")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
The Cumulative Effect of Gene–Gene Interactions Between GSTM1, CHRNA3, CHRNA5 and SOD3 Gene Polymorphisms Combined with Smoking on COPD Risk
Authors Ganbold C, Jamiyansuren J , Tumurbaatar A, Bayarmaa A, Enebish T, Dashtseren I, Jav S
Received 1 June 2021
Accepted for publication 27 September 2021
Published 18 October 2021 Volume 2021:16 Pages 2857—2868
DOI https://doi.org/10.2147/COPD.S320841
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Chimedlkhamsuren Ganbold,1,* Jambaldorj Jamiyansuren,1,2,* Ariuntungalag Tumurbaatar,3 Agarzandan Bayarmaa,4 Tseepil Enebish,5 Ichinnorov Dashtseren,6 Sarantuya Jav1
1Department of Molecular Biology and Genetics, School of Biomedicine, MNUMS, Ulaanbaatar, Mongolia; 2Department of Biochemistry, School of Medicine, International University of Health and Welfare, Narita, Japan; 3Department of Pulmonology, The General Hospital for State Special Servants, Ulaanbaatar, Mongolia; 4Department of Biology, School of Biomedicine, MNUMS, Ulaanbaatar, Mongolia; 5Department of Pulmonology, The Second General Hospital, Ulaanbaatar, Mongolia; 6Department of Pulmonology, School of Medicine, MNUMS, Ulaanbaatar, Mongolia
*These authors contributed equally to this work
Correspondence: Sarantuya Jav
Department of Molecular Biology and Genetics, School of Biomedicine, Mongolian National University of Medical Sciences, S. Zorig Street - 3, Ulaanbaatar, 14210, Mongolia
Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) is a multifactorial disorder which is affected by external and internal risk factors. People with no external risk factors may be significantly affected and develop pulmonary disease. The study aimed to define gene–gene and gene–environmental effects on COPD.
Methods: A case control study involved 181 COPD patients and 292 healthy individuals, with peripheral blood sampling and adequate questionnaires. Genotyping was done with various types of PCR design for GSTM1 (null del), GSTT1 (null del), EPHX1 (rs2234922 and rs1051740), GSTP1 (rs1695 and rs1138272), CHRNA3 (rs1051730 and rs12914385), CHRNA5 (rs16969968 and rs17486278), and SOD3 (rs1799895 and rs699473) gene polymorphisms. Gene–gene and gene–environmental interactions were investigated using multidimensional regression analysis.
Results: Frequency of risk alleles of rs1051730 (p = 0.001), rs16969968 (p < 0.001), and rs1799895 (p < 0.001) polymorphisms were significant in univariate analysis. For gene–gene interaction, GSTM1 null, rs1051730, rs16969968, and rs1799895 polymorphisms independently contributed to risk of COPD and any combinations of the risk genotypes have a higher risk of disease. A cumulative effect of the four risk polymorphisms increased the risk of COPD for the smoking index (cOR = 13.6, p < 0.001), cigarettes per day (cOR = 32.08, p < 0.01), nicotine dependence (cOR = 12.0, p < 0.01), and smoking status (cOR = 17.02, p < 0.01) for gene–environmental interaction.
Conclusion: Several pivotal genes showed distinct effects for COPD, and some synergistic effects affected the disease progression. The development of COPD was synergistically increased with gene–gene and gene–environmental risk factors.
Keywords: COPD, MDR, genotyping, risk factor, smoking
Introduction
Chronic obstructive pulmonary disease (COPD) is the most prevalent non-communicable respiratory disease and the third leading cause of mortality worldwide.1 According to WHO estimates, there were 3.23 million COPD deaths in 2019, corresponding to 6% of all deaths worldwide. More than 80% of COPD deaths occur in low and middle-income countries.2 The prevalence and burden of COPD are expected to increase in the coming decades due to smoking, air pollution, and other factors in low and middle-income countries.3
The development of COPD involves a complex interplay among exposure to environmental factors and genetic susceptibility. Smoking is a leading environmental risk factor for COPD, however, only 10–20% of all chronic smokers will have COPD in their lifetime. Discordance between prevalence of COPD and lower chronic smokers could be explained by genetic susceptibility. Few studies defined the relevance of smoking with genetic factors.4–7 Researchers found additive interactions between rs8004738 SNP of serine protein inhibitor A1 (SERPINA1) gene and smoking for COPD in the Chinese population.6 As for the polygenic disease, it is clear that one gene cannot be responsible for the COPD progress. Two- and three-way gene–gene interaction models for lung function-related quantitative traits use multifactor dimensionality reduction (MDR) analysis. Microsomal epoxide hydrolase (EPHX1) and glutathione-S-transferase (GST) P1, SERPINA2 and transforming growth factor beta 1 (TGFβ1) two-way gene–gene interaction for forced expiratory volume in the first second (FEV1)/forced vital capacity (FVC) and FEV1.8
Genetic variations on multiple genes, such as GSTM1, GSTP1, EPHX1, GSTT1 and superoxide dismutase (SOD3), which are encoding the proteins that detoxify xenobiotic products found in cigarette smoke, may contribute to the development of COPD.9 Recent studies suggested that several polymorphisms of EPHX1, GSTP1 and deletion (null) of GSTM1, GSTT1 genes affect enzymatic activity or its efficiency. Reduced or lacked excretion of glutation S transferase may lead to dysfunction of the detoxifying process, resulting in an excess of oxidants and free radicals in lung tissue. It promotes the inflammation of airway tissue, which may cause bronchitis, emphysema, and COPD. Chinese researchers found that GSTM1, GSTP1, EPHX1 gene polymorphisms are associated with COPD development, especially with FEV1 trait. In 2011, the GSTM1 null genotype is significantly associated with increased risk of COPD in the Indian population.10 And meta-analysis of other populations repeated the result as GSTM1, GSTT1 null genotypes significantly increase the risk of COPD.11
A meta-analysis of EPHX1 gene polymorphisms was associated with COPD susceptibility. Therefore, studies have shown that the polymorphisms in cholinergic receptor nicotinic alpha 3, 5 (CHRNA3, CHRNA5) genes, encoding the subunits of nicotinic receptors, associated with the number of cigarettes per day, nicotine dependence and COPD risk. And rs1051730 and rs16969968 polymorphisms were related to higher risk of COPD.12 Also, association has been found between above-mentioned polymorphisms of CHRNA3, CHRNA5 genes and number of cigarettes per day (CPD) with nicotine addiction.13 Currently, there is no evidence for the synergism of the genetic variations and cigarette smoking on the development of COPD. We hypothesized combined or positive interactions between GSTM1, GSTP1, EPHX1, GSTT1, CHRNA3, and CHRNA5 gene polymorphisms and cigarette smoking, which may increase the risk of COPD.
This study aimed to investigate interaction of polymorphisms of GSTM1 (null del), GSTT1 (null del), EPHX1 (rs2234922, rs1051740), GSTP1 (rs1695, rs1138272), CHRNA3 (rs1051730, rs12914385), CHRNA5 (rs16969968, rs17486278), and SOD3 (rs1799895, rs699473) genes and to assess their potential interactions with cigarette smoking in the risk of COPD among the Mongolian population.
Materials and Methods
Study Population
All participants approved and signed the written informed consent to participate in the study. This study was approved by the Research Ethics Committee of Mongolian National University of Medical Science in accordance with the Declaration of Helsinki. A case-control study conducted with 181 patients for the case group and enrolled from October 2016 to February 2019, who had been referred to the First, Second, and Third Central Hospital of Ulaanbaatar, Mongolia and diagnosed with COPD. Inclusion criteria for the COPD group were applied in line with the Global Initiative for Chronic Obstructive Lung Disease, 2015 (GOLD). The criteria were as follows: a chronic or recurrent cough or sputum production for 3 months, ≥40 age, an FEV1 <70% of predicted, an FEV1/FVC ratio of <0.70 and an increase in FEV1 of <12% 15 min after the inhalation of 400 μg Ventolin (albuterol sulfate).
A total of 292 unrelated, age-matched healthy volunteers, who had no known medical illness or family disorders and were taking no medications, enrolled for the control group. Exclusion criteria were that patients had been previously or currently diagnosed with any other disease of the respiratory system, such as asthma, lung cancer, sarcoidosis, tuberculosis, and lung fibrosis, which may affect lung function.
The CDQ and FTND questionnaires were fulfilled and spirometer was performed for all subjects. Ex-smokers were excluded from the study. Tobacco consumption was estimated in the group of current smokers with cigarettes smoked per day (CPD). Current smokers were divided into two groups: heavy smokers who smoked 20 or more cigarettes per day and light smokers who smoked fewer than 20 cigarettes per day. Age at onset of daily smoking was evaluated retrospectively among ever-smoking participants who were dichotomized into early-onset (at age 16 or younger) and late-onset (at age 17 or older). Nicotine dependence (ND) was assessed in the group of current smokers with the Fagerström Test for Nicotine Dependence (FTND) score range of 0–10. Current smokers were divided into low-dependence (0–3 scores) and high-dependence (4–10 scores) according to this scale.14
SNP Genotyping
Genomic DNA was extracted, purified from whole blood using a DNeasy Blood and Tissue Kit (QIAGEN, Germany) according to the manufacturer’s protocol and used for PCR directly. Samples were kept at −20 °C for long-term storage. The null genotypes of GSTM1 and GSTT1 were detected by multiplex polymerase chain reaction (PCR), which was performed as previously described.14 Single nucleotide variations rs1051740 and rs2234922 of EPHX1 gene, rs1695 of GSTP1 gene, were determined by RFLP as previously described.15,16 List of the primers, the restriction enzymes and size of products are shown in Supplementary Table 1. PCR reactions were carried out using the AccuPower® PCR PreMix Kit (BioNeer Corporation, Korea) according to the manufacturer`s protocol. The products were analyzed by electrophoresis with an agarose gel (Promega Corporation, USA) and visualized with ethidium-bromide staining.
Statistical Analysis
Analyses were performed using STATA 13.0 (StataCorp, USA) and Microsoft Excel (Microsoft Corporation, USA) software. Comparisons of numerical variables including age, body mass index (BMI), FEV1, FVC, and FEV1/FVC ratio were analyzed by the Student’s t-test, ANOVA, or Mann–Whitney U-test. A Pearson’s chi-squared test (χ2) for 2x2, 2x3, or 2×4 contingency tables and the Fisher’s exact test were used to analyze the distribution of allele and genotype frequency. For all univariate analysis, a p-value of 0.05 was considered statistically significant. The Online Encyclopedia for Genetic Epidemiology calculator was used to test the Hardy–Weinberg equilibrium. Selection among the genetic models was performed by a four-model strategy described by Horita and Kaneko.17 MDR analysis was performed using MDR 3.0.2 software to identify the gene–gene combined effect on the COPD risk. To reduce the chance of false positives, data were generated using a 10-fold cross-validation procedure. The best model was selected based on maximum cross-validation consistency (CVC), training balance accuracy (TrBA) and testing balance accuracy (TeBA). Possible additive interactions between gene–environment or environment–environment in association with COPD were examined by relative excess risk due to interaction (RERI), synergy index (S) and the proportion attributable of interaction (AP), formulas described by Knol et al.18 A logistic regression analysis was performed to detect the association between COPD risk and each potential factors. Crude (cOR) for the univariate model and adjusted odds ratios (aOR) for the multivariate model with a 95% confidence interval (CI) were calculated by logistic regression. P values for multivariate model was corrected by Bonferronni correction. The statistical power was calculated by post-hoc test, to estimate the level of association.
Results
General Characteristics of Study Participants
A total of 473 participants (181 COPD patients and 292 controls) were included in the analysis. The baseline demographic data of the study groups are summarized in Table 1. No significant differences were observed for age, gender, BMI, education level, occupational exposure to dust, and smoking years between groups. On the other hand, the number of current or never smokers, cigarettes per day, pack-years of smoking, spirometer measurements were significantly different between COPD patients, and control group individuals.
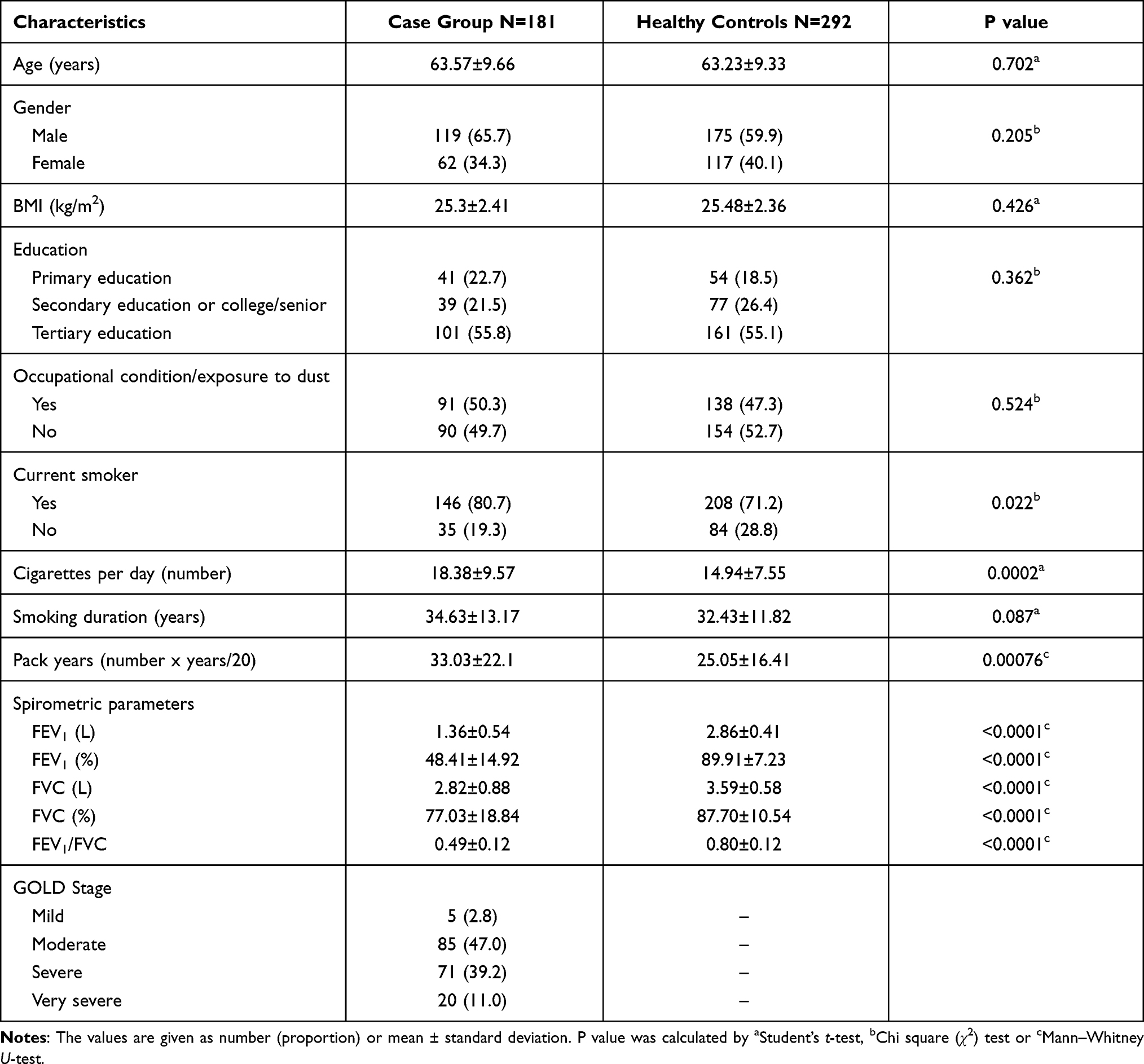 |
Table 1 Baseline Characteristics of the Groups |
All participants were divided into three or four subgroups by smoking status, pack years of smoking, cigarettes per day, nicotine dependence, and age at onset of daily smoking, to examine significant differences between the groups (Table 2). The univariate model showed a significantly higher risk of COPD, for a current smoker (cOR = 1.68; 95% CI, 1.08–2.64, p = 0.02), participants who had early onset of daily smoking (cOR = 2.19; 95% CI, 1.17–4.09, p = 0.014) compared with never smokers. Pack years of smoking (cOR = 2.85; 95% CI, 1.62–5.01, p = 0.0002), ND (cOR = 2.32; 95% CI, 1.43–3.79, p = 0.0006), and CPD (cOR = 2.9; 95% CI, 1.73–4.88, p <0.001) were also significantly different in groups. The multivariate-logistic regression analysis has shown that heavy smokers (≥20 CPD, aOR = 2.87; 95% CI, 1.67–4.94, p = 0.002), who had pack years ≥40 (aOR = 3.02; 95% CI, 1.64–5.57, p = 0.004), had 3-fold higher risk for COPD compared with never-smokers. In addition, participants who were assessed by FTND as having nicotine high-dependency, have a higher risk (aOR = 2.18; 95% CI, 1.32–3.59, p = 0.031) for COPD.
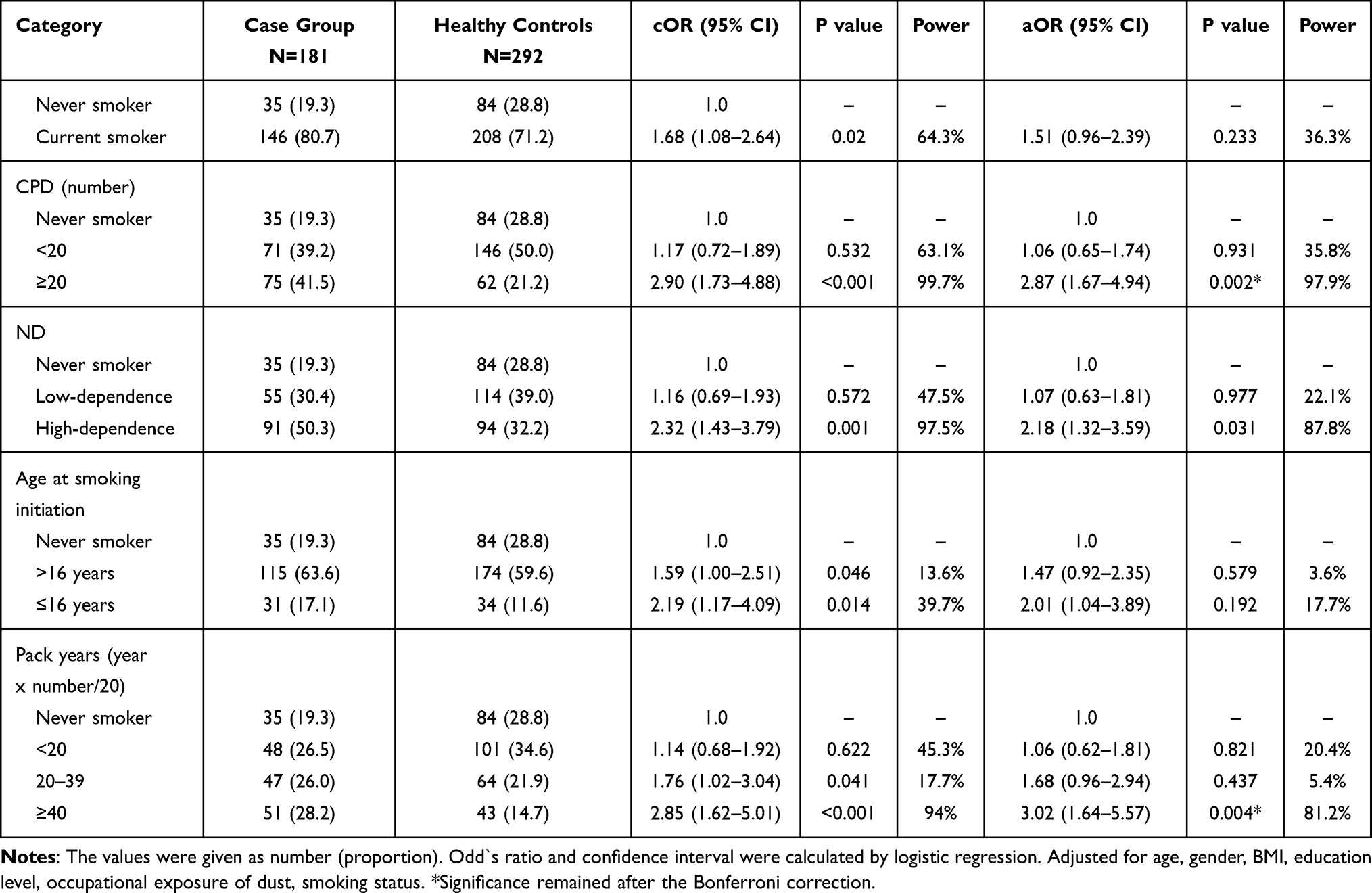 |
Table 2 Association Between Cigarette Smoking-Related Phenotypes and COPD Risk |
Alleles and Genotypes of SNP Polymorphisms
Distribution of the genetic polymorphisms among all subjects was found in accordance with those expected by Hardy–Weinberg equilibrium (p >0.05). Prevalence of alleles and genotypes of rs2234922, rs1051740, rs1695, rs1138272, rs12914385, rs17486278, and rs699473 did not differbetween COPD patients and controls. Frequency of risk alleles of rs1051730 (cOR = 1.91; 95% CI, 1.30–2.81, p = 0.001), rs16969968 (cOR = 1.89; 95% CI, 1.32–2.70, p <0.001) and rs1799895 (cOR = 2.36; 95% CI, 1.60–3.47, p <0.001) polymorphisms were significant in univariate analysis (Table 3).
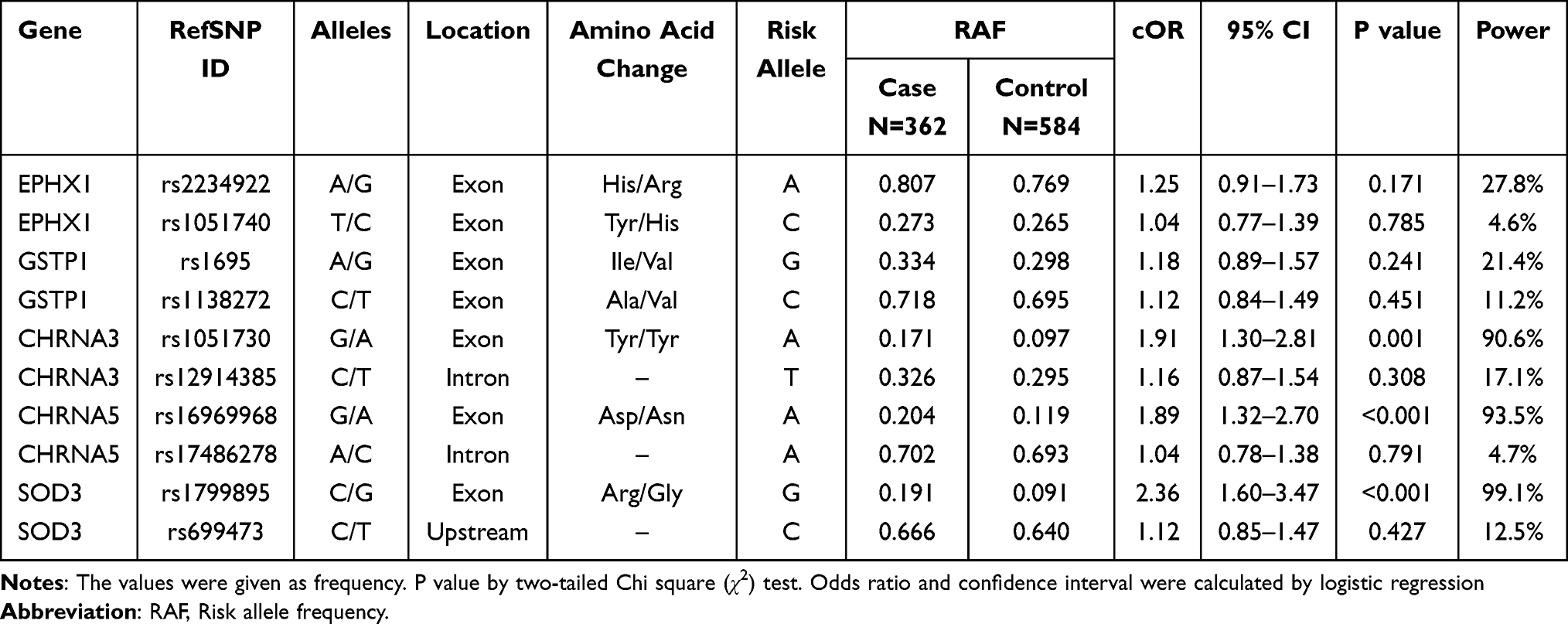 |
Table 3 Association Between Allele Frequencies of SNPs and COPD |
For GSTM1 gene, null genotype was significantly the most prevalent genotype among COPD patients (cOR = 2.43; 95% CI, 1.66–3.56, p <0.001). In multivariate analysis, the same significance was shown for GSTM1 null genotype (aOR = 2.19; 95% CI, 1.41–3.39, p <0.001). GSTT1 null genotype was more frequent in the case group but a statistical difference was not observed between groups for the genotype. The genotype distribution in genetic models showed that rs1799895 (G/G+G/C vs C/C, aOR = 2.87; 95% CI, 1.71–4.80, p = 0.0006) and rs16969968 (A/A+G/A vs G/G, aOR = 2.24; 95% CI, 1.40–3.57, p = 0.0046) were significant to increased risk of COPD in the dominant model. Comparisons of all genotype frequencies between the groups are shown in Table 4.
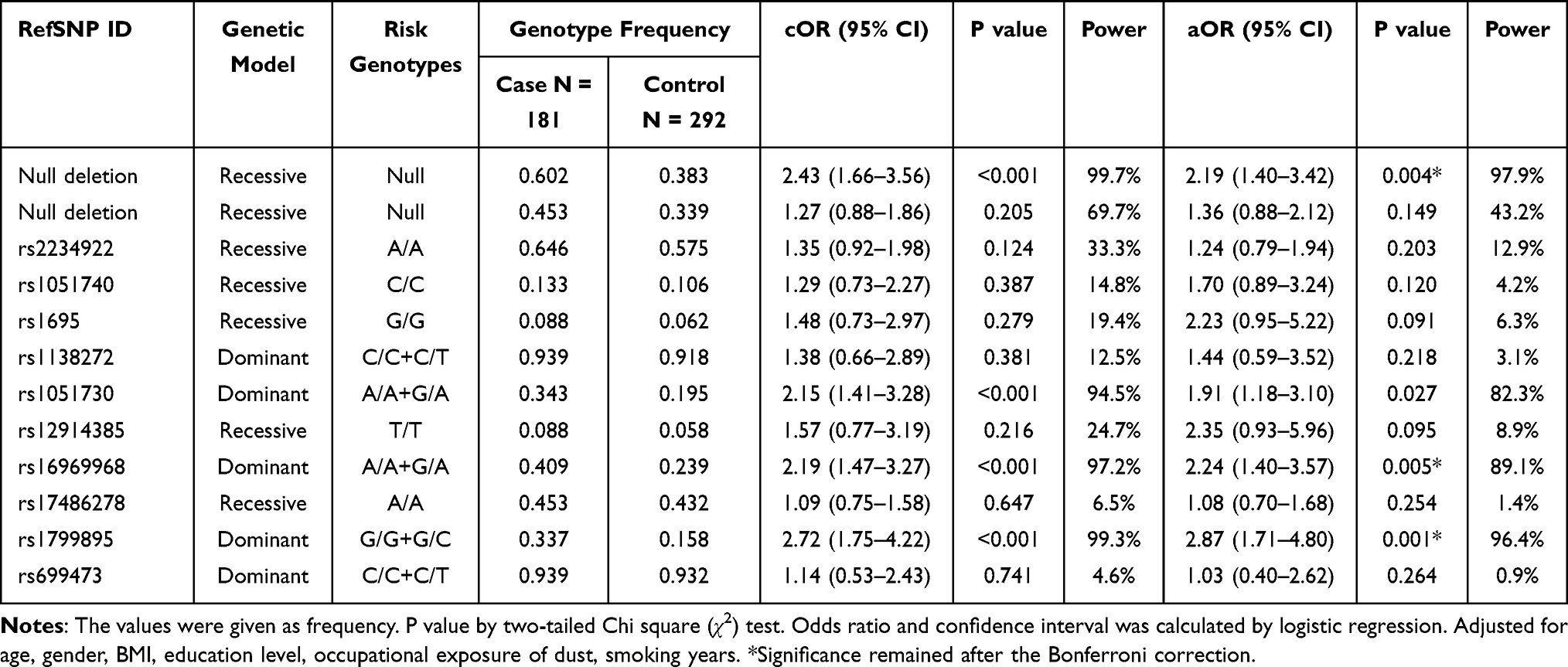 |
Table 4 Genotype Frequencies of SNPs in Selected Genetic Models Among Groups |
Gene–Gene Interactions
The entropy-based gene–gene interaction network is shown in Figure 1. The GSTM1 null (3.29%), rs1799895 (3.07%), rs16969968 (2.27%), and rs1051730 (1.93%) polymorphisms were found to contribute the highest independent effect among all genetic factors. A high degree of synergistic interaction was detected between rs2234922 and rs1695. Also, moderate synergistic interaction was found between rs1138272 and rs17486278, whereas interactions of GSTM1, CHRNA5, and CHRNA3 gene polymorphisms were detected as redundancy.
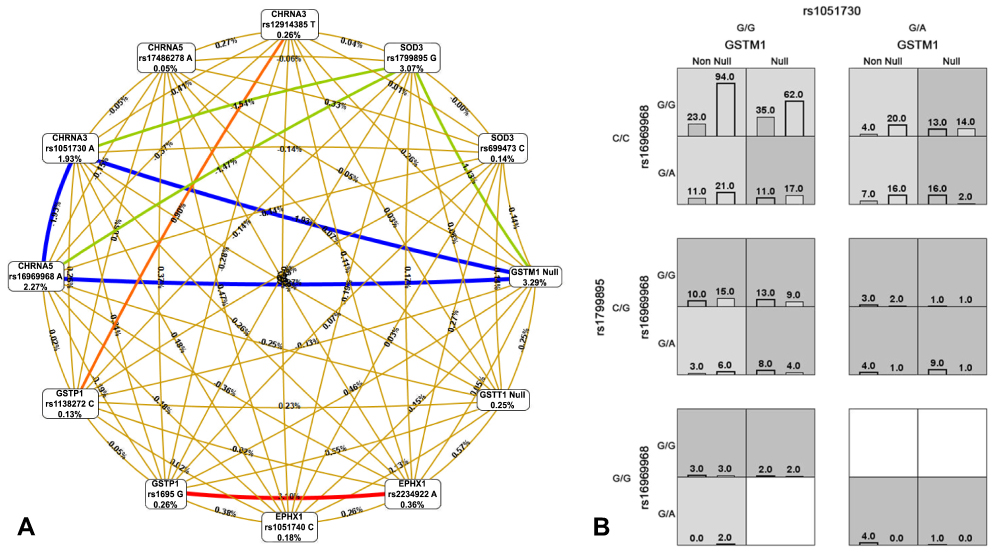 |
Figure 1 (A) Entropy-based SNP-SNP interaction network of 12 polymorphisms of the genes in case and control subjects. The percent of the entropy for independent factors as well as their interactions are represented in the graph where positive percentage of entropy denotes synergistic interaction while negative percentage denotes redundancy. The best MDR model for gene–gene interaction. Here, the red and orange colors indicate synergistic interaction, gold color denotes the mid-point, green color represents moderate redundancy while blue color denotes the highest. (B) The best model is composed of GSTM1 null deletion, rs1051730, rs16969968, and rs1799895. In each cell, the left bar represents a positive score, and the right bar a negative score. High-risk cells are indicated by gray shading, low-risk cells by light shading, and empty cells by no shading. The patterns of high-risk and low-risk cells differ across each of the different multi-locus dimensions, presenting evidence of epistasis. |
Gene–gene interaction analysis was performed among only GSTM1 null, rs1051730, rs16969968, and rs1799895 polymorphisms, which were associated with COPD risk. Best interaction models identified MDR from 10-fold cross-validation for COPD, are listed in Table 5. Significant associations were found for the combined genotype frequencies of GSTM1 and SOD3 genes between the groups (shown in Table 6). Participant who carried null (aOR = 2.24; 95% CI, 1.32–3.82, p = 0.041), non CC (aOR = 2.84; 95% CI, 1.36–5.92, p = 0.001) genotypes or both of them (aOR = 6.50; 95% CI, 2.89–14.64, p <0.001) have a higher risk of COPD compared with participants without any of the risk genotypes (Table 6).
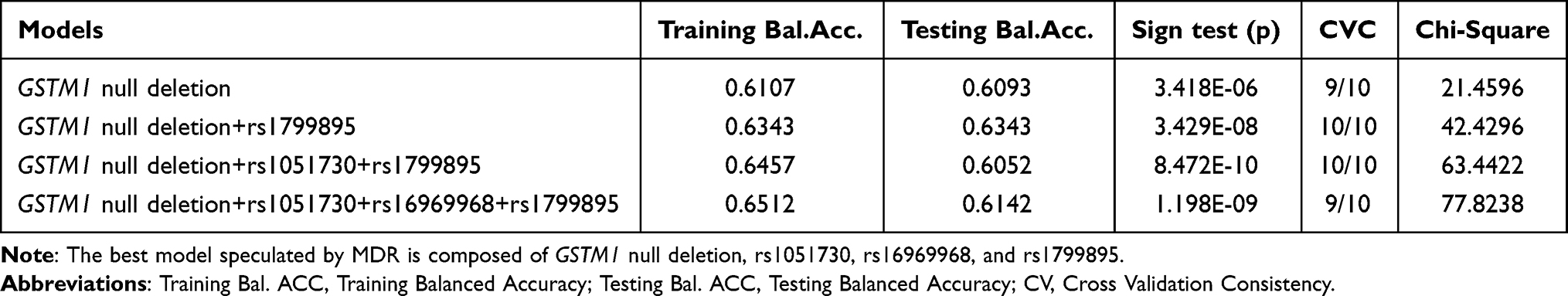 |
Table 5 Best Models of Gene–Gene Interactions Among the Four COPD Associated Polymorphisms |
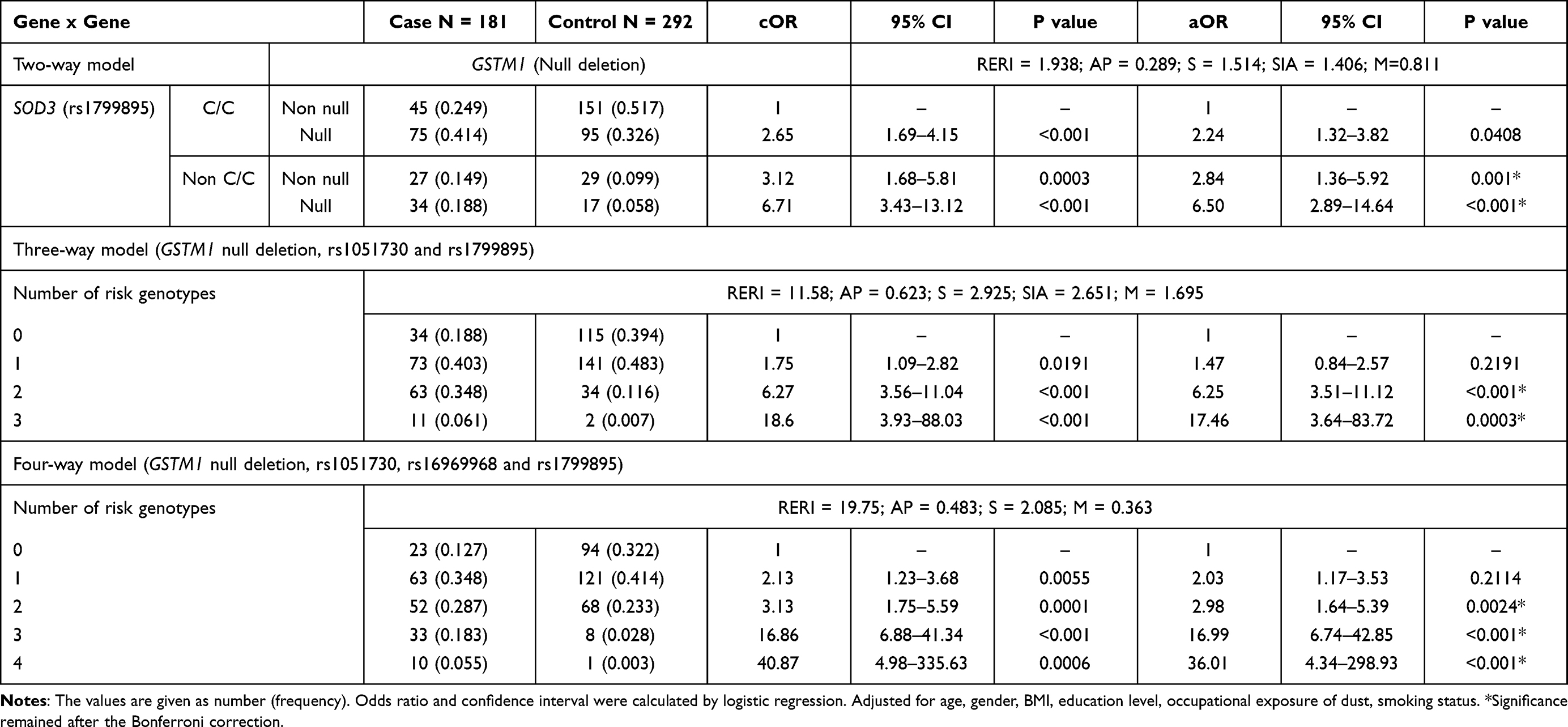 |
Table 6 Cumulative Effect of Best Models of Gene–Gene Interactions on COPD |
The three-gene cumulative effect of GSTM1 null, rs1051730, and rs1799895 polymorphisms indicates that participants who carry any combination of risk genotypes have an extremely higher risk of COPD (Table 6). We found that the frequency of the combination that was null for GSTM1, G/A for rs1051730, G/G or G/C for rs1799895, was significantly different between the study groups in univariate (cOR = 18.6; 95% CI, 3.93–88.03, p <0.001) and multivariate (aOR = 17.46; 95% CI, 3.64–83.72, p = 0.0003) analysis. It shows that carriers of these three risk genotypes together had a significantly higher risk of COPD compared with participants without any of these risk genotypes.
According to the four-gene interaction model, as shown in Table 6, we compared the combinations of risk genotypes of four genes among cases and controls. We found that men who carried a combination of four-risk genotypes have an extremely higher risk of COPD (aOR = 36.01; 95% CI, 4.34–298.93, p = 0.0003). Our findings showed that GSTM1 null, rs1051730, rs16969968, and rs1799895 polymorphisms independently contributed to the risk of COPD. However, any combinations of the risk genotypes have a higher risk of COPD, which indicates that additive interactions exist among the polymorphisms.
Gene–Environmental Interactions
This result suggests that there is some positive-additive interaction existing between of GSTM1, CHRNA3, CHRNA5 genes, and cigarette smoking-related factors for COPD risk. The stepwise analyses were focused on interactions between genetic polymorphism and smoking-related phenotypic measures in COPD. As a result, we found some significant interactions between the risk factors. Sample number for estimation of gene–smoking was reduced due to lack of smoking information for some individuals. Among GSTM1 null genotype carriers, smokers with 20 or more pack-years, had a higher risk of COPD (cOR = 4.02; 95% CI, 2.17–7.57, p <0.001, RERI = 1.562; AP = 0.389, S = 2.072). The result has shown that heavy smokers, who were carrying null genotype of GSTM1, had nearly 7-fold higher risk for COPD compared with light smokers (cOR = 6.56; 95% CI, 3.32–12.97, p <0.001, RERI = 4.122; AP = 0.628, S = 3.86). As shown in Table 7, risk of COPD was 4-fold higher for heavy smokers with G/A genotype of CHRNA3 than light smokers with G/G genotype (cOR = 4.28; 95% CI, 2.18–8.40, p <0.001, RERI = 1.592; AP = 0.372, S = 1.942). In heavy smokers, carriers of rs1799895 non-C/C (C/G or G/G) had 6-fold increased ORs compared with C/C carriers (cOR = 6.43; 95% CI, 3.17–13.06, p <0.001, RERI = 4.078; AP = 0.634, S = 4.015). In participants who are addicted to nicotine, with either G/A genotype of rs1051730, increased COPD risk has been observed (cOR = 4.16, 95% CI, 2.11–8.22, p <0.001). The combination of C/G+G/G alleles of SOD3 gene polymorphism has been shown as a higher risk for the age at onset of smoking after 16 years (cOR = 7.68, 95% CI, 2.49–23.63, p <0.0004) and the same genotype variant has been a high risk for the current smoker (cOR = 3.91, 95% CI, 2.14–7.14, p <0.001).
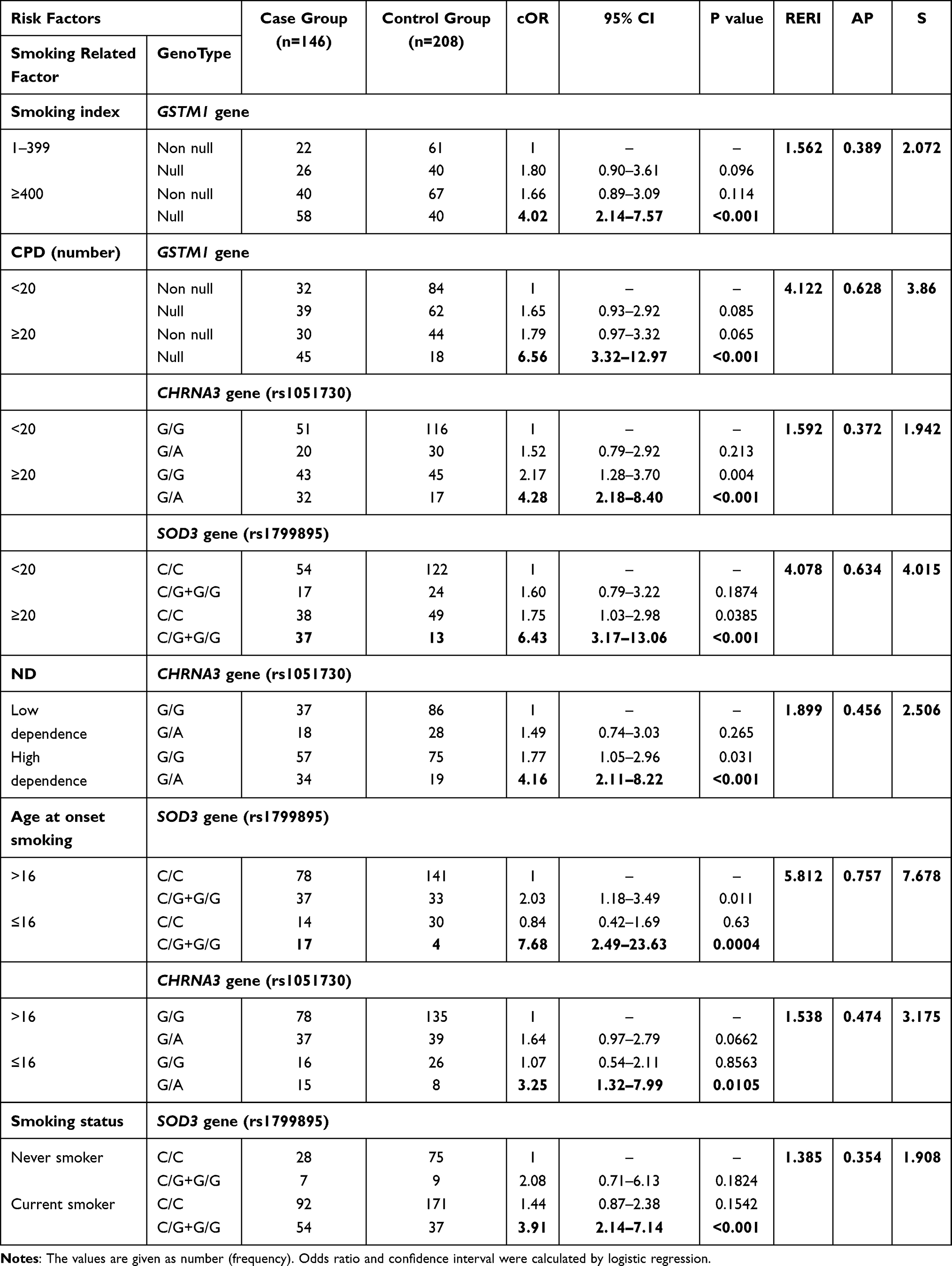 |
Table 7 Possible Interaction of Smoking-Related Factors with GSTM1, CHRNA3, CHRNA5 and SOD3 Genes Polymorphisms in COPD |
A cumulative effect of the four risk polymorphisms has been increased risk of COPD for the smoking index (cOR = 13.6, 95% CI, 1.70–108.56, p <0.001), cigarettes per day (cOR = 32.08, 95% CI, 1.86–551.9, p <0.01) and nicotine dependence (cOR = 12.0, 95% CI, 1.48–97.02, p <0.01) as shown in Table 8.
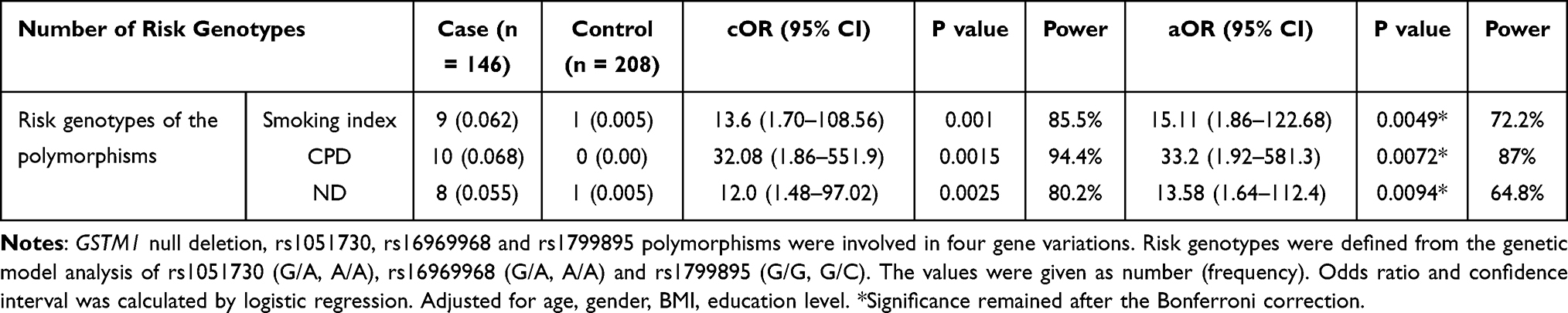 |
Table 8 Cumulative Effect of Four Risk Polymorphisms with Smoking Related Factors on COPD |
Discussion
Polymorphisms of GSTM1, CHRNA3, CHRNA5 and SOD3 genes are well-studied genetic variations as risk factors for COPD. Nevertheless, few studies consider gene-gene or gene-environment interaction with the genetic factors in COPD susceptibility. In this case-control study, we studied 12 polymorphisms among 181 COPD patients and 292 controls. We evaluated association of genetic polymorphism and smoking-related phenotypic factors with COPD risk.
GSTM1, encoding Glutathione S Transferase Mu 1, is the protein class of the highly polymorphic, cytosolic and membrane bound glutathione S-transferase, of which the null variation has been linked to COPD and lung cancer, due to increased susceptibility to toxins and carcinogens.10,19 Our result showed null deletion genotype showed high risk of COPD as described elsewhere. CHRNA3/CHRNA5, encoding alpha 3 or 5 subunit of nicotinic acetylcholine receptor, more likely related to nicotine dependence of smoking.12 Allelic distribution of two polymorphisms on the exon of CHRNA3 and CHRNA5 showed association between case and control groups. Genotype variants of these two SNPs showed higher risk of COPD. SOD3, encoding superoxide dismutase, is the protein that catalyses superoxide radicals, which protects the lung from oxidative stress. GSTM1 null, rs1051730, rs16969968, and rs1799895 polymorphisms contribute to the COPD risk, independently.
The highest gene–gene interaction was observed between EPHX1 and GSTP1 gene polymorphisms indicating that alteration of the combined detoxifying system affects COPD development. Moderate effect of gene–gene interaction between GSTP1 and CHRNA5 is burdensome. However, independently both gene polymorphisms result in a high risk of lung cancer, which may explain the indirect connection of glutathione S transferase and acetyl cholinergic systems. Combined genotype of GSTM1 and SOD3 genes were highest in two-way analysis, indicating a synergist effect of complex detoxification function through cellular membrane. With three and four gene analysis, all polymorphisms were shown to be highly interconnected and increased the risk of COPD in univariate and multivariate analysis.
Cigarette smoking is a preventable common risk factor of non-communicable diseases including COPD, lung cancer, diabetes, and heart diseases. It is important to understand how cigarette use has been measured before getting into details about how we can determine how much of cigarette use is attributed to genes, the environment, and their interactions.20,21 In this study, we used common phenotypic measures of cigarette consumption such as adolescent smoking/early onset of smoking, cigarettes per day, nicotine dependence, and smoking cessation. The findings showed that COPD risk from the smoking phenotypic measures was similar to previously reported data from recent studies. Gene–environmental interaction analysis proved that highly toxic compounds of cigarettes are damaging to lung tissue and interaction between genes of the detoxifying system and nicotine dependence results in a further entangling in COPD. However, while we had chosen causative gene polymorphisms, the limitation of this study was the small sample size that lowers the statistical power of the study.
Conclusion
We suggest the development of COPD can be driven with gene–gene interaction between GSTM1, CHRNA3, CHRNA5, and SOD3 genes. The effect of interaction is believed to be synergistic for all two-, three- and four-way gene models, which states polygenic condition of COPD. Additively, gene–environmental interaction proved that smoking leads to an increased susceptibility to lung disease.
Ethics
This study was approved by the Research Ethics Committee of Mongolian National University of Medical Science.
Acknowledgements
This research received assistance from the Research and Development Foundation, the Mongolian National University of Medical Sciences (Grant No.1/18), and the Research and Development Foundation, the Ministry of Science and Education, Mongolia (Grant no. 2020/48).
Disclosure
The authors declare no conflicts of interest.
References
1. Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–2128. doi:10.1016/S0140-6736(12)61728-0
2. World Health Organization. Chronic obstructive pulmonary disease; June 21, 2021. Available from: https://www.who.int/news-room/fact-sheets/detail/chronic-obstructive-pulmonary-disease-(copd).
3. Mathers CD, Loncar D, Samet J. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. doi:10.1371/journal.pmed.0030442
4. de Jong K, Boezen HM, Hacken N, Postma DS, Vonk JM; the LifeLines cohort s. GST-omega genes interact with environmental tobacco smoke on adult level of lung function. Respir Res. 2013;14(1):83. doi:10.1186/1465-9921-14-83
5. Mehta AJ, Thun GA, Imboden M, et al. Interactions between SERPINA1 PiMZ genotype, occupational exposure and lung function decline. Occup Environ Med. 2014;71(4):234–240. doi:10.1136/oemed-2013-101592
6. Deng X, Yuan C-H, Chang D. Interactions between single nucleotide polymorphism of SERPINA1 gene and smoking in association with COPD: a case-control study. Int J Chron Obstruct Pulmon Dis. 2017;12:259–265. doi:10.2147/COPD.S116313
7. Liao SY, Lin X, Christiani DC. Gene-environment interaction effects on lung function- a genome-wide association study within the Framingham heart study. Environ Health. 2013;12(1):101. doi:10.1186/1476-069X-12-101
8. An L, Lin Y, Yang T, Hua L. Exploring the interaction among EPHX1, GSTP1, SERPINE2, and TGFB1 contributing to the quantitative traits of chronic obstructive pulmonary disease in Chinese Han population. Hum Genomics. 2016;10(1):13. doi:10.1186/s40246-016-0076-0
9. Ishii T, Matsuse T, Teramoto S, et al. Glutathione S-transferase P1 (GSTP1) polymorphism in patients with chronic obstructive pulmonary disease. Thorax. 1999;54(8):693. doi:10.1136/thx.54.8.693
10. Shukla RK, Kant S, Bhattacharya S, Mittal B. Association of genetic polymorphism of GSTT1, GSTM1 and GSTM3 in COPD patients in a north Indian population. COPD. 2011;8(3):167–172. doi:10.3109/15412555.2011.560128
11. Ding Z, Wang K, Li J, Tan Q, Tan W, Guo G. Association between glutathione S-transferase gene M1 and T1 polymorphisms and chronic obstructive pulmonary disease risk: a meta-analysis. Clin Genet. 2019;95(1):53–62. doi:10.1111/cge.13373
12. Pérez-Morales R, González-Zamora A, González-Delgado MF, et al. CHRNA3 rs1051730 and CHRNA5 rs16969968 polymorphisms are associated with heavy smoking, lung cancer, and chronic obstructive pulmonary disease in a Mexican population. Ann Hum Genet. 2018;82(6):415–424. doi:10.1111/ahg.12264
13. Buczkowski K, Sieminska A, Linkowska K, et al. Association between genetic variants on chromosome 15q25 locus and several nicotine dependence traits in Polish population: a case-control study. Biomed Res Int. 2015;2015:350348. doi:10.1155/2015/350348
14. Heatherton TF, Kozlowski LT, Frecker RC, Fagerström KO. The Fagerström test for nicotine dependence: a revision of the Fagerström tolerance questionnaire. Br J Addict. 1991;86(9):1119–1127. doi:10.1111/j.1360-0443.1991.tb01879.x
15. Yim JJ, Park GY, Lee CT, et al. Genetic susceptibility to chronic obstructive pulmonary disease in Koreans: combined analysis of polymorphic genotypes for microsomal epoxide hydrolase and glutathione S-transferase M1 and T1. Thorax. 2000;55(2):121–125. doi:10.1136/thorax.55.2.121
16. Watson MA, Stewart RK, Smith GB, Massey TE, Bell DA. Human glutathione S-transferase P1 polymorphisms: relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis. 1998;19(2):275–280. doi:10.1093/carcin/19.2.275
17. Horita N, Kaneko T. Genetic model selection for a case-control study and a meta-analysis. Meta Gene. 2015;5:1–8. doi:10.1016/j.mgene.2015.04.003
18. Knol MJ, van der Tweel I, Grobbee DE, Numans ME, Geerlings MI. Estimating interaction on an additive scale between continuous determinants in a logistic regression model. Int J Epidemiol. 2007;36(5):1111–1118. doi:10.1093/ije/dym157
19. Lakhdar R, Denden S, Knani J, et al. Combined analysis of EPHX1, GSTP1, GSTM1 and GSTT1 gene polymorphisms in relation to chronic obstructive pulmonary disease risk and lung function impairment. Dis Markers. 2011;30(5):253–263. doi:10.1155/2011/956250
20. Do E, Maes H. Narrative review of genes, environment, and cigarettes. Ann Med. 2016;48(5):337–351. doi:10.1080/07853890.2016.1177196
21. Tan WC, Sin DD, Bourbeau J, et al. Characteristics of COPD in never-smokers and ever-smokers in the general population: results from the CanCOLD study. Thorax. 2015;70(9):822. doi:10.1136/thoraxjnl-2015-206938
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.