")
Back to Journals » OncoTargets and Therapy » Volume 10
The crossroads of breast cancer progression: insights into the modulation of major signaling pathways
Authors Velloso FJ, Bianco AFR , Farias JO, Torres NEC , Ferruzo PY , Anschau V, Jesus-Ferreira HC, Chang TH, Sogayar MC , Zerbini LF, Correa RG
Received 31 July 2017
Accepted for publication 6 October 2017
Published 20 November 2017 Volume 2017:10 Pages 5491—5524
DOI https://doi.org/10.2147/OTT.S142154
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Fernando J Velloso,1,* Arthur FR Bianco,2,* Jessica O Farias,3 Nadia EC Torres,4 Pault YM Ferruzo,3 Valesca Anschau,5 Henrique C Jesus-Ferreira,1 Ted Hung-Tse Chang,6 Mari Cleide Sogayar,1 Luiz F Zerbini,6 Ricardo G Correa7
1Cell and Molecular Therapy Center (NUCEL-NETCEM), School of Medicine, 2Department of Pharmacology, Biomedical Sciences Institute, 3Department of Biochemistry, Institute of Chemistry, 4Departament of Immunology, Biomedical Sciences Institute, 5Department of Genetics and Evolutionary Biology, Institute of Biosciences, University of São Paulo, São Paulo, Brazil; 6Cancer Genomics Group, International Center for Genetic Engineering and Biotechnology (ICGEB), Cape Town, South Africa; 7Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA, USA
*These authors contributed equally to this work
Abstract: Cancer is the disease with highest public health impact in developed countries. Particularly, breast cancer has the highest incidence in women worldwide and the fifth highest mortality in the globe, imposing a significant social and economic burden to society. The disease has a complex heterogeneous etiology, being associated with several risk factors that range from lifestyle to age and family history. Breast cancer is usually classified according to the site of tumor occurrence and gene expression profiling. Although mutations in a few key genes, such as BRCA1 and BRCA2, are associated with high breast cancer risk, the large majority of breast cancer cases are related to mutated genes of low penetrance, which are frequently altered in the whole population. Therefore, understanding the molecular basis of breast cancer, including the several deregulated genes and related pathways linked to this pathology, is essential to ensure advances in early tumor detection and prevention. In this review, we outline key cellular pathways whose deregulation has been associated with breast cancer, leading to alterations in cell proliferation, apoptosis, and the delicate hormonal balance of breast tissue cells. Therefore, here we describe some potential breast cancer-related nodes and signaling concepts linked to the disease, which can be positively translated into novel therapeutic approaches and predictive biomarkers.
Keywords: breast cancer, estrogen receptor, PI3K, MAPK, JAK/STAT, Wnt, TGFβ, NFκB
Introduction
Cancer is one of the main causes of morbidity and mortality, being the second most prominent cause of deaths worldwide, with about 14 million new cases and 8.2 million deaths in 2012,1,2 representing one of the major issues in public health. According to the World Health Organization, breast cancer is the malignancy with the highest incidence among women, accounting for 25% of all female cancers diagnosed. Despite recent improvements in diagnosis and treatment, breast cancer remains the leading cause of cancer mortality among woman, representing 7% of all cancer deaths. The incidence and prevalence of breast cancer is increasing, especially in industrialized countries in North America, Western and Northern Europe, and Oceania.3 From 2006 to 2010, breast cancer accounted for 19.8% of diagnosed cancers in the US.4 For 2017, a total of 252,710 new cases and 40,610 deaths due to female breast cancer are estimated in the US alone.1
The development of the mammary gland encompasses different stages, such as embryonic, prepubertal, pubertal, pregnancy, and lactation. In each of these stages, such processes as epithelial proliferation during pregnancy and secretory alveolar differentiation during lactation take place.5 It has been proposed that breast cancer can develop through different mechanisms, but starts most frequently with imbalanced activity of pathways involved in patterning and morphogenesis of mammary gland developmental stages.
Several breast cancer classifications have been proposed, according to their invasive characteristics, occurrence, histology, or molecular profiling. Based on their site of occurrence, tumors can be classified as lobular (located at breast lobules) or ductal (at breast ducts). Carcinomas may also arise from invasive epithelial cells (medullary carcinoma), mucus-producing cells (mucinous carcinoma, also called colloid carcinoma), or a subtype of invasive ductal carcinoma (tubular carcinoma).6,7 Traditionally, breast cancer has been classified by immunohistochemical detection of hallmark receptors associated with distinct cell functions. Cancers derived from luminal cells are the most common types of breast cancer, expressing hormone receptors for ER, PR, or the amplified HER2/ErbB2 receptor for EGF-like growth factors. HER2 is a transmembrane glycoprotein belonging to a family of receptors that regulate cell growth, proliferation, survival, differentiation, and angiogenesis. Activation of the receptor and its subsequent phosphorylation lead to several signaling pathways, such as those of PI3K, MAPK, and PKC, and also to their pathophysiological functions.7,8 HER2-overexpressing tumors are considered to be more malignant, overexpressing other genes, such as GRB7 and PGAP3, and may display representative TP53 mutation. Each category is usually targeted by a specific drug therapy.
Tumors lacking expression of all three of these receptors (ER, PR, HER2) are referred to as triple-negative breast cancers, which most often derive from cells of basal origin.9 Molecular gene expression profiling has also redefined breast cancer subtypes as luminal A, luminal B, HER2-rich, and basal-like, which roughly parallel the immunohistochemical categories.10–12 In addition, a more recently discovered claudin-low class of breast cancers has been defined.13
Claudins are a family of tight junction proteins that are expressed exclusively in epithelial cells.6,7,9 Claudin-low tumors, which lack or display low levels of E-cadherin and claudin 3, 4, and 7, are more heterogeneous than basal and luminal A subtypes, in addition to displaying more extensive lymphocytic infiltrates, larger tumors, and a high expression of mesenchymal markers as their main features. This tumor subtype is also associated with young age of onset and lower survival rates when compared with the luminal A subtypes.14,15 A summary of histological and molecular classification of the different breast cancer subtypes is presented in Table 1.
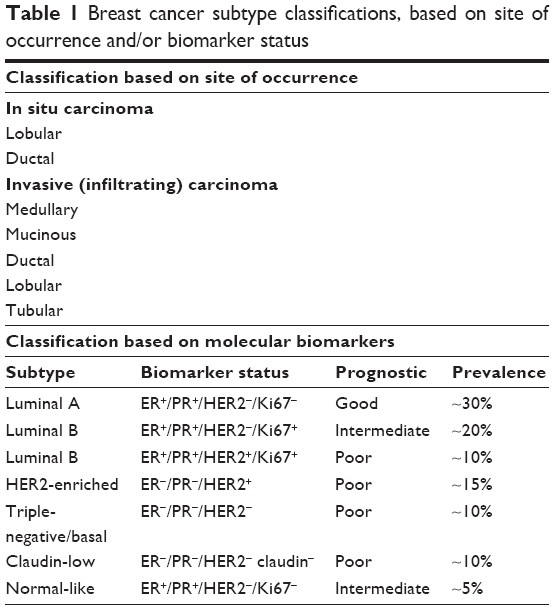 | Table 1 Breast cancer subtype classifications, based on site of occurrence and/or biomarker status |
Understanding the development of breast cancer, applying appropriate (and often patient-specific) treatment, and making an adequate prognosis is frequently mixed by the diverse histological patterns and biological/morphological features of the disease, as well as its clinical behavior and molecular characteristics. Additionally, the onset and progression of the disease are often related to lifestyle, race,10 geographical variation, age at menarche or menopause, and family history,16 attributable to high penetrance susceptibility genes such as BRCA1 and BRCA2.17,18 As a result, a combination of these factors leads to deregulation of signaling pathways that modulate the normal function and development of the mammary gland. Therefore, unraveling the molecular network that is behind this disease should provide guidance toward early (and more accurate) diagnosis, as well as better targets for possible therapy intervention. Here, we describe the roles of major signaling pathways in the normal development of the mammary gland and in breast cancer progression. A summary of the most important molecular alterations associated with breast cancer along these pathways is presented in Table 2.
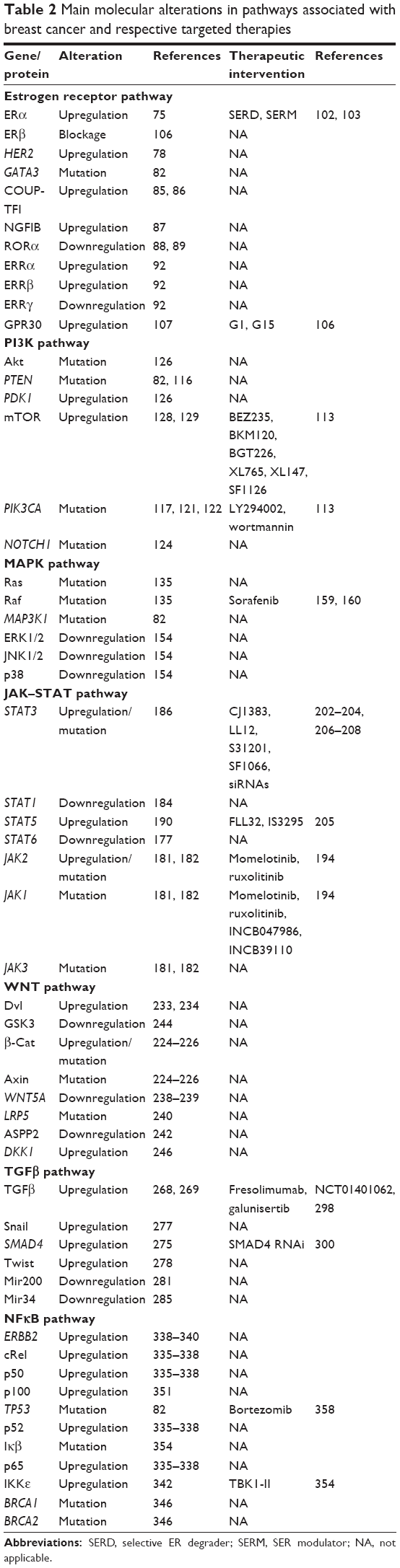 | Table 2 Main molecular alterations in pathways associated with breast cancer and respective targeted therapies |
Estrogen receptor
The ER pathway
Estrogen comprises a set of steroid hormones involved in the development and maintenance of reproductive, cardiovascular, bone morphogenetic, and immune and central nervous systems.19 ERs belong to the nuclear hormone receptor superfamily (or nuclear receptor family), which are highly conserved throughout evolution. These receptors are mostly present in the nucleoplasm, but may also be bound to the plasma membrane. Their early appearance in evolution contributed to the wide range of effects triggered by this pathway, such as ER regulating lipid and glucose metabolism in the liver,20 whereas in cardiac tissue estrogen prevents apoptosis and necrosis of cardiac and endothelial cells.21
The main mechanism of ER activation is through estrogen (E2) binding to its receptor (ER). E2–ER complexes translocate to the nucleus and bind to EREs present in the DNA, regulating transcription of a subset of ERE-modulated genes. Typically, an ERE encompasses a 15 bp palindromic RE containing two PuGGTCA motifs, separated by 3 bp.22 Binding of the E2–ER complex also results in recruitment of coactivator complexes, with histones being further acetylated, leading to chromatin remodeling and finally to recruitment of the basal transcription machinery.23 More than 230 estrogen-stimulated human genes with conserved and unconserved elements have been identified, and more than 70,000 potential ERE motifs have been found through genome wide screenings.24 In humans, the main estrogen hormone is 17β-estradiol (E2), which binds to the ligand binding domain of the ER, a globular domain that harbors a hormone binding site and a dimerization interface and also contains coactivator–corepressor interaction capabilities. However, ERs display a ligand cavity slightly wider than their ligands, allowing them to be activated by a set of steroid hormones, environmental contaminants, xenoestrogens, phytoestrogens, and synthetic compounds with diverse structures.25,26
An alternative mechanism involving activation of ER in the absence of estrogen binding has been described,27–29 in which ER activation occurs through phosphorylation by cellular kinases cross-coupling with ER signaling. Phosphorylation of ERs on both serine30 and tyrosine31 residues has been observed, with serine phosphorylation of the N-terminal transcriptional activation function (AF)-1 domain (which influences the receptor’s transactivation activity) and phosphorylation of ER’s serine 118 (necessary for EGF response involving the MAPK pathway).32 Further signaling studies revealed a new membrane-coupled receptor, namely GPR30, which can mediate a rapid response. GPR30 (also named GPER1) is a G-protein-coupled receptor located in the cell membrane and endoplasmic reticulum that stimulates intracellular calcium mobilization, activation of phosphoinositide kinase signaling, and inhibition of cell migration.33,34
ER variants
Two ERs are expressed in human cells: ERα (ERα66), encoded by the ESR1 gene, and ERβ (ERbβ1), encoded by ESR2. ERα displays six distinct domains, termed “A”–“F”, whereas ERβ has the same distribution but lacks the first “A” domain. In most cells, ERα and ERβ localize to the plasma membrane and to cytoplasmic organelles, such as mitochondria and the endoplasmic reticulum, exhibiting overlapping tissue distribution and functions under normal conditions.35 However, ERα and ERβ integrate the estrogen pathway with opposing functions for some genes involved in cell proliferation.36 For instance, in HeLa cells, estrogen activates ERα, whereas ERβ is activated by antiestrogens.37 Additionally, ERα and ERβ have opposite actions at the cyclin D1 promoter,36 where ERβ is able to inhibit cyclin D1 expression even when ERα is activated by saturating concentrations of 17β-estradiol. These opposing functions are purportedly carried out by the activation of functional domains (AF1) on both receptors. ERβ does not contain a strong AF1 domain, but contains a repressor domain that may inhibit the overall transcriptional activity of the receptor.38 These studies support a potential modulatory role for ERβ in proliferation of mammary tissue induced by estrogen.36
Although ERα and ERβ receptors have similar structures, their splice variants are much more diverse. In ERα, for instance, exon skipping generates isoforms with highly variable lengths and motif composition.39 The variants generated by alternative splicing could play an important role in cancer development.40 Many of these variants are translated as ERs with diverse responses to hormone stimulation. For example, ERβ increases ERα degradation, while the isoform ERβ2 is strongly correlated with high grade prostatic cancer and poor prognosis. Currently, a great deal of information linking splicing variants to susceptibility to disease is available, in addition to their roles in maintaining normal physiology. Different types of variants can exert diverse effects: some variants can fail to bind ligands, others display altered subnuclear localization and capacity for transcriptional activation by dimerization, while others bind to cofactors more weakly and fail to bind ERE to start transcription.41
Genomic and nongenomic pathways
There are two main types of ER signaling pathways. The first, named genomic or classical mechanism, is initiated by activation of the ER in response to a given stimulus. ERs can be activated through binding of their specific ligand (E2) or by phosphorylation, independently of the ligand, prompting transcription of a group of genes, either by direct binding to promoter sequences or by activating complexes of transcription factors that bind to non-ERE sequences. The E2–ER complex recruits coregulatory proteins to promoter sequences, activating or inhibiting transcription, the result being determined by the type of ligand, the type of complex formed, and even by the presence of some splicing variants. Activation of this pathway from stimulus to response may take several hours.42
The second type of ER signaling pathway, nongenomic mechanism, triggers much faster responses, ie, within seconds to minutes. In this mechanism, ER activation on the plasma membrane or adjacent to receptors is mediated by various protein kinase cascades, generating such responses as nitric oxide (NO) release, increases in ion flux across membranes, and activation of RTK and the protein lipid kinase pathway.34 Also, E2 stimulates Gβγ subunit protein-dependent transactivation of the EGFR–ERK signaling axis, through release of pro-HBEGF and suppression of the EGFR–ERK pathway.43 The proposed mechanism involves membrane-localized GPR30, ERα, and ERβ associated with G-protein complexes, generating the earliest rapid signals.44–46 Specific G-proteins or proteins physically associated with ER can activate growth factor receptors, such as EGFR, which triggers secondary signals, like ERK and PI3K activation, calcium, PKC, or Src kinase.47–49 Most of this membrane protein complex is associated with scaffold anchor proteins, such as caveolin 1, striatin, and SHC, generally in caveolae.50–52 The outcome of these signaling pathways is the modulation of cell adhesion, migration, survival, and proliferation, thus playing an import role in the onset of cancer53 (Figure 1).
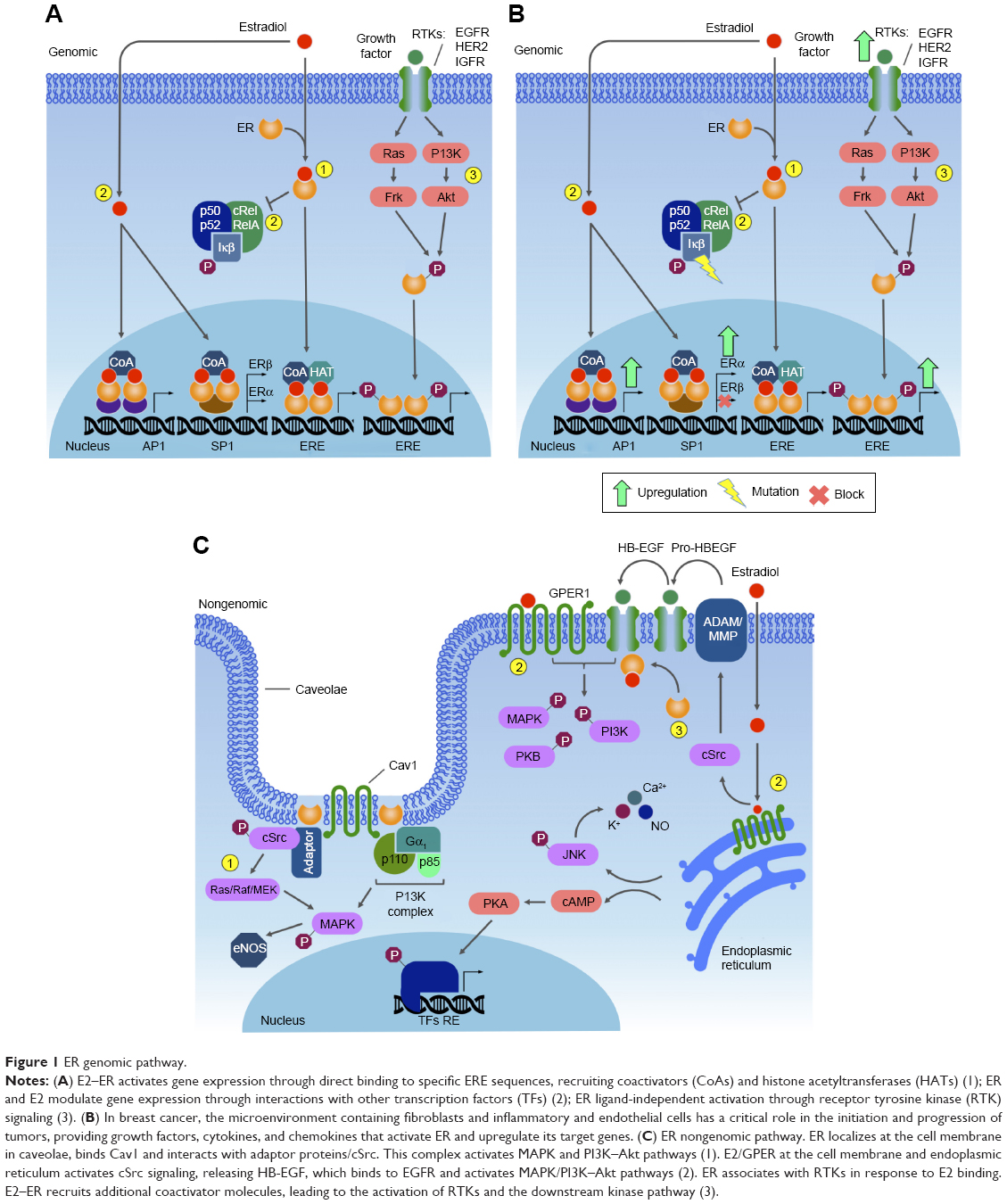 | Figure 1 ER genomic pathway. |
ER in mammary tissue development
The E2–ER complex has essential roles in morphogenesis in many female tissues, such as uterus,54,55 ovary,56 and mammary glands,57 as well as male tissues like gonads58 and the prostate.59,60 In normal mammary gland tissue, cell division starts during puberty, proceeding with cyclic cell proliferation, controlled by endocrine regulation through ovarian secretion of E2 and also by GH and IGF1 to complete branching morphogenesis.61,62 In adult tissue, progesterone plays an important role in mammary SC expansion.63 E2, GH, and IGF1 receptors mediate pathways regulating ductal growth and morphogenesis.64
The human breast tissue epithelium is arranged hierarchically, involving the participation of SCs, which do not express ERα or PR, differentiated hormone-sensing ERα+ cells, and myoepithelial cells. Morphologically, two main types of epithelial cell types may be distinguished: luminal cells lining the central lumen, where the majority of ER+ cells are found, and the myoepithelial cells, which lay adjacently to the basement membrane.65,66
A possible mechanism for the estrogen sensitivity of the mammary tissue is that ERα+ cells initiate proliferation by signaling in a paracrine manner. Therefore, ERα+ cells would be required to initiate proliferation,67,68 such that after estrogen stimulation, EGFs, such as Areg, are released by ERα+ cells and trigger cell proliferation.69,70 Areg is strongly induced by estrogen in mammary epithelial cells at puberty, binding to EGFR, which is located on stromal cells. This protein requires induction of the ADAM17 metalloproteinase to be released from mammary epithelial cells and reach the stromal cells, a mechanism that allows activation of stromal EGFR.71
In the suggested model, GH induces the production of IGF1 in both the liver and locally in mammary stroma and epithelium. As such, E2 and IGF1 generate the first signal to trigger proliferation, required for ductal morphogenesis. The effect of E2 on ERα+ sensing cells then induces the release of Areg, which acts through stromal cells, recruiting additional growth factors that contribute to the rapid growth of mammary glands, and stimulating terminal end bud formation and ductal branching, which occurs at puberty.72 Therefore, a paracrine circuitry model consisting of estrogen response via cross talk with the stroma and IGF1 as the primary effector downstream of E2 and GH73,74 is responsible for the sensitivity of breast tissue to estrogen. In adult mammary gland tissue, progesterone has a proliferative effect, which is activated via a paracrine mechanism. However, estrogen also upregulates PRs, playing a key role in maintaining differentiated ERα+ cells and the presence of populations of SCs in the adult mammary gland.63
ER and breast cancer
In most cases of breast cancer, it is well known that the E2–ER complex is involved in malignant transformation and progression. Estrogen is important in the maintenance of breast tissue; however, the mechanism by which this hormone renders quiescent epithelial ERα+ cells into highly proliferative cells during tumorigenesis is still poorly understood. The answer may lie in the cross talk between epithelial ERα+ and stromal EGFR signaling, since some studies show that both ERα and Areg are upregulated in early hyperplastic precursors of breast cancer, which together may initiate tumorigenesis.75 On the other hand, estrogen withdrawal ensues during menopause. The suppressive activity of ERα on some growth factor receptors in the normal mammary epithelium is affected by the decrease in estrogen level during menopause, allowing for expression of growth factor receptors on ER-positive cells, thus rendering these cells potentially proliferative and cancerous. Once this occurs, ER may be activated by growth factor-stimulated TKs, and normal regulation of cell proliferation is then lost.76,77
Breast cancer may arise by different mechanisms. Overexpression of HER2 in 12%–20% of breast cancers is often due to gene amplification.78 In some types of breast cancers, there are specific translocations, such as those between chromosomes 12 and 15 (t[12;15]), commonly detected in secretory carcinomas of mammary tissue,79 as well as that between chromosomes 11 and 19 (t[11;19][q21;p13]), which is characteristic of breast mucoepidermoid carcinoma.80 In some subtypes of ER+ breast cancer, somatic mutations have been identified by whole genome sequencing. These mutations are present in a set of genes that impact important signaling pathways, which possibly affect ER function.81 For instance, the luminal A breast cancer subtype displays mutations in PIK3CA (49%), MAP3K1 (14%), GATA3 (14%), TP53 (12%), and MAP2K4 (12%) and loss of PTEN (13%), among others. The luminal B breast cancer subtype has mutations in TP53 (32%), PIK3CA (32%), MAP3K1 (5%) and other genes.82 These data reinforce the concept that breast cancer is etiologically diverse, with the current classification based on histology and morphology reflecting the heterogeneity of this disease.
An emerging group of nuclear receptors involved in key processes of mammary tissue development are orphan nuclear receptors.83 These receptors share functional domains with the ER, wherein their AF1 sites are ligand-independent84 and thus constitutively active. Several studies have demonstrated a correlation between the clinical outcomes of different types of breast cancer with the expression of these receptors, such as COUP-TFI,85,86 NGFIB,87 rRORα,88,89 ERRα, ERRβ, and ERRγ. Due to their high degree of homology with ERs, more emphasis has been given to the study of ERRs and particularly to ERRα, a master regulator of cellular energy metabolism in both normal and cancer cells.90–92 Interestingly, high ERRα expression positively correlates with HER2 status and poor outcome in breast tumors, suggesting a tentative possibility for a new prognostic biomarker.93,94 Recent studies found that ERRα is part of the AMPK–PGC1α–ERRα axis, a key regulator in reprogramming of cellular metabolism and cellular adaptation to metabolic stress.95,96 PGC1α–ERRα is a known repressor of folate metabolism97 and one of the main pathways upregulated in cancer cells; however, several studies have associated the PGC1α–ERRα transcriptional axis with increased tumor growth in breast cancer.97,98 One possible explanation for this apparent contradiction is the number of metabolic pathways regulated by the PGC1α–ERRα axis,99–101 acting in conjunction and cross talking to achieve energy balance in response to stress and several signals in the tumor microenvironment. These results highlight the use of ERRα as a putative biomarker in breast cancer, and reinforce the idea that directing specific drugs to the folate pathway in tumors overexpressing PGC1α/ERRα is a powerful tool to improve patient prognosis.83
Clinical targeting of the ER pathway
Overexpression and activation of ERα increase cell proliferation and malignant transformation of luminal-type breast cancers,75 prompting the development of antiestrogen treatments. The most successful therapies for ERα+ breast cancer have relied on synthetic molecules designed to block mainly ERα, such as selective ER modulators like tamoxifen, raloxifene, and toremifene, aromatase inhibitors, and selective ER degraders, such as fulvestrant. Though highly effective, these drugs have unwanted side effects in nontarget tissues, with approximately 50% of patients acquiring resistance and developing further metastases.102,103 Combined drug therapies, such as tamoxifen and aromatase inhibitors, have been shown to improve disease-free survival substantially.104 Expression of ER, PRs, and HER2 determines the tumor hormone receptor status. In fact, these molecular markers are valuable to determine prognosis and predict response to anti-ERα therapy.105
In normal breast tissue, the predominant ER is ERβ. However, ERβ levels are reduced in breast tumors, compromising the potential efficacy of targeted therapies.106 Most compounds that selectively target ERβ elicit anti-inflammatory effects, but show no therapeutic effect in cancer. Alternative targets are also currently under development, such as G1 (agonist) and G15 (antagonist), specifically targeting GPR30. Although drugs targeting ER are widely used, caution should be applied, since the mechanisms by which these drugs operate, their effects on specific tissues and cell types, and their specificity toward each receptor type, including GPR30, remain to be determined.107
Phosphatidylinositide-3 kinase
The PI3K pathway
PI3K signaling is central for a number of cellular processes related to cell growth, proliferation, motility, survival, and apoptosis. This pathway mediates metabolism by modulating both glycolytic flux and fatty acid synthesis. Activation of PI3K signaling consistently leads to cell mass accumulation and cell proliferation, as well as differentiation events by promoting protein synthesis, cell cycle progression, and actin rearrangement.108 PI3K signaling also modulates autophagy, a nonapoptotic programmed cell death mechanism, as well as protein and organelle recycling into metabolic intermediates through lysosomes.108–110
PI3Ks are lipid-based protein kinases, usually divided into three distinct classes (I, II, and III). Class I is subdivided into IA and IB, according to their structural characteristics and substrate specificity. Class IA consists of heterodimers with regulatory and catalytic subunits, the former mediating activation of the latter by RTKs, G-protein-coupled receptors, and adaptor proteins, such as the Ras oncoprotein.111–113 Upon activation, PI3K phosphorylates PIP2 in the plasma membrane, converting it into PIP3. Dephosphorylation of PIP3 is an important modulatory step in this pathway, due to the activity of the PTEN tumor suppressor, which acts as a negative regulator of this pathway.114 PIP3 recruits and activates Akt, also known as PKB. This activation stimulates the catalytic activity of Akt, resulting in phosphorylation of other downstream proteins that regulate cell growth, survival, and entry into the cell cycle. The Akt protein has three isoforms: Akt1, Akt2, and Akt3. During breast cancer progression, Akt1 promotes cell proliferation by S6 and cyclin D1 upregulation, while Akt2 promotes cell migration and invasion through F-actin and vimentin induction.108,113 Akt3 is the less studied isoform, but is known to be mainly involved in triple-negative breast cancers, in which it may play a role in migration, invasion, and tumor growth.115
One of the major downstream effectors of the PI3K signaling pathway is a serine/threonine protein kinase complex called mTOR. mTOR is subdivided into two complexes – mTORC1 and mTORC2 – which are structurally similar but functionally distinct.116,117 Autophagy is regulated by mTORC1, which can either phosphorylate or dissociate the ULK1 complex to block or initiate autophagy, respectively. When nutrients are limited, mTORC1 dissociates from the ULK1 complex, initiating autophagy, in which PIP3 plays an important role, establishing the autophagosomes and translocating the cytoplasmic material to lysosomes for degradation.109 mTORC2 organizes the cellular actin cytoskeleton and regulates Akt phosphorylation116 (Figure 2).
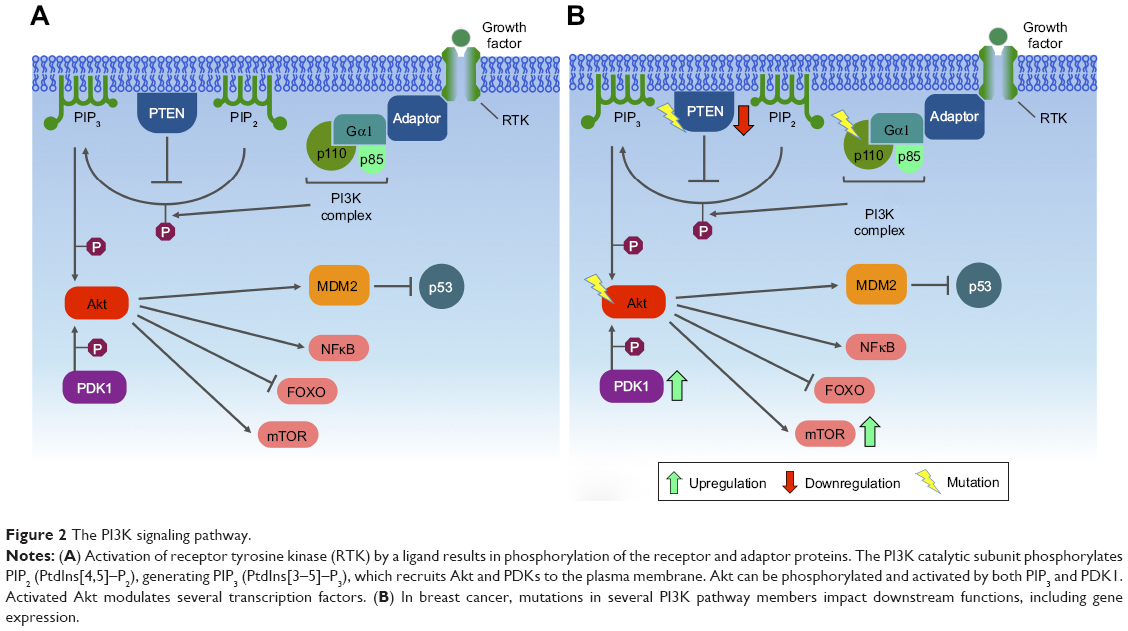 | Figure 2 The PI3K signaling pathway. |
PI3K in mammary tissue development
In vivo studies suggest that the p110 catalytic isoform p110α, the catalytic subunit of class IA PI3K, is involved in duct outgrowth and branching during puberty, postpartum lactation, EGF signaling in mammary epithelial cells, and mammary gland development. The p110β isoform may play a regulatory role in refinement of PI3K downstream signaling, responding to insulin and EGF, while its absence induces a moderate hypermorphic mammary gland phenotype.118
mTORC1 requires RAPTOR as a main cofactor, while mTORC2 requires RICTOR.119 With mammary epithelial cells, it has been observed that Akt inhibition decreases branching and colony size, suggesting that Akt is involved in signaling growth control and branching morphogenesis dependent on RICTOR. Moreover, inhibition of mTORC2 reduces ductal branching and lengthening. Finally, it has been suggested that the mTORC2–RICTOR complex is necessary for PKCα signaling through the small GTPase Rac1, in order to trigger mammary morphogenesis cell survival, cell junctions, and motility.120
PI3K and breast cancer
Mutations in PIK3CA, the gene encoding the p110α catalytic subunit of class IA PI3K, are among the most common alterations observed in human malignancies, found in approximately 25% of breast cancers. This mutation confers a gain of function to p110α and may render HER2-based therapy ineffective.117,121 Most of the mutations in PIK3CA occur in three sites: two in the helical domain (E542 and E545) and one in the kinase domain (H1047).122 Other mutations along the PI3K pathway are frequently found in human cancers, especially breast cancer. PTEN mutations are frequent in basal-like (67%) and HER2+ (22%) breast cancers, while AKT1 mutations are often found in luminal/HR+ (2.6%) tumors.116
Notch activation may render breast cancer cells resistant to PI3K-mTORC1 inhibition, and loss of PTEN may lead to resistance to Notch inhibition.123 The Notch signaling pathway is evolutionarily conserved, and mainly regulates cell fate determination during development and maintains adult tissue homeostasis.124 In mammals, four Notch proteins are present (Notch1, Notch2, Notch3, And Notch4), which are defined by a large extracellular domain and a cytoplasmic domain. In particular, Notch1 is involved in breast cancer tumorigenesis and metastasis, apparently triggering EMT in epithelial cancer cells.124
With regard to the function of Akt during breast tumorigenesis, Akt1 and Akt2 regulate cell migration and invasion in different manners. While Akt2 promotes cell migration and invasion through F-actin and vimentin induction, Akt1 inhibits cell migration and invasion through downregulation of β1 integrin and FAK.125 Akt may be directly phosphorylated and activated by PIP3 or by the putative PDPK1/PDK1. Activation of PIP3 provides a docking site (containing pleckstrin homology domains) for both Akt and PDK1.113 In breast cancer, PDK1 is overexpressed and amplified in approximately 20% of tumors. PDK1 phosphorylates Akt1 at Thr308, suggesting that another protein kinase, namely PDK2, is responsible for Ser473 phosphorylation. However, it is still unclear whether PDK2 is an alternative kinase or a modification of PDK1 by the PDK1-interacting fragment.126
Clinical targeting of the PI3K pathway
Several preclinical and initial clinical trials have shown great potential in targeting the PI3K–Akt pathway in cancer-related conditions.127 Inhibition of the PI3K pathway has been associated with improved antitumor T-cell response and tumor angiogenesis, since: 1) the activity of p110β is involved in tumors displaying PTEN loss; 2) p110α drives angiogenesis; 3) p110γ, p110δ, and p110β have important functions in inflammatory cells in the tumor microenvironment; and 4) p110δ and mTOR control important aspects of adaptive immunity, which include lymphocyte activation and differentiation.128,129 For breast cancer treatment, PI3K inhibitors can be very effective, especially when combined with HER2 inhibitors, since PIK3CA mutations seem to be associated with resistance to these drugs. Therefore, a combinatorial treatment by inhibiting PI3K, in addition to HER2 blockade, may constitute a powerful therapy.121 Targeting mutated kinases and RTKs may be interesting for drug development, considering that similar therapies have demonstrated promising results in other cancer types, such as non-small cell lung cancer.122,130 LY294002 and wortmannin comprise the first generation of PI3K inhibitors and, thus lack selectivity for particular PI3K isoforms and present toxic effects. Some other drugs, such as BEZ235, BKM120, and BGT226 (from Novartis), XL765 and XL147 (from Exelixis), or SF1126 (from Semaphore), which inhibit nonselective PI3K/mTOR, have been employed in preclinical or in clinical trials. Other drugs, such as PX316 (from Pro1X Pharmaceuticals), GSK690693 (from GlaxoSmithKline), Akti1/2 and MK2206 (both from Merck), and XL418 (from Exelixis), inhibit Akt activity. Another viable target in the PI3K pathway is the mTOR node, blocked by the anticancer drugs rapamycin (also known as sirolimus [Rapamune, from Pfizer]), CCI779 (Torisel, from Pfizer), RAD001 (Afinitor, from Novartis), and AP23573 (from Merck/Ariad).113
Mitogen-activated protein kinase
The MAPK pathway
MAPKs are phosphoproteins stimulated by mitogens.131 Most of the signals directly involving MAPKs are evolutionarily conserved and biologically versatile, and found in animals, plants, fungi, and protists.132–135 MAPK activity is regulated by a cascade composed of a MAPK, MAPKK/MKK/MEK, and MAPKKK or MEKK. The MAPK pathway may be activated by STE20 kinase or small GTP-binding proteins,136 and is responsible for controlling cell survival and adaptation in response to chemical and physical stress.135,136
MAPKs are key components in the control of embryogenesis, cell differentiation, growth, proliferation, migration, and apoptosis.135,137 Six groups of MAPKs have been characterized in mammals: 1) ERK1/2, 2) ERK3/4, 3) ERK5, 4) ERK7/8, 5) JNK1/2/3, and 6) the isoforms of p38 a/b/g/d.135,138 MAPKs are composed of 80 kDa homodimers containing a threonine/x/tyrosine domain, where the X represents glutamate (E), proline (P), or glycine (G), in ERK, JNK, or p38, respectively,139–141 with N- and C-terminal regions of different lengths.138
Generally, the initial stimulus in this cascade comes from an extracellular signal, such as a peptide growth factor, binding to its receptor protein embedded in the plasma membrane. This binding induces the receptor, such as RTKs, to initiate an autophosphorylation process that elicits the adaptor protein SHC, which in turn binds RTK and becomes self-phosphorylated. The receptor SHC complex then recruits the GRB2 adapter protein and the SOS guanine nucleotide exchange factor, catalyzing the conversion of GDP-Ras to GTP-Ras, which leads to MAPKKK phosphorylation. MAPKKK phosphorylates MAPKK in two serine residues, Ser218 and Ser222, thereby rendering it active and capable of phosphorylating MAPK in a threonine and tyrosine residue.132–135,142–146 Depending on the stimulus, phosphorylation of MAPKKK may be carried out by other small GTPases, such as Rac, CDC42, and Rho, which in turn modulate MAPK activation status downstream138,147 (Figure 3).
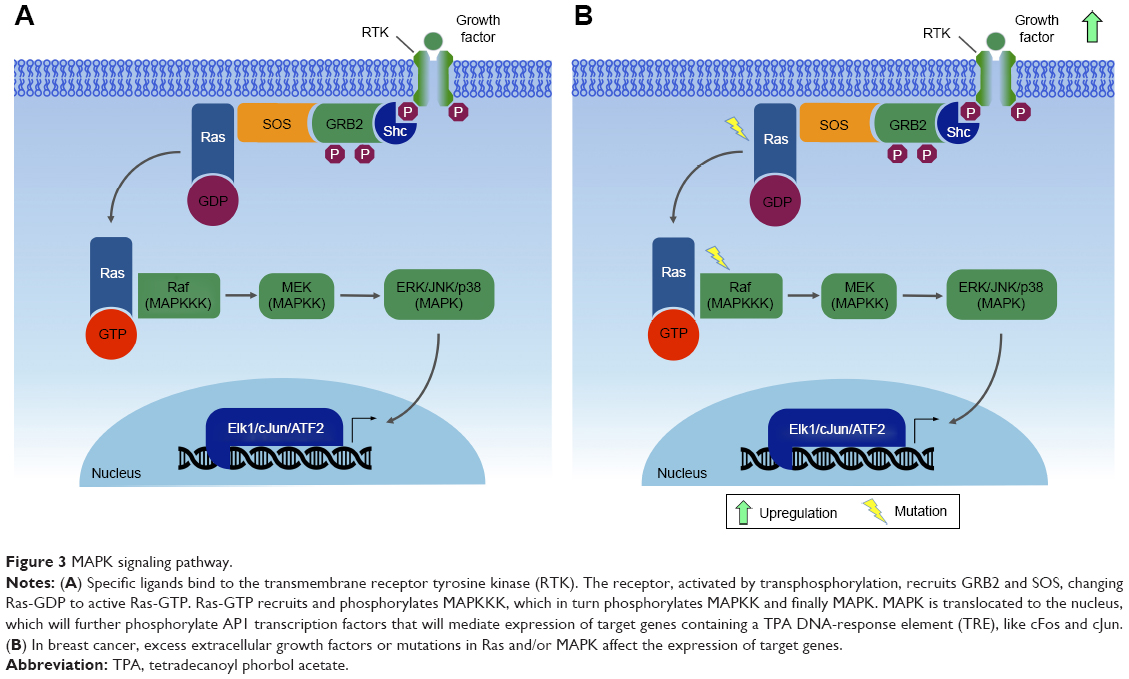 | Figure 3 MAPK signaling pathway. |
A single MAPK cascade may lead to different cellular responses. Every MAPK is activated by specific MAPKKs (MEK1/2 for ERK1/2, MKK3/6 for p38, MKK4/7 [JNKK1/2] for the JNKs, and MEK5 for ERK5); however, each MAPKK may be activated by more than one MAPKKK.136 For instance, the cascade comprising Raf1 or Mos (MAPKKK), MEK1/2 (MAPKKs), and ERK1/2 (MAPKs)133,142,148 may induce mitogenesis, transdifferentiation (PC12 cells) and even activation of the CDC2 cyclin B complex (oocytes), depending on the cell type and the stimuli.133,149
The Raf–MEK–ERK-MAPK cascade, present in various animal cell types, can be activated by the GTPase Ras.133,142 Activated Ras induces the protein kinase activity of Raf kinases. The Raf family of protein kinases is composed of A-Raf, B-Raf, and Raf1,150 and each of the isoforms contains three conserved regions: CR1, CR2, and CR3. The CR3 region corresponds to the kinase domain, whereas the CR1 and CR2 regions are located in the amino-terminal end, being involved in regulation of the catalytic domain. Deletion of CR1 and CR2 generates constitutively active Raf1 mutants, which may be activated independently of Ras.150,151
MAPK and breast cancer
Mutations in RAS have been observed in many tumors.152 These mutations are mainly associated with the constitutive activation of ERK1/2, thus promoting tumor cell proliferation. Abnormalities in MAPK signaling affect most of the cellular processes commonly associated with the development of cancers:135 independence from proliferation checkpoints, evasion of apoptotic signals, unlimited replicative potential, invading and metastatic capacities, and the ability to attract and sustain angiogenesis. In particular, recent studies have reported higher frequency of modified cells with activated MAPK in breast cancer.153
Mutations along the MAPK pathway, which may lead to the development of cancer, are often found in Ras and B-Raf, whereas JNK and p38 appear to be poorly related to malignant transformation.135 The ERK pathway has been the best studied to date, with deregulations being described in approximately a third of human cancer cases. In cancer cells, the constitutive activation of ERK signaling occurs in the early steps of the pathway, and may be due to a series of factors, such as overexpression of RTK, activating mutations in TK receptors, autocrine or sustained paracrine production of activating ligands, and mutations of RAS and BRAF. However, this amplification and/or deregulation may also occur in targeted nuclear transcription factors, such as c-Myc and AP1.135
Due to the high frequency of activating mutations, the Ras–Raf axis is suggested as a regulatory node of the pathway. Mathematical modeling predicts that activation of this node leads to general activation in the cell.135 Ras GTPases control the activity of various signaling pathways, and mutations in KRAS and NRAS have already been described for several types of cancers, since they lead to inefficient GTP hydrolysis, causing Ras to be in an active state and leading to continuous activation of the MAPK route. This deregulated Ras activation can also recruit scaffold proteins, such as KSR and SUR8/SHOC2, which modulate Raf activation by Ras.135
Human breast cancer may be subdivided into estrogen-dependent and -independent types. Estrogen typically activates growth factors which increase the levels of MAPK activity. In estrogen-independent tumors, MAPK stimulates growth upon activation by other peptide hormones.153 The functions of MAPK in breast cancer appear to be complex, owing to several cellular responses that they modulate and also their interaction with different pathways (ie, ER and HER2). Generally, MAPKs are associated with good prognosis across most classifications of breast cancer, notably in those with positive ER.154
Studies demonstrate that ERK1/2 and phospho-ERK1/2 are associated with good clinicopathological status and that good prognosis may be related to their roles in inducing apoptosis. Phosphorylated JNK1/2 has also been shown to be stimulated by stress or growth factors, activating apoptosis and even augmenting cell death signaling in MCF7 breast cancer-derived cells under the influence of high estradiol levels. Finally, growing evidence suggests a role for phospho-p38 in inducing apoptosis in breast cancer. Also, MAPKs have been associated with longer survival of ER+ patients.154
Activated MAPK can also phosphorylate ER, either directly or indirectly, and increase its transcriptional efficiency (an important feature of hormone-dependent breast cancer). A recent report also suggested that RSK (downstream target of MAPK) can phosphorylate ER, an effect that increases its own transcriptional efficiency.155,156 Additionally, MAPK can phosphorylate PR at sites that act as ubiquitination signals, leading to degradation by the 26S proteasome.157
In the absence of estrogen and progesterone, breast cancer cells could be stimulated by growth factors (ie, EGF, EGF1, insulin, prolactin, TGFα, or TGFβ), increasing MAPK activation. Cells overexpressing ErbB2 (a type of RTK) exhibit increased activated MAPK, which is mediated through ErbB2 interaction with endogenous ligands, such as EGF and neuregulin.153 The tumor suppressor PTEN, with dual-specificity phosphatase activity, blocks phosphorylation of the insulin-stimulated MAPK in MCF7 cells by inhibiting IRS1 phosphorylation and inhibiting the formation of IRS1–GRB2–SOS complexes. Consequently, cell growth is suppressed and cyclin D1 downregulated, halting cell cycle progression.158
MAPKs have an important role in cell cycle arrest and sequestration of ERK in the cytoplasm. Feedback to upstream mediators is common in inhibitory signals mediated by mutated RAS and RAF. Further stimulation through this pathway is omitted by HDM2 and FOXO3. The MAPK cellular function is context-specific and cell type-specific, in order to mediate signals that can lead to diverse cellular functions. Furthermore, the function of MAPKs is affected by their cross talk and interaction with other pathways, which can influence their behavior.154
Clinical targeting of the MAPK pathway
Since MAPK signaling regulates both physiological and pathological processes, upstream inhibition is not advised as a good method for treatment. Each MAPK could be involved in several responses; therefore, blocking an MAPK for treatment of a specific disease could be deleterious for another physiological process, while inhibiting an upstream protein could cause negative downstream effects. Sorafenib, a drug that can inhibit the Raf kinase, is approved for treatment of several cancer types.159,160 However, no targeted therapy is available for other MAPKs. Many p38 blocking drugs have been tested, but so far none has reached approval for use, due to their low efficiency and/or hepatoxicity.161
Janus kinase/signal transducer and activator of transcription
The JAK–STAT pathway
Several extracellular signals may cause rapid changes in the expression of specific genes. The JAK–STAT pathway is one of example of how cells are able to recognize signals to generate rapid and accurate responses. Studies of interferon-induced intracellular signaling have led to the discovery of this pathway, which relates to the major signaling mechanism used by cytokines and growth factors.162 Among the main cytokines that activate this pathway are interferons, interleukins, and colony-stimulating factors. Among growth factors, such hormones as prolactin, leptin, erythropoietin, and thrombopoietin are the main activators.163 Activation of this pathway stimulates important cellular events, such as proliferation, differentiation, migration, and apoptosis, as well as critical processes, such as hematopoiesis, immunodevelopment, mammary gland development, and lactation.164,165
This pathway is relatively simple, comprising a few major components: cell surface receptors, JAKs (TKs that are constitutively associated with the receptor), and STATs (latent cytoplasmic transcription factors).162 JAKs are cytoplasmic TKs that participate in downstream signaling of various cell surface receptors that have no intrinsic TK activity. In mammals, four members of the JAK family are known: JAK1, JAK2, JAK3, and Tyk2.166,167 JAKs share seven domains (JH1–JH7), including: the four-point-one protein–ezrin–radixin–moesin (FERM) domain, which is important for association of JAKs with receptors; the SH2 motif, with scaffold function; the pseudokinase motif, which regulates JAK kinase activity; and the TK motif, which is catalytically active, being important for phosphorylation of receptors and STATs.168
STATs are latent cytoplasmic transcription factors, which are activated upon recruitment to an activated receptor complex. To date, seven members of the STAT family have been identified in mammals: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6.163 Six well-defined domains compose STAT structure, including: a conserved N-terminal domain involved in regulation of STAT activity, such as the formation of STAT tetramers and tyrosine dephosphorylation;169 a coiled-coil domain, involved in receptor binding and association with regulatory proteins;170 a DNA binding domain; the SH2 domain, which is important for recruitment of STATs to the activated receptor complex; and a variable C-terminal transactivation domain, which is important in transcriptional modulation of target genes. Interaction among the different types of JAK and STAT proteins will depend on the cytokine receptor that has been activated.171
Binding of cytokines or growth factors to their specific receptors on the cell surface leads to receptor dimerization and subsequent activation of JAKs by transphosphorylation (ie, JAK molecules phosphorylate each other). Activated JAKs have the ability to phosphorylate specific tyrosine residues present in the receptor, which will then serve as anchoring sites for STATs. Anchored STATs are phosphorylated by JAKs, leading to their homodimerization or heterodimerization. Phosphorylated and dimerized STATs leave the receptor and translocate to the nucleus by an importin α5-dependent mechanism and the Ran nuclear import pathway, where they bind DNA at specific regulatory regions in promoters, activating transcription of target genes (Figure 4).172,173
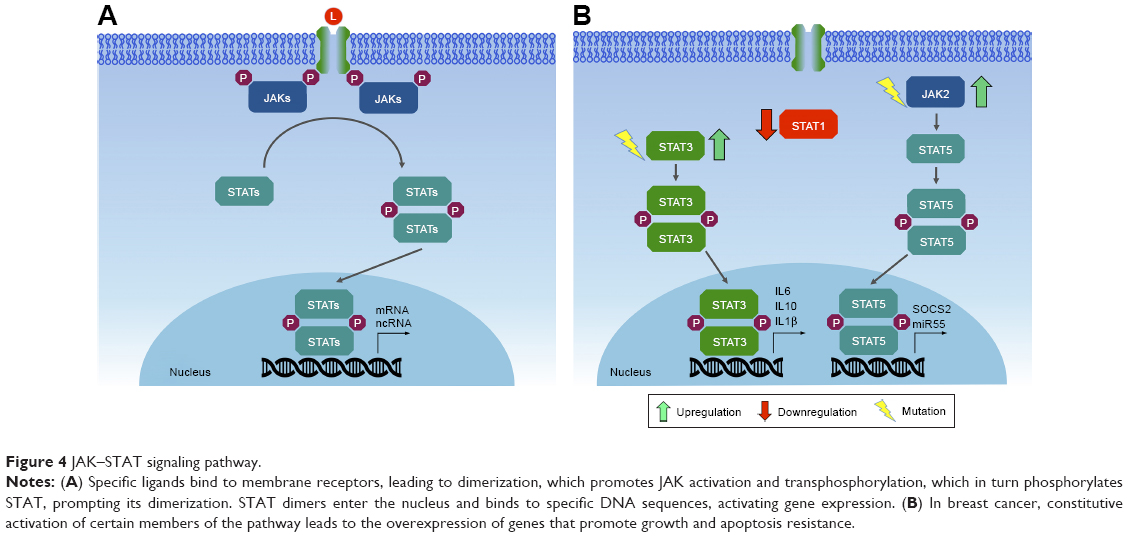 | Figure 4 JAK–STAT signaling pathway. |
JAK/STAT in mammary tissue development
The STAT family of transcription factors participates significantly in the development of the mammary gland, since its members play important roles in the regulation of cell proliferation and apoptosis. Deletion of STAT3 results in fetal lethality, preventing further investigation into the development of the gland, although some authors believe that STAT3 is important in the self-renewal of mammary SCs.174 Other members of the STAT family (1, 3, 5, and 6) have important functions in the adult mammary gland. For instance, STAT1 is phosphorylated in the virgin gland (a gland that did not undergo physiological changes) and late involution during remodeling of the gland, but not during pregnancy and lactation. STAT3 is a mediator of cell death and inflammatory signaling in gland involution, becoming phosphorylated on the day of birth and in the first 10 days of involution. STAT5 is essential for lobuloalveolar development and expression of milk protein genes, being predominantly activated during pregnancy. Finally, STAT6 induces the expression of cytokines and important transcription factors in the maintenance of luminal alveolar cells.175
During lactation, mammary gland cells maintain upregulated growth to produce milk through activation of STAT5. However, upon weaning, the mammary glands undergo an involution process. During involution, STAT5 is inactivated and STAT3 triggers the apoptosis process to reduce cell numbers.175 One of the transcriptional targets of STAT3 is the PI3K regulatory subunits p55α and p50α, which in turn inhibit or downregulate PI3K/Akt survival signaling to promote apoptosis. In addition, STAT3 downregulation of Akt, which blocks activation of STAT5, might represent an additional mechanism of mammary gland involution.176–178
JAK/STAT and breast cancer
Due to the high number of cytokines and growth factors that activate this pathway, gain-of-function, loss-of-function mutations, and polymorphisms in JAK and/or STAT genes are associated with several human diseases. Constitutive activation of this pathway may arise by several mechanisms, including production of autocrine/paracrine cytokines, activating mutations (point mutations that generate amino-acid substitution) of receptors, JAKs and/or other upstream oncogenes, which then activate STATs.179 Due to the important roles in regulating cell proliferation and survival, as well as in inflammation, it is not surprising that most JAKs and STATs are also implicated in tumorigenesis of breast tissues, acting either as oncogenes or as tumor suppressors.
Somatic mutations in members of the JAK family are rare in cancer.180 Despite this, somatic mutations in JAK1, JAK2, and JAK3 have already been described in breast cancer samples.181,182 Recent research shows the possible involvement of JAK2 and TYK2 in the development of breast cancer. Studies on epithelial mammary cells show that constitutive activation of JAK2 caused by the V617F point mutation generates hyperactivation of STAT5, leading to increased proliferation and resistance to cell death. Transgenic mice expressing JAK2V617F presented an accelerated tumorigenic process.182 Tyk2−/− mice inoculated with breast cancer cells show increased tumor growth and metastasis compared to wild-type mice, suggesting a possible tumor suppressor role, mediated in part by myeloid-derived suppressor cells.183
A great deal of evidence indicates the involvement of STAT1, STAT3, STAT5, and STAT6 in the formation, progression, prognosis and prediction of breast cancer.177 Depending on the physiological context, STAT1 may act as an oncogene or as a tumor suppressor. Approximately 45% of ER+ and 22% of ER− breast cancer cases exhibit low levels of STAT1 in neoplastic cells, while high levels of STAT1 are detected in benign breast tissue adjacent to the tumor. This may indicate that STAT1 is repressed during breast tumor progression.184 Conversely, high STAT1 expression is associated with metastasis and drug resistance.185 It appears that in postmenopausal ER+ breast tumors, STAT1 acts as a tumor suppressor, while in ER− tumors or ER+ premenopausal malignancies, STAT1 promotes tumor progression.177
STAT3 is constitutively activated in a wide range of solid tumors, including breast cancer.186 Its aberrant activation may promote invasion and metastasis, in addition to regulating the inflammatory response in breast tumorigenesis. Human studies have shown that activation of STAT3 is frequently observed in primary breast cancers, being associated with poor prognosis and tissue invasion.187,188 Constitutive activation of STAT3 appears to affect the tumor microenvironment through secretion of various cytokines, such as IL-10, IL-6, IL-1β, by tumor cells, which stimulate nontumor cells, T-helper (TH)-17 cells, and tumor associated macrophages to secrete even more cytokines, thereby creating a positive feedback loop. This promotes growth and differentiation of tumor cells. STAT3-directed secretion of IL-10 by tumor cells also results in inhibition of antitumor immunity, eg, by inhibiting maturation of dendritic cells.189
Like STAT3, STAT5 is also constitutively activated in breast cancer, but is considered weakly oncogenic in breast cancer mouse models. According to the data currently available, activated STAT5 may promote tumorigenesis, but it is not a genuine proto-oncogene, at least in breast cancer. Activated STAT5 promotes tumorigenesis by expanding the population of mammary alveolar cells, which have been suggested to be especially susceptible to tumorigenesis.190 Finally, STAT6, which is important in the balance between TH1 and TH2 cells, also affects tumor progression, facilitating evasion of the immune system by tumor cells.189 T lymphocytes can differentiate into either TH1 cells or TH2 cells, depending on the cytokines secreted in their environment. TH1 cells identify tumor antigens and trigger an immunoresponse, whereas TH2 cells display oncogenic potential, promoting invasion and tumor metastasis. STAT6 is essential for the differentiation of T lymphocytes in TH2 cells mediated by IL4.177
Clinical targeting of the JAK–STAT pathway
Mounting data showing the relevance of the JAK–STAT pathway in diseases related to the immune system and various types of cancers render the members of this pathway attractive as therapeutic targets. The effectiveness of targeting JAK–STAT signaling has been demonstrated in clinical trials in patients with solid tumors.
JAK inhibitors
The first studies with JAK inhibitors (ie, tyrphostin AG490) led to inhibition of recurrent leukemia B-cell growth in vitro and in vivo.191 In the early 2000s, another JAK inhibitor (pyridone 6) was introduced by Merck, with activity against all members of the JAK family in vitro; however, this compound did not present significant results in vivo.192 Analysis of its crystallographic structure showed that pyridone 6 binds to adenosine triphosphate pocket in the JH1-kinase domain of the JAK2 active conformation. This information served as the basis for the development of various JAK inhibitors described thus far.193
Several selective JAK inhibitors are currently in clinical trials for the treatment of solid tumors. These inhibitors focus mostly on two members of the JAK family: JAK1 and JAK2. Ruxolitinb (Novartis), which targets both JAK1 and JAK2, is one of the most promising compounds, used in various clinical trials (Phase I, II, and III) for a wide variety of solid tumors (breast, lung, gastric, colon, colorectal, and pancreatic cancer). Another interesting inhibitor targeting JAK1 and JAK2 (momelotinib; Gilead Sciences) has also been tested for lung, colon, and pancreatic cancers. The INCB047986 and INCB39110 inhibitors (Incyte Corp.), which specifically target JAK1 by blocking its phosphorylation, are also in advanced phases of clinical trials.194
STAT inhibitors
Constitutive activation of STATs is well described in several types of cancer.195–197 STATs are important transcription factors for pathology progression, by controlling genes with key roles in cell proliferation and modulation of the tumor microenvironment.198 Therefore, the STAT family is an increasingly attractive therapeutic target in cancer. However, great challenges persist in developing STAT inhibitors, since their blockade is much more complex than blocking kinases, such as JAKs.179 One of the challenges lies in target specificity, which may generate off-target effects, resulting in undesired side effects.199 Another challenge is redundancy in the action of different STATs. For example, selective STAT3 blockade, which is critical for IL-6 downstream signaling, may not be fully effective since IL-6 can also act through STAT1 signaling.200
Despite these limitations, a wide range of molecules that mainly inhibit STAT3 and STAT5 have been developed and tested both in vivo and in vitro. These include peptidomimetics, small-molecule inhibitors, and oligonucleotides.201 As an example of peptidomimetics, CJ1383, a STAT3 inhibitor, has shown promising results in two breast cancer cell lines with high levels of phosphorylated STAT3.202 Several small inhibitory molecules targeting the SH2 domain in STAT3 and STAT5 should also be mentioned. LLL12, S31201 (both from Biovision, Inc.) and SF-1066, which inhibit STAT3 activity in different ways, have shown important results using the MDA-MB-231 breast cancer line as a model.203,204 FLL32 and IS3295, which inhibit STAT5, have also shown promising in vitro results in several breast cancer cell lines, such as MDA-MB-231, SUM159, and SK-BR-3.205 Finally, as an example of nucleotide-based inhibitors, siRNAs and G-quartets (guanine-rich oligonucleotides that form inter- or intramolecular four-stranded structures) against STAT3 have shown positive results, based on studies using the MCF7 and MDA-MB-231 breast cancer cell lines.206–208
Wingless-type MMTV-integration-site family
The Wnt pathway
Human and most mammalian genomes harbor 19 WNT genes, falling into 12 evolutionarily conserved WNT subfamilies.209 Wnt proteins are secreted ~40 kDa cysteine-rich glycoprotein ligands, activating a complex mechanism of signal transduction via multiple pathways: the canonical β-catenin-dependent pathway and several noncanonical β-catenin-independent pathways.210 These Wnt cascades play an important role in embryonic development processes, as well as in carcinogenesis.211 The noncanonical Wnt signaling pathway, which operates independently of β-catenin-mediated transcription (Figure 5A), is separated into the Wnt planar cell polarity and Wnt/Ca2+ signaling branches.212 Moreover, this pathway appears to function independently of transcription (Figure 5B and C).
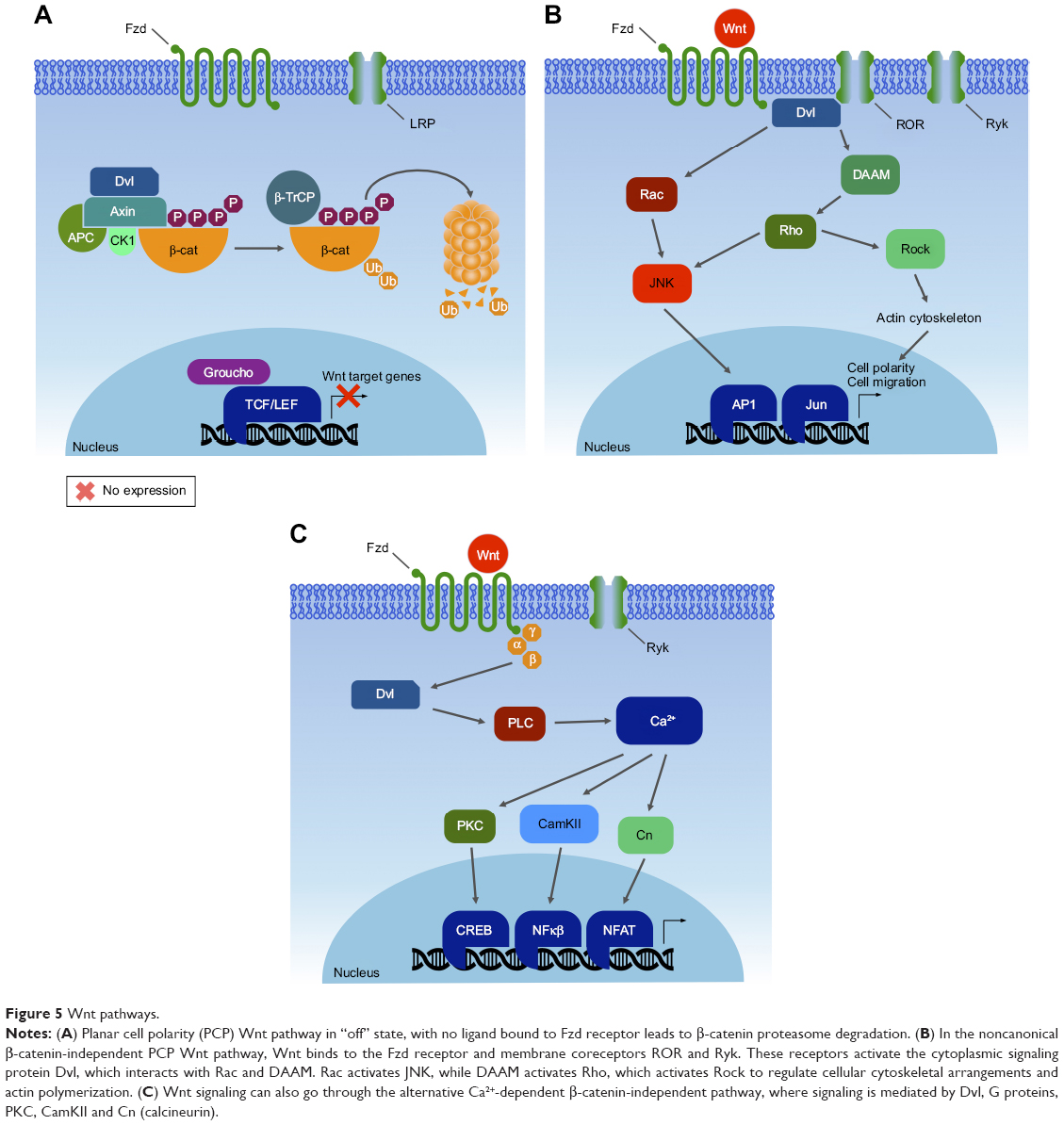 | Figure 5 Wnt pathways. |
The canonical pathway was first identified and delineated from genetic screens in Drosophila melanogaster, and intensive studies in worms, frogs, fish, and mice have led to the identification of a basic molecular signaling framework.213 Briefly, the canonical Wnt pathway is defined by the translocation of β-catenin into the nucleus, where it acts as a coactivator of transcription. Wnt ligands bind to an Fzd and a member of coreceptors LRP5/6. Intracellular signaling proceeds via the Axin and Dvl proteins (Figure 6A). These in turn lead to inactivation of the β-catenin destruction complex, which causes β-catenin to be stabilized and relocated into the nucleus. Within the nucleus, β-catenin forms a complex with the TCF/LEF transcription factors, triggering the expression of specific target genes.214,215 This transcriptional activity determines cell fate decisions, survival, and proliferation. The precise targets may vary among cell types, but in some cases can include the oncogenes c-Myc and cyclin D1.216 In the new model, Axin stabilizes the destruction complex in both the presence and absence of WNT, and β-catenin is degraded through phosphorylation-mediated recognition by β-TrCP in the intact complex.212 This allows newly synthesized β-catenin to accumulate in the cytosol before nuclear translocation (Figure 6B). Despite the fact that major pathway components have been characterized, the function of Wnt signaling within the context of cancer biology is intriguingly complex and remains only partially understood.
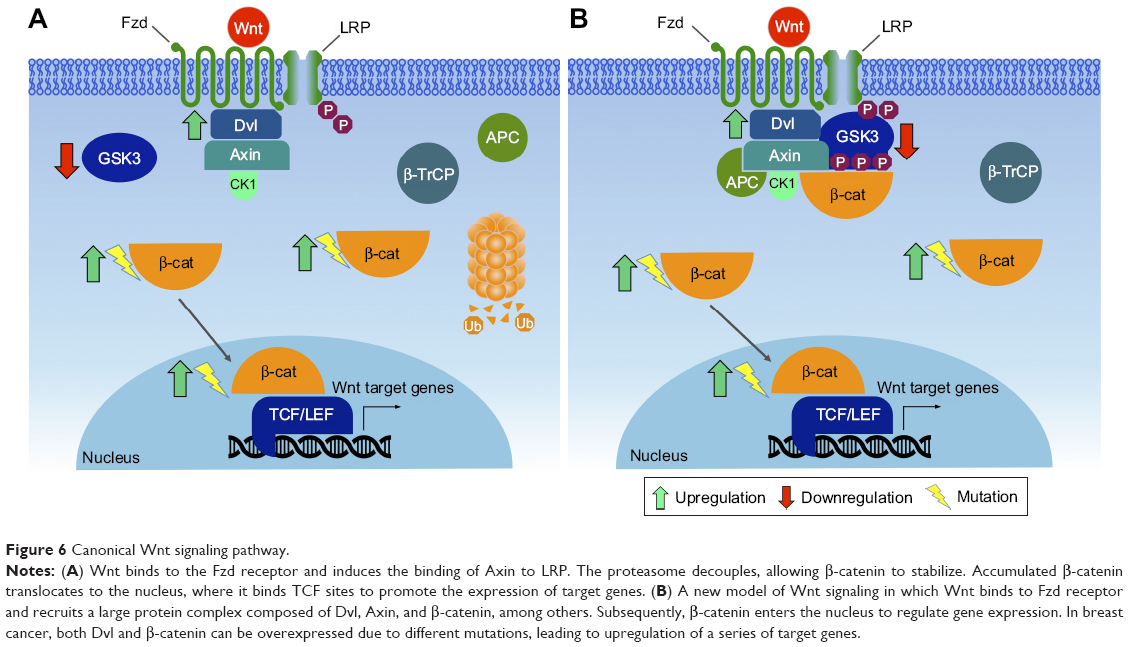 | Figure 6 Canonical Wnt signaling pathway. |
Wnt in mammary tissue development
Several components of Wnt signaling have been documented during various stages of morphogenesis of the mammary gland, including a wide range of Wnt ligands, receptors, downstream effectors, and DNA-binding proteins.212 Functionally, it is clear that canonical Wnt signaling is necessary for the initiation of mammary development. Dkk1 expression prevents localized expression of all mammary placode markers.217 Wnt6 is initially expressed on the surface ectoderm as a broader band surrounding the Wnt10-expressing mammary lines.209,218 In addition, Wnt4 mediates progesterone-induced ductal side branching in early pregnancy.219 Contrarily, the expression of Axin, which acts to destabilize β-catenin, causes defective alveolar formation during pregnancy.220 β-Catenin also plays important roles in mammary development, such as cell adhesion, signal transduction, and regulation of gene expression, which are essential for mammary SC biology during mammary development.221
Noncanonical Wnt signaling has also been shown to take part in the negative regulation of mammary epithelial outgrowths, adding another level of complexity.209 For example, loss of Wnt5a during other polarized morphogenic events increases cytoplasmic and nuclear β-catenin and accelerates ductal outgrowth.222 Importantly, Wnt5a is an essential mediator of TGFβ, suggesting that low thresholds of β-catenin signaling are maintained during pubertal ductal morphogenesis through TGFβ and Wnt5a antagonism.
Wnt and breast cancer
Given the importance of Wnt signaling for adult SC biology, it is not surprising that Wnt pathway mutations are observed frequently in cancer, most notably in tissues that normally depend on Wnt for self-renewal or repair.223 Mutations including β-catenin, Axin, or other Wnt pathway components, which result in β-catenin accumulation, are found in breast tumorigenesis.224–226
Wnt signaling is activated in over 50% of breast cancer and linked to reduced overall survival.227 Deregulated Wnt signaling leads to increased SC renewal, proliferation, migration, and survival.228 Studies have shown that molecular subtypes of breast cancer exhibit different levels of Wnt pathway activity.229 Solid human tumors, including those of the breasts, prostate, liver, and ovaries, have increased levels of cytoplasmic and nuclear β-catenin, indicating overexpression of Wnt signaling.230,231 In mice, overexpression of Wnt1, Wnt3, or Wnt10 leads to mammary carcinomas.209,232 Increased expression of Dvl1, a cytosolic positive regulator of Wnt signaling, is prominent in primary breast cancers.233,234
Investigations have shown that noncanonical Wnt members, such as Wnt5a receptors, are connected to breast cancer metastases,235 as well as to shorter overall survival of breast cancer patients.183,236,237 Immunohistochemistry expression profiling in 90 triple-negative breast cancer specimens has shown that low levels of Wnt5a expression are associated with positive lymph node metastasis, enhanced cell motility, Ki67 proliferation, and significantly worse recurrence-free survival.238,239 Modulation of receptor activation could be another factor for hyperactive Wnt signaling in breast cancer.209 An alternative splicing of LRP5, which removes the coding region of LRP5 that interacts with the secreted Wnt signaling repressor Dkk1, has been reported in parathyroid and breast cancer.240
Metastasis is a trademark of late-stage cancer and a main challenge to therapy. A main adaptive change of tumors during therapy is EMT.241 EMT is a known example of epithelial plasticity that is important in cancer metastasis.242 During tumor progression, EMT allows benign tumor cells to infiltrate the surrounding tissue and metastasize to distant sites.243 Activation of canonical Wnt signaling stabilizes Snai2 by inhibiting GSK3βactivity and initiates EMT transcriptional programs in breast cancer cells.244 Another candidate gene that regulates EMT is ASPP2, which encodes a protein that binds and inhibits the N-terminal phosphorylation of β-catenin, leading to its stabilization. Decreased expression of ASPP2 leads to EMT and is correlated with low survival in hepatocellular and breast cancer.242
Identification of novel biomarkers would be a promising approach for developing new diagnostic and therapeutic strategies. Markers that can predict the site of metastasis are scarce, and there are few reports on the utility of evaluating Wnt signaling members in the diagnosis of cancer.245 Research has suggested Dkk1 as a diagnostic biomarker for a wide variety of cancers, including breast cancer, implicating the therapeutic potential of anti-Dkk1 antibody to neutralize the activity of Dkk1 function for cancer cell invasion and growth.246 The majority of cancer patients presented elevated Dkk1 levels compared to healthy controls, and thus confirmed previous data supporting the usefulness of Dkk1 as a serological biomarker of cancer.
Wnt1-expressing mouse mammary epithelial cells show transcriptional upregulation of COX2; however, it is not clear whether this is due to direct regulation of the COX2 promoter by β-catenin.247 β-catenin is expressed in the nucleus, cytoplasm, and/or mesenchyme of 36.1% of breast cancer patients.248 Therefore, β-catenin status might serve as a predictive biomarker in breast cancer.
Clinical targeting of the Wnt pathway
Despite significant effort, there are no drugs currently approved for clinical use in breast cancer, which mainly target members of the Wnt pathway. However, a few small-molecule groups that target modulators of Wnt-related genes has emerged. Among these, the leading group are the so-called Porcupine inhibitors (LGK974 [Novartis]).249 Porcupine is a membrane-bound O-acetyltransferase that is required for palmitoylation of Wnt ligands, a necessary step in the processing and secretion of Wnt ligands.249 LGK974 is now in clinical Phase I trials for melanoma, breast cancer (triple-negative), pancreatic adenocarcinoma, colorectal cancer, and head and neck cancers.
Transforming growth factor beta
The TGFβ pathway
TGFβ was first isolated in 1982 in a search for secreted autocrine growth factors capable of transforming normal fibroblasts into malignant cells, which can proliferate in the absence of normal growth controls.250,251 The purified factor promoted wound healing by stimulating production of extracellular matrix factors and vascularization. At the same time, growth inhibitory effects of purified TGFβ were clearly demonstrated,252 leading to the paradox of TGFβ playing a role in both promoting and inhibiting cell growth.253 TGFβ is expressed in nearly all tissue, and the cellular response to TGFβ is highly dependent on the different contexts. The role of TGFβ has been most often studied in models for immunosuppression and inflammation, extracellular remodeling, cell proliferation, differentiation, survival, and invasion, all of which are paramount in cancer onset and progression.254
TGFβ signal transduction is carried by canonical and noncanonical pathways. In the canonical pathway, the TGFβ extracellular ligand binds the TGFβ2-membrane receptor complex, which contains a cytoplasmic serine/threonine kinase domain that in turn phosphorylates receptor-regulated SMADs (homologue of Drosophila MAD), notably SMAD2 and SMAD3, to transduce the activating signal. SMAD2/3 dimerizes with the common mediator SMAD4, and this activated complex translocates into the nucleus to participate as a coactivator or corepressor of targeted gene transcription,255 leading to the propagation of a diverse variety of TGFβ-induced responses in their specific contexts. TGFβ also triggers the Snail family of transcriptional repressors, which includes Snai1, Snai2/Slug, Twist1, ZEB1, and ZEB2.256
TGFβ in mammary tissue development
Because the ubiquitously expressed TGFβ provokes many different phenotypes in different tissues, attributing specific outcomes to TGFβ is a complex exercise that largely depends on cell context and timing. Much of the insight into the role of TGFβ in mammary gland development has been achieved through experiments using ectopic TGFβ treatment of explants and transgenic mouse models. Early experiments showed that implanted slow-release pellets of TGFβ inhibited mammary ductal branching and proliferation at the mammary end buds.257,258 Consistent with this observation, transgenic mice expressing TGFβ driven by an MMTV enhancer/promoter showed suppressed mammary ductal tree development, though lactation was unaffected and no spontaneous tumors emerged in this model.259 The converse inhibition of TGFβ using dominant-negative transgenic mice resulted in increased ductal tree side branching and proliferation.260 In heterozygous TGFβ mice, which express <10% of normal TGFβ expression levels, mammary ductal and alveolar development is accelerated,261 consistent with a fundamentally inhibitory role for TGFβ during mammary development. When TGFβ expression was directed specifically to alveolar cells by WAP gene regulatory elements, lobuloalveolar development was impaired and milk production inhibited.262 Transgenic transplantation experiments revealed that the mammary epithelium itself was defective, ruling out possible trans-signaling effects.263 Mammary explants treated with TGFβ also show suppression of milk production.264 After lactation, the involution process of the mammary gland correlates with TGFβ expression and other apoptotic regulatory genes.265,266 An inhibitory role for TGFβ in ductal morphogenesis and alveolar development and function and a promotional role for TGFβ in mammary gland involution and remodeling have been clearly established; however, mechanisms and downstream intracellular factors remain unclear.
TGFβ and breast cancer
In cultured cells, TGFβ is a potent inducer of EMT, a prerequisite for metastasis and cancer progression. EMT precedes invasion of the vasculature, transport in circulation, penetration of the basement membrane of a distal tissue, and finally metastatic tumor formation. During the complex, stepwise process of EMT, changes in tissue architecture correspond to changes in cellular functions. Epithelial cells lose cell–cell adherens junctions and apical–basal polarity. The resulting mesenchymal cells acquire a spindle-like morphology, with increased migration and invasive potential. Expression of specific structural and differentiation genes changes as well, such as from cytokeratins and E-cadherin in epithelial cells to vimentin and N-cadherin in mesenchymal cells.
Exposure of mammary carcinoma cells to exogenous TGFβ in the absence of inhibitory antibodies increases cell invasion potential in migration assays and lads to lung metastasis in rodents.267 Immunohistopathological analysis of human breast carcinomas reveals higher levels of extracellular TGFβ protein, especially at the advancing edges of infiltrating mammary duct carcinomas and in lymph node metastases.268,269
TGFβ has been shown directly to promote EMT in breast cancer in both cell cultures assays and mouse models. Exogenous TGFβ induces Ras-transformed mammary epithelial cells into EMT and secretion of TGFβ in these cells in an autocrine loop, and maintains the mesenchymal state.270 TGFβ1 and TGFβ3 ligands have been identified in a screen for genes that cooperate with ErbB2/HER2 in activating EMT in a mammary epithelial cell migration assay. Addition of ectopic TGFβ recapitulated this migration in a dose-dependent manner.271 Transgenic mouse models expressing both activated ErbB2/HER2 and TGFβ have also revealed modulation of the invasiveness and metastasis of mammary tumors in vivo that was dependent on the expression or repression of TGFβ.272–274
TGFβ affects EMT via downstream canonical SMAD-dependent pathways, as well as through noncanonical signaling and cross talk with other pathways.255 In mammary epithelial cells, the TWIST1 and SNAI1 genes are induced by activated SMADs, and subsequently repress transcription of key genes involved in maintaining epithelial identity and integrity, notably E-cadherin, provoking an EMT transition.275,276 Snail protein expression is found in duct-infiltrating breast carcinomas, presenting lymph node metastases and is inversely correlated with the grade of tumor differentiation.277 Knocking down TWIST inhibits metastases of mammary carcinomas to the lung, and high levels of Twist expression are found in highly invasive infiltrating lobular carcinomas: breast cancers that have lost E-cadherin expression.278
A clear role for noncoding RNA in regulating TGFβ-induced EMT in breast cancer has been established.279,280 Several members of the miR200 family (miR-200f) play a tumor suppressor role by inhibiting the Snail family transcriptional repressors ZEB1 and ZEB2.281–283 High expression of miR200f sequences targets the TGFβ pathway genes ZEB1/ZEB2, SMAD2, SMAD5, SNAI1, and others, increases E-cadherin expression, reduces cell motility, and restores an epithelial phenotype to mammary carcinoma cells in vitro.282,284 Conversely, ZEB1 and ZEB2 are capable of suppressing miR200f clusters, creating a negative feedback loop with TGFβ promoting EMT and miR-200 antagonizing.281 A similar feedback loop has been established between miR34 and Snail in a breast carcinoma cell line.285
Breast carcinomas exhibit a spectrum of both epithelial and mesenchymal phenotypes, and this nonbinary mix of features in tumors has given rise to the notion of partial EMT, describing an intermediate state where conversion to either epithelial or mesenchymal states is possible and reversible.286 Such plasticity between EMT states complicates our understanding of in vivo cellular processes during metastatic invasion, as the context of each specific tumor microenvironment often determines the outcome. The advent of molecular gene expression profiling has led to a narrowing identification of the cells of origin of breast cancer and suggested new ways to classify them. This research has intersected with studies on EMT, mammary SCs, and TGFβ.
A subpopulation of breast cancer cells with increased tumorigenic potential has been isolated by cell sorting using cell surface markers. These CD44+/CD24− cells were called tumor-initiating cells, due to their higher potency in initiating tumors when xenografted into immunocompromised mice.287 A gene expression profile revealed that the TGFβ pathway genes were specifically expressed and activated in this CD44+/CD24− cell population. Furthermore, the gene expression signature of CD44+/CD24− cells resembled that of normal mammary SCs and putative breast cancer SCs (CSCs) more than they did other subpopulations of sorted breast cancer cells.288
Gene expression signatures from the CD44+/CD24− tumor-initiating cell population and a mammary SC population were compared in cells forced to undergo EMT, and found to share specific expression of SC markers, as well as EMT mesenchymal markers. SC-like properties, such as ability to undergo multipotent tissue differentiation, were observed in both populations.289 An autocrine–paracrine feedback loop maintains the mesenchymal, stemlike state in these tumor-initiating cells.290 From this point forward, the concept of a breast CSC or breast tumor-initiating cell (BTIC) was proposed, describing a distinct subpopulation of cells with SC-like self-renewing properties, expressing mesenchymal markers, including those involved in TGFβ signaling, which are capable of effecting tumor progression through invasion, migration, intravasation, and metastasis.
Sorting of cell populations by flow cytometry and grouping by functional gene signature profiles has allowed researchers to begin reconciling apparent contradictions in the growth-promoting vs -inhibiting effects of TGFβ signaling. Instead of a temporal switch from a cytostatic role of TGFβ toward a tumorigenic role in EMT and metastasis, both activities can take place in different cells of the same tumor cell population. CD44+/CD24−/claudin-low subpopulations of tumor and normal mammary epithelial cells respond selectively to exogenous TGFβ treatment by expansion of their BTIC or basal/SC population, whereas the BTICs of other sorted populations remain static or are reduced.291 In ER+ breast cancers, differing TGFβ/SMAD3-driven gene expression signatures are generated that are able to uncouple the tumor suppressive effects of TGFβ in patient cohorts with good clinical outcomes from the tumor-promoting activities of TGFβ with poorer outcomes.292 In the near future, greater knowledge about specific gene signatures from individual tumors will be informative for the choice of therapy, such as employing TGFβ antagonists or not. The epigenetic landscape will play an increasingly important role in determining TGFβ-induced phenotypes within specific cell contexts and cancer subtypes. The epigenomes of BTIC-promoting vs BTIC-suppressing breast cancer cells differentially determine the subset of target genes that can be bound and activated by TGFβ/SMAD3, adding another layer of complexity to context-dependent signaling by TGFβ (Figure 7).293
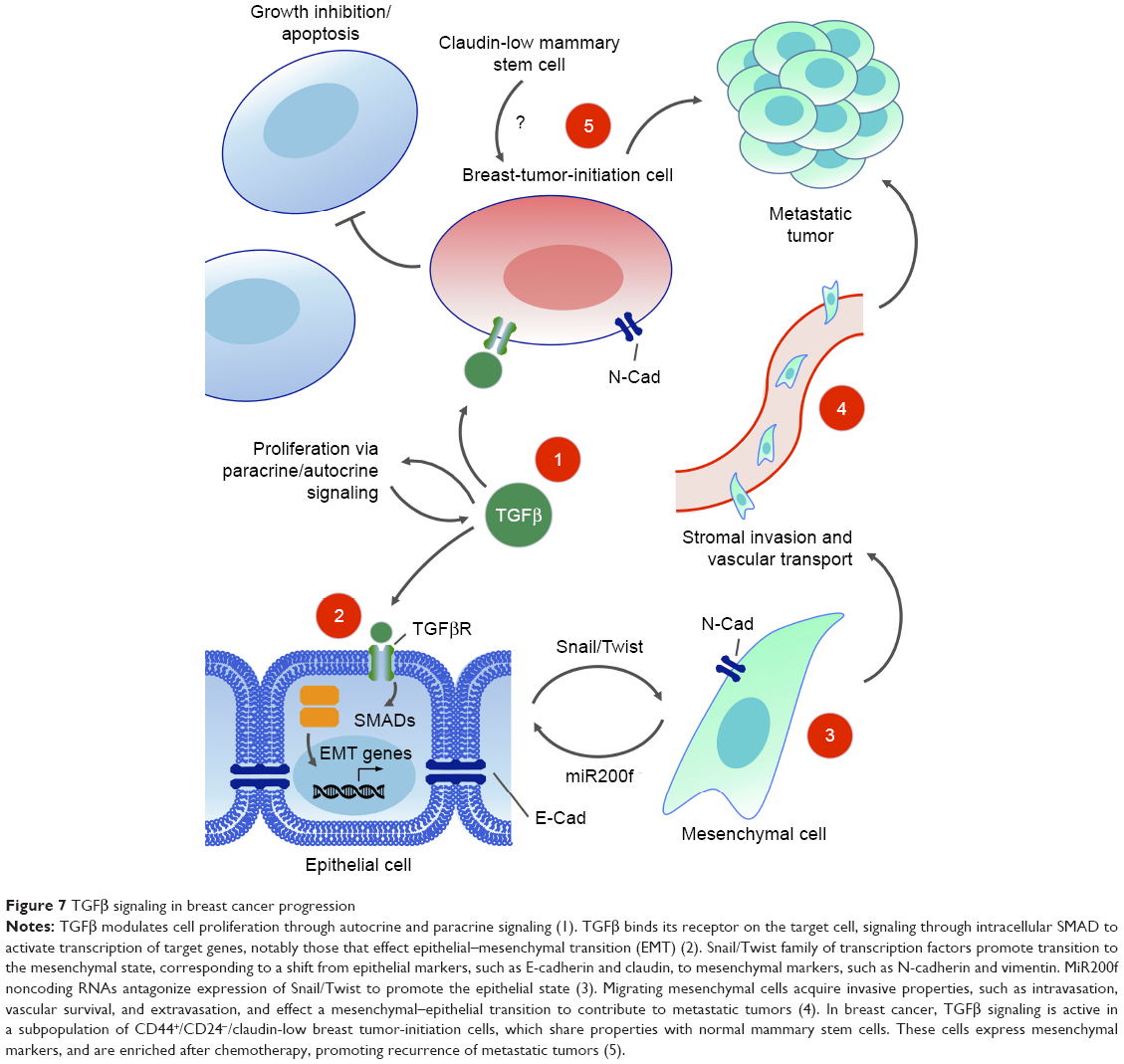 | Figure 7 TGFβ signaling in breast cancer progression |
Clinical targeting of the TGFβ pathway
TGFβ has become a popular target for drug development in cancer therapy. In addition to EMT and growth inhibition, TGFβ signaling is also involved in normal tissue homeostasis, extracellular matrix regulation, and immunoresponse modulation. With such highly pleiotropic activities of TGFβ, greatly influenced by specific contexts of cells, tissues, and architecture in complex processes, developing specific therapies to target TGFβ for a single disease presents a formidable challenge.294 The attraction of targeting TGFβ would be to antagonize its EMT-promoting activities and BTIC-promoting properties, as well (perhaps) as to reduce interstitial fluid pressure to improve the efficiency of drug delivery.295 Reducing TGFβ ligands or TGFβ signaling is seen as the goal, rather than direct ablation of TGFβ-inducing or -responding cells, so the efficacy of a therapy can only be assessed in a systemic in vivo setting, rather than via cytotoxicity screens on cells in vitro.
Two therapies against metastatic breast cancer have entered clinical trials. Fresolimumab, a humanized inhibitory antibody directed against the TGFβ ligand, is currently in a Phase II trial (NCT01401062) and administered in conjunction with radiotherapy. Eli Lilly has developed the drug LY2157299 (galunisertib), which targets the TGFβ1 receptor.296 This small-molecule inhibitor is also administered in conjunction with radiotherapy, and is currently recruiting participants (NCT02538471). LY2157299 has been able to inhibit TGFβ signaling and consequently tumor formation in a human-derived mouse model of glioblastoma.297 In another promising study, the TGFβ inhibitor LY2157299 was used in parallel with inhibitory antibodies against TGFβ2 receptors and SMAD4 RNAi. Following chemotherapy with the taxane paclitaxel, these TGFβ inhibitors blocked tumor reinitiation in a mouse model of triple-negative breast cancer by suppressing the expansion of BTICs.298 Such therapies combining TGFβ inhibitors with chemotherapeutic drugs show the most promise, as TGFβ activation in BTICs may promote drug resistance and cytotoxic chemotherapeutic drugs may enrich drug-resistant TGFβ-dependent BTIC populations, which contribute to tumor reinitiation.299–301
Nuclear factor-κ light-chain enhancer of activated β-cells
The NFκB pathway
NFκB was originally discovered as a nuclear factor that specifically binds to a 10-base-pair DNA sequence (5′-GGGACTTTCC-3′) within the enhancer of the Igκ light chain of activated β-cells.302 Currently, NFκB is largely known to be involved in cell cycle regulation, immunoresponse, inflammation, proliferation, and cell death. In mammals, the NFκB transcription factor family is composed of five members, divided in two classes based on the sequences of the C-terminal domain. Members of one class – the NFκB proteins NFκB1/p105 and NFκB2/p100 – have long C-terminal domains that contain multiple copies of ankyrin repeats, which inhibit these molecules until they are processed by either limited proteolysis or arrested translation into p50 and p52, respectively.303 Although p52 and p50 lack the transcription activation domains, they can positively regulate transcription through heterodimerization with members of the second class: the Rel subfamily, composed of cRel, RelB, and RelA (p65), which contain transcription activation domains that confer the ability to initiate transcription.304 They all share an Rel homology region, which mediates specific DNA binding to the NFκB consensus sequence and dimerization and interaction with IκB inhibitory molecules.305–308
Prior to stimuli, NFκB dimers are bound to inhibitory molecules of the IκB protein family (eg, IκBα, IκBβ, IκBγ, IκBε), which keep NFκB complexes inactive in the cytoplasm and block their binding capacity toward DNA.309 The classical (or canonical) NFκB pathway is induced by inflammatory cytokine signaling, DNA damage, or infectious agents. This stimulation leads to activation of the IκB kinase (IKK) complex (composed of IKKα/IKK1 and IKKβ/IKK2 and the scaffolding protein IKKγ/NEMO),310,311 which phosphorylates Iκα and then allows the E3 ubiquitin ligase βTrCP to promote polyubiquitination and further proteasome-dependent degradation of IκB.312–314 As a result, liberated NFκB rapidly enters the nucleus, where it can activate the expression of specific genes that contain DNA binding sites for NFκB (κB enhancer sites).315 An alternative (or noncanonical) pathway of NFκB activation is activated by signals transmitted by a subset of immunorelated receptors, including BAFFR, LTβR, CD40, RANKL, TNFR2, and Tweakr.316 This alternative induction mechanism leads to activation of NFκB inducing kinase (NIK), which predominantly phosphorylates and activates IKKα. Activated IKK phosphorylates p100, resulting in its ubiquitination and partial processing to p52.317 Following processing, alternate NFκB dimers (mainly composed of cRel and p52) are released to translocate further to the nucleus and act as transcription factors, thereby promoting expression of NFκB-related target genes. Nevertheless, binding of NFκB to the respective κB sites does not ensure transcriptional initiation, which in fact requires the interaction of NFκB with a few mediators, such as transcriptional adaptors and coactivator proteins like CBP and its paralogue p300, for assembly of the transcription machinery318 (Figure 8).
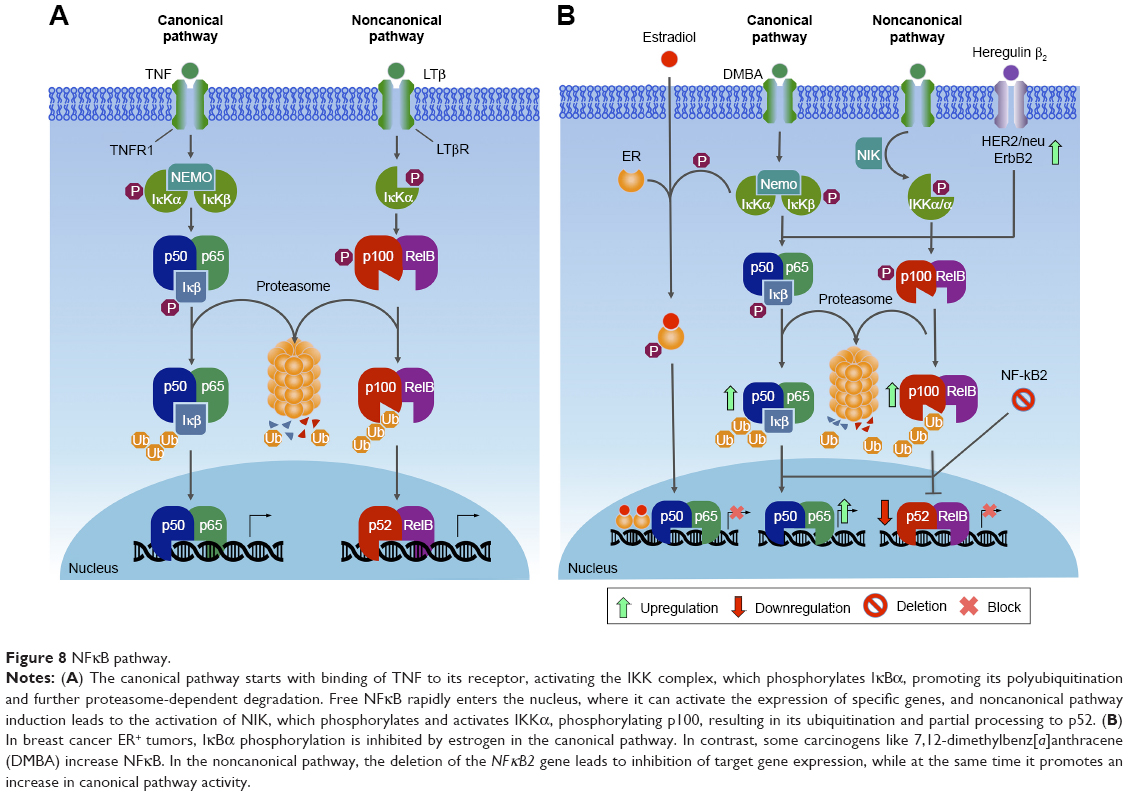 | Figure 8 NFκB pathway. |
NFκB in mammary tissue development
Expression of the NFκB subunits, such as p50, p52, p65 (RelA), and RelB, as well as the prototypical inhibitor IκBα, follows a consistent pattern during mouse mammary tissue development, being typically high during pregnancy, diminished during lactation, and elevated again in breast involution.319–323 NFκB acts as a negative regulator of β-casein gene expression during pregnancy in mice by interfering with tyrosine phosphorylation of STAT5, blocking a premature differentiation of the gland.320 Ablation of IκBα in the mammary epithelium results in hyperplasia and increased lateral ductal branching in virgin mice, suggesting that NFκB modulates proliferation, branching, and normal structural development of the mammary epithelium during early postnatal morphogenesis.324 IKKαAA/AA mice bearing a serine–alanine mutation that prevents its activation, fail to develop lobuloalveolar tissue properly, which is caused by impaired RANKL-mediated induction of cyclin D1, which is encoded by an NFκB target gene.325 Furthermore, a deletion of the gene for IKKβ in the mammary gland reduces apoptosis and delays involution, suggesting a proapoptotic role for IKKβ,326 whereas its constitutive expression leads to a reduction β-casein levels, accompanied by detection of cleaved caspase 3 during lactation and involution.327 In immortalized cultures of KIM2 cells, lactogenic hormone deprivation leads to increased NFκB activity, suggesting a survival role for NFκB in mammary involution.321 The p65–p300 complex is associated with modulation of inflammatory responses 48 hours after weaning.328 High levels of nitric oxide (NO) are detected during the first hours of involution, when NFκB binds to the NOS2 promoter to modulate its transcription.329 Collectively, these data demonstrate the importance of NFκB signaling in the development of normal mammary glands.
NFκB and breast cancer
Abnormally high NFκB activity is a clinical hallmark of chronic inflammation, being detected in a vast number of cancers. However, this information has to be taken with caution, since NFκB may act either as a tumor suppressor or a tumor promoter, depending on the type of cancer.330,331 The role for NFκB as a tumor promoter arises from its aberrant activation and nuclear localization as a result of defects in regulation of the pathway, loss of negative feedback mechanisms, resistance to cell death, and influence of the tumor microenvironment.331 However, other observations suggest that NFκB can also inhibit tumor growth. Tumor suppressor proteins, such as p53 and ARF, can induce the association of NFκB subunits with HDAC1 corepressor complexes.332 This results in NFκB-dependent repression of target gene expression, providing a model through which NFκB subunits can facilitate apoptosis and cell cycle arrest and thus function as tumor suppressors themselves. This suggests a dual function for NFκB during tumor progression, inhibiting tumor growth, but as further mutations lead to a loss of tumor suppressor expression, the oncogenic functions of NFκB become unleashed, contributing to tumorigenesis.330 The tumor inflammatory microenvironment maintains NFκB constitutively active in most tumor cells, and NFκB has been found to control multiple cellular processes in cancer, such as inflammation, transformation, proliferation, angiogenesis, invasion, metastasis, chemoresistance, and radioresistance. Therefore, NFκB suppression may have a positive impact by inhibiting the growth of tumor cells.
Though NFκB is required for normal mammary gland morphogenesis, its deregulated activation has been linked to tumor progression via stimulation of cell proliferation and survival and angiogenesis pathways and metastasis, ultimately driving breast carcinogenesis. In vivo and in vitro experiments in female rats, mice, and human mammary epithelial cell lines treated with a carcinogen, such as 7,12-dimethylbenz[a]anthracene, showed elevated activity of NFκB prior to malignant transformation and tumor formation.333,334 Elevated NFκB expression has been widely reported in human breast cancer cell lines and tissues, with abnormal constitutive expression of both canonical and noncanonical NFκB subunits, such as cRel, p65, p50, and p52.335–338 Several reports have observed a positive correlation between NFκB activation and the ErbB2 receptor, also known as HER2/Neu.338–340 In addition, NFκB is predominantly activated in ER− and ER−/HER2+ breast cancers, suggesting its importance in specific classes of breast cancers.335,340 Progression of ER-dependent to ER-independent breast cancer phenotype is correlated with an increase in NFκB activity. In an E2-independent breast ductal carcinoma cell line (MCF7/LCC1) model, constitutive activation of NFκB was observed prior to the loss of ER expression.341 In ErbB2-expressing cell lines, administration of the ErbB2 activator heregulin β1 led to increased NFκB activation, whereas herceptin, an anti-ErbB2 monoclonal antibody, blocked this activation and led to apoptosis.340 Phosphorylation of a tumor suppressor protein that regulates NFκB activation (CYLD) by the breast cancer oncogene kinase IKBKE342 promotes cell transformation.343 In vitro assays have revealed that constitutive NFκB activity in breast cancer cells stimulates osteoclastogenesis by upregulating GM-CSF. Blocking NFκB suppressed osteolytic lesion development through osteoclastic bone resorption in vivo.344
Together with oncogenic RAS, NFκB activity is required for protection of mammary epithelial cells from TGFβ-induced apoptosis. This protection is a prerequisite for these cells to undergo EMT toward an invasive, metastatic tumor phenotype.345 In a triple transgenic model in which NFκB activity can be inhibited by doxycycline treatment in defined windows during PYMT-induced tumorigenesis, inhibition of NFκB signaling leads to increased tumor latency and decreased tumor burden.327
BRCA1-deficient breast tumors exhibit aggressive behavior and are associated with poor survival.346 Within BRCA1-deficient mammary glands, there is a subset of cells named luminal progenitor cells, which can form 3D colonies in a hormone-independent fashion.347 In these cells, constitutive NFκB (cRel/p52) activation due to DNA damage caused by progesterone leads to hormone-uncoupled proliferation, increasing the risk of genomic instability and transformation.348
Self-renewing breast CSCs are key players in perpetuating tumor maintenance and in treatment resistance and relapse. Interestingly, in HER2-dependent tumors, the proportion of CD44+ cells (marker for SC population) dramatically decreased upon NFκB suppression, indicating that this transcription factor is important in the maintenance of progenitor cell expansion.349 Another study showed that repression of CD44, a cell surface glycoprotein, via NFκB inhibition consequently decreased proliferation and invasiveness of breast cancer cells.350 Silencing of NFκB2/p100 in mammary cancer cell lines led to loss of p100 and p52 and to context-dependent effects on tumorigenicity of these cells.351 In 4T1 mammary carcinoma cells, increased activity of NFκB (which induces EMT and CSCs) was a result of p100 loss, whereas in N202.1A mammary carcinoma cells, CSCs and tumorigenicity were positively regulated by p52, suggesting an opposite role for this protein in regulation of breast CSCs, and indicating that inhibiting p100 processing may be a potential therapeutic strategy to suppress CSC activity in a subset of breast tumors.351
Clinical targeting of the NFκB pathway
Targeting the NFκB pathway seems to be a reasonable objective in view of the numerous implications of this pathway in diverse diseases, including breast cancer. Combinatorial therapeutic strategies, targeting members of the PI3K/Akt and MAPK pathways that lead to NFκB activation, have already been employed in several clinical trials.352 Association between ErbB2 expression and NFκB activation and the inverse correlation found between the latter and ER status may be exploited in human breast cancer therapy.353 Other agents, such as specific IKK inhibitors, are under investigation. Inhibition of TBK1, a noncanonical IKK, with TBK1-II, a drug that efficiently inhibits TBK1 and IKKε, suppresses growth of human HER2+ breast cancer cells and induced cellular senescence.354 Inhibition of IKKs sensitizes cells to the cytotoxic effect of doxorubicin in different breast cancer cell lines.355 In addition, inhibition of NFκB DNA binding is a good putative approach to inhibit NFκB activity, since this would prevent transactivation of prosurvival and antiapoptotic downstream targets, being highly selective. More than 780 compounds described to have NFκB inhibitory activity356 could be used in combinatorial therapy for breast cancer. Compounds such as the proteasome inhibitor bortezomib have undergone clinical trials in breast cancer patients with some success.357,358 However, unselective pathway disruption potentially leads to adverse effects, such as immunosuppression, which could be particularly harmful in cancer patients, who are likely to be immunocompromised due to previous treatments.359
Signaling integration (crosstalk) within breast cancer
Breast cancer is heterogeneous in nature, due to a slew of aberrations at the genomic and molecular levels affecting various signaling pathways. To understand the integrative role of these pathways in different physiological and pathological conditions, it is necessary to take into consideration their complex cross talk. The best example of cross talk in the ER signaling pathway is the signalosome, a large protein complex present at the membrane. Upon activation by HER2, ERK1/2 phosphorylates ER, thus increasing its sensitivity for its ligands or leading to ligand-independent activation. In addition, ER coregulators can be modified by kinases, thereby indirectly affecting ER activity.107,360,361 Also GPR30, which colocalizes with ERα in the membrane, collaborates in transmission of signals to downstream kinase cascades.45
The importance of signaling cross talk in cancer is also illustrated by the influence of estrogen signaling in EMT. Cross talk between ERα and several EMT regulators, such as Snail and Slug, leads to increased EMT.362 Additionally, ER signaling can mediate delocalization of E-cadherin and cytoskeleton reorganization via the ER–Src–PELP1–PI3K–ILK1 pathway. Therefore, this cross talk can affect breast cancer progression, leading to metastasis via EMT and contributing to breast cancer cell motility.102 Most of the nongenomic mechanisms in the ER pathway are implicated in breast cancer metastasis, with ERK and Akt phosphorylation being involved in breast cancer cell migration and Src and ILK1 kinases playing critical roles in cell invasion and metastatis.361
PI3K signaling also exerts downstream control on other transcriptional factors, which include members of the FOX protein family (FOXO1, FOXO2, FOXO4, and FOXO6) and NFκB. In addition, PI3K signaling can inhibit p53 activity by a mechanism that involves the E3 ubiquitin protein ligase MDM2, regulating the cell cycle, inflammation, apoptosis, and DNA repair.122 LKB1, a kinase located upstream of AMPK, negatively regulates mTORC1 signaling by tuberous sclerosis 1 or 2.116,117 PKC (activated in the PI3K pathway) has the potential to phosphorylate Raf1. PAK1 stimulated by PI3K and/or CDC42 pathways can also phosphorylate Raf1. Upon stimulation by PI3K, Akt can phosphorylate and inhibit both B-Raf and Raf1.153,363
NFκB-dependent transcription is not only tightly controlled by positive and negative regulatory mechanisms but also closely coordinated with other signaling pathways. Since activation depends on IκB degradation, the IKK complex is the gatekeeper of NFκB signaling, representing a critical node for interaction within parallel signaling pathways. Other upstream molecules, such as receptor interacting proteins and TNFR, are critical to IKK activation, but also signal other pathways. Thus, TNFR-associated factors represent a central point of divergence for activation of NFκB and AP1 transcription factor pathways.364
MAPKs, JNK, ERK1/ERK2, and p38 promote activation of AP1, and activated AP1 affects cell survival, apoptosis, and stress responses. Another molecule, RIP1, has been described as being involved in activation of the PI3K–Akt pathway through not only NFκB-dependent negative regulation of expression of the mTOR kinase but also through NFκB-independent downregulation of the PI3K antagonist PTEN.365 The IKKβ-induced degradation of p105 may potentially influence several additional pathways, since IKKβ can also phosphorylate Dok1, an Ras GAP-associated TK substrate and cell growth inhibitor, leading to inhibition of ERK1 and ERK2, demonstrating the context dependence of IKKβ-mediated MAPK regulation.366 JAKs can phosphorylate MAPK, PIK3, and TGFβ associated receptors, thereby creating a site for anchoring proteins displaying SH2 domains in their structure. Upon phosphorylation, these proteins are able to stimulate a signaling cascade in their respective pathways.367
It has been shown that ER activation can inhibit TGFβ-induced transcriptional activity and cell migration. Several lines of investigation have suggested that ERα is a major modifier of the TGFβ signaling pathway, with ER and TGFβ pathways being able to promote breast cancer metastasis.368 Moreover, it is important to consider that the cross talk between Notch, Wnt, and SHH signaling pathways, as well as their role in regulating TICs, may also modulate breast cancer onset and progession.369
Conclusion
Clearly, the risk for breast cancer is sexually dimorphic. In fact, breast cancer is about 100 times less common among men than in women. Still, the molecular role of sex hormones modulating breast cancer risk remains elusive. Elucidation of the function of ER in breast cancer cells should provide critical new insights into the function of these receptors. Expanding the vision of the entire network involving ER may lead to better design of specific therapies to treat breast cancer, with fewer side effects. It should also provide a better understanding of the mechanisms behind antiestrogen resistance. In vitro and in vivo models, clinical trials, and innovative treatments should contribute to better understanding of the function of each type of hormone receptor.
Besides the classic hormone signaling mechanisms, the pathways discussed in this review are shown to modulate central cellular functions that are critical in the onset and progression of breast cancer. The PI3K signaling pathway is important for mammary morphogenesis and mammary gland involution; however, alterations in the PI3K pathway are very common in several diseases, including Parkinson’s, obesity, type 2 diabetes, and different cancer types. Since several dysregulations in key nodes of this pathway are known to be associated with various disease states, the ability to pinpoint a specific alteration and to understand its functional relevance would allow for more accurate choice of treatment, with minimal side effects. Direct and indirect involvement of the MAPK pathway has been demonstrated for the development of breast cancer, participating in promotion of cell proliferation or rendering other pathways more active. JAK–STAT is one of the main signaling pathways that promotes communication of the extracellular medium with the cell nucleus. Wnt signaling regulates cell differentiation, proliferation, and SC pluripotency, and thus deregulation of Wnt signaling may lead to cancer development. TGFβ plays a dual role in breast cancer progression by either its tumor suppressing or tumor enhancing effects. NFκB represents a central factor in inflammation, stress response, and cell death, being recognized as an important element in several steps of cancer initiation and progression, activated by a large variety of stimuli, and displaying several downstream targets, thereby generating broad and complex feedback loops.
Several aspects of regulation of these pathways remain to be clarified, especially regarding their cross talk with other pathways, feedback, tumor microenvironment interactions, cell metabolism, risk factors, and response to drug therapy. However, it is clear that all of these pathways act as a large orchestrated network, and that a broad scope needs to be taken in planning future therapy strategies and designing prognostic tools. In this context, an increasingly detailed study of these pathways becomes even more important, since it may generate relevant and transformative knowledge regarding the nature of cellular communication, as well as the molecular basis for gene expression regulation in both normal and cancer cells. Therefore, identifying the range of mutations and/or molecular changes occurring in these major signaling pathways and how they interact across pathways is crucial for understanding breast cancer development, as well as for implementation of molecular-based approaches for continuous improvement in diagnostics, prognostics, and treatment of breast cancer patients.
Abbreviations
AF-1, N-terminal activation function 1 domain; AKT, Serine/threonine protein kinase 1; AMPK, AMP-activated protein kinase; AR, Androgen receptor; Areg, Amphiregulin; ARF, ADP ribosylation factor; ASPP2, Apoptosis-stimulating of p53 protein 2; ATP, Adenosine triphosphate; BAFF-R, B-cell activation factor; BRCA1, Breast cancer 1 supressor gene; BRCA2, Breast cancer 2 supressor gene; BTIC, Breast tumor initiating cell; CBP, CREB-binding protein; CDC42, Cell division cycle 42; COUP-TFI, Chicken ovalbumin upstream promoter-transcription factor-1; COX2, Cyclooxygenase-2; CSC, Cancer stem cell; CSF, Colony stimulating factor; CYLD, Cylindromatosis human gene; DAAM, Dvl-associated activator of morphogenesis; DKK1, Dickkopf-related protein 1; DMBA, 7,12-Dimethylbenz[a]anthracene; DNA, Deoxyribonucleic acid; dsDNA, Double-stranded DNA; Dvl, Dishevelled; E2, Estradiol:17β-estradiol; EGF, Epidermal growth factor; EGFR, Epidermal growth factor receptor; EMT, Epithelial-mesenchymal transition; ER, Estrogen receptor; ErbB2, Human epidermal growth factor receptor 2; ERE, Estrogen response element; ERK, Extracellular signal–regulated kinase; ERRα, Estrogen-related receptor alpha; ERRβ, Estrogen-related receptor beta; ERRγ, Estrogen-related receptor gamma; FAK, Focal adhesion kinase; FERM, Four-point-one motif, ezrin, radixin, moesin; FN14, Fibroblast growth factor-inducible 14; FOXO, Forkhead box O; FZD, Frizzled receptor; GATA3, GATA biding protein 3; GH, Growth hormone; GM-CSF, Granulocyte macrophage colony-stimulating factor; GOF, Gain-of-function; GPCR, G protein-coupled receptor; GPER1, G protein-coupled ER 1; GRB-2, Growth factor receptor-bound protein 2; GRB-7, Growth factor receptor-bound protein 7; GSK3β, Glycogen synthase kinase 3β, Gβγ, G beta-gamma complex; HDAC1, Histone deacetylase 1; HDM2, Human double minute 2 protein; IFN, Interferon; IGF1, Insulin-like growth factor 1; IKK, IκB kinase complex; IKKα, Inhibitor of nuclear factor kappa-B kinase subunit alpha; IKKβ, Inhibitor of nuclear factor kappa-B kinase subunit beta; IL-10, Interleukin 10; IL-1β, Interleukin 1 beta; IL-6, Interleukin 6; IRS-1, Insulin receptor substrate 1; IκB, Inhibitor of kappa B; JAK, Janus kinase; JNK, Jun amino-terminal kinase; LBD, Ligand-binding domain; LOF, Loss-of-function; LRP, LDL-related receptor protein; LRP5/6, Lipoprotein receptor-related protein; LTβR, Lymphotoxin β receptor; MAPK, Mitogen-activated protein kinase; MAPKKK, Mitogen-activated protein kinases kinases kinase; MCF7/LCC1, Breast ductal carcinoma cell line; MEK, Mitogen-activated protein kinase kinase (or MAPKK, MKK); MMTV, Mouse mammary tumor virus; mTOR: Mammalian target of rapamycin; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cell; NGFI-B, Nerve growth factor induced-B; NIK, NF-κB inducing kinase; NO, Nitric oxide; NOS-2, Nitric oxide synthase, inducible; NR, Nuclear receptor family; PAK1,P21 (RAC1) activated kinase 1; PCP, Planar cell polarity; PDK, Putative 3-phosphoinositide-dependent kinase (or PDPK1); PGAP3, Post-GPI attachment to proteins 3; PI3K, Phosphatidylinositol-4,5-bisphosphate 3-kinase; PIK3CA, Phosphoinositide 3-kinase catalytic subunit α; PIP2, Phosphatidylinositol-4,5-bisphosphate; PIP3, Phosphatidylinositol-3,4,5-trisphosphate; PKB, Protein kinase B; PKC, Protein kinase C; PR, Progesterone receptor; proHB-EGF, Membrane-anchored heparin-binding EGF like growth factor; PTEN, Phosphatase and tensin homologue; PyMT, Polyomavirus middle T-antigen oncogene; RANK, Receptor activator for nuclear factor kappa B; RAPTOR, Regulatory-associated protein of mammalian target of rapamycin; RHR, Rel homology region; RIP1,Receptor Interacting Serine/Threonine Kinase 1; ROCK, Rho-associated kinase; RORα: Retinoic acid-related orphan receptor alpha; RTK, Receptor tyrosine kinase; SC, stem cell; SERD, Selective estrogen receptor degrader; SERM, Selective estrogen receptor modulator; SH2, Src homology-2; SHC, Src Homology 2 domain-containing-transforming protein C1; siRNA: Small interfering RNA; SMAD: Contraction of SMA (small body size) and MAD (Mothers against decapentaplegic); SNAI2, SNAIL family transcriptional repressor 2; SOS, Son of sevenless; SRC,V-Src avian sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog; STAT, Signal transducer and activator of transcription; TAD, Transcription activation domain; TBK1, TANK-binding kinase 1; TCF/LEF, T-cell factor/lymphoid enhancer factor; TGFβ, Transforming growth factor β; Th17, Helper T cells 17; TNBC, Triple-negative breast cancer; TNFR2, Tumor necrosis factor receptor 2; TP53, tumor protein 53 (or p53); TRAF, TNFR-associated factor; TSC, Tuberous sclerosis; TWEAK-R, TWEAK receptor; TWEAK, tumor necrosis factor-like weak inducer of apoptosis; TYK2, Tyrosine kinase 2 or tyrosine-protein kinase 2; ULK1, Unc-51-like kinase 1; WNT, Wingless (wg)/integration 1 gene (int); ZEB1, Zinc finger E-box-binding homeobox 1; ZEB2, Zinc finger E-box-binding homeobox 2; β-TRCP, Beta-transducin repeat containing E3 ubiquitin protein ligase.
Acknowledgments
We are grateful to the Department of Biochemistry from the Institute of Chemistry (IQ) of the University of São Paulo (USP, São Paulo, Brazil) for the academic support and opportunity to write this review. We thank the following Brazilian research funding agencies for the financial support: FAPESP (São Paulo State Foundation for Research), CNPq (National Research Council), and CAPES (Federal Agency for Superior Education and Training). MCS was additionally supported by grants from BNDES (Brazilian National Bank for Economic and Social Development), FINEP (Financiadora de Estudo e Projetos), MCTI (Science, Technology, and Innovation Ministry), MS-DECIT (Science and Technology Department of the Health Ministry), and Ouro Fino Saúde Animal Ltd. LFZ was supported by the International Centre for Genetic Engineering and Biotechnology (ICGEB). RGC was supported by a Special Visiting Researcher (PVE) grant from the “Science Without Borders” program (CAPES).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
World Health Organization. Cancer. 2017. Available from: http://www.who.int/cancer/en. Accessed July 14, 2017. | ||
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108. | ||
DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64(1):52–62. | ||
Medina D. The mammary gland: a unique organ for the study of development and tumorigenesis. J Mammary Gland Biol Neoplasia. 1996;1(1):5–19. | ||
Sharma GN, Dave R, Sanadya J, Sharma P, Sharma KK. Various types and management of breast cancer: an overview. J Adv Pharm Technol Res. 2010;1(2):109–126. | ||
Malhotra GK, Zhao X, Band H, Band V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol Ther. 2010;10(10):955–960. | ||
Iqbal N, Iqbal N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol Biol Int. 2014;2014:852748. | ||
Dai X, Li T, Bai Z, et al. Breast cancer intrinsic subtype classification, clinical use and future trends. Am J Cancer Res. 2015;5(10):2929–2943. | ||
Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295(21):2492–2502. | ||
Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. | ||
Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. | ||
Prat A, Parker JS, Karginova O, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. | ||
Sabatier R, Finetti P, Guille A, et al. Claudin-low breast cancers: clinical, pathological, molecular and prognostic characterization. Mol Cancer. 2014;13:228. | ||
Dias K, Dvorkin-Gheva A, Hallett RM, et al. Claudin-low breast cancer: clinical and pathological characteristics. PLoS One. 2017;12(1):0168669. | ||
McPherson K, Steel CM, Dixon JM. ABC of breast diseases – breast cancer: epidemiology, risk factors, and genetics. BMJ. 2000;321(7261):624–628. | ||
Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71. | ||
Easton D, Ford D, Peto J. Inherited susceptibility to breast cancer. Cancer Surv. 1993;18:95–113. | ||
Gustafsson JA. What pharmacologists can learn from recent advances in estrogen signalling. Trends Pharmacol Sci. 2003;24(9):479–485. | ||
Shen M, Shi H. Sex hormones and their receptors regulate liver energy homeostasis. Int J Endocrinol. 2015;2015:294278. | ||
Knowlton AA, Lee AR. Estrogen and the cardiovascular system. Pharmacol Ther. 2012;135(1):54–70. | ||
Sanchez R, Nguyen D, Rocha W, White JH, Mader S. Diversity in the mechanisms of gene regulation by estrogen receptors. Bioessays. 2002;24(3):244–254. | ||
McDonnell DP, Norris JD. Connections and regulation of the human estrogen receptor. Science. 2002;296(5573):1642–1644. | ||
Bourdeau V, Deschênes J, Métivier R, et al. Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol Endocrinol. 2004;18(6):1411–1427. | ||
Bolger R, Wiese TE, Ervin K, Nestich S, Checovich W. Rapid screening of environmental chemicals for estrogen receptor binding capacity. Environ Health Perspect. 1998;106(9):551–557. | ||
Ruff M, Gangloff M, Wurtz JM, Moras D. Estrogen receptor transcription and transactivation: structure-function relationship in DNA- and ligand-binding domains of estrogen receptors. Breast Cancer Res. 2000;2(5):353–359. | ||
McBryan J, Young L. Ligand-independent signalling through estrogen receptor pathways in breast cancer. In: Larionov A, editor. Resistance to Aromatase Inhibitors in Breast Cancer. Heidelberg: Springer; 2015:115–144. | ||
Weigel NL, Zhang Y. Ligand-independent activation of steroid hormone receptors. J Mol Med. 1998;76(7):469–479. | ||
Cenni B, Picard D. Ligand-independent activation of steroid receptors: new roles for old players. Trends Endocrinol Metab. 1999;10(2):41–46. | ||
Ali S, Metzger D, Bornert J, Chambon P. Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. EMBO J. 1993;12(3):1153–1160. | ||
Migliaccio A, Di Domenico M, Castoria G, et al. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 1996;15(6):1292–1300. | ||
Bunone G, Briand PA, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996;15(9):2174–2183. | ||
Bologa CG, Revankar CM, Young SM, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2(4):207–212. | ||
Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307(5715):1625–1630. | ||
Teng J, Wang ZY, Jarrard DF, Bjorling DE. Roles of estrogen receptor α and β in modulating urothelial cell proliferation. Endocr Relat Cancer. 2008;15(1):351–364. | ||
Liu MM, Albanese C, Anderson CM, et al. Opposing action of estrogen receptors α and β on cyclin D1 gene expression. J Biol Chem. 2002;277(27):24353–24360. | ||
Paech K. Differential ligand activation of estrogen receptors ER and ER at AP1 sites. Science. 1997;277(5331):1508–1510. | ||
Hall JM, McDonnell DP. The estrogen receptor β-isoform (ERβ) of the human estrogen receptor modulates ERα transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology. 1999;140(12):5566–5578. | ||
Poola I, Koduri S, Chatra S, Clarke R. Identification of twenty alternatively spliced estrogen receptor alpha mRNAs in breast cancer cell lines and tumors using splice targeted primer approach. J Steroid Biochem Mol Biol. 2000;72(5):249–258. | ||
Herynk MH, Fuqua SA. Estrogen receptor mutations in human disease. Endocr Rev. 2004;25(6):869–898. | ||
Taylor SE, Martin-Hirsch PL, Martin FL. Oestrogen receptor splice variants in the pathogenesis of disease. Cancer Lett. 2010;288(2):133–148. | ||
Li C, Briggs MR, Ahlborn TE, Kraemer FB, Liu J. Requirement of Sp1 and estrogen receptor α interaction in 17β-estradiol-mediated transcriptional activation of the low density lipoprotein receptor gene expression. Endocrinology. 2001;142(4):1546–1553. | ||
Filardo EJ, Quinn JA, Frackelton AR, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16(1):70–84. | ||
Kumar P, Wu Q, Chambliss KL, et al. Direct interactions with Gαi and Gβγ mediate nongenomic signaling by estrogen receptor α. Mol Endocrinol. 2007;21(6):1370–1380. | ||
Vivacqua A, Bonofiglio D, Recchia AG, et al. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17β-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol Endocrinol. 2006;20(3):631–646. | ||
Prossnitz ER, Arterburn JB, Sklar LA. GPR30: a G protein-coupled receptor for estrogen. Mol Cell Endocrinol. 2007;265–266:138–142. | ||
Marquez DC, Lee J, Lin T, Pietras RJ. Epidermal growth factor receptor and tyrosine phosphorylation of estrogen receptor. Endocrine. 2001;16(2):73–81. | ||
Razandi M, Pedram A, Park ST, Levin ER. Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem. 2003;278(4):2701–2712. | ||
Song RX, Zhang Z, Santen RJ. Estrogen rapid action via protein complex formation involving ERα and Src. Trends Endocrinol Metab. 2005;16(8):347–353. | ||
Balogh P, Szabó A, Katz S, Likó I, Patócs A, Kiss AL. Estrogen receptor alpha is expressed in mesenteric mesothelial cells and is internalized in caveolae upon Freund’s adjuvant treatment. PLoS One. 2013;8(11):e79508. | ||
Chaudhri RA, Schwartz N, Elbaradie K, Schwartz Z, Boyan BD. Role of ERα36 in membrane-associated signaling by estrogen. Steroids. 2014;81:74–80. | ||
Shaul PW. Regulation of endothelial nitric oxide synthase: location, location, location. Annu Rev Physiol. 2002;64(1):749–774. | ||
Davis NM, Sokolosky M, Stadelman K, et al. Deregulation of the EGFR/PI3K/PTEN/Akt/mTORC1 pathway in breast cancer: possibilities for therapeutic intervention. Oncotarget. 2014;5(13):4603–4650. | ||
Murphy LJ, Ghahary A. Uterine insulin-like growth factor-1: regulation of expression and its role in estrogen-induced uterine proliferation. Endocr Rev. 1990;11(3):443–453. | ||
Cooke PS, Buchanan DL, Young P, et al. Stromal estrogen receptors mediate mitogenic effects of estradiol on uterine epithelium. Proc Natl Acad Sci U S A. 1997;94(12):6535–6540. | ||
Bai W, Oliveros-Saunders B, Wang Q, Acevedo-Duncan ME, Nicosia SV. Estrogen stimulation of ovarian surface epithelial cell proliferation. In Vitro Cell Dev Biol Anim. 2000;36(10):657–666. | ||
Mallepell S, Krust A, Chambon P, Brisken C. Paracrine signaling through the epithelial estrogen receptor α is required for proliferation and morphogenesis in the mammary gland. Proc Natl Acad Sci U S A. 2006;103(7):2196–2201. | ||
Aschim EL, Sæther T, Wiger R, Grotmol T, Haugen TB. Differential distribution of splice variants of estrogen receptor β in human testicular cells suggests specific functions in spermatogenesis. J Steroid Biochem Mol Biol. 2004;92(1–2):97–106. | ||
Chen M, Yeh CR, Shyr CR, Lin HH, Da J, Yeh S. Reduced prostate branching morphogenesis in stromal fibroblast, but not in epithelial, estrogen receptor α knockout mice. Asian J Androl. 2012;14(4):546–555. | ||
Prins GS, Birch L, Couse JF, et al. Estrogen imprinting of the developing prostate gland is mediated through stromal estrogen receptor α: studies with αERKO and βERKO mice. Cancer Res. 2001;61(16):6089–6097. | ||
Mueller SO. Mammary gland development in adult mice requires epithelial and stromal estrogen receptor. Endocrinology. 2002;143(6):2357–2365. | ||
Ruan W, Kleinberg LD. Insulin-like growth factor I is essential for terminal end bud formation and ductal morphogenesis during mammary development. Endocrinology. 1999;140(11):5178–5184. | ||
Joshi PA, Jackson HW, Beristain AG, et al. Progesterone induces adult mammary stem cell expansion. Nature. 2010;465(7299):803–807. | ||
Russo J, Ao X, Grill C, Russo IH. Pattern of distribution of cells positive for estrogen receptor a and progesterone receptor in relation to proliferating cells in the mammary gland. Breast Cancer Res Treat. 1999;53(3):217–227. | ||
Lim E, Vaillant F, Wu D, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15(8):907–913. | ||
Anderson E, Clarke RB, Howell A. Estrogen responsiveness and control of normal human breast proliferation. J Mammary Gland Biol Neoplasia. 1998;3(1):23–35. | ||
Cunha GR, Young P, Hom YK, Cooke PS, Taylor JA, Lubahn DB. Elucidation of a role for stromal steroid hormone receptors in mammary gland growth and development using tissue recombinants. J Mammary Gland Biol Neoplasia. 1997;2(4):393–402. | ||
Zeps N, Bentel JM, Papadimitriou JM, D’Antuono MF, Dawkins HJ. Estrogen receptor-negative epithelial cells in mouse mammary gland development and growth. Differentiation. 1998;62(5):221–226. | ||
Luetteke NC, Qiu TH, Fenton SE, et al. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development. 1999;126(12):2739–2750. | ||
Wilson CL, Sims AH, Howell A, Miller CJ, Clarke RB. Effects of oestrogen on gene expression in epithelium and stroma of normal human breast tissue. Endocr Relat Cancer. 2006;13(2):617–628. | ||
Sternlicht MD, Sunnarborg SW, Kouros-Mehr H, Yu Y, Lee DC, Werb Z. Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development. 2005;132(17):3923–3933. | ||
Kleinberg DL, Ruan W. IGF-I, GH, and sex steroid effects in normal mammary gland development. J Mammary Gland Biol Neoplasia. 2008;13(4):353–360. | ||
Hovey R, Trott J, Vonderhaar B. Establishing a framework for the functional mammary gland: from endocrinology to establishing a framework for the functional mammary gland: from endocrinology to morphology. J Mammary Gland Biol Neoplasia. 2002;7(1):17–38. | ||
Rowson AR, Daniels KM, Ellis SE, Hovey RC. Growth and development of the mammary glands of livestock: a veritable barnyard of opportunities. Semin Cell Dev Biol. 2012;23(5):557–566. | ||
Lee S, Medina D, Tsimelzon A, et al. Alterations of gene expression in the development of early hyperplastic precursors of breast cancer. Am J Pathol. 2007;171(1):252–262. | ||
El-Ashry D, Miller D, Kharbanda S, Lippman ME, Kern F. Constitutive Raf-1 kinase activity in breast cancer cells induces both estrogen-independent growth and apoptosis. Oncogene. 1997;15(4):423–435. | ||
Pietras RJ, Arboleda J, Reese DM, et al. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene. 1995;10(12):2435–2446. | ||
Rakha EA, Pinder SE, Bartlett JM, et al. Updated UK recommendations for HER2 assessment in breast cancer. J Clin Pathol. 2015;68(2):93–99. | ||
Tognon C, Knezevich SR, Huntsman D, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2(5):367–376. | ||
Tonon G, Modi S, Wu L, et al. t(11;19)(q21;p13) Translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat Genet. 2003;33(2):208–213. | ||
Troester MA, Herschkowitz JI, Oh DS, et al. Gene expression patterns associated with p53 status in breast cancer. BMC Cancer. 2006;6:276. | ||
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumors. Nature. 2012;490(7418):61–70. | ||
Benoit R, Cooney A, Giguere V, Ingraham H, Lazar M, Muscat G. International Union of Pharmacology – LXVI: orphan nuclear receptors. Pharmacol Rev. 2006;58(4):798–836. | ||
Wärnmark A, Treuter E, Wright AP, Gustafsson JA. Activation functions 1 and 2 of nuclear receptors: molecular strategies for transcriptional activation. Mol Endocrinol. 2003;17(10):1901–1909. | ||
Boudot A, Kerdivel G, Lecomte S, et al. COUP-TFI modifies CXCL12 and CXCR4 expression by activating EGF signaling and stimulates breast cancer cell migration. BMC Cancer. 2014;14:407. | ||
Xu M, Qin J, Tsai SY, Tsai M. The role of the orphan nuclear receptor COUP-TFII in tumorigenesis. Acta Pharmacol Sin. 2015;36(1):32–36. | ||
Hedrick E, Lee SO, Kim G, et al. Nuclear receptor 4A1 (NR4A1) as a drug target for renal cell adenocarcinoma. PLoS One. 2015;10(6):e0128308. | ||
Du J, Xu R. RORα, a potential tumor suppressor and therapeutic target of breast cancer. Int J Mol Sci. 2012;13(12):15755–15766. | ||
Xiong G, Wang C, Evers BM, Zhou BP, Xu R. RORα suppresses breast tumor invasion by inducing SEMA3F expression. Cancer Res. 2012;72(7):1728–1739. | ||
Hong EJ, Levasseur MP, Dufour CR, Perry MC, Giguère V. Loss of estrogen-related receptor α promotes hepatocarcinogenesis development via metabolic and inflammatory disturbances. Proc Natl Acad Sci U S A. 2013;110(44):17975–17980. | ||
Giguère V. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev. 2008;29(6):677–696. | ||
Deblois G, Giguère V. Oestrogen-related receptors in breast cancer: control of cellular metabolism and beyond. Nat Rev Cancer. 2012;13(1):27–36. | ||
Ariazi EA, Clark GM, Mertz JE. Estrogen-related receptor α and estrogen-related receptor γ associate with unfavorable and favorable biomarkers, respectively, in human breast cancer. Cancer Res. 2002;62(22):6510–6518. | ||
Suzuki S, Takagi K, Miki Y, et al. Nucleobindin 2 in human breast carcinoma as a potent prognostic factor. Cancer Sci. 2012;103(1):136–143. | ||
Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–262. | ||
Audet-Walsh E, Papadopoli DJ, Gravel SP, et al. The PGC-1α/ERRα axis represses one-carbon metabolism and promotes sensitivity to anti-folate therapy in breast cancer. Cell Rep. 2016;14(4):920–931. | ||
Deblois G, St-Pierre J, Giguère V. The PGC-1/ERR signaling axis in cancer. Oncogene. 2013;32(30):3483–3490. | ||
Bianco S, Sailland J, Vanacker JM. ERRs and cancers: effects on metabolism and on proliferation and migration capacities. J Steroid Biochem Mol Biol. 2012;130(3–5):180–185. | ||
Ziello JE, Jovin IS, Huang Y. Hypoxia-inducible factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med. 2007;80(2):51–60. | ||
Klimcakova E, Chénard V, McGuirk S, et al. PGC-1α promotes the growth of ErbB2/neu-induced mammary tumors by regulating nutrient supply. Cancer Res. 2012;72(6):1538–1546. | ||
St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127(2):397–408. | ||
Chakravarty D, Nair SS, Santhamma B, et al. Extranuclear functions of ER impact invasive migration and metastasis by breast cancer cells. Cancer Res. 2010;70(10):4092–4101. | ||
Garcia-Becerra R, Santos N, Diaz L, Camacho J. Mechanisms of resistance to endocrine therapy in breast cancer: focus on signaling pathways, miRNAs and genetically based resistance. Int J Mol Sci. 2013;14(1):108–145. | ||
Leary A, Dowsett M. Combination therapy with aromatase inhibitors: the next era of breast cancer treatment? Br J Cancer. 2006;95(6):661–666. | ||
Hammond ME, Hayes DF, Dowsett M, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch Pathol Lab Med. 2010;134(7):e48–e72. | ||
Hayashi SI, Eguchi H, Tanimoto K, et al. The expression and function of estrogen receptor α and β in human breast cancer and its clinical application. Endocr Relat Cancer. 2003;10(2):193–202. | ||
Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. 2011;7(12):715–726. | ||
Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329–341. | ||
Shanware NP, Bray K, Abraham RT. The PI3K, metabolic, and autophagy networks: interactive partners in cellular health and disease. Annu Rev Pharmacol Toxicol. 2013;53(1):89–106. | ||
DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. | ||
Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–5510. | ||
Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. | ||
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–644. | ||
Baselga J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist. 2011;16(Suppl 1):12–19. | ||
Chin YR, Yoshida T, Marusyk A, Beck AH, Polyak K, Toker A. Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res. 2014;74(3):964–973. | ||
Lee JJ, Loh K, Yap Y. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol Med. 2015;12(4):342–354. | ||
Paplomata E, O’Regan R. The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. Ther Adv Med Oncol. 2014;6(4):154–166. | ||
Utermark T, Rao T, Cheng H, et al. The p110α and p110β isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev. 2012;26(14):1573–1586. | ||
Rosner M, Hengstschläger M. Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components Rictor and sin1. Hum Mol Genet. 2008;17(19):2934–2948. | ||
Morrison MM, Young CD, Wang S, et al. mTOR directs breast morphogenesis through the PKC-α-Rac1 signaling axis. PLoS Genet. 2015;11(7):e1005291. | ||
Rexer BN, Chanthaphaychith S, Dahlman KB, Arteaga CL. Direct inhibition of PI3K in combination with dual HER2 inhibitors is required for optimal antitumor activity in HER2+ breast cancer cells. Breast Cancer Res. 2014;16(1):R9. | ||
Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5(12):921–929. | ||
Muellner MK, Uras IZ, Gapp BV, et al. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7(11):787–793. | ||
Bolós V, Mira E, Martínez-Poveda B, et al. Notch activation stimulates migration of breast cancer cells and promotes tumor growth. Breast Cancer Res. 2013;15(4):R54. | ||
Riggio M, Perrone MC, Polo ML, et al. AKT1 and AKT2 isoforms play distinct roles during breast cancer progression through the regulation of specific downstream proteins. Sci Rep. 2017;7:44244. | ||
Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346(3):561–576. | ||
Massacesi C, di Tomaso E, Urban P, et al. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. Onco Targets Ther. 2016;9:203–210. | ||
Okkenhaug K, Graupera M, Vanhaesebroeck B. Targeting PI3K in cancer: impact on tumor cells, their protective stroma, angiogenesis, and immunotherapy. Cancer Discov. 2016;6(10):1090–1105. | ||
Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13(2):140–156. | ||
Paez JG. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. | ||
Rossomando AJ, Payne DM, Weber MJ, Sturgill TW. Evidence that pp42, a major tyrosine kinase target protein, is a mitogen-activated serine/threonine protein kinase. Proc Natl Acad Sci U S A. 1989;86(18):6940–6943. | ||
Kosako H, Gotoh Y, Matsuda S, Ishikawa M, Nishida E. Xenopus MAP kinase activator is a serine/threonine/tyrosine kinase activated by threonine phosphorylation. EMBO J. 1992;11(8):2903–2908. | ||
Ferrell JE Jr. Tripping the switch fantastic: how a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem Sci. 1996;21(12):460–466. | ||
Huang CY, Ferrell JE Jr. Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1996;93(19):10078–10083. | ||
Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290. | ||
Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. | ||
Pearson G, Robinson F, Gibson TB, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22(2):153–183. | ||
Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2001;75(1):50–83. | ||
Payne DM, Rossomando AJ, Martino P, et al. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J. 1991;10(4):885–892. | ||
Ahn NG, Campbell JS, Seger R, Jensen AL, Graves LM, Krebs EG. Metabolic labeling of mitogen-activated protein kinase kinase in A431 cells demonstrates phosphorylation on serine and threonine residues. Proc Natl Acad Sci U S A. 1993;90(11):5143–5147. | ||
Robbins DJ, Zhen E, Owaki H, et al. Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J Biol Chem. 1993;268(7):5097–5106. | ||
Crews CM, Alessandrini A, Erikson RL. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science. 1992;258(5081):478–480. | ||
Wu J, Harrison JK, Vincent LA, et al. Molecular structure of a protein-tyrosine/threonine kinase activating p42 mitogen-activated protein (MAP) kinase: MAP kinase kinase. Proc Natl Acad Sci U S A. 1993;90(1):173–177. | ||
Alessi DR, Saito Y, Campbell DG, et al. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 1994;13(7):1610–1619. | ||
Zheng CF, Guan KL. Activation of MEK family kinases requires phosphorylation of two conserved Ser/Thr residues. EMBO J. 1994;13(5):1123–1131. | ||
Ferrell JE Jr, Bhatt RR. Mechanistic studies of the dual phosphorylation of mitogen-activated protein kinase. J Biol Chem. 1997;272(30):19008–19016. | ||
Liu Y, Shepherd EG, Nelin LD. MAPK phosphatases: regulating the immune response. Nat Rev Immunol. 2007;7(3):202–212. | ||
Kyriakis JM, App H, Zhang XF, et al. Raf-1 activates MAP kinase-kinase. Nature. 1992;358(6385):417–421. | ||
Zheng CF, Guan KL. Cloning and characterization of two distinct human extracellular signal-regulated kinase activator kinases, MEK1 and MEK2. J Biol Chem. 1993;268(15):11435–11439. | ||
Morrison DK, Cutler RE. The complexity of Raf-1 regulation. Curr Opin Cell Biol. 1997;9(2):174–179. | ||
Whitehurst CE, Owaki H, Bruder JT, Rapp UR, Geppert TD. The MEK kinase activity of the catalytic domain of RAF-1 is regulated independently of Ras binding in T cells. J Biol Chem. 1995;270(10):5594–5599. | ||
Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129(7):1287–1292. | ||
Santen RJ, Song RX, McPherson R, et al. The role of mitogen-activated protein (MAP) kinase in breast cancer. J Steroid Biochem Mol Biol. 2002;80(2):239–256. | ||
Ahmad DA, Negm OH, Alabdullah ML, et al. Clinicopathological and prognostic significance of mitogen-activated protein kinases (MAPK) in breast cancers. Breast Cancer Res Treat. 2016;159(3):457–467. | ||
Kato S, Endoh H, Masuhiro Y, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270(5241):1491–1494. | ||
Joel PB, Smith J, Sturgill TW, Fisher TL, Blenis J, Lannigan DA. PP90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol Cell Biol. 1998;18(4):1978–1984. | ||
Lange CA, Shen T, Horwitz KB. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci U S A. 2000;97(3):1032–1037. | ||
Weng LP, Smith WM, Brown JL, Eng C. PTEN inhibits insulin-stimulated MEK/MAPK activation and cell growth by blocking IRS-1 phosphorylation and IRS-1/Grb-2/Sos complex formation in a breast cancer model. Hum Mol Genet. 2001;10(6):605–616. | ||
Kim DH, Sim T. Novel small molecule Raf kinase inhibitors for targeted cancer therapeutics. Arch Pharm Res. 2012;35(4):605–615. | ||
Matsuda Y, Fukumoto M. Sorafenib: complexities of Raf-dependent and Raf-independent signaling are now unveiled. Med Mol Morphol. 2011;44(4):183–189. | ||
Genovese MC. Inhibition of p38: has the fat lady sung? Arthritis Rheum. 2009;60(2):317–320. | ||
Darnell J, Kerr I, Stark G. Jak-STAT pathways and transcriptional activation in response. Science. 1994;264(5164):1415–1421. | ||
Ward AC, Touw I, Yoshimura A. The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood. 2000;95(1):19–29. | ||
Igaz P, Tóth S, Falus A. Biological and clinical significance of the JAK-STAT pathway: lessons from knockout mice. Inflamm Res. 2001;50(9):435–441. | ||
O’Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109 Suppl:S121–S131. | ||
Taniguchi T. Cytokine signaling through nonreceptor protein-tyrosine kinases. Science. 1995;268(5208):251–255. | ||
Ihle JN. Signaling by the cytokine receptor superfamily in normal and transformed hematopoietic cells. Adv Cancer Res. 1996;68:23–65. | ||
Jatiani SS, Baker SJ, Silverman LR, Reddy EP. Jak/STAT pathways in cytokine signaling and myeloproliferative disorders: approaches for targeted therapies. Genes Cancer. 2010;1(10):979–993. | ||
Shuai K, Liao J, Song MM. Enhancement of antiproliferative activity of gamma interferon by the specific inhibition of tyrosine dephosphorylation of Stat1. Mol Cell Biol. 1996;16(9):4932–4941. | ||
Kisseleva T, Bhattacharya S, Braunstein J. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285(1–2):1–24. | ||
Schindler C. Cytokines and JAK-STAT signaling. Exp Cell Res. 1999;253(1):7–14. | ||
Darnell JE. STATs and gene regulation. Science. 1997;277(5332):1630–1635. | ||
Levy DE, Darnell JE Jr. STATs: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3(9):651–662. | ||
Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12(13):2048–2060. | ||
Hughes K, Watson CJ. The spectrum of STAT functions in mammary gland development. JAKSTAT. 2012;1(3):151–158. | ||
Abell K, Bilancio A, Clarkson RW, et al. Stat3-induced apoptosis requires a molecular switch in PI(3)K subunit composition. Nat Cell Biol. 2005;7(4):392–398. | ||
Haricharan S, Li Y. STAT signaling in mammary gland differentiation, cell survival and tumorigenesis. Mol Cell Endocrinol. 2014;382(1):560–569. | ||
Clarkson RW, Boland MP, Kritikou EA, et al. The genes induced by signal transducer and activators of transcription (STAT)3 and STAT5 in mammary epithelial cells define the roles of these STATs in mammary development. Mol Endocrinol. 2006;20(3):675–685. | ||
O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66(1):311–328. | ||
Greenman C, Stephens PR, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–158. | ||
Jeong EG, Kim MS, Nam HK, et al. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin Cancer Res. 2008;14(12):3716–3721. | ||
Caffarel MM, Zaragoza R, Pensa S, Li J, Green AR, Watson CJ. Constitutive activation of JAK2 in mammary epithelium elevates Stat5 signalling, promotes alveologenesis and resistance to cell death, and contributes to tumourigenesis. Cell Death Differ. 2012;19(3):511–522. | ||
Zhang Y, Toy KA, Kleer CG. Metaplastic breast carcinomas are enriched in markers of tumor-initiating cells and epithelial to mesenchymal transition. Mod Pathol. 2012;25(2):178–184. | ||
Chan SR, Vermi W, Luo J, et al. STAT1-deficient mice spontaneously develop estrogen receptor α-positive luminal mammary carcinomas. Breast Cancer Res. 2012;14(1):R16. | ||
Weichselbaum RR, Ishwaran H, Yoon T, et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A. 2008;105(47):18490–18495. | ||
Watson CJ, Miller WR. Elevated levels of members of the STAT family of transcription factors in breast carcinoma nuclear extracts. Br J Cancer. 1995;71(4):840–844. | ||
Diaz N, Minton S, Cox C, et al. Activation of Stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated Src and survivin expression. Clin Cancer Res. 2006;12(1):20–28. | ||
Charpin C, Secq V, Giusiano S, et al. A signature predictive of disease outcome in breast carcinomas, identified by quantitative immunocytochemical assays. Int J Cancer. 2009;124(9):2124–2134. | ||
Hynes NE, Watson CJ. Mammary gland growth factors: roles in normal development and in cancer. Cold Spring Harb Perspect Biol. 2010;2(8):a003186. | ||
Henry MD, Triplett AA, Oh KB, Smith GH, Wagner K. Parity-induced mammary epithelial cells facilitate tumorigenesis in MMTV-neu transgenic mice. Oncogene. 2004;23(41):6980–6985. | ||
Meydan N, Grunberger T, Dadi H, et al. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996;379(6566):645–648. | ||
Thompson JE, Cubbon RM, Cummings RT, et al. Photochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitor. Bioorganic Med Chem Lett. 2002;12(8):1219–1223. | ||
Lucet IS, Fantino E, Styles M, et al. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood. 2006;107(1):176–183. | ||
Buchert M, Burns CJ, Ernst M. Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene. 2015;35(8):939–951. | ||
Garcia R, Bowman TL, Niu G, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20(20):2499–2513. | ||
Kumar J, Fraser FW, Riley C, Ahmed N, McCulloch DR, Ward AC. Granulocyte colony-stimulating factor receptor signalling via Janus kinase 2/signal transducer and activator of transcription 3 in ovarian cancer. Br J Cancer. 2014;110(1):133–145. | ||
Haddad BR, Gu L, Mirtti T, et al. STAT5A/B gene locus undergoes amplification during human prostate cancer progression. Am J Pathol. 2013;182(6):2264–2275. | ||
Catlett-Falcone R, Landowski TH, Oshiro MM, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10(1):105–115. | ||
Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. 2017;77(5):521–546. | ||
Costa-Pereira AP, Tininini S, Strobl B, et al. Mutational switch of an IL-6 response to an interferon-γ-like response. Proc Natl Acad Sci U S A. 2002;99(12):8043–8047. | ||
Furqan M, Akinleye A, Mukhi N, Mittal V, Chen Y, Liu D. STAT inhibitors for cancer therapy. J Hematol Oncol. 2013;6:90. | ||
Chen J, Bai L, Bernard D, et al. Structure-based design of conformationally constrained, cell-permeable STAT3 inhibitors. ACS Med Chem Lett. 2010;1(2):85–89. | ||
Liu A, Liu Y, Jin Z, et al. XZH-5 inhibits STAT3 phosphorylation and enhances the cytotoxicity of chemotherapeutic drugs in human breast and pancreatic cancer cells. PLoS One. 2012;7(10): e46624. | ||
Siddiquee K, Zhang S, Guida WC, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104(18):7391–7396. | ||
Turkson J, Zhang S, Mora LB, Burns A, Sebti S, Jove R. A novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J Biol Chem. 2005;280(38):32979–32988. | ||
Jing N, Li Y, Xiong W, Sha W, Jing L, Tweardy DJ. G-quartet oligonucleotides: a new class of signal transducer and activator of transcription 3 inhibitors that suppresses growth of prostate and breast tumors through induction of apoptosis. Cancer Res. 2004;64(18):6603–6609. | ||
Kunigal S, Lakka SS, Sodadasu PK, Estes N, Rao JS. Stat3-siRNA induces Fas-mediated apoptosis in vitro and in vivo in breast cancer. Int J Oncol. 2009;34(5):1209–1220. | ||
Yang Z, Cai JH, Xie SJ, et al. Therapeutic effects of signal transducer and activator of transcription 3 siRNA on human breast cancer in xenograft mice. Chin Med J (Engl). 2011;124(12):1854–1861. | ||
Yu QC, Verheyen EM, Zeng YA. Mammary development and breast cancer: a Wnt perspective. Cancers (Basel). 2016;8(7):1–26. | ||
Tanaka K, Kitagawa Y, Kadowaki T. Drosophila segment polarity gene product porcupine stimulates the posttranslational N-glycosylation of Wingless in the endoplasmic reticulum. J Biol Chem. 2002;277(15):12816–12823. | ||
Kim YM, Kahn M. The role of the Wnt signaling pathway in cancer stem cells: prospects for drug development. Res Rep Biochem. 2014;4:1–12. | ||
Pohl SG, Brook N, Agostino M, Arfuso F, Kumar AP, Dharmarajan A. Wnt signaling in triple-negative breast cancer. Oncogenesis. 2017;6(4):e310. | ||
Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4(2):68–75. | ||
Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of β-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10(12):1443–1454. | ||
Jang G, Kim J, Cho S, et al. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci Rep. 2015;5:12465. | ||
Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14(15):1837–1851. | ||
Boras-Granic K, Wysolmerski JJ. Wnt signaling in breast organogenesis. Organogenesis. 2008;4(2):116–122. | ||
Veltmaat JM, Van Veelen W, Thiery JP, Bellusci S. Identification of the mammary line in mouse by Wnt10b expression. Dev Dyn. 2004;229(2):349–356. | ||
Brisken C, Heineman A, Chavarria T, et al. Essential function of Wnt-4 in mammary gland development downstream of progesterone signaling service. Genes Dev. 2000;(617):650–654. | ||
Hsu W, Shakya R, Costantini F. Impaired mammary gland and lymphoid development caused by inducible expression of Axin in transgenic mice. J Cell Biol. 2001;155(6):1055–1064. | ||
Incassati A, Chandramouli A, Eelkema R, Cowin P. Key signaling nodes in mammary gland development and cancer: β-catenin. Breast Cancer Res. 2010;12(6):213. | ||
Roarty K, Serra R. Wnt5a is required for proper mammary gland development and TGF-β-mediated inhibition of ductal growth. Development. 2007;134(21):3929–3939. | ||
Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192–1205. | ||
Howe LR, Brown AM. Wnt signaling and breast cancer. Cancer Biol Ther. 2004;3(1):36–41. | ||
Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH. Wnt/β-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am J Pathol. 2010;176(6):2911–2920. | ||
Yang W, Yan HX, Chen L, et al. Wnt/β-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008;68(11):4287–4295. | ||
Lin SY, Xia W, Wang JC, et al. β-Catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci U S A. 2000;97(8):4262–4266. | ||
MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. | ||
Smid M, Wang Y, Zhang Y, et al. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008;68(9):3108–3114. | ||
Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17(1):45–51. | ||
Clevers H. Wnt/β-catenin signaling in development and disease. Cell. 2006;127(3):469–480. | ||
Brown AM. Wnt signaling in breast cancer: have we come full circle? Breast Cancer Res. 2001;3(6):351–355. | ||
Nagahata T, Shimada T, Harada A, et al. Amplification, up-regulation and over-expression of DVL-1, the human counterpart of the Drosophila disheveled gene, in primary breast cancers. Cancer Sci. 2003;94(6):515–518. | ||
Prasad CP, Gupta SD, Rath G, Ralhan R. Wnt signaling pathway in invasive ductal carcinoma of the breast: relationship between β-catenin, dishevelled and cyclin D1 expression. Oncology. 2007;73(1–2):112–117. | ||
Klemm F, Bleckmann A, Siam L, et al. β-Catenin-independent WNT signaling in basal-like breast cancer and brain metastasis. Carcinogenesis. 2011;32(3):434–442. | ||
Nishita M, Enomoto M, Yamagata K, Minami Y. Cell/tissue-tropic functions of Wnt5a signaling in normal and cancer cells. Trends Cell Biol. 2010;20(6):346–354. | ||
Henry C, Quadir A, Hawkins NJ, et al. Expression of the novel Wnt receptor ROR2 is increased in breast cancer and may regulate both β-catenin dependent and independent Wnt signalling. J Cancer Res Clin Oncol. 2014;141(2):243–254. | ||
Zeng R, Huang J, Zhong MZ, et al. Multiple roles of WNT5A in breast cancer. Med Sci Monit. 2016;22:5058–5067. | ||
Zhong Z, Shan M, Wang J, Liu T, Shi Q, Pang D. Decreased Wnt5a expression is a poor prognostic factor in triple-negative breast cancer. Med Sci Monit. 2016;22:1–7. | ||
Björklund P, Svedlund J, Olsson AK, Akerström G, Westin G. The internally truncated LRP5 receptor presents a therapeutic target in breast cancer. PLoS One. 2009;4(1):e4243. | ||
Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461–1473. | ||
Wang Y, Bu F, Royer C, et al. ASPP2 controls epithelial plasticity and inhibits metastasis through β-catenin-dependent regulation of ZEB1. Nat Cell Biol. 2014;16(11):1092–1104. | ||
Gujral TS, Chan M, Peshkin L, Sorger PK, Kirschner MW, MacBeath G. A noncanonical Frizzled 2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell. 2014;159(4):844–856. | ||
Wu ZQ, Li XY, Hu CY, Ford M, Kleer CG, Weiss SJ. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic breast cancer 1, early onset (BRCA1) repression. Proc Natl Acad Sci U S A. 2012;109(41):16654–16659. | ||
Pishvaian MJ, Byers SW. Biomarkers of WNT signaling. Cancer Biomark. 2007;3(4–5):263–274. | ||
Sato N, Yamabuki T, Takano A, et al. Wnt inhibitor Dickkopf-1 as a target for passive cancer immunotherapy. Cancer Res. 2010;70(13):5326–5336. | ||
Nuñez F, Bravo S, Cruzat F, Montecino M, de Ferrari GV. Wnt/β-catenin signaling enhances cyclooxygenase-2 (COX2) transcriptional activity in gastric cancer cells. PLoS One. 2011;6(4):e18562. | ||
Wang Z, Zhang H, Hou J, et al. Clinical implications of β-catenin protein expression in breast cancer. Int J Clin Exp Pathol. 2015;8(11):14989–14994. | ||
Liu J, Pan S, Hsieh MH, et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110(50):20224–20229. | ||
Anzano MA, Roberts AB, Meyers CA, et al. Synergistic interaction of two classes of transforming growth factors from murine sarcoma cells. Cancer Res. 1982;42(11):4776–4778. | ||
Moses HL, Roberts AB, Derynck R. The discovery and early days of TGF-β: a historical perspective. Cold Spring Harb Perspect Biol. 2016;8(7):a021865. | ||
Tucker RF, Shipley GD, Moses HL, Holley RW. Growth inhibitor from BSC-1 cells closely related to platelet type β transforming growth factor. Science. 1984;226(4675):705–707. | ||
Roberts AB, Wakefield LM. The two faces of transforming growth factor β in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100(15):8621–8623. | ||
Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–630. | ||
Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19(23):2783–2810. | ||
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. | ||
Daniel CW, Robinson SD. Regulation of mammary growth and function by TGF-β. Mol Reprod Dev. 1992;32(2):145–151. | ||
Silberstein GB, Daniel CW. Reversible inhibition of mammary gland growth by transforming growth factor-β. Science. 1987;237(4812):291–293. | ||
Pierce DF Jr, Johnson MD, Matsui Y, et al. Inhibition of mammary duct development but not alveolar outgrowth during pregnancy in transgenic mice expressing active TGF-β1. Genes Dev. 1993;7(12A):2308–2317. | ||
Bottinger EP, Jakubczak JL, Haines DC, Bagnall K, Wakefield LM. Transgenic mice overexpressing a dominant-negative mutant type II transforming growth factor β receptor show enhanced tumorigenesis in the mammary gland and lung in response to the carcinogen 7,12-dimethylbenz-[a]-anthracene. Cancer Res. 1997;57(24):5564–5570. | ||
Ewan KB, Shyamala G, Ravani SA, et al. Latent transforming growth factor-β activation in mammary gland: regulation by ovarian hormones affects ductal and alveolar proliferation. Am J Pathol. 2002;160(6):2081–2093. | ||
Jhappan C, Geiser AG, Kordon EC, et al. Targeting expression of a transforming growth factor β1 transgene to the pregnant mammary gland inhibits alveolar development and lactation. EMBO J. 1993;12(5):1835–1845. | ||
Kordon EC, McKnight RA, Jhappan C, Hennighausen L, Merlino G, Smith GH. Ectopic TGFβ1 expression in the secretory mammary epithelium induces early senescence of the epithelial stem cell population. Dev Biol. 1995;168(1):47–61. | ||
Robinson SD, Roberts AB, Daniel CW. TGFβ suppresses casein synthesis in mouse mammary explants and may play a role in controlling milk levels during pregnancy. J Cell Biol. 1993;120(1):245–251. | ||
Strange R, Li F, Saurer S, Burkhardt A, Friis RR. Apoptotic cell death and tissue remodelling during mouse mammary gland involution. Development. 1992;115(1):49–58. | ||
Bierie B, Gorska AE, Stover DG, Moses HL. TGF-β promotes cell death and suppresses lactation during the second stage of mammary involution. J Cell Physiol. 2009;219(1):57–68. | ||
Welch DR, Fabra A, Nakajima M. Transforming growth factor β stimulates mammary adenocarcinoma cell invasion and metastatic potential. Proc Natl Acad Sci U S A. 1990;87(19):7678–7682. | ||
Dalal BI, Keown PA, Greenberg AH. Immunocytochemical localization of secreted transforming growth factor-β1 to the advancing edges of primary tumors and to lymph node metastases of human mammary carcinoma. Am J Pathol. 1993;143(2):381–389. | ||
Xie W, Mertens JC, Reiss DJ, et al. Alterations of Smad signaling in human breast carcinoma are associated with poor outcome: a tissue microarray study. Cancer Res. 2002;62(2):497–505. | ||
Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E. TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996;10(19):2462–2477. | ||
Seton-Rogers SE, Lu Y, Hines LM, et al. Cooperation of the ErbB2 receptor and transforming growth factor β in induction of migration and invasion in mammary epithelial cells. Proc Natl Acad Sci U S A. 2004;101(5):1257–1262. | ||
Muraoka RS, Dumont N, Ritter CA, et al. Blockade of TGF-β inhibits mammary tumor cell viability, migration, and metastases. J Clin Invest. 2002;109(12):1551–1559. | ||
Muraoka RS, Koh Y, Roebuck LR, et al. Increased malignancy of Neu-induced mammary tumors overexpressing active transforming growth factor β1. Mol Cell Biol. 2003;23(23):8691–8703. | ||
Siegel PM, Shu W, Cardiff RD, Muller WJ, Massagué J. Transforming growth factor β signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A. 2003;100(14):8430–8435. | ||
Tan EJ, Thuault S, Caja L, Carletti T, Heldin CH, Moustakas A. Regulation of transcription factor Twist expression by the DNA architectural protein high mobility group A2 during epithelial-to-mesenchymal transition. J Biol Chem. 2012;287(10):7134–7145. | ||
Thuault S, Tan EJ, Peinado H, Cano A, Heldin CH, Moustakas A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J Biol Chem. 2008;283(48):33437–33446. | ||
Blanco MJ, Moreno-Bueno G, Sarrio D, et al. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene. 2002;21(20):3241–3246. | ||
Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. | ||
Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601. | ||
Gulei D, Mehterov N, Ling H, Stanta G, Braicu C, Berindan-Neagoe I. The “good-cop bad-cop” TGF-β role in breast cancer modulated by non-coding RNAs. Biochim Biophys Acta. 2017;1861(7):1661–1675. | ||
Bracken CP, Gregory PA, Kolesnikoff N, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68(19):7846–7854. | ||
Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283(22):14910–14914. | ||
Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22(7):894–907. | ||
Perdigao-Henriques R, Petrocca F, Altschuler G, et al. miR-200 promotes the mesenchymal to epithelial transition by suppressing multiple members of the Zeb2 and Snail1 transcriptional repressor complexes. Oncogene. 2016;35(2):158–172. | ||
Kim NH, Kim HS, Li XY, et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol. 2011;195(3):417–433. | ||
Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21–45. | ||
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. | ||
Shipitsin M, Campbell LL, Argani P, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11(3):259–273. | ||
Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. | ||
Scheel C, Eaton EN, Li SH, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145(6):926–940. | ||
Bruna A, Greenwood W, Le Quesne J, et al. TGFβ induces the formation of tumour-initiating cells in claudinlow breast cancer. Nat Commun. 2012;3:1055. | ||
Sato M, Kadota M, Tang B, et al. An integrated genomic approach identifies persistent tumor suppressive effects of transforming growth factor-β in human breast cancer. Breast Cancer Res. 2014;16(3):R57. | ||
Vidakovic AT, Rueda OM, Vervoort SJ, et al. Context-specific effects of TGF-β/SMAD3 in cancer are modulated by the epigenome. Cell Rep. 2015;13(11):2480–2490. | ||
Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790–811. | ||
Salnikov AV, Roswall P, Sundberg C, Gardner H, Heldin NE, Rubin K. Inhibition of TGF-β modulates macrophages and vessel maturation in parallel to a lowering of interstitial fluid pressure in experimental carcinoma. Lab Invest. 2005;85(4):512–521. | ||
Herbertz S, Sawyer JS, Stauber AJ, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-β signaling pathway. Drug Des Devel Ther. 2015;9:4479–4499. | ||
Anido J, Saez-Borderias A, Gonzalez-Junca A, et al. TGF-β receptor inhibitors target the CD44high/Id1high glioma-initiating cell population in human glioblastoma. Cancer Cell. 2010;18(6):655–668. | ||
Bhola NE, Balko JM, Dugger TC, et al. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123(3):1348–1358. | ||
Bandyopadhyay A, Wang L, Agyin J, et al. Doxorubicin in combination with a small TGFβ inhibitor: a potential novel therapy for metastatic breast cancer in mouse models. PLoS One. 2010;5(4):e10365. | ||
Brunen D, Willems SM, Kellner U, Midgley R, Simon I, Bernards R. TGF-β: an emerging player in drug resistance. Cell Cycle. 2013;12(18):2960–2968. | ||
Fischer KR, Durrans A, Lee S, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472–476. | ||
Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46(5):705–716. | ||
Gilmore TD. The Rel/NF-κB signal transduction pathway: introduction. Oncogene. 1999;18(49):6842–6844. | ||
Module S, Huxford T, Ghosh G. A structural guide to proteins of the NF-κB signaling module. Cold Spring Harb Perspect Biol. 2009;1(3):a000075. | ||
Ghosh G, Van Duyne G, Ghosh S, Sigler PB. Structure of NF-κB p50 homodimer bound to a κB site. Nature. 1995;373(6512):303–310. | ||
Müller CW, Rey FA, Sodeoka M, Verdine GL, Harrison SC. Structure of the NF-kappa B p50 homodimer bound to DNA. Nature. 1995;373(6512):311–317. | ||
Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature. 1998;391(6665):410–413. | ||
May M, Ghosh S. Rel/NF-κB and IκB proteins: an overview. Cancer Biol. 1997;8(2):63–73. | ||
Chen F, Ghosh G. Regulation of DNA binding by Rel/NF-κB transcription factors: structural views. Oncogene. 1999;18(49):6845–6852. | ||
Chen Z, Parent L, Maniatis T. Site-specific phosphorylation of IκBα a by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84(6):853–862. | ||
Yamaoka S, Courtois G, Bessia C, et al. Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell. 1998;93(7):1231–1240. | ||
Yaron A, Gonen H, Alkalay I, et al. Inhibition of NF-κB cellular function via specific targeting of the IκB-ubiquitin ligase. EMBO J. 1997;16(21):6486–6494. | ||
Yaron A, Hatzubai A, Davis M, et al. Identification of the receptor component of the IκBa-ubiquitin ligase. Nature. 1998;396(6711):590–594. | ||
Ben-Neriah Y. Regulatory functions of ubiquitination in the immune system. Nat Immunol. 2002;3(1):20–26. | ||
Sheppard K, Rose D, Haque Z, et al. Transcriptional activation by NF-κB requires multiple coactivators. Mol Cell Biol. 1999;19(9):6367–6378. | ||
Sun SC. Non-canonical NF-κB signaling pathway. Cell Res. 2010;21(1):71–85. | ||
Xiao G, Harhaj E, Sun SC. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol Cell. 2001;7(2):401–409. | ||
Mukherjee SP, Behar M, Birnbaum HA, Hoffmann A, Wright PE, Ghosh G. Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-κB-driven transcription. PLoS Biol. 2013;11(9):e1001647. | ||
Brantley DM, Yull FE, Muraoka RS, Hicks DJ, Cook CM, Kerr LD. Dynamic expression and activity of NF-κB during post-natal mammary gland morphogenesis. Mech Dev. 2000;97(1–2):149–155. | ||
Geymayer S, Doppler W. Activation of NF-κB p50/p65 is regulated in the developing mammary gland and inhibits STAT5-mediated β-casein gene expression. FASEB J. 2000;14(9):1159–1170. | ||
Clarkson RW, Heeley JL, Chapman R, et al. NF-κB inhibits apoptosis in murine mammary epithelia. J Biol Chem. 2000;275(17):12737–12742. | ||
Demicco E, Kavanagh K, Romieu R, et al. RelB/p52 NF-κB complexes rescue an early delay in mammary gland development in transgenic mice with targeted superrepressor IκB-α expression and promote carcinogenesis of the mammary gland. Mol Cell Biol. 2005;25(22):10136–10147. | ||
Connelly L, Robinson C, Chont M, et al. A transgenic model reveals important roles for the NF-κB alternative pathway (p100/p52) in mammary development and links to tumorigenesis. J Biol Chem. 2007;282(13):10028–10035. | ||
Brantley D, Chen C, Muraoka R, et al. Nuclear factor-κB (NF-κB) regulates proliferation and branching in mouse mammary epithelium. Mol Biol Cell. 2001;12(5):1445–1455. | ||
Cao Y, Bonizzi G, Seagroves T, et al. IKKα provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell. 2001;107(6):763–775. | ||
Baxter FO. IKK/2 induces TWEAK and apoptosis in mammary epithelial cells. Development. 2006;133(17):3485–3494. | ||
Connelly L, Barham W, Onishko HM, et al. Inhibition of NF-κB activity in mammary epithelium increases tumor latency and decreases tumor burden. Oncogene. 2011;30(12):1402–1412. | ||
Torres L, Serna E, Bosch A, et al. NF-κB as node for signal amplification during weaning. Cell Physiol Biochem. 2011;28(5):833–846. | ||
Zaragozá R, Miralles VJ, Rus AD, et al. Weaning induces NOS-2 expression through NF-κB modulation in the lactating mammary gland: importance of GSH. Biochem J. 2005;391(Pt 3):581–588. | ||
Perkins N. NF-κB: tumor promoter or suppressor? Trends Cell Biol. 2004;14(2):64–69. | ||
Karin M, Cao Y, Greten FR, Li ZW. NF-κB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–310. | ||
Rocha S, Martin AM, Meek DW, Neil D, Perkins ND. P53 represses cyclin D1 transcription through down regulation of Bcl-3 and inducing increased association of the p52 NF-κB subunit with histone deacetylase 1. Mol Cell Biol. 2003;23(13):4713–4727. | ||
Kim DW, Sovak MA, Zanieski G, et al. Activation of NF-κB/Rel occurs early during neoplastic transformation of mammary cells. Carcinogenesis. 2000;21(5):871–879. | ||
Pratt MA, Tibbo E, Robertson SJ, et al. The canonical NF-κB pathway is required for formation of luminal mammary neoplasias and is activated in the mammary progenitor population. Oncogene. 2009;28(30):2710–2722. | ||
Nakshatri H, Bhat P, Martin D, Goutlet R, Sledge G. Constitutive activation of NF-κB during progression of breast cancer to hormone-independent growth. Mol Cell Biol. 1997;17(7):3629–3639. | ||
Sovak M, Bellas R, Kim D, et al. Aberrant nuclear factor-κB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 1997;100(12):2952–2960. | ||
Cogswell PC, Guttridge DC, Funkhouser WK. Selective activation of NF-κB subunits in human breast cancer: potential roles for NF-κB2/p52 and for Bcl-3. Oncogene. 2000;19(9):1123–1131. | ||
Zhou Y, Eppenberger-Castori S, Eppenberger U, Benz C. The NFκB pathway and endocrine-resistant breast cancer. Endocr Relat Cancer. 2005;12 Suppl 1:S37–S46. | ||
Hou MF, Lin SB, Yuan SS, et al. The clinical significance between activation of nuclear factor κB transcription factor and overexpression of HER-2/neu oncoprotein in Taiwanese patients with breast cancer. Clin Chim Acta. 2003;334(1–2):137–144. | ||
Biswas DK, Shi Q, Baily S, et al. NF-κB activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci U S A. 2004;101(27):10137–10142. | ||
Liu H, Lee E, Gajdos C, et al. Apoptotic action of 17β-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst. 2003;95(21):1586–1597. | ||
Boehm J, Zhao J, Yao J, Kim S. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129(6):1065–1079. | ||
Hutti JE, Shen RR, Abbott DW, et al. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKε promotes cell transformation. Mol Cell. 2010;34(4):461–472. | ||
Park B, Zhang H, Zeng Q, et al. NF-κB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nat Med. 2007;13(1):62–69. | ||
Huber MA, Azoitei N, Baumann B, et al. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114(4):569–581. | ||
Turner NC, Reis-Filho JS. Basal-like breast cancer and the BRCA1 phenotype. Oncogene. 2006;25(43):5846–5853. | ||
Shehata M, Teschendorff A, Sharp G, et al. Phenotypic and functional characterisation of the luminal cell hierarchy of the mammary gland. Breast Cancer Res. 2012;14(5):R134. | ||
Sau A, Lau R, Cabrita MA, et al. Persistent activation of NF-κB in BRCA1-deficient mammary progenitors drives aberrant proliferation and accumulation of DNA damage. Cell Stem Cell. 2016;19(1):52–65. | ||
Shostak K, Chariot A. NF-κB, stem cells and breast cancer: the links get stronger. Breast Cancer Res. 2011;13(4):214. | ||
Smith SM, Lyu YL, Cai L. NF-κB affects proliferation and invasiveness of breast cancer cells by regulating CD44 expression. PLoS One. 2014;9(9):e106966. | ||
Yeo SK, French R, Spada F, Clarkson R. Opposing roles of NFκB2 gene products p100 and p52 in the regulation of breast cancer stem cells. Breast Cancer Res Treat. 2017;162(3):465–477. | ||
Godwin P, Baird AM, Heavey S, Barr MP, O’Byrne KJ, Gately K. Targeting nuclear factor-κB to overcome resistance to chemotherapy. Front Oncol. 2013;3:120. | ||
Ahmed KM, Cao N, Li JJ. HER-2 and NF-κB as the targets for therapy-resistant breast cancer. Anticancer Res. 2006;26(6B):4235–4243. | ||
Jiang Z, Liu JC, Chung PE, Egan SE, Zacksenhaus E. Targeting HER2+ breast cancer: the TBK1/IKKε axis. Oncoscience. 2014;1(2):2–4. | ||
Tapia MA, González-Navarrete I, Dalmases A, et al. Inhibition of the canonical IKK/NFκB pathway sensitizes human cancer cells to doxorubicin. Cell Cycle. 2007;6(18):2284–2292. | ||
Gilmore T, Herscovitch M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene. 2006;25(51):6887–6899. | ||
Orlowski RZ, Dees EC. The role of the ubiquitination-proteasome pathway in breast cancer: applying drugs that affect the ubiquitin-proteasome pathway to the therapy of breast cancer. Breast Cancer Res. 2003;5(1):1–7. | ||
Awada A, Albanell J, Canney PA, et al. Bortezomib/docetaxel combination therapy in patients with anthracycline-pretreated advanced/metastatic breast cancer: a phase I/II dose-escalation study. Br J Cancer. 2008;98(9):1500–1507. | ||
Wakefield A. Role of Bcl3 in the normal and neoplastic mammary gland. 2011. Available from: http://orca.cf.ac.uk/55135. Accessed October 12, 2017. | ||
Björnström L, Sjöberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19(4):833–842. | ||
Roy SS, Vadlamudi RK. Role of estrogen receptor signaling in breast cancer metastasis. Int J Breast Cancer. 2012;2012:654698. | ||
Li Y, Wu Y, Abbatiello T, et al. Slug contributes to cancer progression by direct regulation of ERα signaling pathway. Int J Oncol. 2015;46(4):1461–1472. | ||
Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9(2):180–186. | ||
Chung JY, Park YC, Ye H, Wu H. All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci. 2002;115(Pt 4):679–688. | ||
Park S, Zhao D, Hatanpaa KJ, et al. RIP1 activates PI3K-Akt via a dual mechanism involving NF-κB-mediated inhibition of the mTOR-S6K-IRS1 negative feedback loop and down-regulation of PTEN. Cancer Res. 2009;69(10):4107–4111. | ||
Lee S, Andrieu C, Saltel F, et al. IκB kinase phosphorylates Dok1 serines in response to TNF, IL-1, or radiation. Proc Natl Acad Sci U S A. 2004;101(50):17416–17421. | ||
Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117(8):1281–1283. | ||
Band AM, Laiho M. Crosstalk of TGF-β and estrogen receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2011;16(2):109–115. | ||
Kamdje AH, Etet PF, Vecchio L, et al. New targeted therapies for breast cancer: a focus on tumor microenvironmental signals and chemoresistant breast cancers. World J Clin cases. 2014;2(12):769–786. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.