS, et al. Next generation delivery system for proteins and genes of therapeutic purpose: why and how? Biomed Res Int. 2014;2014.
3. Templeton NS, editor. Gene and Cell Therapy: Therapeutic Mechanisms and Strategies. Crc Press; 2008.
4. Vermezovic J, Stergiou L, Hengartner MO, Di Fagagna FD. Differential regulation of DNA damage response activation between somatic and germline cells in Caenorhabditis elegans. Cell Death Differ. 2012;19(11):1847–1855. doi:10.1038/cdd.2012.69
5. Reis RL. Encyclopedia of Tissue Engineering and Regenerative Medicine. Academic Press; 2019.
6. Colella P, Ronzitti G, Mingozzi F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol Ther Methods Clin Dev. 2018;8:87–104. doi:10.1016/j.omtm.2017.11.007
7. Merten O-W, Charrier S, Laroudie N, et al. Large-scale manufacture and characterization of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum Gene Ther. 2011;22(3):343–356. doi:10.1089/hum.2010.060
8. Ginn SL, Amaya AK, Alexander IE, Edelstein M, Abedi MR. Gene therapy clinical trials worldwide to 2017: an update. J Gene Med. 2018;20(5):e3015.
9. Sung YK, Kim SW. The practical application of gene vectors in cancer therapy. Integrat Cancer Sci Therap. 2018;5:1–5.
10. Suleiman AA, Saedi WY, Muhaidi MJ. Widely used gene editing strategies in cancer treatment a systematic review. Gene Rep. 2020;100983.
11. Zhang HX, Zhang Y, Yin H. Genome editing with mRNA encoding ZFN, TALEN, and Cas9. Mol Therapy. 2019;27(4):735–746. doi:10.1016/j.ymthe.2019.01.014
12. Ernst MP, Broeders M, Herrero-Hernandez P, Oussoren E, van der Ploeg AT, Pijnappel WP. Ready for repair? Gene editing enters the clinic for the treatment of human disease. Mol Therapy Methods Clin Dev. 2020.
13. Sallmyr A, Tomkinson AE. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J Biol Chem. 2018;293(27):10536–10546. doi:10.1074/jbc.TM117.000375
14. Zaman QU, Li C, Cheng H, Hu Q. Genome editing opens a new era of genetic improvement in polyploid crops. Crop J. 2019;7(2):141–150. doi:10.1016/j.cj.2018.07.004
15. Palpant NJ, Dudzinski D. Zinc finger nucleases: looking toward translation. Gene Ther. 2013;20(2):121–127. doi:10.1038/gt.2012.2
16. Cassandri M, Smirnov A, Novelli F, et al. Zinc-finger proteins in health and disease. Cell Death Discovery. 2017;3(1):1–2. doi:10.1038/cddiscovery.2017.71
17. Sun N, Zhao H. Transcription activator‐like effector nucleases (TALENs): a highly efficient and versatile tool for genome editing. Biotechnol Bioeng. 2013;110(7):1811–1821. doi:10.1002/bit.24890
18. Nakano C, Kitabatake Y, Takeyari S, et al. Genetic correction of induced pluripotent stem cells mediated by transcription activator-like effector nucleases targeting ALPL recovers enzyme activity and calcification in vitro. Mol Genet Metab. 2019;127(2):158–165. doi:10.1016/j.ymgme.2019.05.014
19. Hille F, Richter H, Wong SP, Bratovič M, Ressel S, Charpentier E. The biology of CRISPR-Cas: backward and forward. Cell. 2018;172(6):1239–1259.
20. Rath D, Amlinger L, Rath A, Lundgren M. The CRISPR-Cas immune system: biology, mechanisms and applications. Biochimie. 2015;117:119–128. doi:10.1016/j.biochi.2015.03.025
21. Terns RM, Terns MP. CRISPR-based technologies: prokaryotic defense weapons repurposed. Trends Genetics. 2014;30(3):111–118. doi:10.1016/j.tig.2014.01.003
22. Li Y, Glass Z, Huang M, Chen ZY, Xu Q. Ex vivo cell-based CRISPR/Cas9 genome editing for therapeutic applications. Biomaterials. 2020;234:119711. doi:10.1016/j.biomaterials.2019.119711
23. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–1278. doi:10.1016/j.cell.2014.05.010
24. Mali S. Delivery systems for gene therapy. Indian J Hum Genet. 2013;19(1):3. doi:10.4103/0971-6866.112870
25. Sung YK, Kim SW. Recent advances in the development of gene delivery systems. Biomaterials Res. 2019;23(1):8. doi:10.1186/s40824-019-0156-z
26. Humphreys IR, Sebastian S. Novel viral vectors in infectious diseases. Immunology. 2018;153(1):1–9. doi:10.1111/imm.12829
27. Bouard D, Alazard‐Dany N, Cosset FL. Viral vectors: from virology to transgene expression. Br J Pharmacol. 2009;157(2):153–165. doi:10.1038/bjp.2008.349
28. Goswami R, Subramanian G, Silayeva L, et al. Gene therapy leaves a vicious cycle. Front Oncol. 2019;9:297.
29. Pilgrim S, Stritzker J, Schoen C, et al. Bactofection of mammalian cells by Listeria monocytogenes: improvement and mechanism of DNA delivery. Gene Ther. 2003;10(24):2036–2045. doi:10.1038/sj.gt.3302105
30. Johnson SA, Ormsby MJ, McIntosh A, Tait SW, Blyth K, Wall DM. Increasing the bactofection capacity of a mammalian expression vector by removal of the f1 ori. Cancer Gene Ther. 2019;26(7):183–194. doi:10.1038/s41417-018-0039-9
31. Celec P, Gardlik R. Gene therapy using bacterial vectors. Front Biosci. 2017;22:81–95. doi:10.2741/4473
32. Wang W, Li W, Ma N, Steinhoff G. Non-viral gene delivery methods. Curr Pharm Biotechnol. 2013;14(1):46–60.
33. Ramamoorth M, Narvekar A. Non viral vectors in gene therapy-an overview. J Clin Diagnostic Res. 2015;9(1):GE01. doi:10.7860/JCDR/2015/10443.5394
34. Buck J, Grossen P, Cullis PR, Huwyler J, Witzigmann D. Lipid-based DNA therapeutics: hallmarks of non-viral gene delivery. ACS Nano. 2019;13(4):3754–3782. doi:10.1021/acsnano.8b07858
35. Yang R, Chen F, Guo J, Zhou D, Luan S. Recent advances in polymeric biomaterials-based gene delivery for cartilage repair. Bioactive Mater. 2020;5(4):990–1003. doi:10.1016/j.bioactmat.2020.06.004
36. Van Bruggen C, Hexum JK, Tan Z, Dalal RJ, Reineke TM. Nonviral gene delivery with cationic glycopolymers. Acc Chem Res. 2019;52(5):1347–1358. doi:10.1021/acs.accounts.8b00665
37. Darr JA, Zhang J, Makwana NM, Weng X. Continuous hydrothermal synthesis of inorganic nanoparticles: applications and future directions. Chem Rev. 2017;117(17):11125–11238. doi:10.1021/acs.chemrev.6b00417
38. Riley MK, Vermerris W. Recent advances in nanomaterials for gene delivery—a review. Nanomaterials. 2017;7(5):94. doi:10.3390/nano7050094
39. Alsaggar M, Liu D. Physical Methods for Gene Transfer. In Advances in Genetics. Vol. 89. Academic Press; 2015:1–24.
40. Li X, Ruddy B, Taberner A. Characterization of needle-assisted jet injections. J Controlled Release. 2016;243:195–203. doi:10.1016/j.jconrel.2016.10.010
41. Suda T, Liu D. Hydrodynamic gene delivery: its principles and applications. Mol Therapy. 2007;15(12):2063–2069. doi:10.1038/sj.mt.6300314
42. Herrero MJ, Sendra L, Miguel A, Aliño SF. Physical Methods of Gene Delivery. In Safety and Efficacy of Gene-Based Therapeutics for Inherited Disorders. Cham: Springer; 2017:113–135.
43. Du X, Wang J, Zhou Q, et al. Advanced physical techniques for gene delivery based on membrane perforation. Drug Deliv. 2018;25(1):1516–1525. doi:10.1080/10717544.2018.1480674
44. Smolders S, Kessels S, Smolders SM, et al. Magnetofection is superior to other chemical transfection methods in a microglial cell line. J Neurosci Methods. 2018;293:169–173. doi:10.1016/j.jneumeth.2017.09.017
45. Li T, Kang G, Wang T, Huang H. Tumor angiogenesis and anti-angiogenic gene therapy for cancer. Oncol Lett. 2018;16(1):687–702. doi:10.3892/ol.2018.8733
46. Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, et al. Current clinical applications of in vivo gene therapy with AAVs. Mol Ther. 2021;29(2):464–488. doi:10.1016/j.ymthe.2020.12.007
47. Tristán-Manzano M, Justicia-Lirio P, Maldonado-Pérez N, Cortijo-Gutiérrez M, Benabdellah K, Martin F. Externally-controlled systems for immunotherapy: from bench to bedside. Front Immunol. 2020;11.
48. Bommareddy PK, Patel A, Hossain S, Kaufman HL. Talimogene laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. Am J Clin Dermatol. 2017;18(1):1–5. doi:10.1007/s40257-016-0238-9
49. Sostoa JD, Dutoit V, Migliorini D. Oncolytic viruses as a platform for the treatment of malignant brain tumors. Int J Mol Sci. 2020;21(20):7449. doi:10.3390/ijms21207449
50. Zhang -W-W, Li L, Li D, et al. The first approved gene therapy product for cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum Gene Ther. 2018;29(2):160–179. doi:10.1089/hum.2017.218
51. Xia Y, Li X, Sun W. Applications of recombinant adenovirus-p53 gene therapy for cancers in the clinic in China. Curr Gene Ther. 2020;20(2):127–141. doi:10.2174/1566523220999200731003206
52. Shahryari A, Saghaeian Jazi M, Mohammadi S, Razavi Nikoo H, Nazari Z, Hosseini ES. Development and clinical translation of approved gene therapy products for genetic disorders. Front Genet. 2019;10:868. doi:10.3389/fgene.2019.00868
53. Russell L, Peng K-W. The emerging role of oncolytic virus therapy against cancer. Chine Clin Oncol. 2018;7(2):16. doi:10.21037/cco.2018.04.04
54. Osman AE, Luke JJ. The impact of the fecal microbiome on cancer immunotherapy. BioDrugs. 2019;33(1):
55. Chawla SP, Bruckner H, Morse MA, Assudani N, Hall FL, Gordon EM. A Phase I-II study using rexin-g tumor-targeted retrovector encoding a dominant-negative cyclin g1 inhibitor for advanced pancreatic cancer. Mol Therapy Oncolytics. 2019;12:56–67. doi:10.1016/j.omto.2018.12.005
56. Gordon EM, Hall FL. Rexin-G, a targeted genetic medicine for cancer. Expert Opin Biol Ther. 2010;10(5):819–832. doi:10.1517/14712598.2010.481666
57. Bb Pettitt D, Arshad Z, Smith J, Stanic T, Holländer G, Brindley D. CAR-T cells: a systematic review and mixed methods analysis of the clinical trial landscape. Mol Therapy. 2018;26(2):342–353. doi:10.1016/j.ymthe.2017.10.019
58. Oluwole OO, Davila ML. At The Bedside: clinical review of chimeric antigen receptor (CAR) T cell therapy for B cell malignancies. J Leukoc Biol. 2016;100(6):1265–1272. doi:10.1189/jlb.5BT1115-524R
59. Ali S, Kjeken R, Niederlaender C, et al. The European Medicines Agency Review of Kymriah (Tisagenlecleucel) for the Treatment of Acute Lymphoblastic Leukemia and Diffuse Large B-Cell Lymphoma. oncologist. 2020;25(2):e321. doi:10.1634/theoncologist.2019-0233
60. Gill S, Maus MV, Porter DL. Chimeric antigen receptor T cell therapy: 25years in the making. Blood Rev. 2016;30(3):157–167. doi:10.1016/j.blre.2015.10.003
61. Liu Y, Chen X, Han W, Zhang Y. Tisagenlecleucel, an approved anti-CD19 chimeric antigen receptor T-cell therapy for the treatment of leukemia. Drugs Today. 2017;53(11):597. doi:10.1358/dot.2017.53.11.2725754
62. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Eng J Med. 2019;380(1):45–56. doi:10.1056/NEJMoa1804980
63. Bouchkouj N, Kasamon YL, de Claro RA, et al. FDA approval summary: axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin Cancer Res. 2019;25(6):1702–1708. doi:10.1158/1078-0432.CCR-18-2743
64. Jacobson CA, Farooq U, Ghobadi A. Axicabtagene ciloleucel, an Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy for Relapsed or Refractory Large B-cell lymphoma: practical implications for the community oncologist. Oncologist. 2020;25(1):e138. doi:10.1634/theoncologist.2019-0395
65. Mohty M, Labopin M, Velardi A, et al. Allogeneic genetically modified T Cells (HSV-TK) as adjunctive treatment in haploidentical hematopoietic stem-cell transplantation (haplo-HSCT) of adult patients with high-risk hematological malignancies: a pair-matched analysis from the acute leukemia working party of EBMT. 2016. 672.
66. Hoogendoorn KH. Advanced Therapies: clinical, Non-clinical and Quality. Pharm Biotechn. 2019;357.
67. Parashar D, Rajendran V, Shukla R, Sistla R. Lipid-based nanocarriers for delivery of small interfering RNA for therapeutic use. Eur J Pharm Sci. 2020;142:105159. doi:10.1016/j.ejps.2019.105159
68. Takeshita F, Ochiya T. Therapeutic potential of RNA interference against cancer. Cancer Sci. 2006;97(8):689–696. doi:10.1111/j.1349-7006.2006.00234.x
69. Hu B, Weng Y, Xia X-H, Liang X-J, Huang Y. Clinical advances of siRNA therapeutics. J Gene Med. 2019;21(7):e3097. doi:10.1002/jgm.3097
70. Hu B, Zhong L, Weng Y, et al. Therapeutic siRNA: state of the art. Signal Transduction Targeted Ther. 2020;5(1):1–25.
71. Hossen MN, Wang L, Chinthalapally HR, et al. Switching the intracellular pathway and enhancing the therapeutic efficacy of small interfering RNA by auroliposome. Sci Adv. 2017;22(1):eaba5379. doi:10.1126/sciadv.aba5379
72. Düzgüneş N. Origins of Suicide Gene Therapy. In Suicide Gene Therapy. New York, NY: Humana Press; 2019:1–9.
73. Dey D, Evans GR. Suicide gene therapy by herpes simplex virus-1 thymidine kinase (HSV-TK). Targets Gene Therapy. 2011;65.
74. Sonabend AM, Ulasov IV, Lesniak MS. Gene therapy trials for the treatment of high-grade gliomas. Gene Ther Mol Biol. 2007;11(A):79.
75. Aučynaitė A, Rutkienė R, Tauraitė D, Meškys R, Urbonavičius J. Discovery of bacterial deaminases that convert 5-fluoroisocytosine into 5-fluorouracil. Front Microbiol. 2018;9:2375. doi:10.3389/fmicb.2018.02375
76. Zarogoulidis P, Darwiche K, Sakkas A, et al. Suicide gene therapy for cancer–current strategies. J Genet Syndr Gene Ther. 2013;4.
77. El‐Kenawi AE, El‐Remessy AB. Angiogenesis inhibitors in cancer therapy: mechanistic perspective on classification and treatment rationales. Br J Pharmacol. 2013;170(4):712–729.
78. Li T, Kang G, Wang T, Huang H. Tumor angiogenesis and anti-angiogenic gene therapy for cancer. Oncol Lett. 2018;16(1):687–702.
79. Ma CC, Wang ZL, Xu T, He ZY, Wei YQ. The approved gene therapy drugs worldwide: from 1998 to 2019. Biotechnol Adv. 2020;40:107502.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.