")
Back to Journals » Drug Design, Development and Therapy » Volume 11
The coexpression of multi-immune inhibitory receptors on T lymphocytes in primary non-small-cell lung cancer
Authors Guo WJ, Liu SH, Zhang XL, Chen YT, Qian RL, Zou ZY, Chen X, Luo P
Received 6 August 2017
Accepted for publication 28 September 2017
Published 28 November 2017 Volume 2017:11 Pages 3367—3376
DOI https://doi.org/10.2147/DDDT.S148443
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sukesh Voruganti
Wenjie Guo,1,* Sihan Liu,2,* Xiaoli Zhang,1,* Yating Chen,1 Ruolan Qian,1 Ziyuan Zou,2 Xin Chen,1 Peng Luo1
1Department of Respiratory Medicine, Zhujiang Hospital, 2Department of Respiratory Medicine, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong Province, People’s Republic of China
*These authors contributed equally to this work
Abstract: Non-small-cell lung cancer (NSCLC) is a common disease threatening the health of humankind. It has a low survival rate and a poor prognosis. Under normal circumstances, tumor infiltrating lymphocytes (TILs) play the main role in the antitumor process, but studies in recent years have found that NSCLC is capable of releasing various immunosuppressive factors, inducing the TILs to exhibit high expression of immune inhibitory receptors and relevant immunosuppressive factors. They can not only activate their own signal pathways but also block those of TILs, which causes inefficiency of tumor destruction. Researchers have now developed targeted drugs that specifically bind to immunosuppression receptors. By blocking signal transmission of immune inhibitory receptors, restraint on T lymphocytes can be released to recover antitumor role. Further research and understanding of the immunosuppression signal pathways of NSCLC are of significant importance to promote the development of immune-targeted drugs and the formulation of new treatment plans. This paper summarizes the immunosuppressive mechanisms of multiple important and newly discovered immune inhibitory receptors on T lymphocytes and immunosuppressive factors released by NSCLC cells, and their influence on patients’ survival rate and prognosis. Further laboratory and clinical studies on immune-targeted drugs for primary NSCLC are needed to provide more evidence.
Keywords: NSCLC, TIL, immune inhibitory receptors, immune-targeted drugs
Introduction
Lung cancer is a malignant and threatening disease in the world. It accounts for ~1.82 million newly diagnosed cases and 1.59 million deaths every year (Figures 1 and 2). Among such death cases, 85% are due to non-small-cell lung cancer (NSCLC).1 NSCLC patients also have multiple antitumor mechanisms similar to healthy people. That is to say, tumor infiltrating lymphocytes (TILs) can kill lung cancer cells by releasing various cytokines such as interferon-γ (IFN-γ) and interleukin-2 (IL-2), further differentiate under the stimulation of lung carcinoma cells, and reinforce ability to kill cancer cells.2 Despite its significant role, the survival rate of NSCLC patients up to 5 years is only ~10%.3
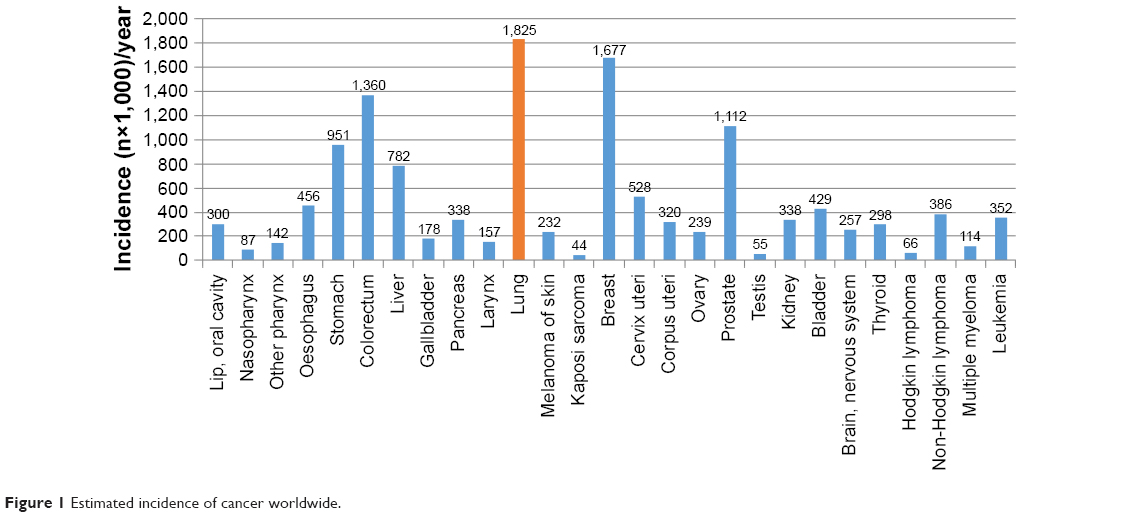 | Figure 1 Estimated incidence of cancer worldwide. |
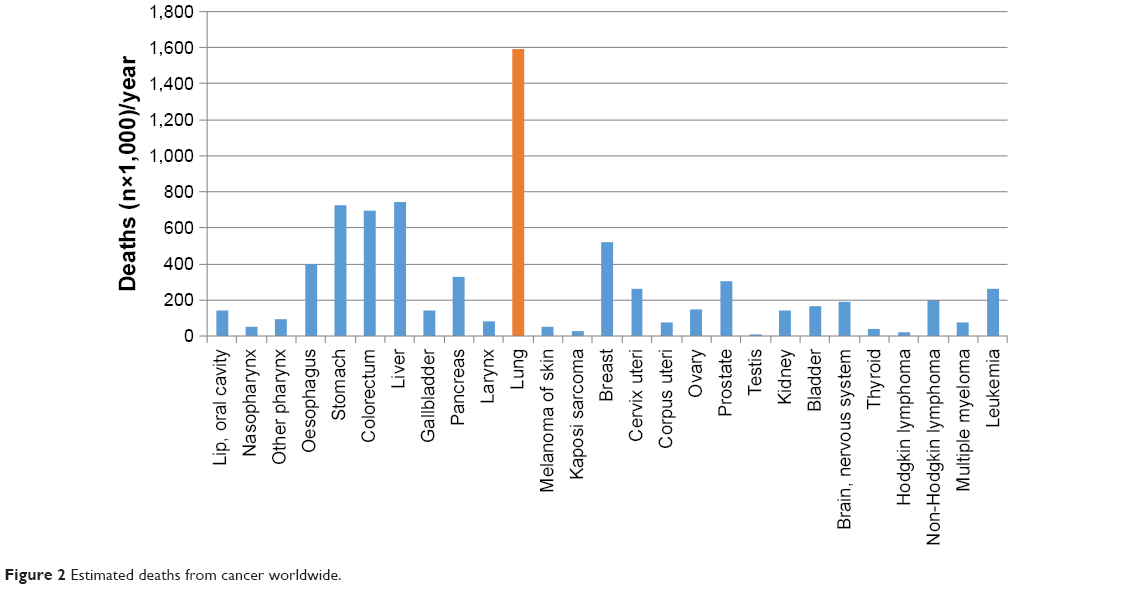 | Figure 2 Estimated deaths from cancer worldwide. |
According to recent research, NSCLC cells are capable of releasing a variety of multiple immunosuppressive factors, such as cyclooxygenase-2, interleukin-10 (IL-10), and transforming growth factor β (TGF-β). Meanwhile, multiple immune inhibitory receptors can be detected on T lymphocytes on the tumor part, including programmed death-1 (PD-1), T-cell immunoglobulin and mucin-domain-containing molecule-3 (TIM-3), cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4), lymphocyte activation gene-3 (LAG-3), and T-cell immunoglobulin immune-receptor tyrosine-based inhibitory motif domain (TIGIT).4 By releasing immunosuppressive factors and binding with corresponding receptors on cytotoxic T lymphocytes, the tumors can activate downstream signal transduction pathways to restrain the cytokine secretion of cytotoxic T lymphocytes and greatly restrain immune function of tumor-specific T lymphocytes. Moreover, immunosuppressive factors released by tumor cells can also bind with receptors on regulatory T lymphocytes, promote the negative immune modulating functions, and restrain proliferation and differentiation of cytotoxic T lymphocytes through IL-10 and IL-35, so it is impossible for patients to generate effective tumor-killing lymphocytes. This is good for proliferation of tumor cells, resulting in progression of disease and death of patients.
Further, aiming to investigate the immune escape mechanism of NSCLC, researchers have developed the antibodies targeting T lymphocytes’ immune inhibitory receptors, which block the immunosuppression signal pathways of cancer cells and restore the antitumor function of T lymphocytes.5 Few studies conducted a comparison between the curative effects of PD-1 receptor antagonists and the traditional chemotherapy drug, Taxol® (paclitaxel; Bristol-Myers Squibb Company, Princeton, NJ, USA) in NSCLC, and found out the overall survival (OS) of patients who took the immune inhibitory receptor antagonists was 12.2–17.3 months and the progression-free survival (PFS) was 5–10.3 months; conversely, the patients taking paclitaxel therapy had the OS of 8.2–9.4 months and the PFS of 4.1–6 months.6–10 Meanwhile, the meta-analysis compared the effects of immune checkpoint inhibitors (ICIs) and traditional chemotherapy drugs in curing squamous carcinoma and nonsquamous carcinoma in NSCLC. The analysis gathered the experimental data of PubMed, MEDLINE, Embase, and ESMO. According to the final results, the PFS value of ICIs in curing squamous carcinoma was significantly higher than that of traditional chemotherapy drugs and the hazard ratio was 0.68. Conversely, the PFS value obtained after curing nonsquamous carcinoma with ICIs was not significantly different from that of traditional chemotherapy drugs and the hazard ratio was 0.88. According to such results, the immune-targeted curative drugs are more effective than traditional chemotherapy drugs and suffer fewer side effects.11,12
In recent years, there have been multiple immune-targeted drugs specific to inhibitory receptors and negative regulatory factors. There is no doubt that immune-targeted therapy will be a major treatment in the future for lung cancer treatment. As a result, in order to better develop immune-targeted drugs, it is necessary to know about categories and corresponding mechanism of multiple immune inhibitory receptors and negative regulatory factors, which have the extremely important significance on developing drugs and formulating therapeutic schedules. We have summarized the mechanisms as to how multiple immune inhibitors bound with regulatory cytokines released by tumor cells affect the differentiation or the production of T lymphocytes in NSCLC patients, hoping to provide reference for the development of immune-targeted therapeutic drugs (Figure 3).
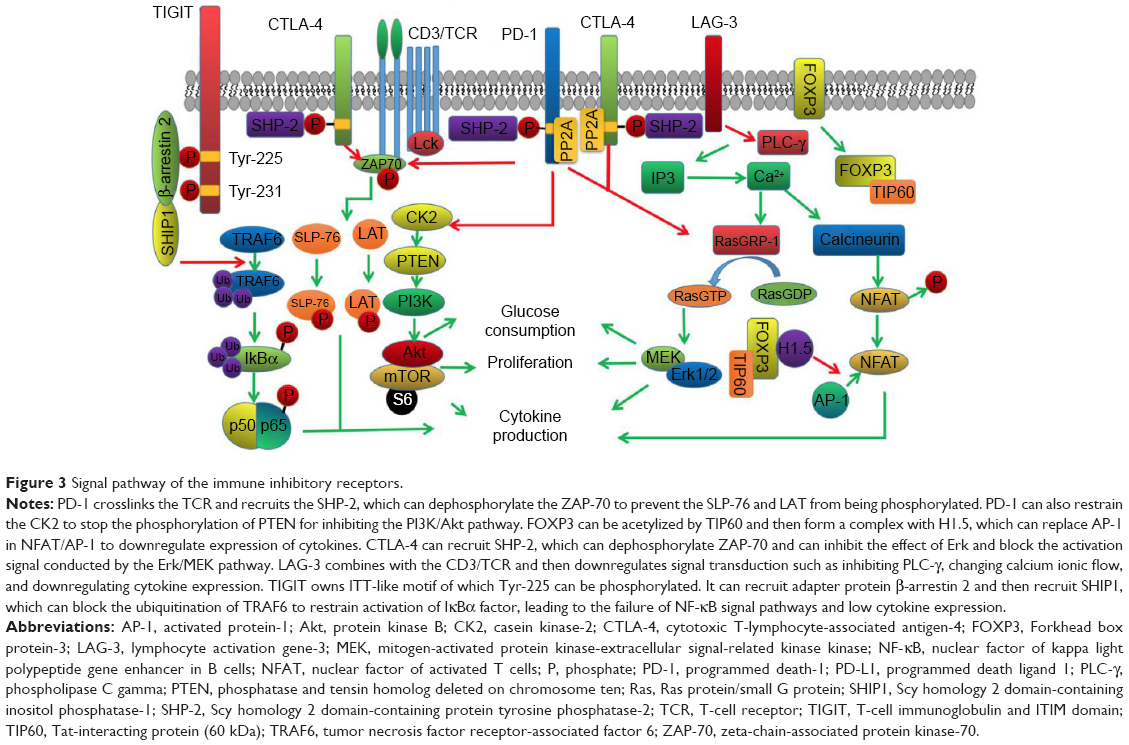 | Figure 3 Signal pathway of the immune inhibitory receptors. |
PD-1
PD-1 belongs to cluster of differentiation 28 (CD28) family, which is coded by chromosome 2q37 segment, and is expressed in the activated T lymphocytes. In normal human tissues, PD-1 binds with programmed death ligand 1 (PD-L1)/CD274 or PD-L2/CD273, restrains the proliferation of T lymphocytes, and promotes the generation of immunosuppressive factors. It also can prevent human body from injury resulting from a strong immunoreaction and autoimmune disease, and it is advantageous for maintaining immune self-stabilization. When PD-1 is highly expressed in TILs, it induces body to generate immune tolerance to tumor cells, resulting in immune escape of tumors.13,14 NSCLC can improve PD-L1 expression on lung cancer cytomembrane through signal transducer and activator of transcription-3 and anaplastic lymphoma kinase signal pathways, so as to further help immune escape of tumor cells.15
Researchers conducted immunohistochemistry (IHC) detection on the lung cancer tissues excised from 139 untreated NSCLC patients. The experimental results showed that PD-1 receptors could be detected on the membrane of TILs, which were extracted from 60 patients’ lung cancer tissues. The experiment also made a comparison on relapse-free survival (RFS) between PD-L1-negative and -positive patients. Results showed that the RFS for PD-L1-negative and -positive patients was 1.85 and 0.97 years, respectively. Existence of PD-1 and PD-L1 had the positive correlation with occurrence and recurrence rate of tumor immune escape.16
Depending on PD-1 and its ligands, NSCLC may restrain immunoreactions through multiple approaches. In the normal immune process, T-cell receptors (TCRs) can specially identify peptide-major histocompatibility complex (MHC) presented by antigen-presenting cells (APCs). Co-receptors (CD4 or CD8) can bind with MHC molecules. The tyrosine kinase connecting to the end of receptors gets close to CD3 cytoplasm segment immunoreceptor tyrosine-based activation motif (ITAM), leads to tyrosine phosphorylation, and launches cascade reaction of kinase activation, so as to provide the first signal of inducing T lymphocytes’ activation. In NSCLC patients’ bodies, after TCRs are activated by antigens, PD-l receptors and TCR form a crosslinking and can be activated through ligand binding, which helps to start recruiting Scy homology 2 domain-containing protein tyrosine phosphatase-1 (SHP-1) and Scy homology 2 domain-containing protein tyrosine phosphatase-2 (SHP-2). SHP-1 and SHP-2 can dephosphorylate the tyrosine residue on immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif on the end of PD-1 receptor in the cytoplasm, so as to activate PD-1 downstream signal pathways, reduce kinase activity inducing T-lymphocyte activation, prevent T lymphocytes from generating immunoreactions on tumor antigens, and induce body to generate immune tolerance to tumor antigens.17,18
When APC surface costimulatory module B7 binds with T-cell surface CD28 to generate second signal inducing T-cell activation and proliferation, PD-1 will dephosphorylate near-end downstream signal modules of signal pathways mediated by CD28, such as lymphocyte-specific protein tyrosine kinase and zeta-chain-associated protein kinase-70 (ZAP70), blocking further transmission of downstream signals. After double-signal pathways are restrained, T cells cannot generate IL-2 as well as high-affinity IL-2R or the level of IL-2 autocrine decreases. Meanwhile, the corresponding receptors fail to combine with ligands efficiently, resulting in the obstruction of signal transmission. Thus, “third signal” activated by T cells is also restrained.19
When costimulatory module B7 from APC binds with T-cell surface CD28 to generate second signal for T-cell activation and proliferation, phosphorylated ITAM of CD3 recruits, phosphorylates, and activates ZAP70, leading to the activation of leukocyte protein Src homology 2 domain-containing leukocyte phosphoprotein of 76 kDa (SLP-76) and integral membrane protein linker for activation of T cells (LAT) signal pathways. Then, they help to induce the transcription and expression of genes for T lymphocytes.20 On the contrary, PD-1 prevents ZAP70 form phosphorylation. After the first and second signals for T lymphocytes’ activation fail to pass down, T cells cannot generate IL-2 as well as high-affinity IL-2R or complete IL-2 autocrine. Thus, third signal for T cells is also restrained.21
In addition to double-signal immune activation mediated by TCR/CD3 and CD28, the activated PD-1 can block another two important immune activation signal pathways: phosphatidylinositol 3 kinase (PI3K)/protein kinase B (Akt) and Ras protein/small G protein (RAS)/mitogen-activated protein kinase-extracellular signal-related kinase kinase (MEK)/extracellular signal-regulated kinase (Erk) signal pathways. After activating normal double-signal pathways, casein kinase-2 (CK2) can phosphorylate phosphatase and tensin homolog deleted on chromosome ten (PTEN) C-terminal serine/threonine to stabilize its structure and restrain activity of phosphatase, so as to be advantageous for signal transmission of PI3K/Akt and promote proliferation and activation of T cells. On the signal pathways mediated by PD-1, it can restrain CK2 to block the phosphorylation on serine/threonine, so as to upregulate phosphatase activity of PTEN and block downstream signal transduction. MEK/Erk signal pathway is a branch in RAS downstream signal pathways. Besides, RAS downstream signal pathways including the MEK/Erk signal pathways activated by RasGRP-1 in downstream also can be blocked by PD-1 (Figure 3).12,22
By studying the NSCLC immune escape mechanism, researchers have developed antibodies targeting PD-1 receptors that are highly expressed on T lymphocytes. By blocking transmission of the PD-1/PD-L1 signal pathways, antitumor ability of T lymphocytes can be reactivated. Some studies showed that, by using the flow cytometry technology, no obvious IFN-γ can be detected on the CD8+ T cells cultured with lung carcinoma in the control group. However, after adding anti-PD-1 antibodies, relatively obvious IFN-γ could be detected in the study group, proving that after adding anti-PD-1 antibodies, antitumor function of T lymphocytes can be recovered to some extent.23
At present, the Food and Drug Administration has already approved three kinds of antibodies against PD-1 as targeted drugs for curing NSCLC, including atezolizumab, nivolumab, and pembrolizumab. Some researchers made a comparison on NSCLC’s therapeutic effects between pembrolizumab and the traditional antitumor drug docetaxel. In the experiment, NSCLC patients whose PD-L1 expression rate of lung carcinoma was >50% participated in experiment. During the experiment, pembrolizumab and docetaxel were used for patients. The results showed that median OS (mOS) of patients with pembrolizumab and of patients taking docetaxel was 10.4–14.9 and 8.2–8.5 months, respectively. And mPFS for patients with pembrolizumab and for patients taking docetaxel was 3.9 and 4.0 months, respectively. The overall response rate (ORR) of tumors to pembrolizumab and docetaxel reached 18%–30% and 8%–9%, respectively, indicating that antitumor action of pembrolizumab was obviously superior to docetaxel (Table 1).24
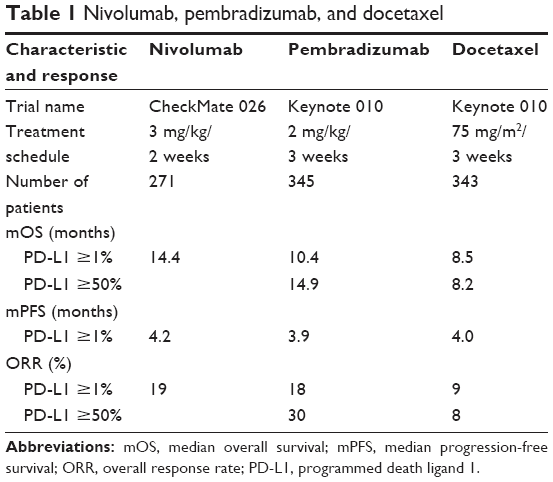 | Table 1 Nivolumab, pembradizumab, and docetaxel |
Forkhead box protein-3 (FOXP3)
In normal bodies, there are mainly two kinds of T lymphocytes, including effector T cell (Te) and regulatory T cell (Treg). Treg is a subtype of CD4+ T cells, with high expression of CD25 and low expression of CD127. The phenotype landmark FOXP3 can restrain immunoreactions through negative regulatory function to prevent human body from autoimmune disease (AD) and excessive inflammatory responses for antigens. The tumor cells of NSCLC can induce Treg to gather on tumor sites and TILs to highly express FOXP3.
FOXP3 structure includes four important components, including N terminal containing proline-rich (PRR) domain, zinc finger, leucine zipper domain, and C terminal used to bind with DNA chain. After FOXP3 on membrane is bound with corresponding ligand, it is activated and enters cells. Tat-interacting protein 60 kDa (TIP60) gets close and binds with N terminal of FOXP3, so that FOXP3 is acetylized. Acetylized FOXP3 polypeptide is considered as the prerequisite for FOXP3 to play a role, and then, FOXP3 can form complex with multiple factors, such as nuclear factor of activated T cells (NFAT). Finally, it is bound on the specific DNA fragment and further regulates expression of relative immune cytokines. To take the mechanism of NFAT as an example, first and second signals activating T cells in normal immune process will activate transcription factors NFAT and activated protein-1 (AP-1). The activated NFAT and AP-1 can form complex and then be transferred to cell nucleus, bind with IL-2 encoding gene, induce expression and secretion of IL-2, and further mediate T-cell activation.25 FOXP3 can recruit histone through PRR and use the binding of leucine zipper domain and histone H1.5 to replace the AP-1 in NFAT/AP-1 complex in cell nucleus (Figure 3).26 Some researchers conducted the contrast experiment. In the experimental group, there were three groups of T lymphocytes differentiated by adding histone, adding FOXP3, and adding FOXP3 and histone. The results found that by comparing with normal T lymphocytes, the IL-2 expression of lymphocyte by adding FOXP3 was reduced by five times. T lymphocytes IL-2 expression by adding FOXP3 and histone H1.5 was reduced by 45 times, showing that FOXP3 activation can greatly restrain IL-2 generation and block T-lymphocyte activation.27
In addition to intervening in NFAT/AP-1 complex, FOXP3 can block the function of Ikaros family members Eos and Runxl-Cbf β for T-cell activation. Researchers extracted Treg in mice, induced Treg to lowly express FOXP3, and injected it into other mice. The results showed that Treg, which lowly expressed FOXP3, has the tendency to transform into other subtype such as Th1 with secretion of IFN-γ and Th2 with secretion of IL-4. It showed the multi-pathway blocking-up function of FOXP3, so as to block further differentiation of T lymphocytes and realize immunosuppressive functions.28 As a consequence, the high proportion of lymphocytes or TILs with high expression of FOXP3 suggests bad prognosis of NSCLC patients. Statistical data found that when ratio of FOXP3-positive CD8+ T lymphocytes to FOXP3-negative CD8+ T lymphocytes in NSCLC patients was changed from low to high, PFS of patients was reduced from 37.8 to 23 months. OS was decreased from 74.3 to 46.4 months. When the expression of FOXP3 was regarded as the single variable, patients with lung cancer whose CD8+ T lymphocytes lowly expressed FOXP3 were compared to the ones that highly expressed FOXP3, showing that PFS in group with high FOXP3 expression and group with low expression was 15.9 and 66.9 months, respectively. OS in group with high FOXP3 expression and group with FOXP3 low expression was 17.4 and 68.8 months, respectively, indicating that FOXP3 had the important significance on prognosis evaluation.12
Though there are no targeted drugs restraining FOXP3 effectively, effective restraint on FOXP3 will be contributed to recovering T cellular immune function for patients, so as to improve prognosis of patients. The study targeting FOXP3 antibodies will be an important study direction in the future immune-targeted therapy.
CTLA-4
CTLA-4 is also a category of inhibitory receptors on T-cell surface. It is mainly expressed in CD8+ T lymphocytes. CTLA-4 and CD28 are homologous, and they owned the common ligands B7-1 (CD80) and B7-6 (CD86). CD28 binds with ligand to develop the role of activating immune reaction. CTLA-4 mediates negative immune regulatory functions.29 Generally, most of the CD28 molecules are found on cell membrane, while most of the CTLA-4 molecules occur in intracellular TGN (trans-Golgi network), endosome, and lysosomes.
In the early immunity, B7 mainly binds with CD28 and provides second signal for T-cell activation until it is activated for 24 hours. Then, CILA-4 replaces CD28 to bind with B7 and generate the negative regulation role in later period. The study shows that this may be related to the tyrosine-rich YVKM (Tyr-Val-Lys-Met, tyrosine-based endocytic motif) on the CTLA-4 terminal. Actually CTLA-4 starts expression as T cell is activated and its affinity with B7 is obviously higher than CD28. In the early stage, unphosphorylated YVKM binds with subunit μ2 in protein complex activated protein-2 (AP-2) and mediates CTLA-4 endocytosis, so that CTLA-4 quantity on the membrane will be reduced.30 Activated T lymphocytes can express a kind of type III transmembrane protein called TCR-interacting molecule (TRIM). With the bonding of antigen and TCR, TRIM is able to bond with p85, a subunit of PI3K and activate PI3K. p85 of PI3K then replaces the subunit μ2 bonding with YVKM. It weakens the endocytosis of CTLA-4 while facilitating CTLA-4 transferring to the cytomembrane and combination with CD80/CD86 to conduct immunosuppression signals.31 In cells, T lymphocyte calcium influx can activate calcium ionophore, making CTLA-4 to transfer to the cell membrane from TGN, and T lymphocyte also can accelerate the transfer of CTLA-4 to the cell membrane via SHP2-interacting transmembrane adapter protein or linker for activation of T cells (LAT). In cells, CTLA-4 can inhibit the effect of Erks through downstream RAS signal channel to finally cutoff the transcription and expression of cytokine and hinder the further activation of T lymphocytes.
The activated CTLA-4 activates SHP-2 by YVKM. Then, SHP-2 phosphorylates the tyrosine residues of CTLA-4, which helps to stabilize the extracellular structure of CTLA-4. When CTLA-4 binds with ligand CD80/CD86 through MYPPPY binding motif, it is able to enhance the activity of T lymphocytes and lead to the failure of the stable combination of T lymphocytes with APC. The ligation between CTLA-4 and CD80/CD86 also hinder the formation of TCR-ZAP70 microclusters, which further obstructs ZAP-70 to phosphorylate SLP-76 and LAT and inhibits the expression of IL-2 and antiapoptosis protein Bcl-X2, resulting in the disorders of T-lymphocyte activation, proliferation, and maturity (Figure 3).32,33
The bonding of CTLA-4 and PI3K can activate phosphoinositide dependent kinase-1 and then phosphorylate protein kinase B (PKB). Activated PKB can phosphorylate Bcl-2 antagonist of cell death (BAD), a kind of proapoptosis factor in cell, to cutoff the bonding between BAD and Bcl-2, thus hindering the apoptosis of T lymphocytes. In this way, tumor can form nonfunctional T-lymphocyte wrapping around part of the lung cancer.34–36
As a result, if CTLA-4 is prevented from binding with ligands, it will be better for CD28 to bind with more ligands and develop the positive immune regulation result. Some findings found that after adding tremelimumab, a kind of antibody against CTLA-4 in T lymphocytes, the amount of CD4+ T lymphocytes secreting IL-2 and CD4+ and CD8+ secreting IFN-γ obviously increased, showing that blocking the binding of CTLA-4 and B7 could effectively promote proliferation of Te and recovery of immunity. Clinical data have proven that antibodies against CTLA-4 exactly could improve survival rate of NSCLC patients: 52 NSCLC patients were individually cured with antibodies against CTLA-4, nivolumab, ORR was 23%, and mOS was 19.4 months. When antibodies against PD-1 and CTLA-4 antibody were combined to use, ORR and 1-year survival rate were significantly improved. In the experiment, results showed that for the combination of two targeted drugs, ORR reached 43%, which was obviously higher than single nivolmab. One-year survival rate reached 83%, proving that CTLA-4 targeted drug contributed to curing NSCLC patients (Table 2).37,38 Also, the combined use of two targeted drugs could have the synergistic effect.39 It will be significant to improve patients’ prognosis and enhance patients’ survival rate.
 | Table 2 Nivolumab and nivolumab plus ipilimumab |
LAG-3
LAG-3 belongs to immunoglobulin superfamily, and it is expressed in natural killer T cell, B lymphocytes, TILs, and dendritic cells. Especially, it is highly expressed in Treg lymphocytes and nonactive T lymphocytes. It also has the negative regulation role on T-cell immune functions.40
LAG-3 is an immunoglobulin composed of 498 amino acids. The LAG-3 genome and CD4 genome are located on the same chromosome. Moreover, the similarity between LAG-3 genomic sequence and CD4 genomic sequence is up to 20%. LAG-3 has the stronger binding capacity with MHC class II (MHC II) of APCs than CD4. LAG-3 and CD4 mediate restraint and activation of immunoreactions, respectively. The research findings showed that LAG-3 is mainly located in the lysosomes of T lymphocytes. When T lymphocytes are stimulated by antigens presented by APC, LAG-3 pulls off glutamic acid–proline repeated sequence under the role of lysosomes and then it is transferred to cytomembrane under the assistance of protein kinase C. After it is transferred to the membrane, LAG-3 binds with CD3/TCR complex and then downregulates signal transduction mediated by CD3/TCR complex, such as inhibiting the PLC-γ, changing calcium ionic flow, and downregulating IL-2 expression. Meanwhile, signal pathways mediated by LAG-3 can further restrain expression of CD3/TCR complex gene, restrain proliferation of T lymphocytes, and reduce immune lethal effects of T lymphocytes on lung cancer cells (Figure 3).41
NSCLC cells can induce T lymphocytes to highly express LAG-3, so as to restrain anti-tumor roles of T lymphocytes. This is advantageous for tumor survival and progression. In the experiment, 139 lung cancer tissues were excised from different NSCLC patients. IHC was used to detect positive rate of LAG-3 on TILs from the tissues, finding that TILs in 36 samples could be detected with LAG-3 expression. Positive rate reached 25.9%. Meanwhile, by comparing RFS and OS of patients with LAG-3-positive TILs and LAG-3-negative TILs, the results showed that the RFS and OS of patients with LAG-3-negative TIL were 1.91 and 3.04 years, respectively. RFS and OS of patients with LAG-3-positive TIL were only 0.87 and 1.08 years, respectively, showing that immunosuppressive action induced by LAG-3 greatly inhibits the antitumor immunoreaction of patients, so as to greatly reduce patients’ survival rate.42
At present, LAG-3-targeted therapy is kept in the study stage. Researchers extracted normal T lymphocytes of mice to culture in vitro and added antibodies against LAG-3. After culturing for several days, the number of T lymphocytes was obviously increased and it was 2.3 times by comparing with the control group. There were multi-directional differentiated T lymphocytes, showing that T lymphocytes started differentiation and maturation after LAG-3 receptors were antagonized.43 The quantity and function of T lymphocytes could be recovered. Anti-Lag-3 is promising to be one of targeted drugs for curing NSCLC in the future.
TIGIT
TIGIT is similar to CTLA-4 and PD-1. All of them belong to a kind of co-suppression receptors. TIGIT is mainly expressed in NK, Treg, CD8+, and CD4+T cells. By binding with ligand CD155, it will restrain pathological reaction caused by autoimmunity and develop a negative regulation role of immunity.44
TIGIT owns immunoglobulin tail tyrosine (ITT)-like motif and an ITIM on the cytosolic terminal. Tyr-225 on ITT-like motif can be phosphorylated, which help recruit adapter protein β-arrestin 2 and then recruit Scy homology 2 domain-containing inositol phosphatase-1 (SHIP1). SHIP1 can block ubiquitination of tumor necrosis factor receptor-associated factor 6 to restrain activation of IκBαl factors which is crucial to activate nuclear factor of kappa light polypeptide gene enhancer in B cells (NF-κB) signal pathways. After NF-κB signal transduction pathway was blocked, production of IFN-γ was reduced (Figure 3). The IFN-γ expression of common NK cells, TIGIT transgenic NK cells, and NK cells knockout TIGIT gene were detected. The experimental results showed that IFN-γ expression in the control group was >300 pg/mL. IFN-γ expression in transgenic group of the experimental group was <100 pg/mL. IFN-γ expression in knockout gene group was >500 pg/mL, which was obviously higher than the control group, implying that TIGIT can restrain NF-κB signal pathways to reduce IFN-γ generation, so as to develop immunosuppression effects.45
In addition, TIGIT-positive Treg cells can highly express and secrete IL-10, which not only can restrain innate immunity in tumor microenvironment and but also can induce generation of tumor antigen’s tolerogenic dendritic cells and interdict presentation of tumor cell antigen, so as to block immunoreactions of lung carcinoma cells. Second, TIGIT-positive Treg cells can regulate different types of immunoreactions selectively, reduce secretion of IL-4, and restrain type II immunoreactions, so as to promote differentiation of tumor-associated macrophage 2, which can secrete lots of IL-10 and TGF-β to restrain T-cell activation and proliferation and restrain generation of T cells’ antitumor immunity.46 In addition, TILs and Tregs gathered at tumor parts of NSCLC can highly express TIGIT receptors and also highly express other types to restrain receptors, so that antitumor ability is obviously weakened.47
At present, the mechanism of TIGIT on immunosuppression effects is still kept in the research stage. By virtue of the extensive inhibiting effects, TIGIT is likely to be the important target spot of curing NSCLC in the future.
Conclusion
The study and analysis of immunosuppression receptors, immunosuppressive factors, and their signal transduction pathways are contributed to clinically producing targeted drugs by targeting various receptors, blocking negative regulation signal transduction pathways, and recovering antitumor immune reactions. Besides, some studies displayed that positive rate of TIM-3 and PD-1 receptors on TIGIT-positive T lymphocytes was higher than TIGIT-negative T lymphocytes, showing that there was mutually promoted relationship among various immune inhibitory receptors. The understanding on various signal pathways of immunosuppression receptors contributes to discovering the relationship between receptors, clinical development of new-type targeted drugs, realizing simultaneous blocking of multiple immune inhibitory signal pathways, and improving therapeutic effects of targeted drugs.
The successful utilization of targeted drugs targeting immune inhibitory receptors offers the new measure to cure NSCLC patients. By blocking the binding between receptors and corresponding ligands, it blocks activation of negative regulation signal pathways and recovers anti-tumor immunoreactions. Clinical test has already showed that multiple targeted drugs have the positive influences on lengthening PFS of patients with lung cancer and improving patients’ survival rate. At present, clinical allowable drugs include anti-CTLA-4 ipilimumab, anti-PD-1 nivolumab and pembrolizumab, and PD-1 ligand antibody atezolizumab. However, there is no clear mechanism of NSCLCs suppressing antitumor immunoreaction, so that anti-immunosuppressive action of immune-targeted drugs is widely applied, resulting in the generation of side effects. Some research analyses indicated that among patients cured by anti-CTLA-4, ipilimumab, occurrence rate of diarrhea and colonitis was up to 32.8% and it often occurred in 6–7 weeks of initial medication. A few patients who had been cured by ipilimumab and nivolumab suffered from hepatitis mediated by autoimmunity. Many patients had no clinical symptoms but had increased aspartate transaminase, alanine transaminase, and total bilirubin.48 A total of 11%–17% of patients who took ipilimumab could develop hypophysitis, thyroiditis, and hypothyroidism because of autoimmunity.49
Among patients taking anti-PD-1 treatment, ~9% of patents might suffer from interstitial pneumonia and alveolar pneumonia mediated by autoimmunity. Patients might have obvious dyspnea, cough, and hemoptysis. Serious pneumonia might result in treatment interruption. In terms of dermal toxicity, the commonest side effects might be skin itch maculopapule. What is more, it will even trigger leucoderma, Steven–Johnson syndrome, toxic epidermolysis, bullous pemphigoid, and so forth.50
By studying immunosuppression signal pathways induced by receptors, researchers have already produced targeted drugs specific to various receptors. They proved that blocking the immunosuppressive signal pathways contributed to curing NSCLC patients. However, it could be observed from above-mentioned side effects that tumor antagonist restrained immune cells and also resulted in unbalance of autoimmune system, so as to result in ADs. On one hand, by focusing on these problems, researchers will further study signal pathways in the future, know about the NSCLC how to induce immune cells to highly express immunosuppression receptors, and block occurrence of immunosuppression from the source. On the other hand, the researchers can study the relationship among various immune inhibitory receptors and invent multiple anti-immunosuppression receptors’ targeted drugs on the basis of the better mechanism comprehension. What is more important: features of immunosuppression signal pathways induced by NSCLC should be studied, so as to increase specificity of targeted drugs for tumor cells and recover T-lymphocyte functions in lung cancer regions without unbalance in immune system and immune diseases after using targeted drugs.
Acknowledgment
This study was supported by the Natural Science Foundation of Guangdong Province, China (2016A030313620).
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. | ||
Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. | ||
Tassi E, Grazia G, Vegetti C, et al. Early effector T lymphocytes coexpress multiple inhibitory receptors in primary non-small cell lung cancer. Cancer Res. 2017;77(4):851–861. | ||
Zhang L, Wang J, Wei F, et al. Profiling the dynamic expression of checkpoint molecules on cytokine-induced killer cells from non-small-cell lung cancer patients. Oncotarget. 2016;7(28):43604–43615. | ||
Champiat S, Ileana E, Giaccone G, et al. Incorporating immune-checkpoint inhibitors into systemic therapy of NSCLC[J]. J Thorac Oncol. 2014;9(2):144–153. | ||
Leprieur EG, Dumenil C, Julie C, et al. Immunotherapy revolutionises non-small-cell lung cancer therapy: results, perspectives and new challenges. Eur J Cancer. 2017;78:16–23. | ||
Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–1639. | ||
Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–1550. | ||
Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387:1837–1846. | ||
Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–1833. | ||
Kim BJ, Kim JH, Kim HS. Survival benefit of immune checkpoint inhibitors according to the histology in non-small-cell lung cancer: a meta-analysis and review. Oncotarget. 2017;8(31):51779–51785. | ||
Jia M, Feng W, Kang S, et al. Evaluation of the efficacy and safety of anti-PD-1 and anti-PD-L1 antibody in the treatment of non-small cell lung cancer (NSCLC): a meta-analysis. J Thorac Dis. 2015;7(3):455–461. | ||
Gray SG, Cuffe S, Finn SP. PD-L1 as a companion biomarker for immune checkpoint inhibitors in NSCLC: should RNA ISH (RISH) be considered? J Thorac Oncol. 2016;11(11):e142–e144. | ||
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | ||
Davies M. New modalities of cancer treatment for NSCLC: focus on immunotherapy. Cancer Manag Res. 2014;6:63–75. | ||
He Y, Rozeboom L, Rivard CJ, et al. PD-1, PD-L1 protein expression in non-small cell lung cancer and their relationship with tumor-infiltrating lymphocytes. Med Sci Moni. 2017;23:1208. | ||
Chen YNP, LaMarche MJ, Chan HM, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535(7610):148–152. | ||
Mueller SN, Vanguri VK, Ha SJ, et al. PD-L1 has distinct functions in hematopoietic and nonhematopoietic cells in regulating T cell responses during chronic infection in mice. J Clin Invest. 2010;120(7):2508–2515. | ||
Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 pathway from discovery to clinical implementation. Front Immunol. 2016;7(1):550. | ||
Huber RG, Fan H, Bond PJ. The structural basis for activation and inhibition of ZAP-70 kinase domain[J]. PLoS Comput Biol. 2015;11(10):e1004560. | ||
El-Osta H, Shahid K, Mills GM, Peddi P. Immune checkpoint inhibitors: the new frontier in non-small-cell lung cancer treatment. Onco Targets Ther. 2016;9:5101–5116. | ||
Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012;5(230):ra46. | ||
Shen M, Ren X. Highlights on immune checkpoint inhibitors in non-small cell lung cancer. Tumour Biol. 2017;39(3):1–16. | ||
Iafolla MAJ, Juergens RA. Update on programmed death-1 and programmed death-ligand 1 inhibition in the treatment of advanced or metastatic non-small cell lung cancer. Front Oncol. 2017;7:67. | ||
Uso M, Jantus-Lewintre E, Bremnes RM, et al. Analysis of the immune microenvironment in resected non-small cell lung cancer: the prognostic value of different T lymphocyte markers. Oncotarget. 2016;7(33):52849–52861. | ||
Wu Y, Borde M, Heissmeyer V, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126(2):375–387. | ||
Song X, Li B, Xiao Y, et al. Structural and biological features of FOXP3 dimerization relevant to regulatory T cell function. Cell Rep. 2012;1(6):665. | ||
Wang Y, Souabni A, Flavell RA, Wan YY. An intrinsic mechanism predisposes Foxp3-expressing regulatory T cells to Th2 conversion in vivo. J Immunol. 2010;185(10):5983–5992. | ||
Ise W, Kohyama M, Nutsch KM, et al. CTLA-4 regulates pathogenicity of antigen-specific autoreactive T cells by cell-intrinsic and -extrinsic mechanisms. Nat Immunol. 2010;11(2):129–135. | ||
Shiratori T, Miyatake S, Ohno H, et al. Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity. 1997;6(5):583–589. | ||
Valk E, Leung R, Kang H, Kaneko K, Rudd CE, Schneider H. T cell receptor-interacting molecule acts as a chaperone to modulate surface expression of the CTLA-4 coreceptor. Immunity. 2006;25:807–821. | ||
Walker LSK. EFIS lecture: understanding the CTLA-4 checkpoint in the maintenance of immune homeostasis. Immunol Lett. 2017;184:43–50. | ||
Schneider H, Smith X, Liu H, Bismuth G, Rudd C. CTLA-4 expression disrupts ZAP-70 microcluster formation, T-cell/APC conjugation and calcium mobilization. Eur J Immunol. 2007;38:40–47. | ||
Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity. 2002;17:201–210. | ||
Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229:12–26. | ||
Walker LS, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol. 2015;270(2):63–70. | ||
Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer[J]. N Engl J Med. 2015;373(2):123–135. | ||
He Y, Rivard CJ, Rozeboom L, et al. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci. 2016;107(9):1193–1197. | ||
Tanvetyanon T, Gray JE, Antonia SJ. PD-1 checkpoint blockade alone or combined PD-1 and CTLA-4 blockade as immunotherapy for lung cancer? Expert Opin Biol Ther. 2017;17(3):305–312. | ||
Dal Bello MG, Alama A, Coco S, Vanni I, Grossi F. Understanding the checkpoint blockade in lung cancer immunotherapy. Drug Discov Today. 2017;22(8):1266–1273. | ||
He Y, Yu H, Rozeboom L, et al. LAG-3 protein expression in non-small cell lung cancer and its relationship with PD-1/PD-L1 and tumor-infiltrating lymphocytes. J Thorac Oncol. 2017;12(5):814–823. | ||
Bae J, Lee SJ, Park CG, Lee YS, Chun T. Trafficking of LAG-3 to the surface on activated T cells via its cytoplasmic domain and protein kinase C signaling. J Immunol. 2014;193(6):3101. | ||
Workman CJ, Vignali DA. Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223). J Immunol. 2005;174(2):688. | ||
Zhang B, Zhao W, Li H, et al. Immunoreceptor TIGIT inhibits the cytotoxicity of human cytokine-induced killer cells by interacting with CD155. Cancer Immunol Immunother. 2016;65(3):1–10. | ||
Liu X, Hou M, Liu Y. TIGIT, a novel therapeutic target for tumor immunotherapy. Immunol Invest. 2017;46(2):172–182. | ||
Li M, Xia P, Du Y, et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-γ production of natural killer cells via β-arrestin 2-mediated negative signaling. J Biol Chem. 2014;289(25):17647. | ||
Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. 2017;276(1):112. | ||
Hofmann L, Forschner A, Loquai C, et al. Cutaneous, gastrointestinal, hepatic, endocrine, and renal side-effects of anti-PD-1 therapy. Eur J Cancer. 2016;60:190–209. | ||
Dine J, Gordon R, Shames Y, Kasler MK, Barton-Burke M. Immune checkpoint inhibitors: an innovation in immunotherapy for the treatment and management of patients with cancer. Asia Pac J Oncol Nurs. 2017;4(2):127. | ||
Howell M, Lee R, Bowyer S, Fusi A, Lorigan P. Optimal management of immune-related toxicities associated with checkpoint inhibitors in lung cancer. Lung Cancer. 2015;88:117–123. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.