")
Back to Archived Journals » Oncolytic Virotherapy » Volume 10
The Current State of Oncolytic Herpes Simplex Virus for Glioblastoma Treatment
Received 5 December 2020
Accepted for publication 18 January 2021
Published 24 February 2021 Volume 2021:10 Pages 1—27
DOI https://doi.org/10.2147/OV.S268426
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tommy Alain
Hong-My Nguyen, Dipongkor Saha
Department of Immunotherapeutics and Biotechnology, Texas Tech University Health Sciences Center, School of Pharmacy, Abilene, TX, 79601, USA
Correspondence: Dipongkor Saha Tel +1 325-6960583
Email [email protected]
Abstract: Glioblastoma (GBM) is a lethal primary malignant brain tumor with no current effective treatments. The recent emergence of immuno-virotherapy and FDA approval of T-VEC have generated a great expectation towards oncolytic herpes simplex viruses (oHSVs) as a promising treatment option for GBM. Since the generation and testing of the first genetically engineered oHSV in glioma in the early 1990s, oHSV-based therapies have shown a long way of great progress in terms of anti-GBM efficacy and safety, both preclinically and clinically. Here, we revisit the literature to understand the recent advancement of oHSV in the treatment of GBM. In addition, we discuss current obstacles to oHSV-based therapies and possible strategies to overcome these pitfalls.
Keywords: glioblastoma, oncolytic herpes simplex virus, armed oHSV, re-targeted oHSV, combination therapy
Introduction
Oncolytic viruses (OVs)1,2 such as oncolytic herpes simplex virus (oHSV) are an emerging treatment strategy for glioblastoma (GBM).3–10 GBM is the most common primary malignant brain tumor that is almost always lethal and has no effective treatments.11,12 OVs selectively replicate in and kill cancer cells and induce immunogenic cell death while stimulating anti-tumor immunity (ie, in situ vaccine effect).13,14 Among all OVs, oHSV is the furthest along in the clinic, with an oHSV talimogene laherparepvec (T-VEC) approved for use in advanced melanoma.15 The highly stable genome, potent cytolytic capability, convenience in genome engineering, and availability of effective anti-herpetic drugs to treat adverse reactions make oHSV an attractive class of anti-cancer therapeutic.16 The first genetically modified oHSV for glioma treatment was reported in 1991.17 Since then, many oHSVs such as unarmed – genetically modified oHSVs but not armed with transgene, re-targeted – genetically altered for tumor receptor-specific viral entry, and armed – engineered to express therapeutic transgene variants, etc., have been developed and tested in GBM preclinically, either alone or in combination with other anti-cancer agents (Table 1–3, Figure 1–3). Some of them are currently under clinical trial evaluation and early oHSV trials confirmed their safety and signs of efficacy in GBM patients (Table 4, figure 1). Despite significant therapeutic advancement, various challenges to oHSV therapy still currently exist (such as suboptimal viral delivery, inefficient viral entry to cancer cells, limited oHSV replication/spread in the tumor microenvironment (TME), host’s anti-viral response, etc.) that compromise oHSV-mediated anti-tumor efficacy (Figure 4).4,18 With the advance in biotechnology and immuno-oncology, many multimodal strategies are budding out for GBM to overcome existing challenges (Figure 4).4,18 In this review, we will first highlight the recent preclinical and clinical studies conducted in GBM using oHSVs (unarmed, re-targeted, or armed), either as monotherapy or in combination. We will then provide a short overview of the current pitfalls of oHSV-based therapies and outline potential strategies to overcome the existing treatment hurdles.
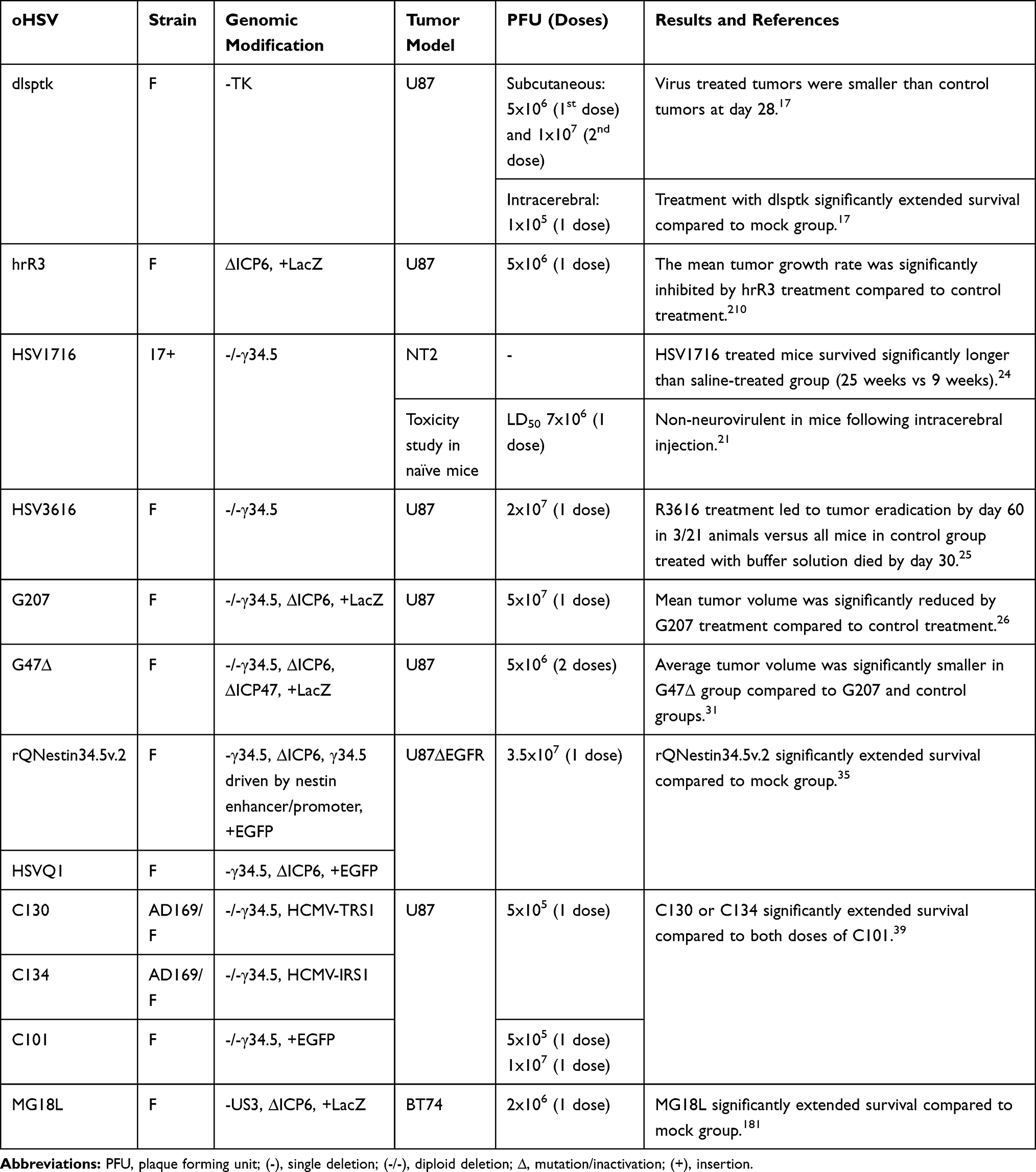 |
Table 1 List of Unarmed oHSVs for GBM Treatment |
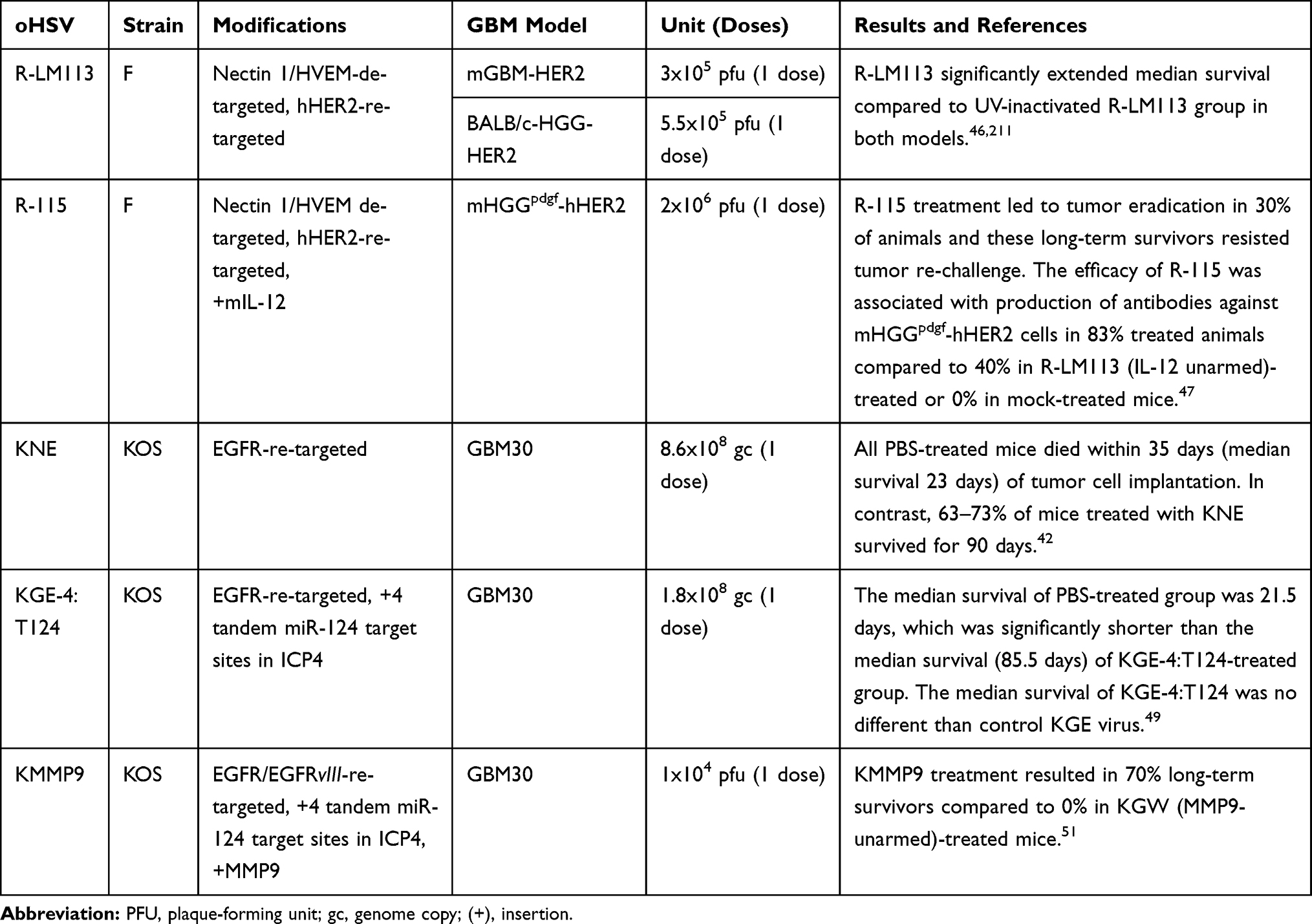 |
Table 2 List of Re-Targeted oHSVs for GBM Treatment |
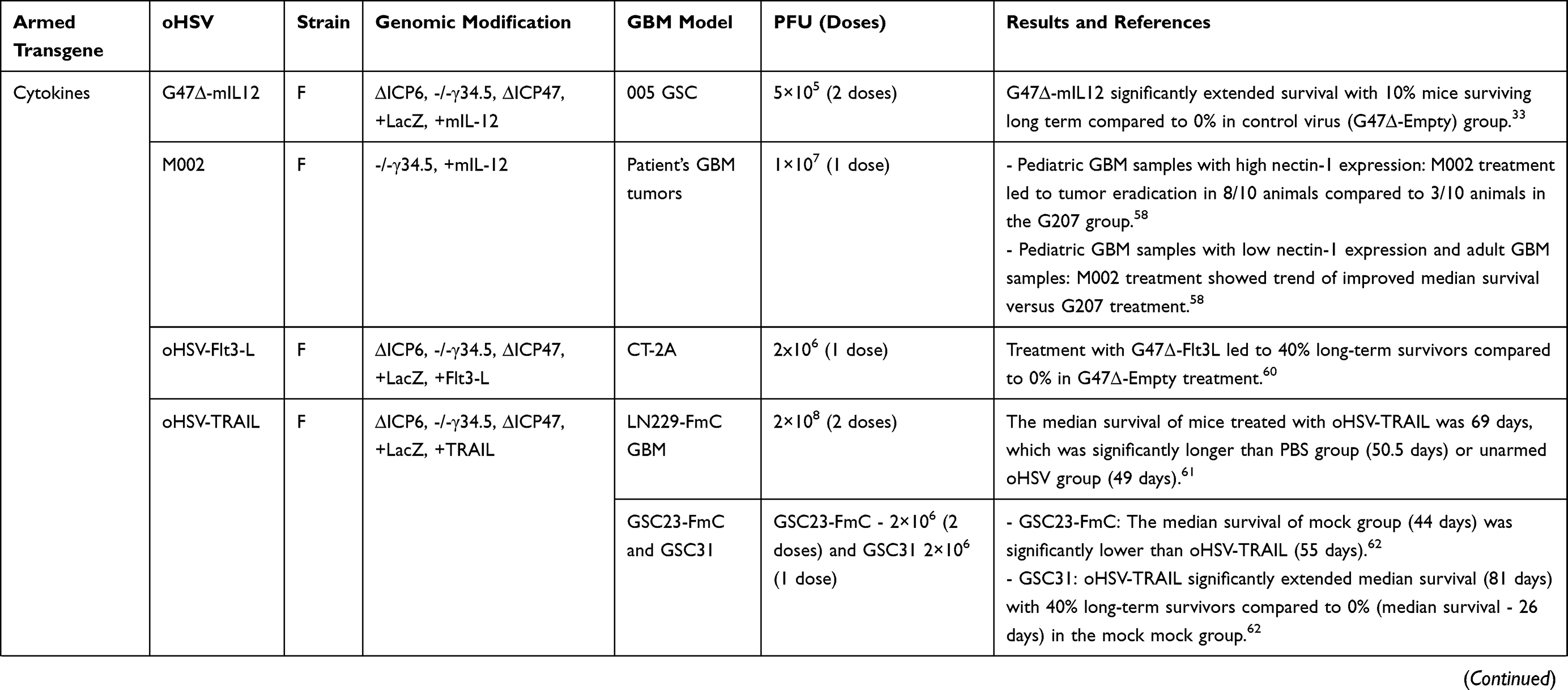 | 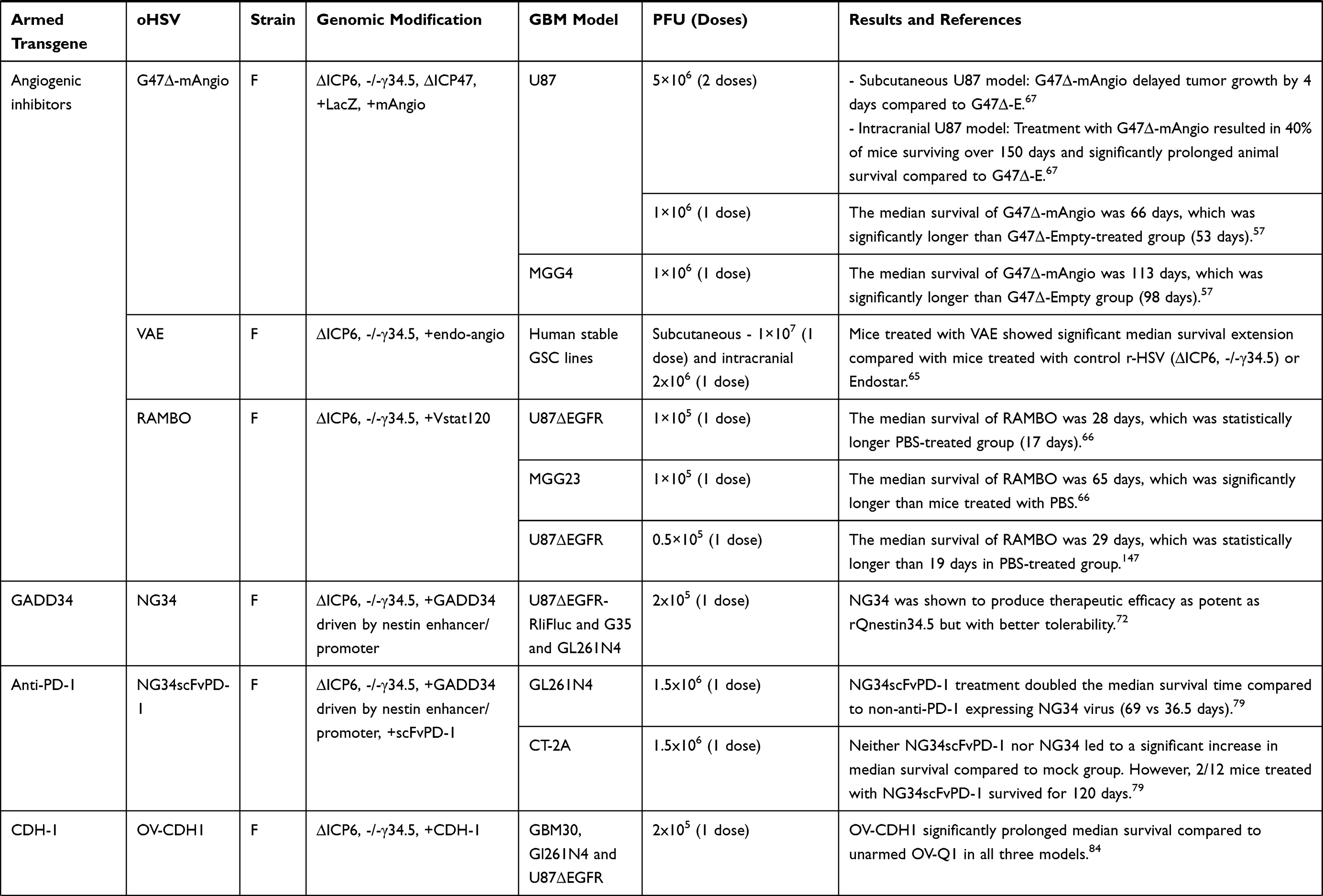 | 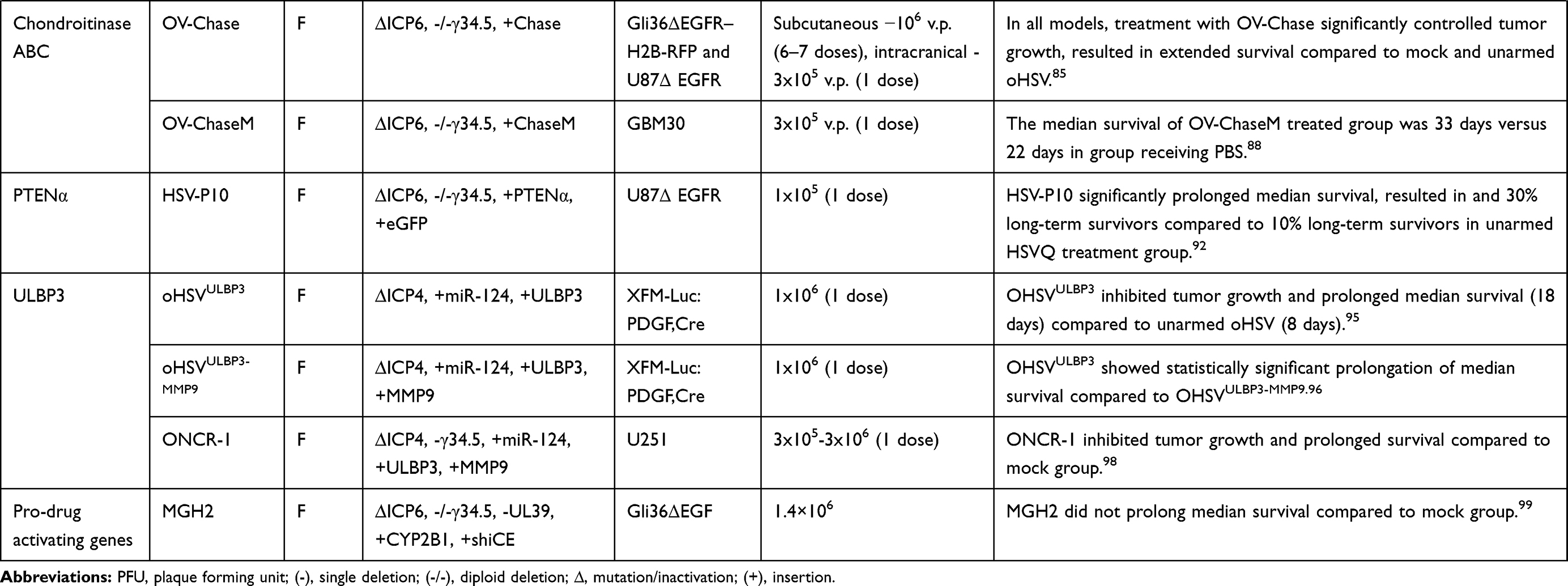 |
Table 3 List of Armed oHSVs for GBM Treatment |
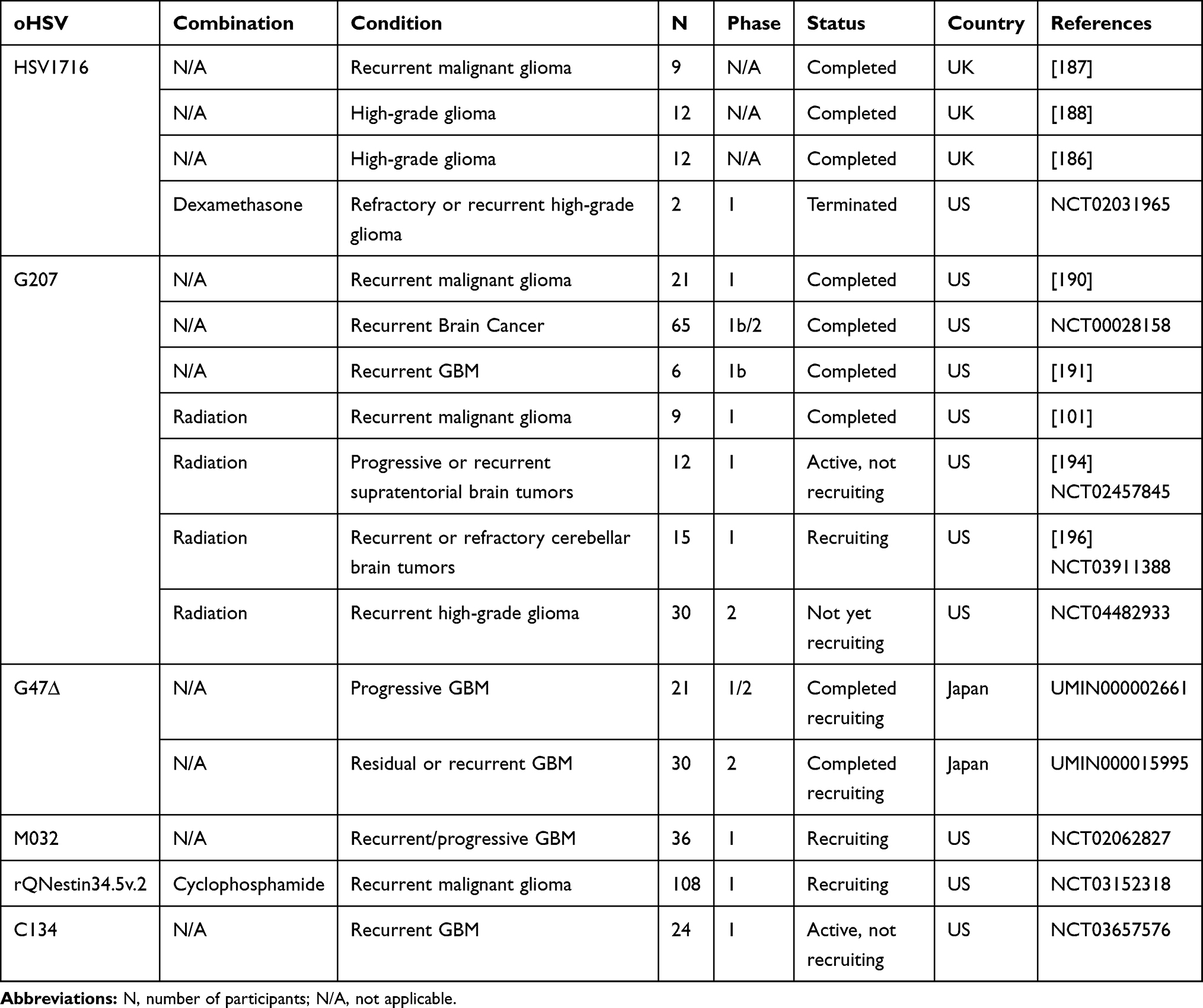 |
Table 4 Clinical Studies with oHSVs in GBM |
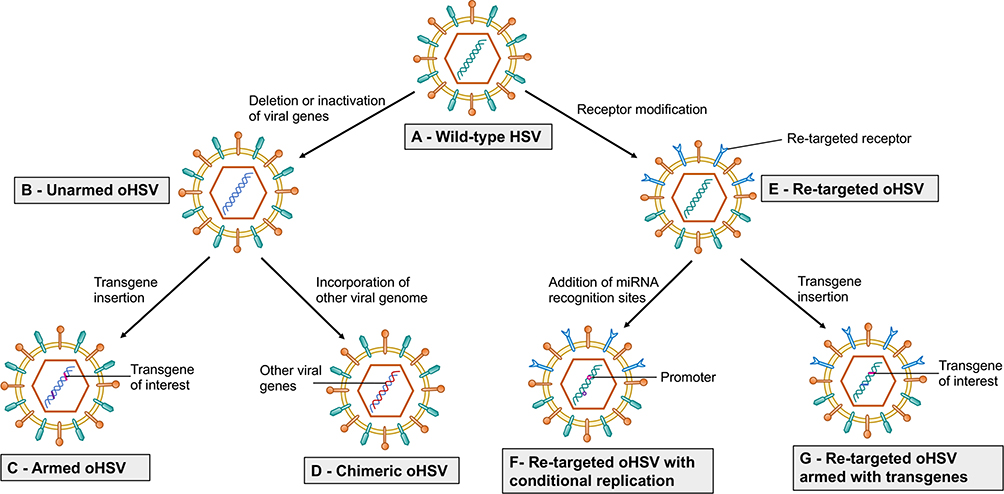 |
Figure 1 Strategy to enhance efficacy of oHSV as monotherapy. This figure represents different strategies to enhance the anti-GBM efficacy of oHSV. (A–B) Wild-type HSV can be genetically modified to remove or inactivate various genes to create unarmed oHSV. This is to ensure virus replication selectively occurs in cancer cells, thus enhancing the safety of oHSV (eg, dlsptk, hrR3, 1716, 3616, G207, G47∆, HSVQ1, rQNestin34.5, MG18L). (C) To further enhance oHSV efficacy, different transgene variants are incorporated into oHSV genome to generate armed oHSV. Transgene expression increases oHSV anti-cancer efficacy through enhancement of viral spread (eg, OV-CDH1, OV-ChaseM) or activation of the host immune response (eg, G47∆-mIL12, αMPD-1 scFv) or other means (See Table 3). (D) In addition, insertion of transgene that has similar function to oHSV genes but originating from other viruses is an alternative approach to enhance oHSV replication/oncolysis. oHSVs that are engineered by this method are defined as chimeric oHSV (eg, C130, C134). (E) Wild-type HSV can also be modified to enhance cancer-specificity, based on cellular surface receptor expression profile. They are known as re-targeted oHSVs, which selectively interact with various receptors that exclusively overexpressed in GBM cells but not in normal neurons/or glial cells (eg, R-LM113, KNE). This approach allows maximal oncolysis because HSV genome remains intact. (F) To enhance safety and avoid off-target effects, re-targeted oHSVs (E) can be further genetically modified to include miRNA recognition binding sites (such as miR-124 in KGE-4:T124) whose expression are distinct in GBM cells compared to healthy neurons/glial cells. These recognition-binding sites are incorporated into the untranslated region (UTR) of important genes responsible for viral pathogenicity. Based on the difference between GBM cells and healthy neurons/glial cells, these recognition binding sites can be occupied and the sequential genes are not translated in normal cells, whereas the recognition binding sites are free and viral translation takes place in GBM cells. In addition, re-targeted oHSVs (E) can be armed with different transgene of interest (G) to further enhance anti-tumor efficacy (eg, R-115, KMMP9). |
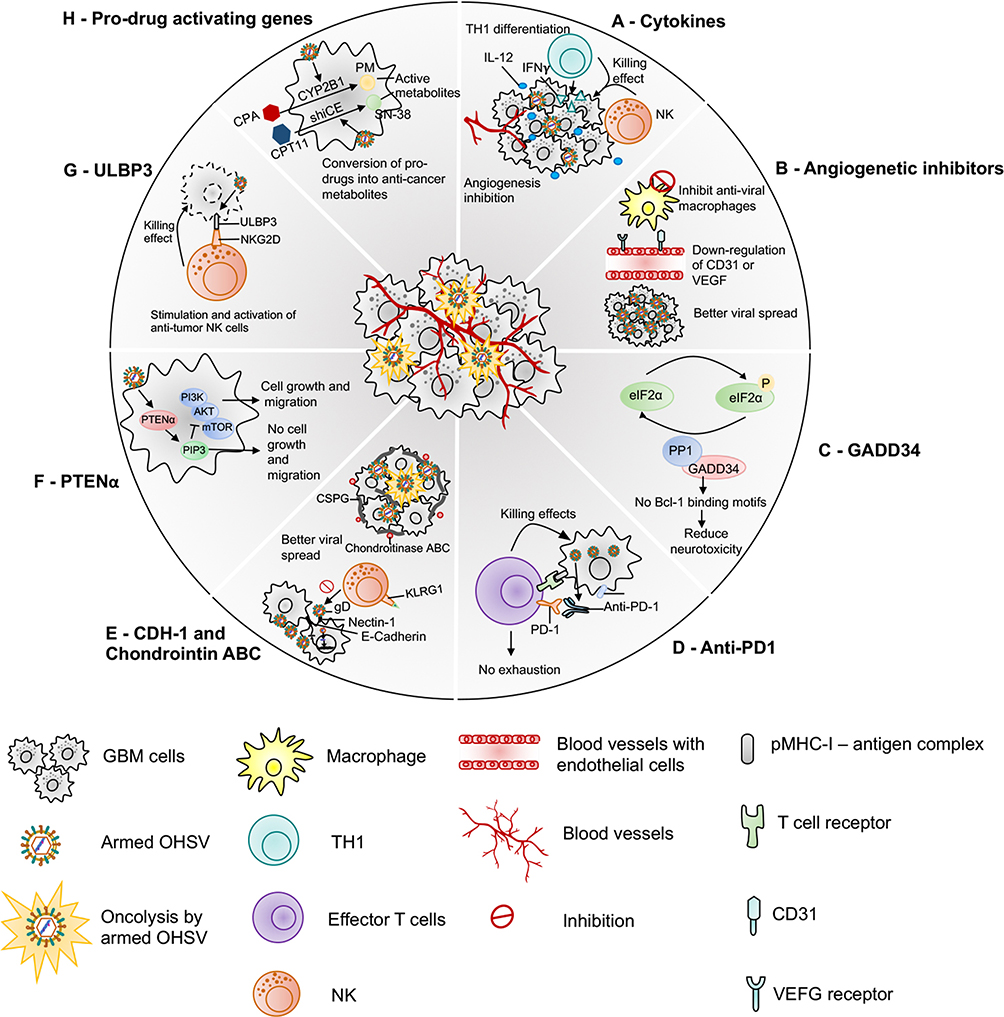 |
Figure 2 Armed oHSVs for GBM treatment. This figure represents different transgene variants that are inserted in the oHSV genome to enhance the anti-tumor efficacy of oHSV. (A) oHSV armed with cytokine such as IL-12, which induces Th1 differentiation, stimulates growth and cytotoxicity of NKs, increases IFN-γ production, and inhibits angiogenesis. (B) oHSV armed with angiogenic inhibitors decrease tumor vascularity (CD31+ vessels) or VEGF, inhibit anti-viral macrophages, and increase viral spread. (C) oHSV can be engineered to replace one copy of γ34.5 with GADD34. GADD34 binds to PP1 and promotes eIF-2α dephosphorylation - a function that corresponds to γ34.5 in HSV. Since GADD34 does not possess beclin-1-binding motifs of γ34.5, it does not produce neurotoxicity as wild-type HSV containing both copies of γ34.5. (D) Localized expression of anti-PD-1 by an oHSV inhibits PD-1/PD-L1 engagement, prevents T cell exhaustion and unleashes anti-tumor immunity. (E) oHSV can be armed with CDH-1 gene, which encodes for E-cadherin – an adhesion molecule and a ligand for an inhibitory receptor expressed on NK cells (KLRG1). E-cadherin can cooperate with nectin-1 and promote cell-to-cell adherent junctions and enhance cell-to-cell oHSV spread. Another strategy to increase viral spread is to create an oHSV that expresses chondroitinase ABC, which removes side chain of CSPG, prevents extracellular space tortuosity, and thus, facilitates viral spread. (F) PTENα expressed by an oHSV metabolizes PIP3 prevents activation of the PI3K/AKT/mTOR signaling pathway and inhibits tumor growth and migration. (G) oHSV armed with ULBP3 enhances anti-tumor immunity. ULBP3 is a ligand for NKG2D. ULBP3-NKG2D interaction augments the anti-tumor activity of NK cells. (H) oHSV can be armed with pro-drug activating genes such as CYP2B1 (which converts CPA to PM) and shiCE (which converts CPT11 to SN-38) to enhance the conversion of these pro-drugs into their anti-cancer active metabolites. Abbreviations: IL-12, interleukin-12; NKs, natural killer cells; IFN-γ, interferon-gamma; VEGF, vascular endothelial growth factor; GADD34, growth arrest and DNA damage-inducible protein 34; Bcl-1, beclin-1; PD-1, programmed death-1; CDH-1, cadherin-1; KLRG1, killer cell lectin-like receptor G1; CSPG, chondroitin sulfate proteoglycan; PTENα, phosphatase and tensin homolog deleted on chromosome 10 alpha; PIP3, phosphatidyl inositol (3,4,5)-trisphosphate; PI3K/AKT/mTOR, phosphoinositide 3-kinase/AKT/mammalian target of rapamycin; ULBP3, UL16-binding protein 3; NKG2D, natural killer group 2; member D; CPA, cyclophosphamide; CPT11, irinotecan; CYP2B1, CPA-activating cytochrome P4502B1; shiCE, CPT11-activating secreted human intestinal carboxylesterase; PM, phosphoramide mustard. |
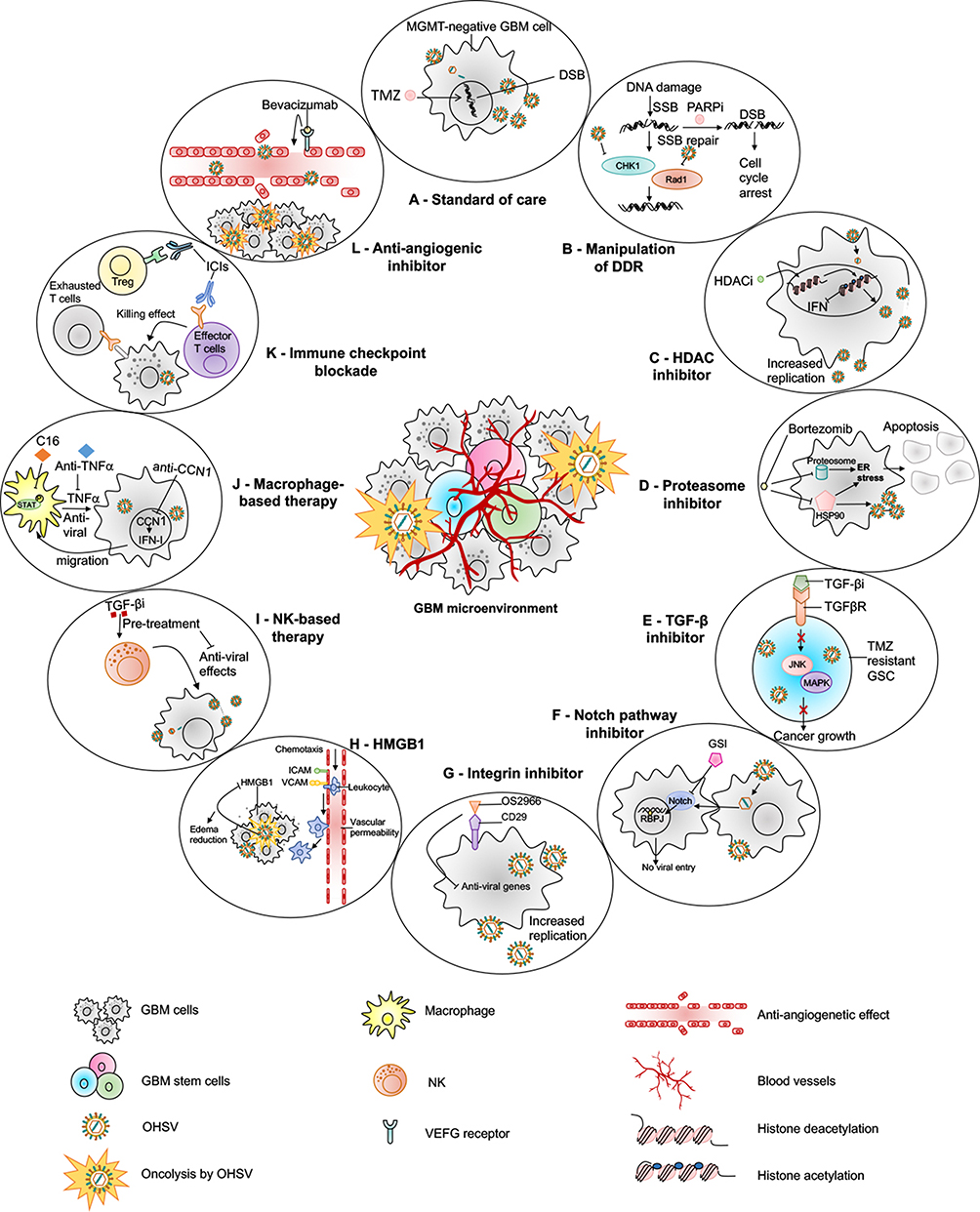 |
Figure 3 oHSV-based combination therapies for GBM. This figure represents strategies to combine oHSV with different anti-cancer therapies for GBM treatment. (A) oHSV can be synergistically combined with TMZ, a DNA-alkylating agent and an immunomodulator, to induce DNA damage response in MGMT-negative GBM cells. (B) oHSV replication promotes degradation of Rad51 and Chk1 whose functions are important for SSB repair mechanism. Inhibition of PARP in DNA-damaged cells facilitates the conversion of SSB to DSB. The combination of oHSV and PAPRi synergistically induces cell cycle arrest in GBM. (C) HDAC is an enzyme that controls cancer cell survival/progression and upregulates IFN genes. Treatment with HDAC inhibitors prior to oHSV infection inhibits induction of anti-viral IFN genes, resulting in increased transcription of viral genes and improved virus replication. (D) Bortezomib is a proteasome inhibitor, which induces unfolded protein response (UPR), characterized by induction of heat-shock proteins (HSP) 40, 70 and 90, and ER stress in cancercells. Bortezomib-induced ER stress and UPR significantly enhance oHSV replication and synergistic killing of GBM cells. (E) TGF-β plays a critical role in GBM pathogenesis and in maintaining the stemness of GSCs. In TMZ-resistant GSC model, the combination of oHSV and TGF-βi synergistically kills TMZ-resistant recurrent GSCs, increases oHSV replication and induces JNK-MAPK signaling blockade, and eventually inhibits tumor progression. (F) oHSV infection of tumor cells leads to activation of notch signaling in adjacent non-infected tumor cells. Notch signaling pathway plays a critical role in cell-cell interaction and viral spread. Inhibition of notch signaling pathway such as GSI results in increased killing of GBM cells after oHSV therapy. (G) ITGB1, also referred as CD29, plays a critical role in tumor cell proliferation and progression. OS2966 (a humanized CD29 blocking antibody) blocks CD29, reduces the expression of anti-viral genes (IFNα, IFNβ, Stat1, OAS1, OAS2, IRF3, IRF9, and PKR), suppresses oHSV-induced macrophage activation, resulting in enhanced oHSV replication and oncolysis. (H) GBM cells that are infected by oHSV upregulate HMGB1. HMGB1 causes upregulation of ICAM and VCAM, increases vascular permeability and PMBC infiltration to the tumor, leading to edema that might cause CNS injuries. Thus, combination of anti-HMGB1 and oHSV increases survival by reducing brain injuries. (I) Recruitment of NKs after oHSV administration can limit oHSV replication and oHSV-mediated anti-tumor efficacy. Transient inhibition of anti-viral effects of NKs by TGF-β inhibitors enhances viral replication and viral yield. (J) Similarly, transient blockade of TNFα, produced from anti-viral macrophages, by TNFα blocking antibodies or inhibition of STAT1/3 phosphorylation by C16 enhances oHSV replication. In addition, virus-infected cells upregulate CCN1, which in turn activates an intracellular type I IFN response and increases infiltration of macrophages to the site of infection. Treatment with anti-CCN1 reduces virus clearance by macrophages, resulting in better anti-tumor efficacy. (K) Administration of immune checkpoint blockade such as anti-PD-1, anti-PD-L1, anti-CTLA4 prevents T cell exhaustion and enhances oHSV-mediated-anti-tumor immunity. (L) Bevacizumab binds to VEGF, reduces tumor vascularization, decreases vascular permeability, and inhibits tumor growth; however, it also induces tumor cell invasion. An oHSV expressing anti-angiogenic vasculostatin that contains an integrin-antagonizing RGD (Arg-Gly-Asp) motif significantly inhibits glioma cell migration/invasion following bevacizumab treatment, leading to a significant extension of survival compared to bevacizumab monotherapy. Abbreviations: TMZ, temozolomide; MGMT, O-6-methylguanine-DNA methyltransferase; DSB, double-strand break; SSB, single-strand break; PARP, poly(ADP-ribose) polymerase; DDR, DNA damage response; HDAC, histone deacetylase; IFN, interferon; UPR, unfolded protein response; ER, endoplasmic reticulum; TNF-α, tumor necrosis factor-alpha; TGF-β, transforming growth factor-beta; GSC, glioma stem cells; RBPJ, recombination signal binding protein for immunoglobulin kappa J region; JNK, c-Jun NH2-terminal kinases; ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule; MAPK, mitogen-activated protein kinase; GSI, gamma-secretase inhibitor; ITGB1, integrin β1; OAS1, 2ʹ-5ʹ-Oligoadenylate Synthetase; IRF, interferon regulatory factor; PKR, protein kinase R; HMGB1, high mobility group box 1; CNS, central nervous system; STAT, signal transducer and activator of transcription; CCN1, cellular communication network factor 1; PD-1, programmed death-1; PD-L1, programmed death ligand-1; CTLA4, cytotoxic T-lymphocyte-associated protein 4. |
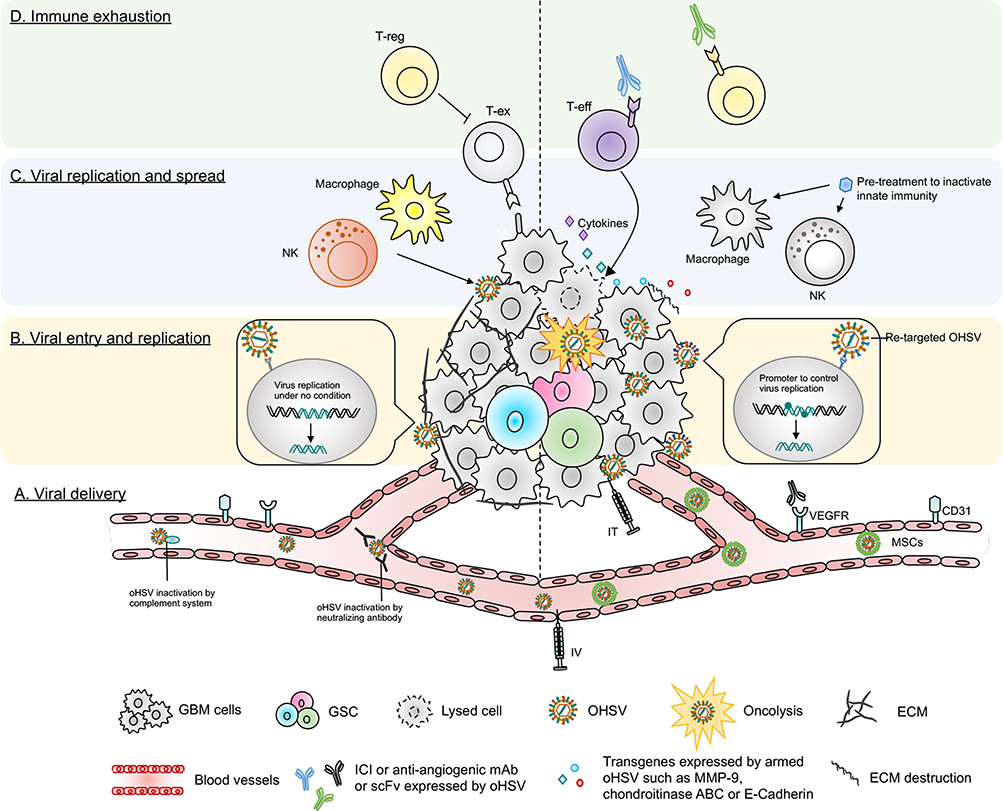 |
Figure 4 Barriers of current oHSV therapy in GBM and their potential solutions. This figure represents various obstacles of oHSV therapy in GBM (left side) and proposed solutions to overcome these hurdles (right side). (A) Viral delivery – oHSV can be delivered systematically (IV) or locally (IT). IT injection can cause backflow of virus solution to the needle or catheter, leading to insufficient viral dose. IV can be another alternative route to deliver oHSV. Systemic administration of naked OVs can be inactivated by host serum/complement and other immune factors. Systemically delivered oHSVs can be protected from serum/immune factors if delivered by carrier cells, such as MSCs. (B) Viral entry and replication – The higher the expression of HSV’s entry surface receptors such as nectin-1, the higher the entry of oHSVs (such as G207, M032) to cancer cells. Genomic modification attenuates virus replication (such as G207) in tumor cells, especially in GSCs. In contrast, receptor (EGFR, HER2, etc.) re-targeting enhances oHSV entry/replication in cancer cells overexpressing these receptors; however, receptor re-targeting can produce off-target effects in healthy cells that also express the same receptors. This issue can be overcome by incorporating miRNA recognition binding sites (such as miR-124 in KGE-4:T124) whose expression are distinct in GBM cells compared to healthy neurons/glial cells. (C) Viral replication and spread – Condensed ECM can limit viral spread in the TME. oHSVs can be engineered to express E-cadherin, chondroitinase ABC, and/or MMP-9 to destroy the ECM components and facilitate viral spread. In addition, anti-viral innate immunity can also limit viral replication/spread in the TME, as characterized by recruitment of macrophage or NKs that clear virus or virus-infected cells. Transient inhibition of anti-viral innate immune response by pre-treatment with different therapies such as TNF-α or TGF-β inhibitors can help to increase oHSV replication/spread. (D) Immune exhaustion – Successful viral delivery, entry, replication and spread will eventually activate the host’s adaptive anti-tumor immunity, leading to recruitment of T cells to the TME. The recruited T cells oftentimes fail to produce anti-cancer effects due to the expression of T cell exhaustion markers. The presence of Tregs and angiogenesis also contribute to GBM immunosuppression. oHSVs can be combined with systemic ICI or systemic anti-angiogenic mAb, or oHSVs can be engineered to locally express scFv of ICI or anti-angiogenic mAb that should overcome GBM immunosuppression. In addition, oHSVs can be armed with different transgene variants (such as cytokines, co-stimulatory ligands) and testing them in combination with ICIs or other immunotherapies such as anti-cancer vaccines (eg, DCs loaded with tumor-associated antigens) to improve anti-tumor efficacy. Abbreviations: IV, intravenous; IT, intra-tumoral; MSCs, mesenchymal stem cells; TME, tumor microenvironment; ECM, extracellular matrix; MMP, matrix metalloproteinase; NK, natural killer cells; TNF-α, tumor necrosis factor-alpha; TGF-β, transforming growth factor-beta; scFv, single-chain fragment variant; ICI, immune checkpoint inhibitor; mAb, monoclonal antibody; VEGF, vascular endothelial growth factor; T-reg, regulatory T cell; T-ex, exhausted T cell; T-eff, effector T cell. |
Unarmed oHSVs for GBM
The HSV genome comprises several essential and non-essential genes that can be modified to ensure selective replication in cancer cells while sparing normal cells.16 The frequently mutated gene to engineer HSV is γ34.5 – a pivotal determinant for viral neuropathogenicity. When HSV infects normal cells, protein kinase R (PKR) phosphorylates eukaryotic translation initiation factor-alpha (eIF2α) and prevents viral protein synthesis. HSV with intact γ34.5 complexes with protein phosphatase 1 alpha (PP1α) to dephosphorylate eIF2α and overcomes anti-viral responses in healthy cells.19 Deletion of γ34.5 in HSV results in no eIF2α dephosphorylation, no viral protein translation, and thus, no viral replication in healthy cells.20 On the other hand, γ34.5-deleted oHSV freely replicates in cancer cells due to the defective anti-viral PKR-eIF2α pathway. HSV1716 and R3616 are the first generation oHSVs with γ34.5 diploid deletion.21,22 Both were safer than their wild-type parent viruses21–23 and demonstrated superior anti-tumor efficacy than the control treatment.24,25 Later, the second generation of oHSV (eg, G207) was engineered, with diploid deletion of γ34.5 combined with inactivation of ICP6 – a larger subunit of ribonucleotide reductase (RR).26 G207 effectively lysed human glioma cells, controlled tumor growth, and prolonged survival of mice bearing orthotopic and subcutaneous brain tumor xenografts.26 Although γ34.5 deletion improves safety, loss of γ34.5 genes severely compromised virus replication in GBM stem cells (GSCs),27 which are oftentimes associated with resistance to standard therapies and immunosuppression.28–30 To counter this issue, the next generation of G207-derivative virus, G47∆, was created with the deletion of an additional gene, ICP47.31 ICP47 deletion complements the loss of γ34.5 and restores oHSV replication in GSCs.32 G47∆ was safe and significantly more efficacious than its parent G207 in controlling tumor growth in vivo in both immunodeficient and immunocompetent brain tumor models.31 Intratumoral G47∆-Empty (G47∆-E, a derivative of G47∆ without transgene expression) treatment resulted in a significant but modest extension of survival of mice bearing orthotopic immunocompetent 005 GSC-derived tumors, which was associated with a significant reduction of regulatory T cells (vs mock) and T cell-dependent anti-tumor efficacy.33
Another strategy being utilized to counteract attenuated oHSV replication is restoring one copy of γ34.5 under the transcriptional control of the nestin promoter/enhancer element. rQNestin34.534 and rQNestin34.5v.235 were engineered using this strategy. Nestin-driven γ34.5 expression exclusively takes place in GBM cells, which have an overwhelmingly higher level of nestin expression compared to healthy cells.34,35 rQNestin34.5 injection in mouse brain was safe and significantly prolonged survival of mice bearing orthotopic U87∆EGFR tumors compared to control.35 Cassady et al described a different approach to improve oHSV replication and generated chimeric oHSVs that express the human cytomegalovirus (HCMV) genes, TRS1 (designated C130) or IRS1 (designated C134).36 TRS1 and IRS1 are PKR evasion genes that evade the PKR-eIF2α translational shutoff pathway,37 and therefore, transfer of these HCMV genes to γ34.5-deleted oHSV resulted in virus replication/production and killing of GBM cells as efficient as wild-type HSV.38 C130 and C134 chimeric oHSVs demonstrated superior anti-glioma efficacy than C101 (a γ34.5-deleted oHSV without TRS1 or IRS1 expression) in an immunodeficient U87 glioma model.39 C134 treatment also elicited a durable and circulating anti-tumor memory response in an immunocompetent neuro2A brain tumor model.40 Intracerebral injections of C134 did not produce toxicity in HSV-susceptible CBA/J mouse and non-human primates.41
Overall, unarmed or chimeric oHSVs are safe but their anti-GBM efficacy is modest at best (see Table 1), and in view of this observation, extensive research efforts have been made (which we have discussed below) to develop novel oHSVs and oHSV-based treatment strategies (such as receptor re-targeting, arming with therapeutic transgenes, combination therapies, etc.) to improve oHSV’s cancer selectivity and anti-GBM efficacy, without compromising safety.
Re-Targeted oHSVs for GBM
oHSVs can be engineered to specifically infect cancer cells based on their receptor expression profile.42 Epidermal growth factor receptor (EGFR) is overexpressed in ~60% of primary GBMs.43 To specifically target EGFR+ GBM, an EGFR-retargeted gB:NT recombinant virus (designated KNE) was generated by replacing 2–24 amino acid residues of HSV glycoprotein D (gD, a natural oHSV receptor) with a single-chain fragment variant (scFv) antibody directed against EGFR.42 KNE significantly controlled tumor growth and prolonged survival of mice in an orthotopic GBM xenograft model.42 Like EGFR, human epidermal growth factor receptor 2 (HER2) is also expressed by ~80% of primary GBMs, but not by neurons or glial cells.44,45 Therefore, prior to KNE, an oHSV-1 re-targeted to HER2 receptor (designated R-LM113) was developed.46 R-LM113 was significantly efficacious against HER2 overexpressing murine GBM xenografts, as demonstrated by 21-day extension of median survival compared to the control group.46 R-115 is another HER2 re-targeted oHSV-1, which is armed with murine IL-12.47 R-115 treatment (vs control) also significantly prolonged the median survival of mice bearing orthotopic poorly immunogenic high-grade gliomas. Most importantly, R-115 treatment led to the eradication of established tumors in about 30% of animals and they were protected lethal tumor re-challenge, indicating a cancer-specific immunological memory response.47 This promising anti-tumor efficacy of R-115 monotherapy was associated with the production of antibodies against implanted tumor cells in 83% treated animals (compared to 40% in IL-12 unarmed R-LM113 group or 0% in mock group) and increased infiltration of CD4+ and CD8+ T cells in the tumor mass.47 Despite re-targeted oHSVs elicited promising oncolytic activity and cancer selectivity, most cancer cell surface markers are shared with normal cells. Therefore, to further enhance cancer specificity, an additional mechanism was explored to better localize re-targeted oHSV replication within GBM cells. GBM has a distinguished microRNA (miRNA) expression profile compared to the normal brain.48 miRNA-124 (miR-124) is specifically expressed in neurons but not in GBM cells.48 To utilize this distinct miR-124 feature, FGE-4:T124, the next generation of EGFR re-targeted KNE virus, was engineered, which contains the insertion of four copies of miR-124 recognition site (T124+) into the 3′UTR of the viral ICP4 gene.49 ICP4 is critically required for transcription of early/late proteins and viral DNA synthesis.50 The normal healthy cells have a high expression of miR-124, which binds to miR-124 recognition sites and inhibits T124+ virus replication. In contrast, the T124+ virus replicated robustly in glioma cells, and lentiviral expression of miR-124 in tumor cells selectively blocked its replication, confirming tumor-specificity of T124+.49 Re-targeted oHSVs can be armed with various transgene variants to further enhance the antitumor efficacy. For instance, KMMP9 is an EGFR/EGFRvIII re-targeted oHSV that has an insertion of repeated miR-124 recognition site into the viral ICP4 gene and armed with matrix metalloproteinase 9 in the UL3-UL4 intergenic region.51 At a low viral dose, KMMP9 exhibited significantly enhanced anti-tumor efficacy with 70% long-term survivors compared to 0% with re-targeted unarmed KGW virus in a GBM xenograft model.51 The preclinical efficacy of several re-targeted oHSVs in GBM is listed in Table 2. Overall, the re-targeting strategy not only confers tumor specificity/safety but also ensures enhanced oncolytic activity with promising anti-GBM efficacy.
Armed oHSVs for GBM
While OVs possess an inherent ability to selectively replicate and lyse cancer cells, the success of oncolytic virotherapy requires the inevitable contribution of the host immune system.52 Therefore, the focus of OV therapies has recently shifted from their direct oncolytic effect to secondary effects (such as immunomodulation and anti-angiogenesis), which can be better achieved by localized expression of different immunomodulatory or anti-cancer transgenes. The unique ability of OVs to selectively replicate in cancer cells makes them a good candidate to safely deliver anti-cancer agents locally in the TME and reducing the possible systemic side effects. For example, systemic delivery of interleukin 12 (IL-12) as an immunotherapeutic agent induces toxicity,53 but local IL-12 expression by an OV in the TME54 or in the cerebellum55 was found safe and showed better anti-tumor response compared to parental unarmed OVs.54 Research in armed oHSVs and their application in GBM have bloomed in recent years, as indicated by the development of a significant number of oHSVs expressing various anti-cancer agents (Table 3, Figure 2). In this section, we will provide an overview of the pre-clinical therapeutic efficacy of recently developed armed oHSVs for GBM treatment.
Arming with Cytokines
Among many cytokines explored to enhance the anti-tumor efficacy of OVs, IL-12 is the most commonly used cytokine.54,56 IL-12 regulates innate and adaptive immunity, induces Th1 differentiation, stimulates growth and cytotoxicity of natural killer cells (NKs), increases IFN-γ production, and inhibits angiogenesis.33,56 Cheema et al constructed G47∆-mIL12 (an oHSV expressing murine IL-12), which has similar genomic modifications as G47∆33 (Table 3), with the additional insertion of murine IL-12 cDNA in the ICP6 region.33 G47∆-mIL12 treatment significantly inhibited GBM angiogenesis and extended survival (vs unarmed oHSV) in orthotopic U87 and human GSC-derived GBM xenograft models.57 Although G47∆-mIL12 was efficacious in xenograft models, the contribution of the immune system to the efficacy of oHSV or oHSV product (ie, IL-12) could not be evaluated in those models. In immunocompetent orthotopic mouse 005 GSC-derived GBM model, intratumoral G47∆-mIL12 treatment led to a significant extension of median survival with 10% mice surviving long term compared to treatment with unarmed G47∆-Empty (G47∆-E).33 The promising G47∆-mIL12’s efficacy was associated with a significant reduction of tumor cells (GFP+ 005 GSCs) and CD4+FoxP3+ regulatory T cells (Tregs), and inhibition of CD31+ tumor vascularity, and the efficacy was dependent on T cells but not NK cells.33 Another IL-12 expressing oHSV, M002, showed a trend of improved median survival in three patient-derived pediatric or adult xenograft GBM models compared to unarmed G207.58 Like 005 model, M002 was extremely efficient in an intracranial syngeneic 4C8 murine glioma model, since M002 treatment resulted in 80% long-term survivors.59 Like IL-12, other cytokines such as FMS-like tyrosine kinase 3 ligand (Flt3L) or tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) were inserted into the backbone of G47∆ and generated G47∆-Flt3-L60 and oHSV-TRAIL viruses,61 respectively. Both G47∆-Flt3-L and oHSV-TRAIL were efficacious in GBM60–62 (Table 3). Importantly, recently, Jahan et al demonstrated that oHSV-TRAIL can overcome TMZ-resistance in GBM.62 For instance, intracranial oHSV-TRAIL treatment of mice bearing TMZ-insensitive, recurrent human GSC-derived orthotopic tumors led to potent inhibition of tumor growth (examined by in vivo imaging) and extension of survival with a 40% cure rate. The treatment efficacy of oHSV-TRAIL in chemo-resistant tumors was associated with robust induction of tumor apoptosis.62 TRAIL has a short half-life and produces off-target toxicity63 and the clinical trials of TRAIL receptor agonist have been discontinued in the clinic due to insufficient evidence of anti-cancer efficacy.64 Local expression of TRAIL by an oHSV offers a promising strategy to avoid systemic off-target toxicity.
Arming with Angiogenic Inhibitors
Preclinical studies with oHSV expressing various angiogenetic inhibitors consistently exhibited improved survival outcomes compared to mock or unarmed viruses.57,65–67 G47∆-mAngio, an oHSV armed with murine angiostatin (mAngio), controlled tumor burden and significantly extended median survival compared to G47∆-E in two immunodeficient intracranial GBM models.57,67 A closer look at the TME revealed that G47∆-mAngio treatment resulted in significantly increased virus spread (LacZ expression), decreased tumor vascularity (CD31+ vessels) and anti-viral macrophage density, and reduced expression of vascular endothelial growth factor (VEGF) compared to G47∆-E.57
Zhang et al constructed an oHSV expressing endostatin-angiostatin (endo-angio) fusion gene (designated VAE) by deleting both copies of γ34.5, inactivating ICP6, and inserting endo-angio fusion gene into ICP6 region.65 VAE demonstrated a significant improvement in median survival in two (subcutaneous and intracranial) GSC-derived xenograft models compared to γ34.5/ICP6-deleted r-HSV-1 and recombinant human endostatin (Endostar).65 The better response in VAE-treated group was associated with a reduction in mean micro-vessel density (MVD) compared to r-HSV-1 or Endostar.65 Tomita et al recently described antitumor efficacy of RAMBO – an oHSV (γ34.5/ICP6 deleted) expressing vasculostatin (Vstat120) – in athymic mice bearing intracerebral U87∆EGFR tumors.66 The median survival of PBS-treated mice was significantly shorter (17 days) than mice treated with RAMBO (28 days).66 Mechanistically, Vstat120 expression inhibited TNF-α production by blocking brain-specific angiogenesis inhibitor 1 (BAI1)-mediated macrophage response to viral infection, resulting in an increased viral spread.68
Arming with Growth Arrest and DNA Damage-Inducible Protein 34 (GADD34)
GADD34 or protein phosphatase 1 regulatory subunit 15A (PPP1R15A) is a member of a group of genes whose transcript levels are increased by DNA damage, growth factor deprivation, and other forms of cell stress.69 GADD34 binds to PP1 and promotes eIF-2α dephosphorylation,70 a function that corresponds to γ34.5 in HSV.71 Interestingly, while GADD34 shares sequence homology with the C terminus of γ34.5,70 it does not possess the beclin-1-binding motifs of γ34.5 – a feature that is responsible for neurotoxicity by HSV.72 Therefore, the replacement of γ34.5 by GADD34 is thought to reduce neurotoxicity. Based on this principle, NG34, a novel oHSV (γ34.5/ICP6-deleted) expressing GADD34, was created.72 Noticeably, in NG34, GADD34 expression is transcriptionally controlled by a nestin promoter/enhancer; thus, its expression occurs selectively in GBM.72 NG34 demonstrated a similar replication/propagation pattern in GBM cells as the parental unarmed rQNestin34.5 virus.72 In both human xenograft and immunocompetent GBM models, NG34 produced therapeutic efficacy, which was as potent as rQnestin34.5 but with better tolerability.72
Arming with Programmed Death-1 (PD-1) Inhibitor
Immune checkpoint co-inhibitory molecule PD-1 and its ligand PD-L1 play a critical role in regulating immune responses and suppressing anti-tumor immunity. Anti-PD-1 inhibits PD-1/PD-L1 interaction, prevents T cell exhaustion, and unleashes anti-tumor immunity.73,74 Despite being extensively studied, late-phase clinical trials with anti-PD-1 in GBM did not signify its therapeutic benefits.75,76 The inability of anti-PD-1 to cross the blood-brain barrier (BBB) and highly immunosuppressive TME with a low number of tumor-infiltrating lymphocytes (TILs) are probably the major contributors to treatment failure.77 Local oHSV expression of anti-PD-1 may overcome these obstacles. OVH-aMPD-1, an oHSV encoding a single-chain fragment variable antibody against PD-1 (αMPD-1 scFv), was generated that demonstrated a promising anti-tumor efficacy (ie, enhanced antigen presentation, T cell-mediated cytotoxicity, and reduction of tumor burden) in a liver cancer model, but its efficacy has not been tested yet in GBM.78 Recently, Passaro et al constructed another oHSV by inserting an scFv against PD-1 (designated as NG34scFvPD-1) in the previously mentioned NG34 backbone.79 In a syngeneic GL261N4 immunocompetent GBM mouse model, in situ expression of a PD-1 blockade by NG34scFvPD-1 produced a durable response and long-term memory protection, but surprisingly, these anti-tumor effects were statistically similar to that of parental NG34 virus.79
Arming with Cadherin-1 (CDH-1)
The limited virus spread in the TME due to rapid innate immune clearance of the virus is considered as one of the major obstacles of oHSV virotherapy.80 The interaction between viral glycoprotein D (gD) and nectin-1 receptor is an important step for HSV-1 entry.81 CDH-1 gene encodes for E-cadherin – an adhesion molecule and a ligand for killer cell lectin-like receptor G1 (KLRG1), which is an inhibitory receptor expressed on NK cells.82 E-cadherin can cooperate with nectin-1 and promote cell-to-cell adherent junctions,83 and thus, its overexpression can enhance cell-to-cell oHSV spread. Xu and colleagues engineered an oHSV to express CDH-1 (named OV-CDH1).84 In vitro, OV-CDH1 infection resulted in ectopic expression of E-cadherin that enhanced viral spread/entry by promoting cell-to-cell infection and reduced viral clearance by protecting infected GBM cells from KLRG1+ NK cell killing.84 In vivo, OV-CDH1 treatment significantly prolonged median survival compared to unarmed virus (OV-Q1) in two xenografts and one immunocompetent GBM models.84 The better outcome in OV-CDH1-treated group in the immunocompetent GBM model was associated with an improved viral spread rather than inhibition of the activity of NK cells.84
Arming with Chondroitinase ABC
Chondroitinase ABC is a bacteria-derived enzyme that cleaves and removes the side chain of chondroitin sulfate proteoglycans (CSPG).85 CSPG is upregulated in 60–65% GBM cases,86 tortoises the extracellular space, and limits OV spread.85,87 In a mathematical model, chondroitinase ABC enhanced OV spread and anti-tumor efficacy.87 An oHSV expressing chondroitinase ABC (designated OV-Chase) exhibited widespread three-dimensional glioma cultures compared to the control virus (rHSVQ). In vivo, OV-Chase treatment (vs rHSVQ) significantly extended median survival in subcutaneous and intracranial GBM models.85 The humanized version of OV-Chase (designated OV-ChaseM) also enhanced median survival of athymic mice bearing GBM xenograft (33 days vs 22 days in the mock group).88
Arming with Phosphatase and Tensin Homolog Deleted on Chromosome 10 Alpha (PTENα)
PTEN is a tumor suppressor protein phosphatase that metabolizes phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which directly opposes activation of the PI3K/AKT/mTOR signaling pathway.89 Loss or mutation of PTEN, commonly occurred in GBM,89,90 activates the PI3K/AKT pathway and promotes cell growth/survival and migration.91 Local expression of PTENα, an isoform of PTEN, by an oHSV (termed HSV-P10) significantly prolonged median survival with 30% long-term survivors in a U87∆EGFR xenograft model compared to 10% in the unarmed oHSV (HSVQ) group.92
Arming with UL16-Binding Protein 3 (ULBP3)
Human ULBP3, a ligand for NKG2D (natural killer group 2, member D), is overexpressed on certain tumor cells,93 including brain tumors.94 ULBP3-NKG2D interaction augments the anti-tumor activity of NK cells.93 To exploit this anti-tumor mechanism, an oHSV armed with human ULBP3 gene (OHSVULBP3) was created and tested in a genetically engineered IDH wild-type GBM mouse model (XFM-Luc:PDGF, Cre).95 Intra-tumoral oHSVULBP3 treatment inhibited tumor growth and prolonged survival, whereas oHSV lacking ULBP3 did not.95 The survival efficacy was associated with an increased accumulation of activated macrophages and CD8+ T cells in the TME. The same group later reported an increased tumoral expression of VEGF and matrix metalloproteinase 9 (MMP-9) by oHSVULBP3 treatment. A vast portion of virally enhanced VEGF remained trapped in the extracellular matrix96 and cannot fully exert its effects until trapped VEGF is cleaved by MMP-9 and subsequently released.97 To determine the role of MMP-9 on VEGF signaling and its subsequent effect on oHSVULBP3 efficacy, Wirsching et al engineered an oHSVULBP3-MMP9 to co-express ULBP3 and MMP-9.96 Although oHSVULBP3 significantly prolonged survival of mice bearing XFM-Luc:PDGF, Cre GBM tumors compared to mock, MMP-9 expression, ie, oHSVULBP3-MMP9 treatment nearly abolished the survival benefit of oHSVULBP3 to a mock level.96 The use of a VEGF-neutralizing antibody (B20) rescued anti-tumor effects of oHSVULBP3-MMP9, confirming the role of VEGF in abrogating treatment efficacy.96 Another oHSV (designated ONCR-1) was engineered by deleting one copy of γ34.5, inserting miR-124 binding sites into the ICP4 locus and arming with both ULBP3 and MMP-9.98 Although ONCR-1 controlled tumor growth and extended survival in mice bearing subcutaneous or orthotopic human U251 GBM tumors, it was not determined whether the absence of MMP-9 expression would have facilitated a better spread/efficacy of a single transgene (ULBP3)-armed virus.98
Arming with Prodrug-Activating Genes
Oncolytic effects of an oHSV can be enhanced by arming with pro-drug activating genes.99 MGH2 is an oHSV armed with two prodrug-activating genes, cyclophosphamide (CPA)-activating cytochrome P4502B1 (CYP2B1) and CPT11-activating secreted human intestinal carboxylesterase (shiCE).99 CYP2B1 converts cyclophosphamide (CPA) into active anti-cancer DNA-alkylating metabolite, phosphoramide mustard (PM), whereas shiCE converts irinotecan (CPT11) into active topoisomerase I inhibitor, SN-38. In athymic mice harboring human GBM tumor (Gli36ΔEGFR), MGH2 was not effective alone (vs PBS), but in combination with CPA and CPT11, MGH2 displayed a significantly robust anti-tumor efficacy in comparison to controls.99 MGH2 was later modified to exclude a green fluorescent protein (GFP) expression cassette from its genome, and named MGH2.1, for its assessment in clinical trials.100 The intracerebral administration of MGH2.1 was safe and did not affect the endogenous metabolism of CPA and CPT11.100
The aforementioned reported studies illustrate that oHSVs armed with therapeutic transgene(s) in general produce superior anti-tumor efficacy than unarmed oHSVs (Table 3). The therapeutic efficacy of armed oHSVs varied from model to model, and none of the armed oHSVs led to complete tumor eradication in 100% of animals tested compared to unarmed oHSVs or control treatments (Table 3), indicating further research is needed to improve the efficacy of armed oHSVs.
Carrier Cell-Based oHSV Treatment for GBM
Intra-tumoral injection is the primary mode of viral delivery.101–104 Intra-tumoral injection can cause backflow of virus solution to the needle or catheter, leading to significant loss of infused solution, and thus, patients may not receive the required virus dose.104,105 Although this hurdle can be overcome by multiple injections into several sites, each injection increases the risk of intracranial bleeding and influx of cerebrospinal fluid into the resection cavity that could further rinse out the injected virus.5,104 Intravenous administration is another important viral delivery method; however, systemic (even local) administration of naked OVs can be inactivated by host serum/complement and other immune factors,14 and thus, alternative approaches were explored. In 2008, Sonabend et al reported the first study utilizing mesenchymal stem cells (MSCs) as a carrier to deliver OV for GBM treatment.106 In 2014, Duebgen et al loaded human MSCs with oHSVs (MSC-oHSVs) and demonstrated that MSC-oHSVs were effective in producing oHSV progeny and anti-tumor efficacy.107 Intracranial injection of synthetic extracellular matrix-encapsulated MSC-oHSVs resulted in a significant extension of median survival compared to naked oHSV in a preclinical model of GBM resection.107 Similarly, MSC-oHSV-TRAIL (MSC loaded with oHSV-TRAIL)62 effectively induced apoptosis-mediated killing of oHSV- and TRAIL-resistant GBM cells and prolonged median survival of mice bearing orthotopic tumors.107 Although MSC-oHSV and MSC-oHSV-TRAIL were efficacious in immunodeficient GBM models, it is not clear whether they will be similarly effective against syngeneic GBM tumors.108,109 Recently, few more research groups utilized a similar carrier cell-based treatment approach for GBM.110–112 These studies provide an excellent platform to further investigate oHSV-loaded carrier cells in GBM.
oHSV-Based Combination Therapies for GBM
As discussed above, anti-tumor efficacy of unarmed, even armed viruses is limited. Thus, the focus of oHSV research has been shifted from monotherapy to combination therapy. Multimodal approaches were implemented to combine oHSV with chemotherapy, antiangiogenic agents, etc. (Figure 3). Here, we have covered different combinatorial treatment strategies that were developed in recent years.
Combination with Standard Chemotherapy
Temozolomide (TMZ), an oral DNA-alkylating agent and immunomodulator,113 is the first-line chemotherapy in GBM; however, at least 50% of GBM patients do not respond to TMZ.114 Expression of O-6-methylguanine-DNA methyltransferase (MGMT) is primarily associated with TMZ resistance, in addition to mutations in DNA mismatch repair proteins.114,115 OV-ChaseM sensitized glioma cells to TMZ and enhanced survival.88 It was not clear from this study whether OV-ChaseM can overcome MGMT-mediated TMZ resistance. TMZ was also shown to synergize with G47Δ in killing human GSCs in vitro and enhancing survival in human GSC-derived orthotopic tumors in vivo.116 TMZ-oHSV interaction can be immunologically antagonistic since TMZ abrogated the efficacy of G47Δ-mIL12 in an immunocompetent orthotopic 005 GSC-derived GBM model.117 The antagonistic interaction was probably associated with MGMT expression and immune inhibitory effects of TMZ.117 The addition of an MGMT inhibitor, O6-BG, to TMZ did not overcome the negative effects of TMZ.117 Synergy in the human GSC model and antagonistic effects in the mouse model can be explained by the fact that (i) human cancer cells are typically more permissive to oHSV replication than mouse cancer cells;31 (ii) oHSV impairs DNA damage response in human GSCs,116 but it is not known whether this occurs in mouse GSCs; and (iii) the schedule of TMZ treatment can also affect oHSV efficacy, since OV treatment prior to TMZ significantly extended survival in GL261 GBM model,118 whereas antagonism observed when G47Δ-mIL12 and TMZ were concurrently applied in the 005 model.117
Inhibition of Glioma Invasion and Angiogenesis
GBM is a highly vascularized and invasive tumor.119,120 Anti-angiogenic therapy is somewhat effective in GBM121–123 and synergizes with oHSV.57,67,124 Bevacizumab, an anti-VEGF monoclonal antibody, inhibits GBM growth by reducing tumor vascularity;122,123 however, bevacizumab can cause tumor invasion and resistance via several mechanisms including an integrin pathway-mediated invasion.125,126 To overcome bevacizumab-induced/integrin pathway-mediated invasion,66 Tomita et al utilized RAMBO, an oHSV expressing anti-angiogenic vasculostatin (Vstat120) that contains an integrin-antagonizing RGD (Arg-Gly-Asp) motif,127 which significantly inhibited glioma cell migration/invasion following bevacizumab treatment in vitro. In vivo, the combination (RAMBO + Bevacizumab) significantly reduced the depth of tumor invasion and extended survival of mice bearing orthotopic GBM xenograft compared to bevacizumab monotherapy.66 Mechanistically, RAMBO prevented bevacizumab-induced cysteine-rich protein 61 (CCN1) expression and AKT phosphorylation and eventually suppressed glioma invasion.66 Like RAMBO, G47Δ-mAngio67 was also effective in reducing bevacizumab-induced tumor invasion in vivo and extending survival in a U87 glioma model.67 The combination efficacy was associated with inhibition of tumoral VEGF and expression of invasion markers such as matrix metalloproteinases-2 (MMP2), MMP9, and collagen.67 Like the G47Δ-mAngio virus, G47Δ-mIL12 is also highly anti-angiogenic33 due to the presence of anti-angiogenic cytokine IL-12. G47Δ-mIL12 in combination with a VEGFR tyrosine kinase inhibitor axitinib significantly prolonged survival of mice bearing mouse or human GSC-derived GBM tumors compared to monotherapies via multifaceted mechanisms: direct oncolysis, increased macrophage recruitment, reduction of CD34+ tumor vascularity, and inhibition of PDGFR/ERK pathway in an immunodeficient model, and T cell-dependent efficacy in an immunocompetent model.124 No additional benefit was observed when anti-CTLA4 was added to the combination therapy (G47Δ-mIL12+axitinib).124
Manipulation of DNA Damage Response (DDR)
oHSV G47Δ induces DDR and synergizes with DNA-damaging agents such as TMZ.116 Poly (ADP-ribose) polymerase (PARP) plays a critical role in DNA repair. Ning et al128 demonstrated that PARP activity is uniformly inhibited by PARPi (such as Olaparib) in both PARPi-sensitive and PARPi-resistant GSCs. Importantly, oHSV synergistically interacted with PARPi and induced cytotoxic effects against GSCs, irrespective of their sensitivity to PARPi, through proteasomal degradation of DDR proteins, Rad51 and Chk1. The combination (Olaparib + oHSV-MG18L) significantly extended the survival of mice bearing PARPi-sensitive or PARPi-resistant GSC-derived tumors compared to monotherapies. This combination strategy is unique and should also be effective to other solid tumors, irrespective of their sensitivity to PARPi.
Inhibition of Histone Deacetylase (HDAC)
Otsuki et al129 demonstrated that treatment of glioma cells with valproic acid (VPA), a commonly used antiepileptic agent that inhibits HDAC (an enzyme that controls cancer cell survival, progression, and differentiation130), prior to oHSV infection inhibited induction of anti-viral interferon genes, resulting in increased transcription of viral genes and improved virus replication in vitro. VPA pre-treatment also increased oHSV propagation and oHSV-mediated anti-tumor efficacy in vivo in GBM xenograft model.129 VPA can also enhance oHSV efficacy through suppression of anti-viral innate immunity.131 For instance, VPA administration prior to oHSV therapy reduced NK cell activity against oHSV-infected GBM cells through inhibition of STAT5/T-bet signaling and IFN-γ production.131 Like VPA, tubacin (an HDAC6-specific inhibitor) also increased rQNestin34.5 replication in GSCs by counteracting antiviral effects of type I interferons and by altering the post-entry trafficking of oHSV toward nuclei rather than to the lysosome,132 and improved survival in an orthotopic GSC-derived xenograft model.132
Inhibition of Proteasome
Proteasomes are cellular protein complexes that degrade damaged proteins through proteolysis. Proteasome inhibition can result in the induction of unfolded protein response (UPR) and endoplasmic reticulum (ER) stress, and thus, it offers a way to target GBM.133,134 Bortezomib (a proteasome inhibitor) induces ER stress and UPR, which contributed to significantly enhanced oHSV (34.5ENVE) replication and synergistic killing of glioma cells.135 HSP90 was found to be the most critical UPR for increased oHSV replication. In an intracranial human GBM model, the combination therapy dramatically enhanced survival compared to monotherapies.135 In a follow-up study,136 the same group demonstrated that bortezomib pre-treatment of tumor cells enhanced oHSV-induced necroptotic cell death through induction of RIPK1 and mitochondrial reactive oxygen species (ROS). This coincided with an increased secretion of IL1α from combination-treated tumor cells.136 To confirm whether IL-1α sensitizes tumor cells to natural killer (NK) cells,137,138 a co-culture study (treated brain tumor cells and naïve NK cells) was conducted and revealed a significantly increased secretion/expression of killer cytokines (IFN-γ and TNF-α) and apoptotic markers (cleaved caspase 3, 8 and cleaved PARP) in the combination group than single treatment groups. In vitro findings were also reproduced in vivo and it was reported that the addition of an NK cell-based adjuvant therapy to the combination further significantly enhanced the efficacy.136
Blockade of TGF-β Signaling
Transforming growth factor-beta (TGF-β) plays a critical role in GBM pathogenesis and maintaining GSC stemness.139 In a TMZ-resistant GSC-derived recurrent GBM model, the combination of oHSV (MG18L) and TGF-β receptor kinase inhibitor (TGF-βi) synergistically killed GSCs in vitro and produced 60% long-term survivors in vivo (vs 0% with monotherapies).140 In clinical setting, galunisertib (a TGF-βi) in combination with TMZ/radiotherapy was not effective,141 therefore, further pre-clinical research is needed to understand/optimize the therapeutic potential of TGF-βi in the context of combination therapies.
Inhibition of Notch Signaling
Notch signaling pathway is important for cell–cell interactions, tumor progression, GSC maintenance, and treatment resistance.142 oHSV infection of tumor cells activated notch in adjacent tumor cells, and miR-H16 was found to be responsible for notch induction through downregulation of FIH1 (a direct target of miR-H16).143 Notch inhibition, prior to oHSV treatment, by a gamma-secretase inhibitor (GSI) (RO4929097) resulted in increased killing of glioma cells and improved survival compared to monotherapies.143 Several GSIs were evaluated in the clinic, and hence, oHSV + GSI combination has translational significance.
Inhibition of Integrin Signaling
Integrin β1 (ITGB1), also referred to as CD29, plays an important role in tumor progression144 and resistance to bevacizumab.145 Blockade of ITGB1 with OS2966 (a humanized CD29 blocking antibody) overturned bevacizumab-induced resistance.145 Although OS2966 enhanced oHSV replication and GBM cell cytotoxicity in vitro,146 systemic OS2966 delivery did not enhance the anti-tumor efficacy of oHSV (rHSVQ1-IE4/5-Luciferase) in vivo. Insufficient delivery of OS2966 to the brain may have contributed to the failure to obtain a combination effect. To overcome this issue, OS2966 was directly injected intracranially into GBM xenograft 2 days prior to oHSV treatment, and this time, the combination significantly extended survival compared to monotherapies.146 In contrast to OS2966, systemic delivery of cilengitide (another integrin inhibitor) significantly enhanced the anti-glioma efficacy of RAMBO compared to single treatments.147 Cilengitide has been evaluated in GBM patients148 and could be a better choice than OS2966 for combination studies.
Inhibition of NK Cell-Related Anti-Viral Response
Intra-tumoral oHSV treatment recruits NK cells within hours of infection,14 which substantially limits oHSV efficacy.149 Han et al150 demonstrated that TGF-β1 (an immunosuppressive cytokine) pre-treatment impaired NK’s anti-viral activity, leading to significantly increased virus yield from infected glioma cells in a co-culture study. In an orthotopic U87ΔEGFR model, systemic delivery of TGF-β1 prior to intracranial oHSV treatment significantly increased oHSV titers compared oHSV monotherapy, and this TGF-β1-mediated oHSV replication was dramatically decreased following treatment with 1D11 (a TGF-β neutralizing antibody). In a syngeneic 4C8 GBM model, a single dose of TGF-β prior to oHSV therapy inhibited recruitment/activation of NK cells (and macrophages), leading to efficient tumor control and extension of survival.150 NK depletion also enhanced oHSV efficacy, which was similar to the observed efficacy with TGF-β1 + oHSV combination.150 This study did not test whether transient inhibition of innate immunity has had any detrimental impact on adaptive immunity.
Inhibition of Macrophage-Mediated Anti-Viral Immunity
Infiltrating macrophages/resident microglia following HSV infection151,152 induce an early anti-viral response,153 including the production of TNF-α,154 which negatively influences virus replication in glioma cells and oHSV (rQNestin34.5)-induced efficacy in an intracerebral U87ΔEGFR model.151 Transient TNFα blockade abrogated the negative effects of TNF-α, and thereby, increased oHSV replication and anti-GBM efficacy in a xenograft model.151 Pointer et al155 and Yoo et al156 also reported potentiation of oHSV virotherapy in GBM by TNF-α inhibition using a TNF-α blocking antibody or a MEK kinase inhibitor (trametinib), respectively. These studies suggest that FDA-approved TNF-α inhibitors157 can be utilized to improve oHSV efficacy.
Cysteine-rich 61 (CCN1), an extracellular matrix protein, is upregulated in GBM cells in response to oHSV infection.158 Virally enhanced CCN1 activates type I IFN response159 and increases macrophage migration/activation that contributes to viral clearance.160 Anti-CCN1 neutralizing antibody nullified the anti-viral effects of CCN1 and improved oHSV efficacy in vivo, compared to control.160 Besides CCN1 activation, recently, Delwar et al reported limited oHSV replication in macrophages/microglia due to STAT1/3 phosphorylation, which eventually formed a non-permissive barrier and prevented oHSV dissemination in the glioma TME.161 STAT1/3 inhibition by C16, a derivative of oxindole/imidazole, rescued oHSV-1 replication, both in vitro and in vivo.161
Immune Checkpoint Inhibition
GBM is a highly immunosuppressive tumor and does not respond to immune checkpoint inhibitor (ICI).75,162 Immunosuppression in GBM is governed by various mechanisms163 including, but not limited to, T-cell expression of co-inhibitory molecules such as PD-1, PD-L1, CTLA-4, etc.164–168 Inhibition of PD-1/PD-L1 and/or CTLA-4/B-7 interaction can be a critical turning point to improve oHSV efficacy.152,169,170 In ICI-resistant GSC-like GBM models (005 and CT-2A),152,163,171–174 intra-tumoral IL-12 expression by G47Δ-mIL12 dramatically sensitized tumors to ICIs (anti-PD-1 + anti-CTLA-4), leading to 50–89% long-term survivors and memory protection.152 This remarkable therapeutic efficacy was associated with macrophage polarization and total dependence on CD4+ T cells.152 ICI therapy also demonstrated synergy with oHSVULBP3,95 as evidenced by inhibition of contralateral tumor growth and extension of survival of mice bearing bilateral ICI-resistant XFM-Luc:PDGF, Cre IDH wild-type GBM tumors.95
Inhibition of the Mammalian Target of Rapamycin (mTOR)
mTOR is a key mediator of EGFR/PI3K/Akt signaling that regulates cell growth, proliferation, and survival via two functionally and structurally distinct multiprotein complexes known as mTORC1 and mTORC2.175,176 GBM patients treated with rapamycin, a solely allosteric inhibitor of mTORC1, exhibited a high rate of resistance and worse progression due to mTORC1-mediated feedback and mTORC2 activation.175 Interestingly, rapamycin was previously shown to enhance oHSV production and spread in several difficult-to-treat human tumors.177 Recently, a new generation of mTOR inhibitor, ATP-competitive active-site mTORC1 and 2 inhibitors (asTORi) enhanced ICP0-deleted HSV1 infection specifically in transformed cells (such as human GBM cells) via inhibition of cellular type-I IFN responses, while reducing infection in non-transformed cells.178 The promising outcome of this combination study warrants further pre-clinical research in vivo in GBM models and clinical translation in GBM patients.
Other Combination Therapies
In addition to the above-described combination studies, many other oHSV-based combination therapies were described in the past.179 For example, combining oHSV with ionizing radiation,180 PI3K/Akt pathway inhibitors,181 etoposide182 or HMGB1 blockade183 significantly enhances anti-tumor efficacy in preclinical GBM models.
Clinical Evaluation of oHSVs in GBM and Other Brain Malignancies
Six oHSVs (1716, G207, G47∆, M032, C134, and rQNestin34.5v.2) have been or are being tested in patients with brain tumors (Table 4).5,10,184,185 Early trials with HSV1716 demonstrated no evidence of HSV-mediated adverse events or toxicities.186–188 In the very first study, nine recurrent high-grade glioma (HGG) patients received a stereotactic injection of HSV1716, with doses escalating from 103–105 plaque-forming unit (pfu) in 1 mL.187 Four patients survived longer than 14 months following treatment.187 No evidence of shedding or reactivation of HSV was observed.187 In the second study, 12 HGG patients were intratumorally injected with 105 pfu of HSV1716.188 Tumors were resected 4–9 days after treatment. Infectious HSV was recovered from injected sites in two patients. The viral DNA was detected in 10 patients at the injected site and in 4 patients at the distal tumor site.188 An immune response (ie, changes in the levels of IgG and IgM) to HSV1716 was observed in two patients. Overall, this study confirmed the safety and replication of HSV in HGG tumors. In the third study, 12 recurrent or newly diagnosed HGG patients first underwent maximal surgical resection and then received HSV1716 (105 pfu) into 8 to 10 sites in the cavity wall.186 Three patients were clinically stable for 15–22 months following virotherapy. Like the second trial, two of three seronegative patients were seroconverted. Imaging demonstrated a reduction of the residual tumor over a period of 22 months.186 A Phase I trial using HSV1716 in pediatric patients with surgically removable refractory or recurrent HGG was terminated in 2016 and no results of this trial were posted thus far (NCT02031965).
The safety and efficacy of G207 were reported preclinically.189 Four Phase I/Ib clinical studies involving G207, either alone or in combination with radiation, were reported in patients with malignant glioma or recurrent GBM.101,184,190,191 In the first study, which was the first oHSV clinical trial in the US, 21 recurrent HGG patients received an intra-tumoral injection of G207 up to 3 × 109 pfu at five sites.190 G207 was safe with no virus shedding observed in saliva or conjunctival secretions. Four patients survived 7–19 months following virus inoculation.190 Next, a Phase Ib trial was initiated in six recurrent GBM patients who received two doses of G207 totaling 1.15 × 109 pfu, with 13% of this dose injected stereotactically into the tumor, followed by en bloc tumor resection and administration of the remaining virus dose at multiple sites into the resected cavity wall. Replication and antitumor activity (ie radiographic and neuropathologic response) of G207 were reported.191 Markert and Colleague published a case report demonstrating that G207 treatment (1 × 107 pfu in 120 μL volume stereotactically injected into the enhancing region of the resected tumor) with minimal adjunctive chemotherapy in a recurrent GBM patient led to 6 years of disease-free survival, indicating a long-term response.192 G207 was also tested in combination with irradiation in a Phase I clinical trial in recurrent GBM.101 Nine progressive recurrent malignant glioma patients (seven of them bearing GBMs) were stereotactically/intratumorally treated with G207 (1 × 109 pfu) 24 hours prior to single focal 5 Gy radiation. The combination therapy was well tolerated and no patients developed HSV-related encephalitis. Three instances of marked radiographic response to treatment were observed.101 Based on the safety/efficacy of three published G207 studies101,190,191 and the high sensitivity of pediatric brain tumors to killing by G207,193 the same group designed a clinical trial protocol to evaluate G207, either alone or in combination with radiation, in six pediatric brain tumor patients (five GBM and one anaplastic astrocytoma).194 Patients were safely implanted with 3–4 intra-tumoral silastic catheters for delivery of G207.195 Recently, the same group has also designed a first-in-human immunotherapy Phase I clinical trial to treat pediatric recurrent cerebellar malignancies and a Phase II trial to treat pediatric recurrent HGG with G207 combined with single low-dose radiation (5 Gy). No results are available from these studies yet196 (NCT04482933).
In comparison to G207, G47∆ (which is constructed from G207 by adding a deletion of ICP47 gene) is a relatively newer version of unarmed oHSV. G47Δ was found safe in Phase I–IIa clinical trial in 21 patients with recurrent GBM (UMIN000002661).197 In a follow-up Phase II clinical trial in patients with residual or recurrent GBM (UMIN000015995), G47Δ demonstrated anti-tumor efficacy. In this study, G47Δ (1 × 109 pfu as used for G207 in)101 was stereotactically injected into the tumor twice within 2 weeks and every 4 weeks thereafter for a maximum of 6 times. The treatment was well tolerated.197
Like the G47∆ virus, the safety and efficacy of C134 and rQNestin34.5v.2 are also being tested in recurrent GBM patients (NCT03657576 and NCT03152318).5,6 Since M002 (an oHSV expressing murine IL-12) was efficacious in mouse GBM models,198–200 the same group generated M032 virus (the clinical version of M002 but expressing human IL-12) and demonstrated its safety after intracerebral administration to HSV-sensitive non-human primates.55 M032 is currently in a clinical trial in patients with recurrent or progressive GBM.201 The promising safety profile and preliminary signs of efficacy exhibited by several oHSVs in patients warrant a further clinical evaluation in a larger patient cohort.
Current Challenges and Potential Remedies
FDA approval of T-VEC in 2015 and its promising efficacy in non-GBM cancers have fueled the field of oncolytic virotherapy. Since then, oHSV therapy for GBM has evolved rapidly preclinically. Currently, several oHSVs are under clinical trial evaluation and early trial results demonstrated safety and signs of efficacy in GBM patients (Table 4). Despite significant advancement, there are various treatment challenges to oHSV exist, which include but are not limited to: suboptimal viral delivery, insufficient viral entry/replication, limited oHSV spread, and host anti-viral immune response (Figure 4).4,14 As discussed above, although direct oHSV injection into the tumor is the most commonly used viral delivery method, it is not ideal for tumors that are located in an eloquent area of the brain or when the tumor is metastatic. In addition, the risks associated with neurosurgical procedures make repeated virus administration difficult. Systemic intravascular delivery can be useful in these scenarios, but systemic injection has also a significant number of drawbacks, such as (i) loss of virus through liver metabolism; (ii) potential risk of off-target distribution/replication; (iii) serum/complement factors can neutralize oHSVs; (iv) BBB can limit viral entry to the tumor, etc. To overcome such limitations, other approaches were explored, such as intraventricular injection24,190,202 and carrier cell delivery.106,107,110–112 GBM has aberrant vasculature and its outgrowth can destroy BBB.203 Intraventricular method may provide viral access to the tumor without the need for repeated invasive surgical procedures. However, this approach requires further preclinical validation to assess its risk and effectiveness. Delivery of oHSV by MSCs could be another potential treatment approach, which avoids serum inhibitory factors and demonstrated promising efficacy preclinically. Carrier cell-based approach is under at developmental stage and needs to be extensively tested in immunocompetent/immunodeficient GBM models to determine its utility for patients.
Following viral delivery, the next important step for effective treatment response is efficient viral entry/replication. HSV uses heparan sulfate proteoglycans, herpes virus entry mediators, and nectin-1 for its entry into host cells,204 and thus, their expression on tumor cells may predict the treatment efficacy. Recently, Friedman et al demonstrated that sensitivity of pediatric brain tumor cells to oHSVs (G207, M032) was correlated with high nectin-1 expression,58 indicating receptor expression profile is critical for optimal efficacy. Based on the abundance of tumor cell surface receptors (such as nectin-1, EGFR and HER2), re-targeted oHSVs can be used to enhance viral entry to cancer cells.42,46,47,49,51 Although re-targeting strategy looks promising, new oHSVs with advanced combinations of targets will be needed to address intra- or inter-tumoral heterogeneity. In addition, re-targeted oHSVs might produce off-target effects on healthy cells, since the same receptors (EGFR and HER2) can be shared with non-cancerous cells.205,206 To overcome this issue, re-targeted oHSVs can be further modified to allow virus replication selectively in GBM cells that have distinguished miRNA expression profile compared to normal/healthy brain cells.49,51
Another important issue that needs to be addressed before utilizing the full potential of oHSVs is maximizing oHSV replication/spread. The anti-viral innate immunity and condensed extracellular matrix play important role in limiting viral replication/spread. Various approaches have been employed to address these issues such as the development of HCMV/HSV – a chimeric oHSV that replicates in a similar fashion as wild-type virus,36,38–41 or generation of armed oHSVs that express E-cadherin,84 chondroitinase ABC,88 or MMP-996 to enhance cell-cell oHSV spread, or transient inhibition of anti-viral innate immunity prior to oHSV infection to improve virus replication and spread.150,151,155,156,160,161 It is still not completely clear how the host’s innate resistance mechanisms/pathways (eg IFN signaling pathways and cGAS/STING pathway) limit oHSV replication/spread in cancer cells/TME or whether inhibition of innate (macrophage- or NK-related) immune pathways150,151,155,156,160,161 will somewhat compromise adaptive anti-tumor immunity. Further understanding of the host’s anti-viral signaling pathways will help to design new combination strategies for GBM.
Successful viral delivery, entry, replication and spread should eventually activate the host’s adaptive anti-tumor immunity, leading to recruitment of T cells to the TME. However, recruited T cells are often counter-balanced by the expression of the exhaustion markers such as PD-1. Anti-PD-1 monotherapy failed to reach intended treatment goals in GBM (Phase III CheckMate-143),75,207 illustrating that poor tumor immunogenicity and immunosuppression led to treatment-insensitive lesions.207,208 In advanced melanoma patients, T-VEC promoted intratumoral T cell infiltration and sensitized tumors to anti-PD-1.170 A combination of ICIs and oHSV was shown to eradicate the intracranial tumor in a murine GSC model,152 indicating the potential promise of this combination approach for poorly immunogenic GBMs. Even though T-VEC + ICI combination trials are currently running in non-GBM cancers, more research is needed to optimize new viral vectors and design more rationale combination clinical trials for GBM. This may include the generation of new oHSVs expressing immune modulators (eg, IL-2, CD40L, OX40L and 4–1BBL) and testing them in combination with ICIs or other anti-cancer therapies. Viral oncolysis exposes tumor antigens to dendritic cells (DCs); thus, new oHSVs can also be combined with tumor antigen-loaded DC-based vaccine to provoke a tumor-specific T-cell response.209
Acknowledgments
DS is funded by a grant from DOD (W81XWH-20-1-0702) and Dodge Jones Foundation-Abilene.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sostoa J, Dutoit V, Migliorini D. Oncolytic Viruses as a Platform for the Treatment of Malignant Brain Tumors. Int J Mol Sci. 2020;21(20):20. doi:10.3390/ijms21207449
2. Bommareddy PK, Peters C, Saha D, Rabkin SD, Kaufman HL. Oncolytic Herpes Simplex Viruses as a Paradigm for the Treatment of Cancer. Ann Rev Cancer Biol. 2018;2(1):155–173.
3. Liu P, Wang Y, Wang Y, et al. Effects of oncolytic viruses and viral vectors on immunity in glioblastoma. Gene Ther. 2020.
4. Totsch SK, Schlappi C, Kang KD, et al. Oncolytic herpes simplex virus immunotherapy for brain tumors: current pitfalls and emerging strategies to overcome therapeutic resistance. Oncogene. 2019;38(34):6159–6171.
5. Hua L, Wakimoto H. Oncolytic herpes simplex virus therapy for malignant glioma: current approaches to successful clinical application. Expert Opin Biol Ther. 2019;19(8):845–854.
6. Martikainen M, Essand M. Virus-Based Immunotherapy of Glioblastoma. Cancers. 2019;11:2.
7. Rius-Rocabert S, Garcia-Romero N, Garcia A, Ayuso-Sacido A, Nistal-Villan E. Oncolytic Virotherapy in Glioma Tumors. Int J Mol Sci. 2020;21:20.
8. Chiocca EA, Nassiri F, Wang J, Peruzzi P, Zadeh G. Viral and other therapies for recurrent glioblastoma: is a 24-month durable response unusual? Neuro Oncol. 2019;21(1):14–25.
9. Saha D, Martuza RL, Curry WT. Viral oncolysis of glioblastoma. In: Reiss CS, editor. Neurotropic Viral Infections. Vol. 2. New York: Springer; 2016:481–517.
10. Saha D, Ahmed SS, Rabkin SD. Exploring the Antitumor Effect of Virus in Malignant Glioma. Drugs Future. 2015;40(11):739–749.
11. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro Oncol. 2020;22(12Suppl2):iv1–iv96.
12. Stylli SS. Novel Treatment Strategies for Glioblastoma. Cancers. 2020;12:10.
13. Alayo QA, Ito H, Passaro C, et al. Glioblastoma infiltration of both tumor- and virus-antigen specific cytotoxic T cells correlates with experimental virotherapy responses. Sci Rep. 2020;10(1):5095.
14. Saha D, Wakimoto H, Rabkin SD. Oncolytic herpes simplex virus interactions with the host immune system. Curr Opin Virol. 2016;21:26–34.
15. Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol. 2015;33(25):2780–2788.
16. Glorioso JC, Cohen JB, Goins WF, et al. Oncolytic HSV Vectors and Anti-Tumor Immunity. Curr Issues Mol Biol. 2020;41:381–468.
17. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252(5007):854–856.
18. Zhang Q, Liu F. Advances and potential pitfalls of oncolytic viruses expressing immunomodulatory transgene therapy for malignant gliomas. Cell Death Dis. 2020;11(6):485.
19. He B, Gross M, Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A. 1997;94(3):843–848.
20. Cassady KA, Gross M, Roizman B. The second-site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J Virol. 1998;72(9):7005–7011.
21. MacLean AR, Fareed M, Robertson L, Harland J, Brown SM. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ‘a’ sequence. J Gen Virol. 1991;72(Pt 3):631–639.
22. Chou J, Kern ER, Whitley RJ, Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. 1990;250(4985):1262–1266.
23. McKie EA, MacLean AR, Lewis AD, et al. Selective in vitro replication of herpes simplex virus type 1 (HSV-1) ICP34.5 null mutants in primary human CNS tumours–evaluation of a potentially effective clinical therapy. Br J Cancer. 1996;74(5):745–752.
24. Kesari S, Randazzo BP, Valyi-Nagy T, et al. Therapy of experimental human brain tumors using a neuroattenuated herpes simplex virus mutant. Lab Invest. 1995;73(5):636–648.
25. Advani SJ, Sibley GS, Song PY, et al. Enhancement of replication of genetically engineered herpes simplex viruses by ionizing radiation: a new paradigm for destruction of therapeutically intractable tumors. Gene Ther. 1998;5(2):160–165.
26. Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1(9):938–943.
27. Kanai R, Zaupa C, Sgubin D, et al. Effect of gamma34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J Virol. 2012;86(8):4420–4431.
28. Domenis R, Cesselli D, Toffoletto B, et al. Systemic T Cells Immunosuppression of Glioma Stem Cell-Derived Exosomes Is Mediated by Monocytic Myeloid-Derived Suppressor Cells. PLoS One. 2017;12(1):e0169932.
29. Auffinger B, Spencer D, Pytel P, Ahmed AU, Lesniak MS. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev Neurother. 2015;15(7):741–752.
30. Deshors P, Toulas C, Arnauduc F, et al. Ionizing radiation induces endothelial transdifferentiation of glioblastoma stem-like cells through the Tie2 signaling pathway. Cell Death Dis. 2019;10(11):816.
31. Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci U S A. 2001;98(11):6396–6401.
32. Wakimoto H, Kesari S, Farrell CJ, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69(8):3472–3481.
33. Cheema TA, Wakimoto H, Fecci PE, et al. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci U S A. 2013;110(29):12006–12011.
34. Kambara H, Okano H, Chiocca EA, Saeki Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005;65(7):2832–2839.
35. Chiocca EA, Nakashima H, Kasai K, Fernandez SA, Oglesbee M. Preclinical Toxicology of rQNestin34.5v.2: an Oncolytic Herpes Virus with Transcriptional Regulation of the ICP34.5 Neurovirulence Gene. Mol Ther Methods Clin Dev. 2020;17:871–893.
36. Cassady KA. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J Virol. 2005;79(14):8707–8715.
37. Marshall EE, Bierle CJ, Brune W, Geballe AP. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J Virol. 2009;83(9):4112–4120.
38. Friedman GK, Nan L, Haas MC, et al. gamma(1)34.5-deleted HSV-1-expressing human cytomegalovirus IRS1 gene kills human glioblastoma cells as efficiently as wild-type HSV-1 in normoxia or hypoxia. Gene Ther. 2015;22(4):348–355.
39. Shah AC, Parker JN, Gillespie GY, et al. Enhanced antiglioma activity of chimeric HCMV/HSV-1 oncolytic viruses. Gene Ther. 2007;14(13):1045–1054.
40. Ghonime MG, Jackson J, Shah A, et al. Chimeric HCMV/HSV-1 and Deltagamma134.5 oncolytic herpes simplex virus elicit immune mediated antigliomal effect and antitumor memory. Transl Oncol. 2018;11(1):86–93.
41. Cassady KA, Bauer DF, Roth J, et al. Pre-clinical Assessment of C134, a Chimeric Oncolytic Herpes Simplex Virus, in Mice and Non-human Primates. Mol Ther Oncolytics. 2017;5:1–10.
42. Uchida H, Marzulli M, Nakano K, et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol Ther. 2013;21(3):561–569.
43. Xu H, Zong H, Ma C, et al. Epidermal growth factor receptor in glioblastoma. Oncol Lett. 2017;14(1):512–516.
44. Liu G, Ying H, Zeng G, Wheeler CJ, Black KL, Yu JS. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004;64(14):4980–4986.
45. Ahmed N, Salsman VS, Kew Y, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16(2):474–485.
46. Gambini E, Reisoli E, Appolloni I, et al. Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol Ther. 2012;20(5):994–1001.
47. Alessandrini F, Menotti L, Avitabile E, et al. Eradication of glioblastoma by immuno-virotherapy with a retargeted oncolytic HSV in a preclinical model. Oncogene. 2019;38(23):4467–4479.
48. Piwecka M, Rolle K, Belter A, et al. Comprehensive analysis of microRNA expression profile in malignant glioma tissues. Mol Oncol. 2015;9(7):1324–1340.
49. Mazzacurati L, Marzulli M, Reinhart B, et al. Use of miRNA response sequences to block off-target replication and increase the safety of an unattenuated, glioblastoma-targeted oncolytic HSV. Mol Ther. 2015;23(1):99–107.
50. Grondin B, DeLuca N. Herpes simplex virus type 1 ICP4 promotes transcription preinitiation complex formation by enhancing the binding of TFIID to DNA. J Virol. 2000;74(24):11504–11510.
51. Sette P, Amankulor N, Li A, et al. GBM-Targeted oHSV Armed with Matrix Metalloproteinase 9 Enhances Anti-tumor Activity and Animal Survival. Mol Ther Oncolytics. 2019;15:214–222.
52. Lemos Matos A, Franco LS, McFadden G. Oncolytic Viruses and the Immune System: the Dynamic Duo. Mol Ther Methods Clin Dev. 2020;17:349–358.
53. Berraondo P, Etxeberria I, Ponz-Sarvise M, Melero I. Revisiting Interleukin-12 as a Cancer Immunotherapy Agent. Clin Cancer Res. 2018;24(12):2716–2718.
54. Nguyen HM, Guz-Montgomery K, Saha D. Oncolytic Virus Encoding a Master Pro-Inflammatory Cytokine Interleukin 12 in Cancer Immunotherapy. Cells. 2020;9:2.
55. Roth JC, Cassady KA, Cody JJ, et al. Evaluation of the safety and biodistribution of M032, an attenuated herpes simplex virus type 1 expressing hIL-12, after intracerebral administration to aotus nonhuman primates. Hum Gene Ther Clin Dev. 2014;25(1):16–27.
56. Nguyen KG, Vrabel MR, Mantooth SM, et al. Localized Interleukin-12 for Cancer Immunotherapy. Front Immunol. 2020;11:575597.
57. Zhang W, Fulci G, Wakimoto H, et al. Combination of oncolytic herpes simplex viruses armed with angiostatin and IL-12 enhances antitumor efficacy in human glioblastoma models. Neoplasia. 2013;15(6):591–599.
58. Friedman GK, Bernstock JD, Chen D, et al. Enhanced Sensitivity of Patient-Derived Pediatric High-Grade Brain Tumor Xenografts to Oncolytic HSV-1 Virotherapy Correlates with Nectin-1 Expression. Sci Rep. 2018;8(1):13930.
59. Markert JM, Cody JJ, Parker JN, et al. Preclinical evaluation of a genetically engineered herpes simplex virus expressing interleukin-12. J Virol. 2012;86(9):5304–5313.
60. Barnard Z, Wakimoto H, Zaupa C, et al. Expression of FMS-like tyrosine kinase 3 ligand by oncolytic herpes simplex virus type I prolongs survival in mice bearing established syngeneic intracranial malignant glioma. Neurosurgery. 2012;71(3):741–748.
61. Tamura K, Wakimoto H, Agarwal AS, et al. Multimechanistic tumor targeted oncolytic virus overcomes resistance in brain tumors. Mol Ther. 2013;21(1):68–77.
62. Jahan N, Lee JM, Shah K, Wakimoto H. Therapeutic targeting of chemoresistant and recurrent glioblastoma stem cells with a proapoptotic variant of oncolytic herpes simplex virus. Int J Cancer. 2017;141(8):1671–1681.
63. Rozanov DV, Savinov AY, Golubkov VS, et al. Engineering a leucine zipper-TRAIL homotrimer with improved cytotoxicity in tumor cells. Mol Cancer Ther. 2009;8(6):1515–1525.
64. Kretz AL, Trauzold A, Hillenbrand A, et al. TRAILblazing Strategies for Cancer Treatment. Cancers. 2019;11:4.
65. Zhang G, Jin G, Nie X, et al. Enhanced antitumor efficacy of an oncolytic herpes simplex virus expressing an endostatin-angiostatin fusion gene in human glioblastoma stem cell xenografts. PLoS One. 2014;9(4):e95872.
66. Tomita Y, Kurozumi K, Yoo JY, et al. Oncolytic Herpes Virus Armed with Vasculostatin in Combination with Bevacizumab Abrogates Glioma Invasion via the CCN1 and AKT Signaling Pathways. Mol Cancer Ther. 2019;18(8):1418–1429.
67. Zhang W, Fulci G, Buhrman JS, et al. Bevacizumab with angiostatin-armed oHSV increases antiangiogenesis and decreases bevacizumab-induced invasion in U87 glioma. Mol Ther. 2012;20(1):37–45.
68. Bolyard C, Meisen WH, Banasavadi-Siddegowda Y, et al. BAI1 Orchestrates Macrophage Inflammatory Response to HSV Infection-Implications for Oncolytic Viral Therapy. Clin Cancer Res. 2017;23(7):1809–1819.
69. Brush MH, Weiser DC, Shenolikar S. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol Cell Biol. 2003;23(4):1292–1303.
70. Rojas M, Vasconcelos G, Dever TE. An eIF2alpha-binding motif in protein phosphatase 1 subunit GADD34 and its viral orthologs is required to promote dephosphorylation of eIF2alpha. Proc Natl Acad Sci U S A. 2015;112(27):E3466–3475.
71. Peters C, Paget M, Tshilenge KT, et al. Restriction of Replication of Oncolytic Herpes Simplex Virus with a Deletion of gamma34.5 in Glioblastoma Stem-Like Cells. J Virol. 2018;92:15.
72. Nakashima H, Nguyen T, Kasai K, et al. Toxicity and Efficacy of a Novel GADD34-expressing Oncolytic HSV-1 for the Treatment of Experimental Glioblastoma. Clin Cancer Res. 2018;24(11):2574–2584.
73. Wieder T, Eigentler T, Brenner E, Rocken M. Immune checkpoint blockade therapy. J Allergy Clin Immunol. 2018;142(5):1403–1414.
74. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29–39.
75. Filley AC, Henriquez M, Dey M. Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget. 2017;8(53):91779–91794.
76. Reardon DA, Freeman G, Wu C, et al. Immunotherapy advances for glioblastoma. Neuro Oncol. 2014;16(11):1441–1458.
77. Khasraw M, Reardon DA, Weller M, Sampson JH. PD-1 Inhibitors: do they have a Future in the Treatment of Glioblastoma? Clin Cancer Res. 2020;26(20):5287–5296.
78. Lin C, Ren W, Luo Y, et al. Intratumoral Delivery of a PD-1-Blocking scFv Encoded in Oncolytic HSV-1 Promotes Antitumor Immunity and Synergizes with TIGIT Blockade. Cancer Immunol Res. 2020;8(5):632–647.
79. Passaro C, Alayo Q, De Laura I, et al. Arming an Oncolytic Herpes Simplex Virus Type 1 with a Single-chain Fragment Variable Antibody against PD-1 for Experimental Glioblastoma Therapy. Clin Cancer Res. 2019;25(1):290–299.
80. Kim Y, Yoo JY, Lee TJ, et al. Complex role of NK cells in regulation of oncolytic virus-bortezomib therapy. Proc Natl Acad Sci U S A. 2018;115(19):4927–4932.
81. Bhargava AK, Rothlauf PW, Krummenacher C. Herpes simplex virus glycoprotein D relocates nectin-1 from intercellular contacts. Virology. 2016;499:267–277.
82. Muller-Durovic B, Lanna A, Covre LP, Mills RS, Henson SM, Akbar AN. Killer Cell Lectin-like Receptor G1 Inhibits NK Cell Function through Activation of Adenosine 5ʹ-Monophosphate-Activated Protein Kinase. J Immunol. 2016;197(7):2891–2899.
83. Katsunuma S, Honda H, Shinoda T, et al. Synergistic action of nectins and cadherins generates the mosaic cellular pattern of the olfactory epithelium. J Cell Biol. 2016;212(5):561–575.
84. Xu B, Ma R, Russell L, et al. An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat Biotechnol. 2018.
85. Dmitrieva N, Yu L, Viapiano M, et al. Chondroitinase ABC I-mediated enhancement of oncolytic virus spread and antitumor efficacy. Clin Cancer Res. 2011;17(6):1362–1372.
86. Tsidulko AY, Kazanskaya GM, Volkov AM, et al. Chondroitin sulfate content and decorin expression in glioblastoma are associated with proliferative activity of glioma cells and disease prognosis. Cell Tissue Res. 2020;379(1):147–155.
87. Kim Y, Lee HG, Dmitrieva N, Kim J, Kaur B, Friedman A. Chondroitinase ABC I-mediated enhancement of oncolytic virus spread and anti tumor efficacy: a mathematical model. PLoS One. 2014;9(7):e102499.
88. Jaime-Ramirez AC, Dmitrieva N, Yoo JY, et al. Humanized chondroitinase ABC sensitizes glioblastoma cells to temozolomide. J Gene Med. 2017;19:3.
89. Alvarez-Garcia V, Tawil Y, Wise HM, Leslie NR. Mechanisms of PTEN loss in cancer: it’s all about diversity. Semin Cancer Biol. 2019;59:66–79.
90. Han F, Hu R, Yang H, et al. PTEN gene mutations correlate to poor prognosis in glioma patients: a meta-analysis. Onco Targets Ther. 2016;9:3485–3492.
91. Jiang N, Dai Q, Su X, Fu J, Feng X, Peng J. Role of PI3K/AKT pathway in cancer: the framework of malignant behavior. Mol Biol Rep. 2020;47(6):4587–4629.
92. Russell L, Swanner J, Jaime-Ramirez AC, et al. PTEN expression by an oncolytic herpesvirus directs T-cell mediated tumor clearance. Nat Commun. 2018;9(1):5006.
93. Mou X, Zhou Y, Jiang P, et al. The regulatory effect of UL-16 binding protein-3 expression on the cytotoxicity of NK cells in cancer patients. Sci Rep. 2014;4:6138.
94. Haberthur K, Brennan K, Hoglund V, et al. NKG2D ligand expression in pediatric brain tumors. Cancer Biol Ther. 2016;17(12):1253–1265.
95. Wirsching HG, Zhang H, Szulzewsky F, et al. Arming oHSV with ULBP3 drives abscopal immunity in lymphocyte-depleted glioblastoma. JCI Insight. 2019;4:13.
96. Wirsching HG, Arora S, Zhang H, et al. Cooperation of oncolytic virotherapy with VEGF-neutralizing antibody treatment in IDH wildtype glioblastoma depends on MMP9. Neuro Oncol. 2019;21(12):1607–1609.
97. Lee S, Jilani SM, Nikolova GV, Carpizo D, Iruela-Arispe ML. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J Cell Biol. 2005;169(4):681–691.
98. Hicks A, Grandi P, Paglia M, et al. Abstract 4698: ONCR-1, a novel herpes simplex virus expressing MMP9 and ULBP3 transgenes, evokes potent oncolysis and development of antitumor immune responses. Cancer Res. 2018;78(13Supplement):4698.
99. Tyminski E, Leroy S, Terada K, et al. Brain tumor oncolysis with replication-conditional herpes simplex virus type 1 expressing the prodrug-activating genes, CYP2B1 and secreted human intestinal carboxylesterase, in combination with cyclophosphamide and irinotecan. Cancer Res. 2005;65(15):6850–6857.
100. Kasai K, Nakashima H, Liu F, et al. Toxicology and Biodistribution Studies for MGH2.1, an Oncolytic Virus that Expresses Two Prodrug-activating Genes, in Combination with Prodrugs. Mol Ther Nucleic Acids. 2013;2:e113. doi:10.1038/mtna.2013.38
101. Markert JM, Razdan SN, Kuo H-C, et al. A Phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Therapy. 2014;22(5):1048–1055. doi:10.1038/mt.2014.22
102. Kicielinski KP, Chiocca EA, Yu JS, Gill GM, Coffey M, Markert JM. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol Ther. 2014;22(5):1056–1062.
103. Forsyth P, Roldan G, George D, et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008;16(3):627–632.
104. Parker Kerrigan BC, Shimizu Y, Andreeff M, Lang FF. Mesenchymal stromal cells for the delivery of oncolytic viruses in gliomas. Cytotherapy. 2017;19(4):445–457.
105. Chaichana KL, Pinheiro L, Brem H. Delivery of local therapeutics to the brain: working toward advancing treatment for malignant gliomas. Ther Deliv. 2015;6(3):353–369.
106. Sonabend AM, Ulasov IV, Tyler MA, Rivera AA, Mathis JM, Lesniak MS. Mesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial glioma. Stem Cells. 2008;26(3):831–841.
107. Duebgen M, Martinez-Quintanilla J, Tamura K, et al. Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J Natl Cancer Inst. 2014;106(6):dju090.
108. Martinez-Quintanilla J, Shah K. Response. J Natl Cancer Inst. 2015;107(1):370.
109. Krause MN, Sancho-Martinez I, Izpisua Belmonte JC. RE: stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J Natl Cancer Inst. 2015;107(1):368.
110. Duhrsen L, Hartfuss S, Hirsch D, et al. Preclinical analysis of human mesenchymal stem cells: tumor tropism and therapeutic efficiency of local HSV-TK suicide gene therapy in glioblastoma. Oncotarget. 2019;10(58):6049–6061.
111. Pastorakova A, Jakubechova J, Altanerova U, Altaner C. Suicide Gene Therapy Mediated with Exosomes Produced by Mesenchymal Stem/Stromal Cells Stably Transduced with HSV Thymidine Kinase. Cancers. 2020;12:5.
112. Wei D, Hou J, Zheng K, et al. Suicide Gene Therapy Against Malignant Gliomas by the Local Delivery of Genetically Engineered Umbilical Cord Mesenchymal Stem Cells as Cellular Vehicles. Curr Gene Ther. 2019;19(5):330–341.
113. Karachi A, Dastmalchi F, Mitchell D, Rahman M. Temozolomide for Immunomodulation in the Treatment of Glioblastoma. Neuro Oncol. 2018.
114. Lee SY. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016;3(3):198–210.
115. Yip S, Miao J, Cahill DP, et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009;15(14):4622–4629.
116. Kanai R, Rabkin SD, Yip S, et al. Oncolytic virus-mediated manipulation of DNA damage responses: synergy with chemotherapy in killing glioblastoma stem cells. J Natl Cancer Inst. 2012;104(1):42–55.
117. Saha D, Rabkin SD, Martuza RL. Temozolomide antagonizes oncolytic immunovirotherapy in glioblastoma. J Immunother Cancer. 2020;8:1.
118. Kleijn A, van den Bossche W, Haefner ES, et al. The Sequence of Delta24-RGD and TMZ Administration in Malignant Glioma Affects the Role of CD8(+)T Cell Anti-tumor Activity. Mol Ther Oncolytics. 2017;5:11–19.
119. Das S, Marsden PA. Angiogenesis in glioblastoma. N Engl J Med. 2013;369(16):1561–1563.
120. Vollmann-Zwerenz A, Leidgens V, Feliciello G, Klein CA, Hau P. Tumor Cell Invasion in Glioblastoma. Int J Mol Sci. 2020;21:6.
121. Lu L, Saha D, Martuza RL, Rabkin SD, Wakimoto H. Single agent efficacy of the VEGFR kinase inhibitor axitinib in preclinical models of glioblastoma. J Neurooncol. 2015;121(1):91–100.
122. Vredenburgh JJ, Desjardins A, Herndon JE, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13(4):1253–1259.
123. Gilbert MR, Dignam JJ, Armstrong TS, et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N Eng J Med. 2014;370(8):699–708.
124. Saha D, Wakimoto H, Peters CW, Antoszczyk SJ, Rabkin SD, Martuza RL. Combinatorial Effects of VEGFR Kinase Inhibitor Axitinib and Oncolytic Virotherapy in Mouse and Human Glioblastoma Stem-Like Cell Models. Clin Cancer Res. 2018;24(14):3409–3422.
125. Piao Y, Liang J, Holmes L, et al. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol. 2012;14(11):1379–1392.
126. Ishida J, Onishi M, Kurozumi K, et al. Integrin inhibitor suppresses bevacizumab-induced glioma invasion. Transl Oncol. 2014;7(2):292–302.
127. Hardcastle J, Kurozumi K, Dmitrieva N, et al. Enhanced antitumor efficacy of vasculostatin (Vstat120) expressing oncolytic HSV-1. Mol Ther. 2010;18(2):285–294.
128. Ning J, Wakimoto H, Peters C, Martuza RL, Rabkin SD. Rad51 Degradation: role in Oncolytic Virus-Poly(ADP-Ribose) Polymerase Inhibitor Combination Therapy in Glioblastoma. J Natl Cancer Inst. 2017;109(3):1–13.
129. Otsuki A, Patel A, Kasai K, et al. Histone Deacetylase Inhibitors Augment Antitumor Efficacy of Herpes-based Oncolytic Viruses. Mol Ther. 2008;16(9):1546–1555.
130. Gallinari P, Marco SD, Jones P, Pallaoro M, Steinkühler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17(3):195–211.
131. Alvarez-Breckenridge CA, Yu J, Price R, et al. The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J Virol. 2012;86(8):4566–4577.
132. Nakashima H, Kaufmann JK, Wang PY, et al. Histone deacetylase 6 inhibition enhances oncolytic viral replication in glioma. J Clin Invest. 2015;125(11):4269–4280.
133. Areeb Z, Stylli SS, Ware TM, et al. Inhibition of glioblastoma cell proliferation, migration and invasion by the proteasome antagonist carfilzomib. Med Oncol. 2016;33(5):53.
134. Vlachostergios PJ, Voutsadakis IA, Papandreou CN. The shaping of invasive glioma phenotype by the ubiquitin-proteasome system. Cell Commun Adhes. 2013;20(5):87–92.
135. Yoo JY, Hurwitz BS, Bolyard C, et al. Bortezomib-induced unfolded protein response increases oncolytic HSV-1 replication resulting in synergistic antitumor effects. Clin Cancer Res. 2014;20(14):3787–3798.
136. Yoo JY, Jaime-Ramirez AC, Bolyard C, et al. Bortezomib Treatment Sensitizes Oncolytic HSV-1–Treated Tumors to NK Cell Immunotherapy. Clin Cancer Res. 2016;22(21):5265–5276.
137. Marhaba R, Nazarenko I, Knofler D, et al. Opposing effects of fibrosarcoma cell-derived IL-1 alpha and IL-1 beta on immune response induction. Int J Cancer. 2008;123(1):134–145.
138. Mishra R, Polic B, Welsh RM, Szomolanyi-Tsuda E. Inflammatory cytokine-mediated evasion of virus-induced tumors from NK cell control. J Immunol. 2013;191(2):961–970.
139. Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Autocrine MK. TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell. 2009;5(5):504–514.
140. Esaki S, Nigim F, Moon E, et al. Blockade of transforming growth factor-beta signaling enhances oncolytic herpes simplex virus efficacy in patient-derived recurrent glioblastoma models. Int J Cancer. 2017;141(11):2348–2358.
141. Wick A, Desjardins A, Suarez C, et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Invest New Drugs. 2020.
142. Bazzoni R, Bentivegna A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers. 2019;11:3.
143. Otani Y, Yoo JY, Chao S, et al. Oncolytic HSV-Infected Glioma Cells Activate NOTCH in Adjacent Tumor Cells Sensitizing Tumors to Gamma Secretase Inhibition. Clin Cancer Res. 2020;26(10):2381–2392.
144. Huck L, Pontier SM, Zuo DM, Muller WJ. beta1-integrin is dispensable for the induction of ErbB2 mammary tumors but plays a critical role in the metastatic phase of tumor progression. Proc Natl Acad Sci U S A. 2010;107(35):15559–15564.
145. Carbonell WS, DeLay M, Jahangiri A, Park CC, Aghi MK. beta1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 2013;73(10):3145–3154.
146. Lee TJ, Nair M, Banasavadi-Siddegowda Y, et al. Enhancing Therapeutic Efficacy of Oncolytic Herpes Simplex Virus-1 with Integrin beta1 Blocking Antibody OS2966. Mol Cancer Ther. 2019;18(6):1127–1136.
147. Fujii K, Kurozumi K, Ichikawa T, et al. The integrin inhibitor cilengitide enhances the anti-glioma efficacy of vasculostatin-expressing oncolytic virus. Cancer Gene Ther. 2013;20(8):437–444.
148. Weller M, Nabors LB, Gorlia T, et al. Cilengitide in newly diagnosed glioblastoma: biomarker expression and outcome. Oncotarget. 2016;7(12):15018–15032.
149. Alvarez-Breckenridge CA, Yu J, Price R, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med. 2012;18(12):1827–1834.
150. Han J, Chen X, Chu J, et al. TGFbeta Treatment Enhances Glioblastoma Virotherapy by Inhibiting the Innate Immune Response. Cancer Res. 2015;75(24):5273–5282.
151. Meisen WH, Wohleb ES, Jaime-Ramirez AC, et al. The Impact of Macrophage- and Microglia-Secreted TNFalpha on Oncolytic HSV-1 Therapy in the Glioblastoma Tumor Microenvironment. Clin Cancer Res. 2015;21(14):3274–3285.
152. Saha D, Martuza RL, Rabkin SD. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell. 2017;32(2):253–267.
153. Prionisti I, Buhler LH, Walker PR, Jolivet RB. Harnessing Microglia and Macrophages for the Treatment of Glioblastoma. Front Pharmacol. 2019;10:506.
154. Sergerie Y, Rivest S, Boivin G. Tumor necrosis factor-alpha and interleukin-1 beta play a critical role in the resistance against lethal herpes simplex virus encephalitis. J Infect Dis. 2007;196(6):853–860.
155. Pointer KB, Zhang RR, Kuo JS. Oncolytic Herpes Simplex Virus Glioblastoma Therapy is Potentiated by Tumor Necrosis Factor-alpha Inhibition. Neurosurgery. 2015;77(2):N18–20.
156. Yoo JY, Swanner J, Otani Y, et al. Oncolytic HSV therapy increases trametinib access to brain tumors and sensitizes them in vivo. Neuro Oncol. 2019;21(9):1131–1140.
157. Gerriets V, Bansal P, Khaddour K. Tumor Necrosis Factor (TNF) Inhibitors. In: StatPearls [Internet]. StatPearls Publishing; 2019.
158. Kurozumi K, Hardcastle J, Thakur R, et al. Oncolytic HSV-1 infection of tumors induces angiogenesis and upregulates CYR61. Mol Ther. 2008;16(8):1382–1391.
159. Haseley A, Boone S, Wojton J, et al. Extracellular matrix protein CCN1 limits oncolytic efficacy in glioma. Cancer Res. 2012;72(6):1353–1362.
160. Thorne AH, Meisen WH, Russell L, et al. Role of cysteine-rich 61 protein (CCN1) in macrophage-mediated oncolytic herpes simplex virus clearance. Mol Ther. 2014;22(9):1678–1687.
161. Delwar ZM, Kuo Y, Wen YH, Rennie PS, Jia W. Oncolytic Virotherapy Blockade by Microglia and Macrophages Requires STAT1/3. Cancer Res. 2018;78(3):718–730.
162. Omuro A, Vlahovic G, Lim M, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018;20(5):674–686.
163. Saha D, Martuza RL, Rabkin SD. Oncolytic herpes simplex virus immunovirotherapy in combination with immune checkpoint blockade to treat glioblastoma. Immunotherapy. 2018;10(9):779–786.
164. Pombo Antunes AR, Scheyltjens I, Duerinck J, Neyns B, Movahedi K, Van Ginderachter JA. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife. 2020;9.
165. Pearson JRD, Cuzzubbo S, McArthur S, et al. Immune Escape in Glioblastoma Multiforme and the Adaptation of Immunotherapies for Treatment. Front Immunol. 2020;11:2703.
166. Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer Cell. 2017;31(3):326–341.
167. Razavi SM, Lee KE, Jin BE, Aujla PS, Gholamin S, Li G. Immune Evasion Strategies of Glioblastoma. Front Surg. 2016;3:11.
168. Hodges TR, Ott M, Xiu J, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol. 2017;19(8):1047–1057.
169. Storey KM, Lawler SE, Jackson TL. Modeling Oncolytic Viral Therapy, Immune Checkpoint Inhibition, and the Complex Dynamics of Innate and Adaptive Immunity in Glioblastoma Treatment. Front Physiol. 2020;11:151.
170. Ribas A, Dummer R, Puzanov I, et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell. 2017;170(6):1109–1119.
171. Binello E, Qadeer ZA, Kothari HP, Emdad L, Germano IM. Stemness of the CT-2A Immunocompetent Mouse Brain Tumor Model: characterization In Vitro. J Cancer. 2012;3:166–174.
172. Oh T, Fakurnejad S, Sayegh ET, et al. Immunocompetent murine models for the study of glioblastoma immunotherapy. J Transl Med. 2014;12:107.
173. Seyfried TN. el-Abbadi M, Ecsedy JA, Bai HW, Yohe HC. Influence of host cell infiltration on the glycolipid content of mouse brain tumors. J Neurochem. 1996;66(5):2026–2033.
174. Khalsa JK, Cheng N, Keegan J, et al. Immune phenotyping of diverse syngeneic murine brain tumors identifies immunologically distinct types. Nat Commun. 2020;11(1):3912.
175. Mecca C, Giambanco I, Donato R, Arcuri C. Targeting mTOR in Glioblastoma: rationale and Preclinical/Clinical Evidence. Dis Markers. 2018;2018:9230479.
176. Krakstad C, Chekenya M. Survival signalling and apoptosis resistance in glioblastomas: opportunities for targeted therapeutics. Mol Cancer. 2010;9:135.
177. Fu X, Tao L, Rivera A, Zhang X. Rapamycin enhances the activity of oncolytic herpes simplex virus against tumor cells that are resistant to virus replication. Int J Cancer. 2011;129(6):1503–1510.
178. Zakaria C, Sean P, Hoang HD, et al. Active-site mTOR inhibitors augment HSV1-dICP0 infection in cancer cells via dysregulated eIF4E/4E-BP axis. PLoS Pathog. 2018;14(8):e1007264.
179. Kanai R, Rabkin SD. Combinatorial strategies for oncolytic herpes simplex virus therapy of brain tumors. CNS Oncol. 2013;2(2):129–142.
180. Advani SJ, Markert JM, Sood RF, et al. Increased oncolytic efficacy for high-grade gliomas by optimal integration of ionizing radiation into the replicative cycle of HSV-1. Gene Ther. 2011;18(11):1098–1102.
181. Kanai R, Wakimoto H, Martuza RL, Rabkin SD. A novel oncolytic herpes simplex virus that synergizes with phosphoinositide 3-kinase/Akt pathway inhibitors to target glioblastoma stem cells. Clin Cancer Res. 2011;17(11):3686–3696.
182. Cheema TA, Kanai R, Kim GW, et al. Enhanced antitumor efficacy of low-dose Etoposide with oncolytic herpes simplex virus in human glioblastoma stem cell xenografts. Clin Cancer Res. 2011;17(23):7383–7393.
183. Hong B, Muili K, Bolyard C, et al. Suppression of HMGB1 Released in the Glioblastoma Tumor Microenvironment Reduces Tumoral Edema. Mol Ther Oncolytics. 2019;12:93–102.
184. Foreman PM, Friedman GK, Cassady KA, Markert JM. Oncolytic Virotherapy for the Treatment of Malignant Glioma. Neurotherapeutics. 2017;14(2):333–344.
185. Eissa IR, Bustos-Villalobos I, Ichinose T, et al. The Current Status and Future Prospects of Oncolytic Viruses in Clinical Trials against Melanoma, Glioma, Pancreatic, and Breast Cancers. Cancers (Basel. 2018;10:10.
186. Harrow S, Papanastassiou V, Harland J, et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Therapy. 2004;11(22):1648–1658.
187. Rampling R, Cruickshank G, Papanastassiou V, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Therapy. 2000;7(10):859–866.
188. Papanastassiou V, Rampling R, Fraser M, et al. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Therapy. 2002;9(6):398–406.
189. Bernstock JD, Vicario N, Li R, et al. Safety and efficacy of oncolytic HSV-1 G207 inoculated into the cerebellum of mice. Cancer Gene Ther. 2020;27(3–4):246–255.
190. Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7(10):867–874.
191. Markert JM, Liechty PG, Wang W, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Therapy. 2009;17(1):199–207.
192. Whisenhunt TR, Rajneesh KF, Hackney JR, Markert JM. Extended disease-free interval of 6 years in a recurrent glioblastoma multiforme patient treated with G207 oncolytic viral therapy. Oncolytic Virother. 2015;4:33–38.
193. Friedman GK, Pressey JG, Reddy AT, Markert JM, Gillespie GY. Herpes simplex virus oncolytic therapy for pediatric malignancies. Mol Ther. 2009;17(7):1125–1135.
194. Waters AM, Johnston JM, Reddy AT, et al. Rationale and Design of a Phase 1 Clinical Trial to Evaluate HSV G207 Alone or with a Single Radiation Dose in Children with Progressive or Recurrent Malignant Supratentorial Brain Tumors. Hum Gene Ther Clin Dev. 2017;28(1):7–16.
195. Bernstock JD, Wright Z, Bag AK, et al. Stereotactic Placement of Intratumoral Catheters for Continuous Infusion Delivery of Herpes Simplex Virus −1 G207 in Pediatric Malignant Supratentorial Brain Tumors. World Neurosurg. 2019;122:e1592–e1598.
196. Bernstock JD, Bag AK, Fiveash J, et al. Design and Rationale for First-in-Human Phase 1 Immunovirotherapy Clinical Trial of Oncolytic HSV G207 to Treat Malignant Pediatric Cerebellar Brain Tumors. Hum Gene Ther. 2020;31(19–20):1132–1139.
197. Taguchi S, Fukuhara H, Todo T. Oncolytic virus therapy in Japan: progress in clinical trials and future perspectives. Jpn J Clin Oncol. 2019;49(3):201–209.
198. Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, Markert JM. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci U S A. 2000;97(5):2208–2213.
199. Hellums EK, Markert JM, Parker JN, et al. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro Oncol. 2005;7(3):213–224.
200. Parker JN, Meleth S, Hughes KB, Gillespie GY, Whitley RJ, Markert JM. Enhanced inhibition of syngeneic murine tumors by combinatorial therapy with genetically engineered HSV-1 expressing CCL2 and IL-12. Cancer Gene Ther. 2005;12(4):359–368.
201. Patel DM, Foreman PM, Nabors LB, Riley KO, Gillespie GY, Markert JM. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum Gene Ther Clin Dev. 2016;27(2):69–78.
202. Hunter WD, Martuza RL, Feigenbaum F, et al. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation of intracerebral injection in nonhuman primates. J Virol. 1999;73(8):6319–6326.
203. Malara N, Donato G Blood-Brain Barrier Breakdown by Combined Detection of Circulating Tumor and Endothelial Cells in Liquid Biopsy. In: Liquid Biopsy.2019.
204. Spear PG. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol. 2004;6(5):401–410.
205. Braut T, Krstulja M, Kujundzic M, et al. Epidermal growth factor receptor protein expression and gene amplification in normal, hyperplastic, and cancerous glottic tissue: immunohistochemical and fluorescent in situ hybridization study on tissue microarrays. Croat Med J. 2009;50(4):370–379.
206. Gutierrez C, Schiff R. HER2: biology, detection, and clinical implications. Arch Pathol Lab Med. 2011;135(1):55–62.
207. Adhikaree J, Moreno-Vicente J, Kaur AP, Jackson AM, Patel PM, Mechanisms R. Barriers to Successful Immunotherapy for Treating Glioblastoma. Cells. 2020;9:2.
208. Grabowski MM, Sankey EW, Ryan KJ, et al. Immune suppression in gliomas. J Neurooncol. 2020.
209. Stronen E, Toebes M, Kelderman S, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;352(6291):1337–1341.
210. Mineta T, Rabkin SD, Martuza RL. Treatment of malignant gliomas using ganciclovir-hypersensitive, ribonucleotide reductase-deficient herpes simplex viral mutant. Cancer Res. 1994;54(15):3963–3966.
211. Reisoli E, Gambini E, Appolloni I, et al. Efficacy of HER2 retargeted herpes simplex virus as therapy for high-grade glioma in immunocompetent mice. Cancer Gene Ther. 2012;19(11):788–795.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.