")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 8
The Combination Immunotherapy of TLR9 Agonist and OX40 Agonist via Intratumoural Injection for Hepatocellular Carcinoma
Authors Zhou Z, Lin L, An Y, Zhan M, Chen Y, Cai M, Zhu X, Lu L, Zhu K
Received 28 January 2021
Accepted for publication 9 April 2021
Published 8 June 2021 Volume 2021:8 Pages 529—543
DOI https://doi.org/10.2147/JHC.S301375
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmed Kaseb
Zhimei Zhou,1,* Liteng Lin,1,* Yongcheng An,1,* Meixiao Zhan,2 Ye Chen,1 Mingyue Cai,1 Xiaojing Zhu,1 Ligong Lu,2 Kangshun Zhu1
1Laboratory of Interventional Radiology, Department of Minimally Invasive Interventional Radiology and Department of Radiology, The Second Affiliated Hospital, Guangzhou Medical University, Guangzhou, 510260, People’s Republic of China; 2Zhuhai Interventional Medical Center, Zhuhai Precision Medical Center, Zhuhai People’s Hospital, Zhuhai Hospital Affiliated with Jinan University, Zhuhai, Guangdong Province, 519000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Kangshun Zhu
Laboratory of Interventional Radiology, Department of Minimally Invasive Interventional Radiology and Department of Radiology, The Second Affiliated Hospital, Guangzhou Medical University, 250 Changgang East Road, Guangzhou 510260, People’s Republic of China
Tel +86-20-34152264
Fax + 6-20-34152281
Email [email protected]
Ligong Lu
Zhuhai Interventional Medical Center, Zhuhai Precision Medical Center, Zhuhai People’s Hospital, Zhuhai Hospital Affiliated with Jinan University, 79 Kangning Road, Zhuhai, Guangdong Province 519000, People’s Republic of China
Tel +86 7-56-2222569
Fax +86 7-56-2162086
Email [email protected]
Background: The response rate of immunotherapy via immune checkpoint blockade in hepatocellular carcinoma (HCC) is limited due to multiple immune evasion mechanisms. OX40 is a T cell co-stimulating molecule which suppresses the cancer immune evasion by activating effector T cells (Teffs) and counteracting regulatory T cells (Tregs). TLR9 belongs to the toll-like receptor superfamily which promotes tumour antigen presentation by stimulating the maturation of dendritic cells. Though the combination immunotherapy of TLR9 agonist (CpG) and OX40 agonist (anti-OX40 antibody) has shown encouraging efficacy in various tumours, its effect on HCC remains unknown.
Materials and Methods: Orthotopic and ectopic HCC models were constructed by implanting Hepa1-6 cells at different body sites of the mice. Immune agents were administrated via three ways, including intratumoural injection into one site of the tumour, intraperitoneal injection, and subcutaneous injection. The anti-tumour immune response was evaluated by the regression of both the local treated tumour and distant untreated tumour. The ratio and function of CD4+ T cells, CD8+ T cells, Tregs and myeloid-derived suppressor cells (MDSCs) were analyzed by flow cytometry.
Results: CpG via intratumoural injection remarkably upregulated the weakly expressed OX40 of intratumoural T cells. The combination immunotherapy of CpG and anti-OX40 antibody via intratumoural injection significantly inhibited the growth of local and distant tumours, and also effectively prevented their recurrence. Excitingly, drug administration via intratumoural injection, rather than via intraperitoneal or subcutaneous injections, induced potent anti-tumour immune response. Furthermore, we demonstrated that the combination immunotherapy promoted CD8+ and CD4+ T cells, and inhibited Tregs and myeloid-derived suppressor cells, contributing to the effective inhibition on HCC. Noteworthily, the combination immunotherapy also induced an immune memory response.
Conclusion: The intratumoural administration of combined CpG and anti-OX40 antibody serves as a promising immunotherapy against HCC.
Keywords: hepatocellular carcinoma, immunotherapy, combination therapy, toll-like receptor, intratumoural administration
Introduction
Hepatocellular carcinoma (HCC) is the fifth most frequent tumour and second leading cause of cancer mortality worldwide.1 Despite the application of multimodal treatments involving surgical resection, liver transplantation, radiotherapy, local ablation and chemotherapy, the 5-year survival rates of HCC patients remain very low (18%).2 Currently, immune checkpoint inhibitor (ICI) therapy, particularly blocking the programmed cell death-1 (PD-1)/programmed cell death ligand-1 (PD-L1) pathway, has emerged as a promising therapeutic breakthrough for various tumours. However, enthusiasm around anti-PD-1 therapy for HCC is now becoming tempered with a relatively low objective response rate (15–20%),3 which may primarily be attributable to the unique hepatic microenvironment where immune tolerance develops and merely blocking the immune checkpoint pathway is insufficient to overcome HCC immune evasion.4 HCC immune evasion is a complex pathophysiological process orchestrated by dendritic cells (DCs) with disabled antigen presentation, immunosuppressive regulatory T cells (Tregs), and exhausted effector T cells (Teffs).5–9 In this context, development of innovative immunotherapy that can effectively conquer immune evasion remains an urgent challenge for HCC treatment.
Aside from immune checkpoint blockade, strengthening immune stimulation signals to improve the systemic anti-tumour immune response is emerging as another appealing therapeutic strategy for tumour immunotherapy.10,11 In particular, compelling evidence in both laboratory and clinical studies is uncovering the attractive role of the OX40/OX40L axis as an immune stimulation signal for new anti-tumour drug development. OX40, a T cell co-stimulating molecule belonging to the tumour necrosis factor receptor superfamily, has been demonstrated to interact with its ligand (ie, OX40L) to constitute a central component of the fight against immune evasion of various tumours via facilitating the activation and survival of Teffs, counteracting the immunosuppression of Tregs and promoting the generation of memory T cells.12–20 To date, the effect of activating the OX40/OX40L axis (eg, using agonistic anti-OX40 monoclonal antibody to activate OX40) against HCC immune evasion is unknown. Additionally, we observed that OX40 expression was mainly restricted to the tumour-infiltrating CD4+ T cells of HCC at a relatively low level, such an insufficient OX40 expression has been demonstrated to account for the poor therapeutic outcome of anti-OX40 monoclonal antibody (mAb) for the melanoma.21 Therefore, the development of combination strategies facilitating OX40 expression on T cells has become essential to explore the potential of activating the OX40/OX40L axis to conquer immune evasion for HCC treatment.
The combination application of anti-OX40 mAb and TLR9 agonist (cytosine-phosphate-guanine oligodeoxynucleotide, CpG) has shown encouraging therapeutic efficacy in various tumours, its effect on HCC remains unknown. TLR9, a member of the toll-like receptor (TLR) superfamily that recognizes pathogen-associated molecular patterns, can activate innate and adaptive immune responses against cancer cells.22–25 A recent study demonstrated that activating TLR9 signalling by its agonist (ie, CpG) can significantly upregulate OX40 expression on T cells in lymphoma.26,27 Importantly, such a phenomenon in HCC was also confirmed in our preliminary investigation in the present study. Thus, we hypothesized that combination therapy activating both the OX40/OX40L axis and TLR9 signalling could synergistically enhance anti-tumour immunity. Moreover, TLR9 signalling is known to act as a key stimulator of DC maturation to promote tumour antigen presentation, which is thought to be another pivotal way to overcome HCC immune evasion28–30 and further supports the superior anti-tumour effect of combination therapy. In this study, experiments using orthotopic and ectopic HCC models were performed to evaluate the efficacy of a combination therapy of CpG and anti-OX40 mAb and to explore the mechanism of its anti-tumour effects.
Materials and Methods
Reagents
CpG ODN 1826 (5ʹ-tccatgacgttcctgacgtt-3ʹ) and isotype control were purchased from InvivoGen (San Diego, CA, USA). Anti-OX40 (CD134) monoclonal antibody (clone OX86) and isotype control were purchased from BioXCell (Lebanon, NH, USA). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin solution, phosphate-buffered saline (PBS) and TRIzol reagent were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). The Matrigel was purchased from Corning (Corning, NY, USA). The TUNEL Apoptosis Detection Kit was purchased from KeyGen BioTECH (Nanjing, Jiangsu, China). The T Cell Isolation Kit was purchased from Miltenyi Biotec (Bergisch Gladbach, Germany). Antibodies including CD45-PerCP-Cy5.5, CD3e-BV510, CD4-FITC, CD25-PE, OX40-APC, CD8a-FITC, CD11b-BV421, Gr-1-APC, F4/80-PE, CD8a-APC, CD44-PE-Cy7, CD62L-PE, IFN-γ-PE, and FoxP3-eFluor450 were purchased from BD Biosciences (San Jose, CA, USA) and Biolegend (San Diego, CA, USA).
Cell Lines and Mice
The Hepa1-6 hepatoma cell line was generously provided by Dr Zide Chen (Southern Medical University, Guangzhou, China). The B16-F10 melanoma cell line was generously provided by Dr Zecong Xiao (Guangzhou Medical University, Guangzhou, China). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% foetal bovine serum (FBS) and 100 IU/mL penicillin-streptomycin at 37°C and 5% CO2.
Male C57BL/6 mice and nude mice (6–10 weeks old) were purchased from Guangdong Medical Laboratory Animal Center. Mice were bred and kept in the Animal Facility of the Second Affiliated Hospital of Guangzhou Medical University. The use of the above cell lines and all animal experiments were approved by the Ethics Committee. This study followed the Laboratory animal—Guideline for ethical review of animal welfare (GB/T 35892–2018).
Tumour Model and Treatment
Subcutaneous hepatoma model: Hepa1-6 cells (10×106) were injected subcutaneously into the left or bilateral abdomen of C57BL/6 mice or nude mice.
Orthotopic and Subcutaneous Hepatoma Model
The orthotopic hepatoma model was established as described previously.31 Briefly, after performing a small incision of the midline of the mouse abdomen, Hepa1-6 cells (10×106) mixed with Matrigel (Corning) were injected under the capsule of the median lobe of the liver followed by placing a Gelfoam on the needle track for 5 min. The muscle and skin layer of the abdominal wall were then closed. Next, the same mouse was subcutaneously injected with Hepa1-6 cells (10×106) into the left abdomen.
I Hepa1-6 cells (10×106/site) were subcutaneously implanted on both sides of the abdomen, and B16-F10 cells (0.1×106) were subcutaneously implanted on the right armpit. II B16-F10 cells (0.1×106/site) were subcutaneously implanted on both sides of the abdomen, and 10×106 Hepa1-6 cells were subcutaneously implanted on the right armpit. III Hepa1-6 cells (7×106) and B16-F10 cells (0.05×106) were mixed and subcutaneously implanted in the left abdomen, B16-F10 cells (0.1×106) were subcutaneously implanted in the right abdomen, and Hepa1-6 cells (10×106) were subcutaneously implanted in the right armpit.
When the diameter of the subcutaneous tumour reached 5–7 mm, the mice were randomly divided into different experimental groups. The left subcutaneous tumour was injected with 50 µg CpG, 25 µg anti-OX40 mAb, or 50 µg CpG plus 25 µg anti-OX40 mAb in a total volume of 50 µL every other day for a total of 4 injections. Subcutaneous tumour size (volume = length × width × height) was continuously monitored by a calliper. When the diameter of the tumour reached 15 mm, the mice were sacrificed in accordance with the animal ethics protocol.
In vivo Immune Memory Response Assay
Ninety days after the disappearance of the tumour, mice in the combined treatment group were retransplanted with homogenous Hepa1-6 cells (10×106) on the left abdomen. Age‑matched naive C57BL/6 mice subcutaneously transplanted with Hepa1-6 cells (10×106) on the left abdomen were used as the control group. The growth of the subcutaneous left tumour was continuously monitored by a calliper.
In vivo MRI Scan
Before the beginning of treatment and on day 16 after the first treatment, MRI scans were performed on the orthotopic and left subcutaneous HCC model. Orthotopic tumour size (volume = length × width × height) was detected by MRI with a clinical 3.0 T system (Intera; Philips Medical Systems, Best, Netherlands) with a 50-mm linearly polarized birdcage radiofrequency mouse coil (Chenguang Medical Technologies Co., Shanghai, China). Axial images were obtained using a two-dimensional turbo spin echo T2-weighted sequence (repetition time=3000 ms; echo time=80 ms; flip angle=90°; number of signal averages=3) and a two-dimensional turbo spin-echo T1-weighted sequence (pulse repetition time=500 ms; echo time=33 ms; flip angle=90°; number of signal averages=3). The other parameters for these sequences were as follows: field of view=35 mm; matrix=175×175; section thickness=1.0 mm; no intersection gap.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labelling (TUNEL) Assay
Mice with Hepa1-6 orthotopic tumours were sacrificed after immunotherapy by injecting left subcutaneous tumours. Tumour tissues were fixed and embedded in optimum cutting temperature compound (OCT) and frozen at −80°C. The processed tumour tissues were cut into 5 μm sections and then subjected to in situ TUNEL assays using the TUNEL Apoptosis Detection Kit (KeyGen BioTECH) according to the manufacturer’s protocol. The images were captured by a fluorescence microscope with a digital camera. The percentage of TUNEL-positive cells (apoptotic cells) was quantified using ImageJ software.
Flow Cytometry
Tumour and spleen tissues were enzymatically or mechanically processed into single cell suspensions, respectively. Red cells of the spleen single cell suspensions were lysed with RBC lysis buffer (Biolegend). Single-cell suspensions were stained with viability dye (Fixable Viability Stain 780, BD Biosciences) followed by blocking Fc receptors and staining for surface antigens. If intranuclear proteins were assayed, cells were fixed and permeabilized using a Transcription Factor Buffer Set (BD Biosciences) and stained for intracellular antigens. Anti-mouse antibodies were as follows: CD45-PerCP-Cy5.5, CD3e-BV510, CD4-FITC, CD8a-FITC, CD25-PE, OX40-APC, OX40-PE, CD8a-FITC, CD11b-BV421, Gr-1-APC, F4/80-PE, CD8a-APC, CD4-APC, CD44-PE-Cy7, CD62L-PE, IFN-γ-PE, and FoxP3-eFluor450. Cells were analysed on a Gallios or Navios flow cytometer (Beckman Coulter). Flow cytometric data were analysed with FlowJo X. Dead cells and adhesive cells were excluded first. CD4 T cells were gated as CD45+CD3+CD4+, CD8 T cells were gated as CD45+CD3+CD8+, MDSCs were gated as CD11b+Gr1+, and Treg cells were gated as CD4+CD25+Foxp3+.
Western Blot Analysis
Different doses of CpG were injected into the Hepa1-6 subcutaneous tumours of mice. Tumour tissues were collected from mice 48 hours after treatment, and OX40 protein was detected using antibodies against OX40 (OX86, BioXCell) by Western blotting. The specific method of Western blot analysis was based on a previous study from our group.32,33
IFN-γ Production Assay
Spleens from Hepa1-6 tumour-bearing mice were collected on day 7 after the different immune therapies and mechanically processed into single cell suspensions, whose red cells were lysed with RBC lysis buffer (Biolegend). Splenocytes in each treatment group were cocultured with either media, 1×106 irradiated B16-F10 cells (unrelated control tumour), or Hepa1-6 (homologous tumour) for 24 hours at 37°C and 5% CO2 in the presence of 0.5 µg of anti-mouse CD28 mAb (BD Pharmingen). Monensin (GolgiStop; BD Biosciences) was supplied for the last 6 hours. Next, splenocytes were stained for cell surface antigens (CD45, CD3e, CD8a, and CD44). Then, splenocytes were fixed and permeabilized by the BD Cytofix/Cytoperm Plus Kit and stained for intracellular IFN-γ. The expression of intracellular IFN-γ was assayed by flow cytometry.
Adoptive Immunity Transfer Experiment
Spleens were harvested from Hepa1-6-bearing mice on day 5 after the different immune therapies, at a time when the tumours of combination immunotherapy had regressed. CD3, CD4 or CD8 T cells were negatively selected and collected from single cell suspensions of spleen tissues using a relevant T Cell Isolation Kit (Miltenyi Biotec). The purity was confirmed to be 95% by flow cytometry, and the viability was above 90% by 0.4% Trypan blue (Gibco) staining. These T cells were mixed with Hepa1-6 cells at a ratio of 100:1 and cotransplanted subcutaneously into naive C57BL/6 mice. The growth of tumours in the recipient hosts was monitored.
Statistical Analysis
All results are expressed as mean ± standard deviation (SD). Statistical differences among multiple groups of data were performed using one-way ANOVA followed by Bonferroni correction and the differences between two groups were performed using unpaired Student’s t-test. Survival curves were plotted using the Kaplan-Meier method and analyzed using the Log rank test. All statistical tests were performed using GraphPad Prism 6.0 software (GraphPad Software Inc., USA). P<0.05 was considered statistically significant.
Results
CpG Upregulates the OX40 Expression of Intratumoural CD4+ T Cells in Hepa1-6 Hepatoma
TLR9 is well recognized as an important sensor of the immune system.24 In the present study, we first found that intratumoural injection of CpG oligodeoxynucleotide (the ligand of TLR9) could significantly upregulate expression of the T cell co-stimulating molecule OX40 in HCC tissues, as evidenced by Western blot analysis (Figure 1A). Next, based on previous studies demonstrating that TLR9 signalling can directly promote the proliferation of CD4+ T cells,26,27 we further analysed the effect of CpG on OX40 expression in CD4+ T cells. In mice receiving CpG, flow cytometry analysis showed that the OX40 expression of intratumoural CD4+ T cells increased significantly (Figure 1B). Meanwhile, although CpG showed a mild tendency to enhance the OX40 expression on the intratumoural CD8+ T cells, no statistical significance was obtained (Figure S1). Additionally, we investigated OX40 expression in the CD4+ T cell subpopulation via flow cytometry. As shown in Figure 1C, in mice treated with CpG, intratumoural OX40 expression was remarkably upregulated and was mainly found on effector CD4+ T cells rather than on Treg cells (Figure S2). From these results, we reasoned that CpG could induce the OX40 expression of effector CD4+ T cells in the hepatoma microenvironment, providing a basis for the combined application of CpG and anti-OX40 mAb for HCC therapy.
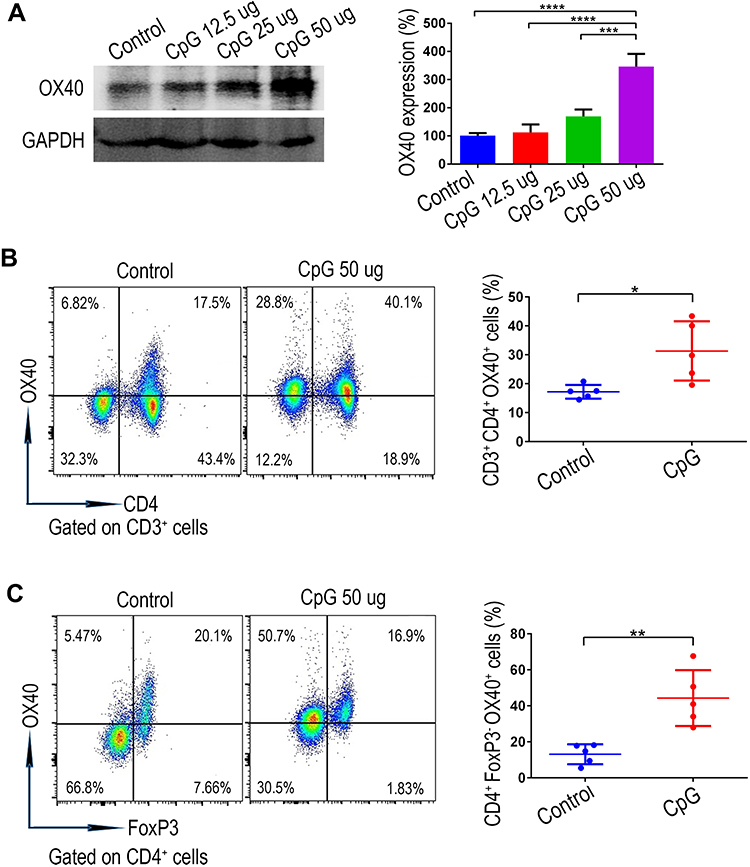 |
Figure 1 The stimulatory effect of CpG on the OX40 expression of intratumoural CD4+ T cells verified using an ectopic liver tumour model. (A) OX40 expression in subcutaneous hepatoma tissues intratumourally injected with different doses of CpG (12.5 µg, 25 µg, 50 µg) was assessed by Western blot analysis (n=3; ***P<0.001, ****P<0.0001). (B) Forty-eight hours after the intratumoural injection of CpG, single cell suspension from the subcutaneous hepatoma tissues was prepared, and the OX40 expression of the CD3+CD4+ T cell subgroup was analyzed by flow cytometry (n=5; *P<0.05). (C) OX40 expression on effector CD4+ T cells (Foxp3-negative) was revealed by flow cytometry (n=5; **P<0.01). |
The Combination of CpG and Anti-OX40 mAb via Intratumoural Injection Achieves Enhanced Immune Responses Against Both Local and Distant Hepa1-6 Hepatoma
The anti-tumour effect of anti-OX40 mAb is limited by insufficient OX40 expression on tumoural T cells.21,34,35 Thus, we hypothesized that CpG treatment might contribute synergistically to the anti-tumour effect of anti-OX40 mAb by increasing OX40 expression on T cells. To verify our hypothesis, mice were subcutaneously inoculated with Hepa1-6 HCC cells on the left and right sides of the abdomen and received different immunotherapies (anti-OX40 mAb, CpG, or the combination of anti-OX40 mAb and CpG) via intratumoural injection only on the left side (Figure 2A). As shown in Figure 2B, neither CpG alone nor anti-OX40 mAb alone prevented tumour growth on either side of the hepatomas. Notably, mice receiving an intratumoural injection with combined CpG and anti-OX40 mAb exhibited significant tumour regression at both sites of the tumours, with tumour-free survival for more than 90 days and long-term survival until the end of the experiment (Figure 2C). Moreover, considering the development of HCC in the unique hepatic environment with immune tolerance, we further evaluated the above findings in an orthotopic HCC model (Figure 2D). We obtained consistent results, demonstrating that intratumoural injection of combined CpG and anti-OX40 mAb induced a remarkably improved immune response against both local and distant Hepa1-6 hepatoma (Figure 2E-G).
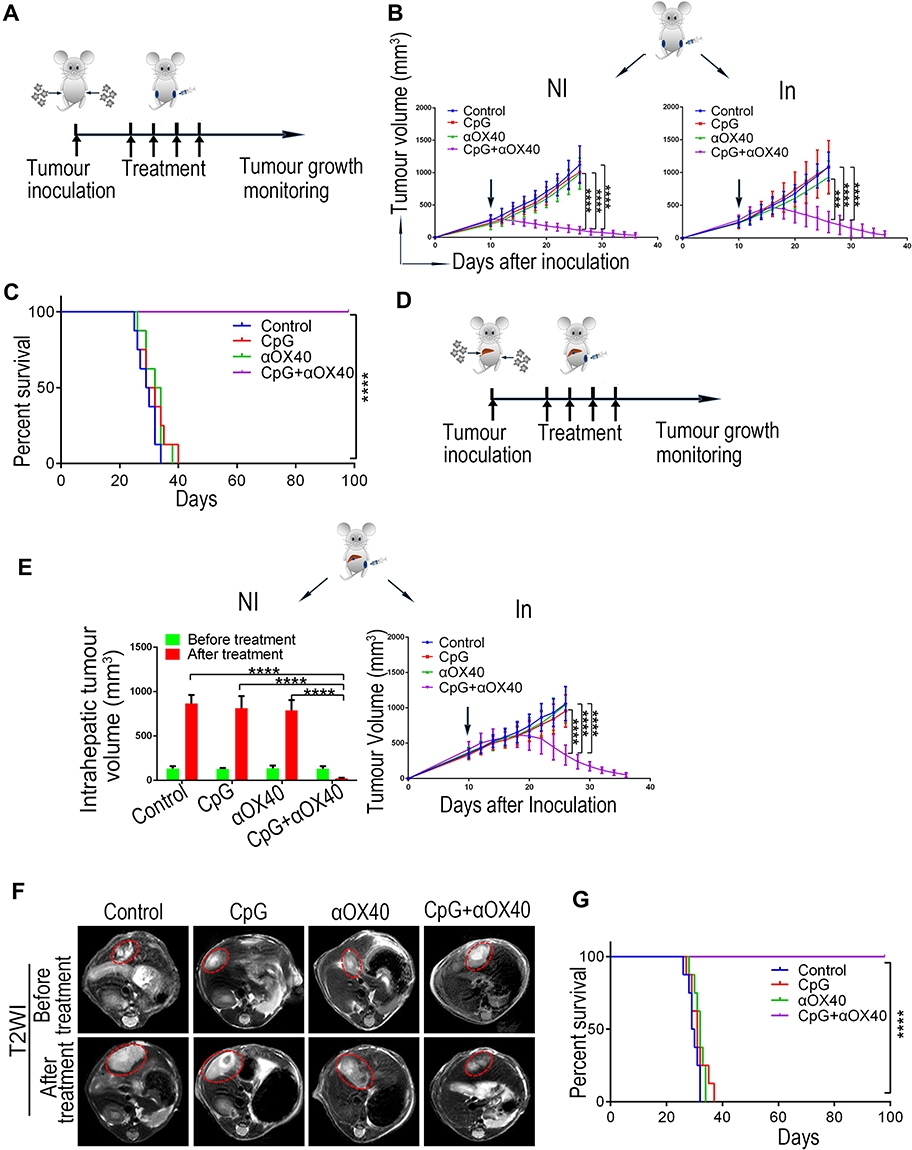 |
Figure 2 The combination of CpG and anti-OX40 mAb via intratumoural injection induces improved immunotherapy against both local and distal Hepa1-6 hepatoma. (A) Schematic diagram of immunotherapy for the ectopic HCC model. C57BL/6 mice were subcutaneously inoculated with Hepa1-6 cells on both sides of the abdomen. When the tumour diameter reached 5–7 mm, the left tumour was injected with different immune drugs (CpG, αOX40 or CpG plus αOX40) every other day for a total of 4 times. The bilateral tumour volumes were continuously measured. (B) Tumour growth of the subcutaneous tumour model. Left: noninjected (NI) tumour volume, right: injected (In) tumour volume; the arrow indicates the beginning of the treatment (n=8; ***P<0.001, ****P<0.0001). (C) Survival curve of the subcutaneous HCC model (n=8; ****P<0.0001). (D) Schematic diagram of immunotherapy for the orthotopic HCC model. C57BL/6 mice were inoculated with Hepa1-6 cells in the subcapsular region of the liver and the subcutaneous site of the left abdomen. The treatment scheme was consistent with (A). (E) Tumour growth of the orthotopic HCC model. Left: the volume of the noninjected (NI) intrahepatic tumour was measured by MRI scan before treatment and on day 16 after the first treatment. Right: the volume of the injected (In) left subcutaneous tumour was continuously measured (n=8; ****P<0.0001). (F) Representative MRI images of the intrahepatic tumours. (G) Survival curve of the orthotopic HCC model (n=8; ****P<0.0001). |
Taken together, these results support our hypothesis that intratumoural injection of CpG and anti-OX40 mAb enhances the local immune response at the tumour site directly receiving immune drugs and also induces a systemic immune response against distal tumours.
The Combined Administration of CpG and Anti-OX40 mAb via Intratumoural Injection Triggers a Tumour-Specific Immune Response
The method of drug administration is known to play an important role in the anti-tumour efficacy of immunotherapy.34,36–38 First, on the basis of the determined synergistic anti-HCC effect of the combined CpG and Anti-OX40 mAb via intratumoural injection, we also investigated the immunotherapy outcome of intraperitoneal drug delivery in the bilateral subcutaneous HCC model (Figure 3A). As shown in Figure 3B, the combination of CpG and anti-OX40 mAb via intraperitoneal administration failed to inhibit tumour growth. And similar results were obtained by the intravenous administration (data not shown). Next, in consideration of the influence of the tumour microenvironment on immunotherapy, we further constructed a unilateral subcutaneous HCC model by inoculating Hepa1-6 cells only in the right abdomen and performed immunotherapy via subcutaneous injection at the site of the tumour-free left abdomen. In contrast to the excellent anti-tumour efficacy of intratumoural drug administration, no clear suppression of tumour growth was observed upon combined immunotherapy via subcutaneous drug delivery.
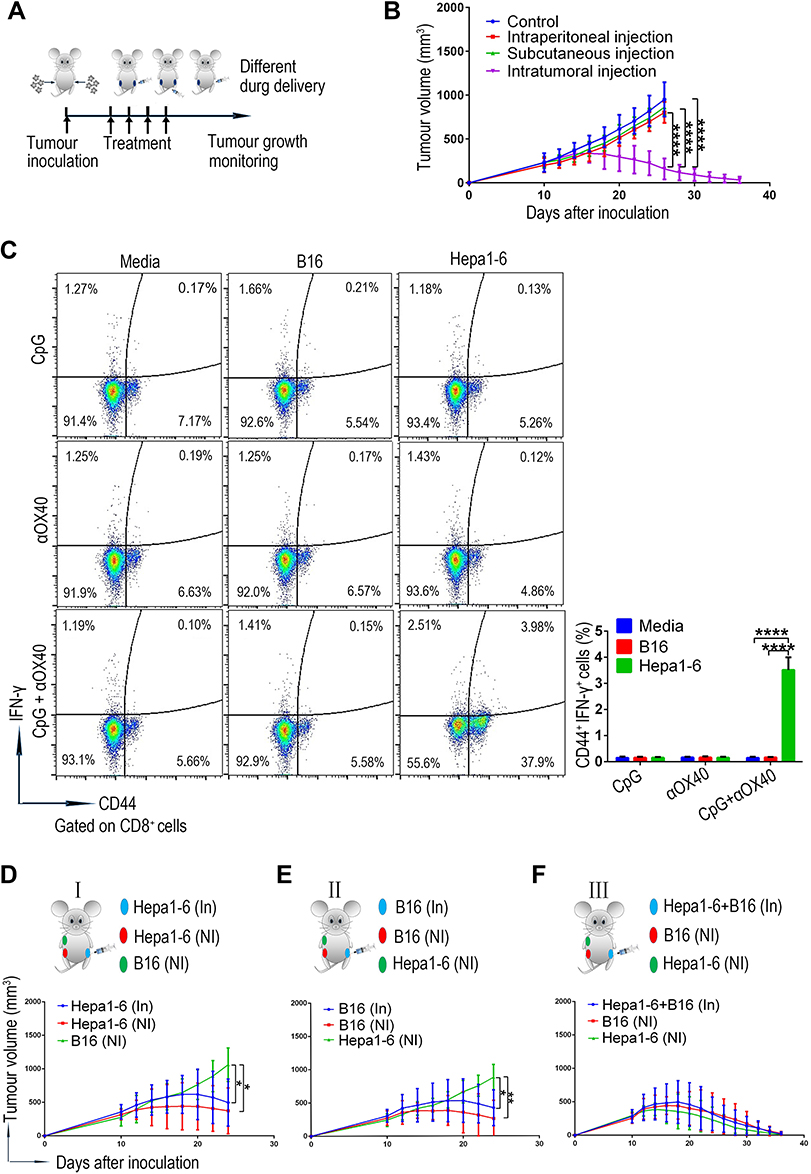 |
Figure 3 The combined CpG and anti-OX40 mAb via intratumoural injection triggers a tumour-specific immune response. (A) Schematic diagram of immunotherapy for the ectopic HCC model inoculated with Hepa1-6 cells. (B) Tumour growth of the subcutaneous HCC model receiving combined CpG and anti-OX40 mAb via different methods of drug delivery. Growth of the right noninjected tumours after treatment was monitored (n=8; ****P<0.0001). (C) On day 7 after treatment, splenocytes derived from ectopic HCC mice receiving different treatments were cocultured with media, B16-F10 cells or Hepa1-6 cells for 24 hours, and the percentage of IFN-γ+CD44+CD8+ T cells was analyzed by flow cytometry (n=5; ****P<0.0001). (D) Tumour growth of the mixed tumour model, with two Hepa1-6 tumours on bilateral abdomen and a B16-F10 tumour on the right armpit (n=8; *P<0.05). (E) Tumour growth of the mixed tumour model, with two B16-F10 tumours on bilateral abdomen and a Hepa1-6 tumour on the right armpit (n=8; *P<0.05, **P<0.01). (F) Tumour growth of the mixed tumour model, with a mixed tumour (Hepa1-6 and B16-F10) on the left abdomen, B16-F10 tumour on the right abdomen and Hepa1-6 tumour on the right armpit. |
The above results provided strong evidence that the systemic immune anti-tumour response of our combined immunotherapy was closely related to the microenvironment of the local tumour. Next, we explored whether such an effect was tumour-specific by evaluating the specific response of T cells via an interferon (IFN)-γ production assay. As evidenced by the flow cytometry results (Figure 3C and Figure S3), the CD8+ T cells, rather than the CD4+ T cells, derived from the mice treated with CpG and OX-40 agonist showed a significantly increased proportion of IFN-γ-positive cells in response to the stimulation of homologous Hepa1-6 cells. Such interesting result indicated that the CpG and OX-40 agonist leaded to an increased anti-tumour response likely by enhancing the role of CD4+ T cells as “helpers” to promote CD8+ T cells, instead of by directly activating the CD4+ T cells to produce IFN-γ. Noteworthily, the tumour-reactive CD8+ T cells in the combination treatment group exerted no response to the stimulation of irrelevant B16-F10 cells, indicating that the systemic immune anti-tumour response of our combined immunotherapy was tumour-specific.
Additionally, we established a mouse model with two types of tumour antigens to further clarify the relationship between the specific anti-tumour effect of our combination therapy and the tumour receiving injection. First, Hepa1-6 cells and B16-F10 cells were subcutaneously inoculated into both sides of the abdomen and the right armpit, respectively (Figure 3D). We observed that the combined treatment with CpG and anti-OX40 mAb on the left Hepa1-6 tumour inhibited the growth of both sides of the two homogenous Hepa1-6 tumours but failed to impede the progression of B16-F10 tumours (Figure 3D). Second, to rule out the interference effect of the graft site of the tumour, we also verified the above findings in mice with an altered tumour distribution and obtained consistent results (Figure 3E). These results suggested that the anti-tumour immune response of the combination therapy was triggered by the tumour at the injection site, whose immunogenicity determined the tumour specificity of the immunotherapy. To further confirm this view, we established a mixed tumour model with mixed tumours (Hepa1-6 and B16-F10) on the left abdomen, B16-F10 tumours on the right abdomen and Hepa1-6 tumours on the right armpit (Figure 3F). As expected, treatment of the mixed tumour via intratumoural injection of CpG and anti-OX40 mAb led to the regression of all the tumours (Figure 3F). Thus, these findings provide clear evidence that the combined administration of CpG and anti-OX40 mAb via intratumoural injection induces a tumour-specific immune response.
The Combined Administration of CpG and Anti-OX40 mAb Synergistically Regulates the Proportion of the Intratumoural Immune Cells
Tumour growth is a complex process involving the mutual interaction between anti-tumour immune factors and immunosuppressive factors in the tumour microenvironment.39 The cellular immune response mediated by T cells plays a leading role in tumour immunity.40 In particular, infiltrating CD8+ T cells behave as key executors via tumour-specific cytotoxicity, while CD4+ T cells act as important “helpers” via their ability to recognize tumour antigens and strengthen CD8+ T cells, collectively facilitating the anti-tumour immune response. In contrast, Tregs and myeloid-derived suppressor cells (MDSCs) are immunosuppressive cells that counteract the anti-tumour immune response in the tumour microenvironment by inhibiting the function of other immune cells.41 Herein, we explored the effects of combination immunotherapy on these immune cells in tumour tissues by flow cytometry. As shown in Figure 4A-D, the tumours in the control group remained relatively immunosuppressive, as evidenced by the lowest proportion of CD8+ and CD4+ T cells, as well as the highest ratio of Tregs and MDSCs. Interestingly, compared to the tumours in the group receiving either CpG or anti-OX40 alone, those in the group receiving combination immunotherapy via intratumoural injection displayed a remarkably increased infiltration of CD8+ and CD4+ T cells and a significantly reduced proportion of Tregs and MDSCs (Figure 4A-D). Consistent with the above findings, the synergistic anti-tumour effect of combined CpG and anti-OX40 mAb was histologically confirmed by a terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labelling (TUNEL) assay, which showed the highest level of tumour necrosis in the group receiving combination immunotherapy (Figure 4E).
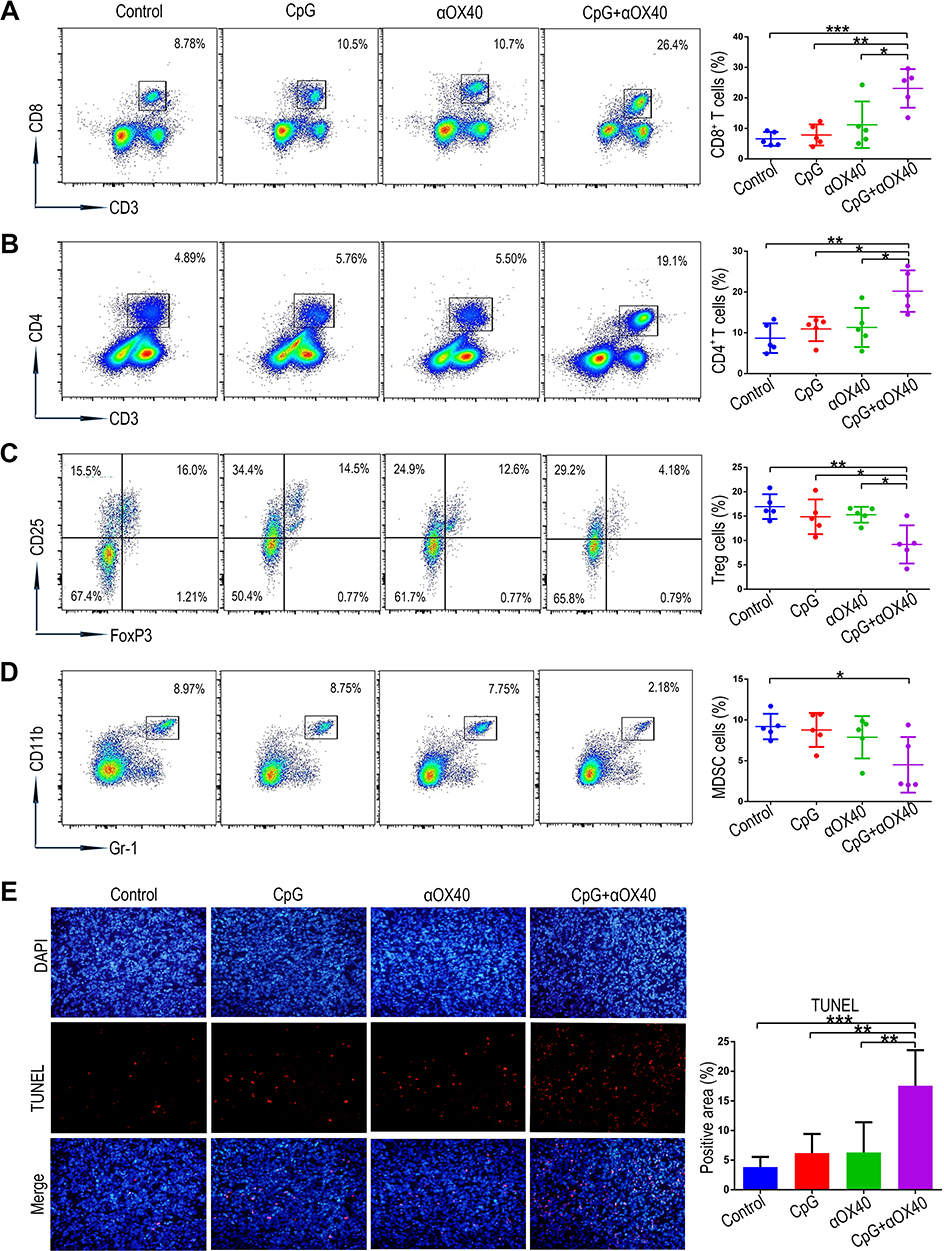 |
Figure 4 The combination immunotherapy regulates the proportion of the intratumoural immune cells and promotes tumour apoptosis. (A-D) Representative flow cytometry quantified the proportion of CD4+ T cells, CD8+ T cells, Tregs and MDSCs in the orthotopic HCC tissues collected from mice on day 16 after the first treatment (n=5; *P<0.05, **P<0.01, ***P<0.001). (E) Tumour apoptosis of orthotopic HCC tissues on day 16 after the first treatment was revealed by TUNEL staining. The location of the cell nucleus was revealed by DAPI staining (n=5; **P<0.01, ***P<0.001). |
The Combined Administration of CpG and Anti-OX40 mAb Exerts T Cell-Dependent Anti-Tumour Effect and Induces a Potent Immune Memory Response to Prevent Tumour Recurrence
Given that the anti-OX40 mAb acts specifically on T cells to strengthen the immune response, we reappraised our combination immunotherapy in nude mice without functional T cells but with abundant B cells to determine whether the anti-tumour efficacy of the combination immunotherapy is dependent on T cells. As expected, the combination therapy was T cell-dependent, as evidenced by its failure to prevent tumour growth in nude mice (Figure 5A). Next, we further explored the anti-tumour effect of different T cell subsets. CD4+ and CD8+ T cells isolated from the spleens of mice with different treatments were mixed with Hepa1-6 tumour cells and then co-transplanted into naive hosts (Figure 5B). As shown in Figure 5C-F, only the combination immunotherapy group including both CD4+ and CD8+ T cells showed a significant anti-tumour effect. These observations suggested that the anti-tumour activity of our combination immunotherapy was T cell-dependent, requiring simultaneous involvement of both CD4+ and CD8+ T cells.
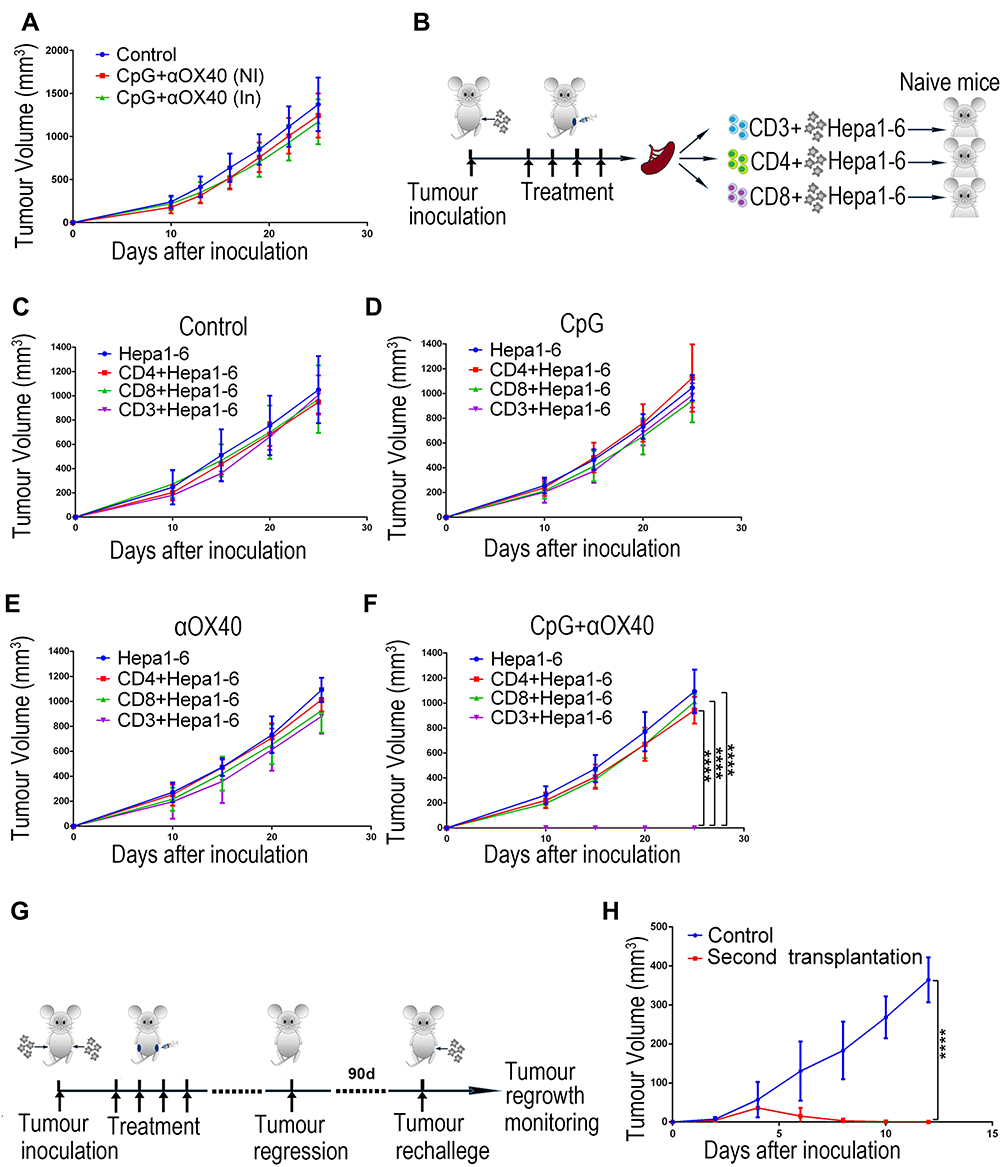 |
Figure 5 The combination immunotherapy exerts T cell-dependent anti-tumour effect and induces effective immune memory response to prevent tumour recurrence. (A) To perform immunotherapy for the ectopic HCC model, Hepa1-6 cells were subcutaneously inoculated into the bilateral abdomen of nude mice, and the left tumours were injected with CpG and anti-OX40 mAb. The tumour growth curves of the nude mice were recorded (NI: non-injected, In: injected; n=8). (B) Schematic diagram of the adoptive transfer experiment. (C-F) Different T cell subsets derived from mice receiving different treatments were mixed with Hepa1-6 cells and subcutaneously implanted into naive C57BL/6 mice. The tumour growth curves of the hosts were recorded (n=6; ****P<0.0001). (G) Schematic diagram of the in vivo immune memory response assay. On day 90 after the primary tumour disappeared, the mice treated with combination immunotherapy were rechallenged with Hepa1-6 cells. (H) The tumour growth curves of the control mice and the mice rechallenged with Hepa1-6 cells. Age-matched mice without any treatment but only transplanted subcutaneously with Hepa1-6 cells were used as controls (n=8; ****P<0.0001). |
Immune memory is a vital component of adaptive immunity that can produce faster and stronger reactivation in response to a secondary attack by the same insult, such as recurrent tumour cells, to protect an organism.42 In order to investigate whether the combination immunotherapy induced effective anti-tumour immune memory, mice receiving the combination immunotherapy were rechallenged by homogenous tumour cells on day 90 after the primary tumour disappeared to determine whether the memory response was sufficient to protect the host from tumour recurrence (Figure 5G). Compared to the control mice that were subcutaneously transplanted with Hepa1-6 cells for the first time, mice that received the combination immunotherapy showed much slower tumour growth. More excitingly, the tumours regressed on day 4 and completely disappeared around day 8 after transplantation in mice that received the combination immunotherapy (Figure 5H). These results indicate that the immune memory response is likely responsible for the long-term anti-tumour effect of the combination immunotherapy by recognizing the homogenous tumour and preventing tumour recurrence. Admittedly, our study lacked additional controls where the re-challenge was performed with the other tumour cell lines. Thus, further work is needed to determine whether the immune memory response induced by our combination immunotherapy is antigen-specific.
Discussion
Compelling evidence has shown the great potential of immunotherapy, particularly the immune checkpoint PD-1 inhibitor which has been approved by the FDA, for the treatment of various malignant tumours. However, immunotherapy via PD-1 inhibition obtained poor objective response rates (15%-20%) in Phase I/II trials and failed to meet the primary end points in Phase III trials for HCC treatment, which might be primarily attributable to the cunning immune evasion induced by the immunosuppressive HCC microenvironment.4,43 Thus, a novel immunotherapy strategy against HCC is urgently warranted. In the present study, the combination of CpG and anti-OX40 mAb aimed to intensify immune stimulation signals and strengthen anti-tumour T cells to provide an effective and practical form of HCC immunotherapy.
In our study, the synergistic anti-tumour effect of CpG and anti-OX40 mAb can be explained by the fact that CpG upregulated OX40 expression on T cells in HCC, which has also been observed in lymphoma according to a previous report.26,27 In this context, our study provided clear evidence that the role of the OX40/OX40L axis as an immune stimulation signal was intensified, leading to a strengthened immune response against HCC by activating immunity-boosting Teffs and inhibiting immunosuppressive Tregs. Additionally, CpG itself, a potent agonist of the TLR9 signal, might serve as an important executor to overcome HCC immune evasion by stimulating DC maturation to promote tumour antigen presentation, which needs to be validated in further work.28–30 Thus, the synergistic anti-tumour effect of the combination immunotherapy depended on the CpG-mediated induction of OX40 expression on T cells, which theoretically enabled tumour antigen presentation and T cell-mediated tumour elimination to overcome HCC immune evasion.
Next, the key executors mediating the anti-HCC effect of the combination immunotherapy by anti-OX40 mAb and CpG were explored. Despite the definite anti-tumour immunity of CD8+ T cells, the anti-tumour immunity of CD4+ T cells remains paradoxical. Morales Kastresana et al found that anti-tumour therapy was still effective after CD4+ T cell depletion. But the anti-tumour immunity of CD4+ T cells was not absolutely excluded, as CD4+ T cell depletion also led to the consumption of Tregs with tumour-promoting effects.44 Herein, our adoptive immunity transfer experiment suggested that the anti-tumour efficiency of the combination immunotherapy of CpG and anti-OX40 mAb required both CD8+ and CD4+ T cells, as CD4+ or CD8+ T cells alone failed to prevent tumour growth. In contrast to CD8+ and CD4+ T cells, Tregs and MDSCs play an immunosuppressive role by inhibiting the function of other immune cells in the tumour microenvironment.41 Herein, our in vivo investigation also demonstrated that the combination immunotherapy remarkably reduced the proportion of Tregs and MDSCs in HCC tissues. Although the exact molecular mechanism mediating such an effect remains to be further identified, it is clear that the anti-tumour effect of the combination immunotherapy by anti-OX40 mAb and CpG is dependent on promoting CD8+ and CD4+ T cells as well as inhibiting Tregs and MDSCs.
Notably, the administration of anti-OX40 mAb and CpG via intratumoural injection, rather than via intraperitoneal, intravenous or subcutaneous injections, induced potent anti-tumour immunity. Such local drug administration, as a therapeutic strategy of in situ tumour vaccination, likely has two unique advantages. On the one hand, drug administration via intratumoural injection can facilitate tumour antigen presentation to break immune tolerance and develop a systemic anti-tumour response.45 As expected, we observed that the combined administration of CpG and anti-OX40 mAb via intratumoural injection simultaneously inhibited the growth of HCC receiving a local injection and distant tumours without injection. These results implied that our combination immunotherapy can induce a systemic immune response, which is of great significance for the anti-tumour effect at primary and metastatic lesions.45 On the other hand, drug administration via intratumoural injection is more liable to present the tumour antigen and intensify anti-tumour immune memory, which is attracting tremendous attention due to its remarkable therapeutic effects for tumour recurrence.46 In the present study, our combination immunotherapy via intratumoural injection was demonstrated to significantly generate immune memory response against the tumour and effectively prevent HCC recurrence, which was consistent with a previous report demonstrating that intratumoural injection, rather than intraperitoneal injection, suppressed lymphoma recurrence by combination immunotherapy (anti-CTLA-4, anti-OX40 and CpG).47 Taken together, given the importance of the method of drug administration to the curative effect, interventional medicine that can easily realize precise intratumoural injection holds great potential to push forward the clinical application of our combination immunotherapy for HCC treatment.
Conclusions
Our study demonstrated that the combination immunotherapy of CpG and anti-OX40 mAb via intratumoural injection could significantly inhibit the growth of local and distant HCC tumours, and also effectively prevent HCC recurrence. Furthermore, we demonstrated that the anti-tumour effect of the combination immunotherapy depended on promoting CD8+ and CD4+ T cells, and inhibiting Tregs and myeloid-derived suppressor cells. And also, the combination immunotherapy induced a potent immune memory response to prevent tumour recurrence. Thus, developing CpG and anti-OX40 mAbs may be promising as combined therapeutic candidates for HCC immunotherapy.
Abbreviations
HCC, hepatocellular carcinoma; ICI, immune checkpoint inhibitor; PD-1, programmed cell death-1; PD-L1, programmed cell death ligand-1; DCs, dendritic cells; Tregs, regulatory T cells; Teffs, effector T cells; TLR, toll-like receptor; MDSCs, myeloid-derived suppressor cells; CpG ODN, cytosine-phosphate-guanine oligodeoxynucleotide; mAb, monoclonal antibody.
Data Sharing Statement
The data used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for Publication
Not applicable.
Ethics Approval and Informed Consent
The use of the cell lines and all animal experiments were approved by the Animal Care and Use Committee of the Second Affiliated Hospital of Guangzhou Medical University. This study followed the Laboratory animal—Guideline for ethical review of animal welfare (GB/T 35892-2018). No human data were included.
Acknowledgments
The authors thank Huang Wensou, Guo Yongjian, Xie Lulu and Wang Xiaobin for assistance with the experiment performance.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (81873920, 81571774, 82001929); National Key Research and Development Program of China (2017YFA0205200); Science and Technology Program of Guangzhou (202002030135); Medical Scientific Research Foundation of Guangdong Province (A2020519). Basic and Applied Basic Research Foundation of Guangdong Province (2020A1515110654). Doctor Initiative Foundation of the Second Affiliated Hospital of Guangzhou Medical University (010G271108, 010G271103).
Disclosure
The authors declare that they have no conflict of interest for this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi:10.3322/caac.21590
3. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, Phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–2502. doi:10.1016/S0140-6736(17)31046-2
4. Nakano S, Eso Y, Okada H, Takai A, Takahashi K, Seno H. Recent advances in immunotherapy for hepatocellular carcinoma. Cancers (Basel). 2020;12:775. doi:10.3390/cancers12040775
5. Lurje I, Hammerich L, Tacke F. Dendritic Cell and T cell crosstalk in liver fibrogenesis and hepatocarcinogenesis: implications for prevention and therapy of liver cancer. Int J Mol Sci. 2020;21:7378. doi:10.3390/ijms21197378
6. Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2015;12:681–700. doi:10.1038/nrgastro.2015.173
7. Yuan CH, Sun XM, Zhu CL, et al. Amphiregulin activates regulatory T lymphocytes and suppresses CD8+ T cell-mediated anti-tumor response in hepatocellular carcinoma cells. Oncotarget. 2015;6:32138–32153. doi:10.18632/oncotarget.5171
8. Jiang R, Tang J, Chen Y, et al. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat Commun. 2017;8:15129. doi:10.1038/ncomms15129
9. Chaoul N, Mancarella S, Lupo L, Giannelli G, Dituri F. Impaired anti-Tumor T cell response in hepatocellular carcinoma. Cancers (Basel). 2020;12:627. doi:10.3390/cancers12030627
10. Omar HA, Tolba MF. Tackling molecular targets beyond PD-1/PD-L1: novel approaches to boost patients’ response to cancer immunotherapy. Crit Rev Oncol Hematol. 2019;135:21–29. doi:10.1016/j.critrevonc.2019.01.009
11. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. 2018;11:39. doi:10.1186/s13045-018-0582-8
12. Willoughby J, Griffiths J, Tews I, Cragg MS. OX40: structure and function - what questions remain? Mol Immunol. 2017;83:13–22. doi:10.1016/j.molimm.2017.01.006
13. Meylan F, Siegel RM. TNF superfamily cytokines in the promotion of Th9 differentiation and immunopathology. Semin Immunopathol. 2017;39:21–28. doi:10.1007/s00281-016-0612-y
14. Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria JC, Zitvogel L, Marabelle A. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer. 2016;52:50–66. doi:10.1016/j.ejca.2015.08.021
15. Ishii N, Takahashi T, Soroosh P, Sugamura K. OX40-OX40 ligand interaction in T-cell-mediated immunity and immunopathology. Adv Immunol. 2010;105:63–98.
16. Fu Y, Lin Q, Zhang Z, Zhang L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm Sin B. 2020;10:414–433. doi:10.1016/j.apsb.2019.08.010
17. Vu MD, Xiao X, Gao W, et al. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110:2501–2510. doi:10.1182/blood-2007-01-070748
18. Webb GJ, Hirschfield GM, Lane PJ. OX40, OX40L and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol. 2016;50:312–332.
19. Zhang X, Xiao X, Lan P, et al. OX40 costimulation inhibits Foxp3 expression and treg induction via BATF3-dependent and independent mechanisms. Cell Rep. 2018;24:607–618. doi:10.1016/j.celrep.2018.06.052
20. Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcγRs, leading to antitumor efficacy. Immunol Cell Biol. 2014;92:475–480. doi:10.1038/icb.2014.26
21. Kjaergaard J, Tanaka J, Kim JA, Rothchild K, Weinberg A, Shu S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res. 2000;60:5514–5521.
22. Javaid N, Choi S. Toll-like receptors from the perspective of cancer treatment. Cancers. 2020;12:297. doi:10.3390/cancers12020297
23. Thomas M, Ponce-Aix S, Navarro A, et al. Immunotherapeutic maintenance treatment with toll-like receptor 9 agonist lefitolimod in patients with extensive-stage small-cell lung cancer: results from the exploratory, controlled, randomized, international Phase II IMPULSE study. Ann Oncol. 2018;29:2076–2084. doi:10.1093/annonc/mdy326
24. Karapetyan L, Luke JJ, Davar D. Toll-like receptor 9 agonists in cancer. Onco Targets Ther. 2020;13:10039–10060. doi:10.2147/OTT.S247050
25. Benbenishty A, Gadrich M, Cottarelli A, et al. Prophylactic TLR9 stimulation reduces brain metastasis through microglia activation. PLoS Biol. 2019;17:e2006859. doi:10.1371/journal.pbio.2006859
26. Sagiv-Barfi I, Czerwinski DK, Levy S, et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med. 2018;10:eaan4488. doi:10.1126/scitranslmed.aan4488
27. Wang D, Jiang W, Zhu F, Mao X, Agrawal S. Modulation of the tumor microenvironment by intratumoral administration of IMO-2125, a novel TLR9 agonist, for cancer immunotherapy. Int J Oncol. 2018;53:1193–1203. doi:10.3892/ijo.2018.4456
28. Longo V, Gnoni A, Casadei Gardini A, et al. Immunotherapeutic approaches for hepatocellular carcinoma. Oncotarget. 2017;8:33897–33910. doi:10.18632/oncotarget.15406
29. Hofmann MA, Kors C, Audring H, Walden P, Sterry W, Phase TU. 1 evaluation of intralesionally injected TLR9-agonist PF-3512676 in patients with basal cell carcinoma or metastatic melanoma. J Immunother. 2008;31:520–527. doi:10.1097/CJI.0b013e318174a4df
30. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi:10.1038/ni1112
31. Reiberger T, Chen Y, Ramjiawan RR, et al. An orthotopic mouse model of hepatocellular carcinoma with underlying liver cirrhosis. Nat Protoc. 2015;10:1264–1274. doi:10.1038/nprot.2015.080
32. Lin L, Li R, Cai M, et al. Andrographolide ameliorates liver fibrosis in mice: involvement of TLR4/NF-κB and TGF-β1/Smad2 signaling pathways. Oxid Med Cell Longev. 2018;2018:7808656. doi:10.1155/2018/7808656
33. Lin L, Gong H, Li R, et al. Nanodrug with ROS and pH dual-sensitivity ameliorates liver fibrosis via multicellular regulation. Adv Sci. 2020;7(7):1903138. doi:10.1002/advs.201903138
34. Marabelle A, Kohrt H, Sagiv-Barfi I, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. 2013;123:2447–2463.
35. Scherwitzl I, Opp S, Hurtado AM, et al. Sindbis virus with anti-OX40 overcomes the immunosuppressive tumor microenvironment of low-immunogenic tumors. Mol Ther Oncolytics. 2020;17:431–447. doi:10.1016/j.omto.2020.04.012
36. Park CG, Hartl CA, Schmid D, Carmona EM, Kim HJ, Goldberg MS. Extended release of perioperative immunotherapy prevents tumor recurrence and eliminates metastases. Sci Transl Med. 2018;10:eaar1916. doi:10.1126/scitranslmed.aar1916
37. Hammerich L, Bhardwaj N, Kohrt HE, Brody JD. In situ vaccination for the treatment of cancer. Immunotherapy. 2016;8:315–330. doi:10.2217/imt.15.120
38. Fransen MF, van der Sluis TC, Ossendorp F, Arens R, Melief CJ. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ T-cell-dependent tumor eradication and decreases risk of toxic side effects. Clin Cancer Res. 2013;19:5381–5389. doi:10.1158/1078-0432.CCR-12-0781
39. Buonaguro L, Mauriello A, Cavalluzzo B, Petrizzo A, Tagliamonte M. Immunotherapy in hepatocellular carcinoma. Ann Hepatol. 2019;18:291–297. doi:10.1016/j.aohep.2019.04.003
40. Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14:717–734. doi:10.1038/nrclinonc.2017.101
41. Fu Y, Liu S, Zeng S, Shen H. From bench to bed: the tumor immune microenvironment and current immunotherapeutic strategies for hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38:396. doi:10.1186/s13046-019-1396-4
42. Xu J, Xu L, Wang C, et al. Near-infrared-triggered photodynamic therapy with multitasking upconversion nanoparticles in combination with checkpoint blockade for immunotherapy of colorectal cancer. ACS Nano. 2017;11:4463–4474. doi:10.1021/acsnano.7b00715
43. Nimmagadda S. Quantifying PD-L1 expression to monitor immune checkpoint therapy: opportunities and challenges. Cancers. 2020;12(11):E3173. doi:10.3390/cancers12113173
44. Morales-Kastresana A, Sanmamed MF, Rodriguez I, et al. Combined immunostimulatory monoclonal antibodies extend survival in an aggressive transgenic hepatocellular carcinoma mouse model. Clin Cancer Res. 2013;19:6151–6162. doi:10.1158/1078-0432.CCR-13-1189
45. Frank MJ, Reagan PM, Bartlett NL, et al. In situ vaccination with a TLR9 agonist and local low-dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discov. 2018;8(10):1258–1269. doi:10.1158/2159-8290.CD-18-0743
46. Qin L, Cao J, Shao K, et al. A tumor-to-lymph procedure navigated versatile gel system for combinatorial therapy against tumor recurrence and metastasis. Sci Adv. 2020;6(36):eabb3116. doi:10.1126/sciadv.abb3116
47. Marabelle A, Kohrt H, Sagiv-Barfi I, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. 2013;123(6):2447–2463.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.