")
Back to Journals » OncoTargets and Therapy » Volume 12
The coexistence of a Wilms’ tumor and renal cell carcinoma in children: a case report and review of the literature
Authors Zou XP, Jiang YY, Liao Y, Dang YW, Chen G , Feng ZB, Ma Y
Received 1 September 2018
Accepted for publication 25 December 2018
Published 30 January 2019 Volume 2019:12 Pages 953—958
DOI https://doi.org/10.2147/OTT.S186102
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjay Singh
Xiao-Ping Zou,1 Yao-Ying Jiang,1 Yan Liao,1 Yi-Wu Dang,1 Gang Chen,1 Zhen-Bo Feng,1 Yun Ma1,2
1Department of Pathology, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi Zhuang Autonomous Region, China; 2Department of Pathology, The Affiliated Tumor Hospital of Guangxi Medical University, Nanning, Guangxi Zhuang Autonomous Region, China
Abstract: A combination of a Wilms’ tumor (WT) and renal cell carcinoma (RCC) is an extremely rare pediatric renal neoplasm. Its prognosis and clinicopathological features remain unclarified. Herein, we describe a case of the coexistence of a WT and an RCC in a male child aged 5 years and 10 months. The child had symptoms of hematuria for more than 1 month. Although his irises were clear, medical imaging revealed a potential malignant tumor in the left kidney. The patient underwent resection of the left kidney. The pathological diagnosis was the coexistence of a WT and papillary RCC. Negative surgical margins were examined. One month following the resection, chemotherapy with vincristine plus dactinomycin (EE-4A regimen) was commenced. At the 69-month follow-up, there was no recurrence or metastasis. The coexistence of a WT and an RCC in the pediatric population is considered a rare pathological event. At present, there is no standard treatment for these renal neoplasms. In this study, the RCC treatment, which was the same as that applied in cases of WTs, was reasonable.
Keywords: papillary renal cell carcinoma, coexistence, renal cell carcinoma, nephroblastoma
Introduction
A Wilms’ tumor (WT) remains one of the most frequently detected kidney malignancies in infants and young children, with a higher morbidity rate among those aged 3–4 years.1,2 WTs are widely regarded as embryonal tumors, which stem from metanephrogenic blastema.3 A renal cell carcinoma (RCC) is a rare malignancy in children. Unlike WTs, most pediatric cases of RCCs are diagnosed in the second decade of life, and the possibility of morbidity increases with age.4 In most adults, the histological type is a clear cell RCC, whereas the histological subtypes of RCCs in children tend to be microphthalmia transcription factor translocation RCCs and papillary RCCs.1,5 To the best of our knowledge, the coexistence of a WT and an RCC is rare, with only a few cases reported in the literature.6–9 In this paper, we describe a case of the coexistence of a WT and an RCC, together with the clinicopathological features, treatment, and prognosis. In addition, a literature review of relevant studies was conducted with the aim of improving our understanding of this rare disease.
Case report
A boy aged 5 years and 10 months was admitted to the hospital on 13 October 2012 with recurring symptoms of hematuria for more than 1 month and weight loss of 1 kg. No urinary irritation symptoms, chills, fever, or rashes were observed. In the neonatal period, the boy had been well nourished. A physical examination revealed no enlargement of the tongue, umbilical hernia, iris loss, genitourinary malformation, or mental dysplasia. The kidneys did not seem to be swollen, and there appeared to be no pain when percussed or pressed. Similarly, pressing on the bladder area induced no pain, and no mass was found. There was no family history of nephroblastomas.
Blood and liver function tests revealed no obvious abnormalities. However, a routine urine test showed proteinuria and hematuria. A computed tomography (CT) scan revealed a 2.9 cm in diameter shadow of round, soft tissue in the mid pole of the left kidney, with an uneven density. Scattered calcifications were also detected (Figure 1). Color ultrasound imaging confirmed the presence of a hyperechoic mass (2.2×1.5 cm) in the mid pole of the kidney, with an intense blood signal (Figure 2). Emission computed tomography (ECT) pointed to the presence of space-occupying lesions in the left kidney, in which the blood supply was increased, suggesting a malignant tumor.
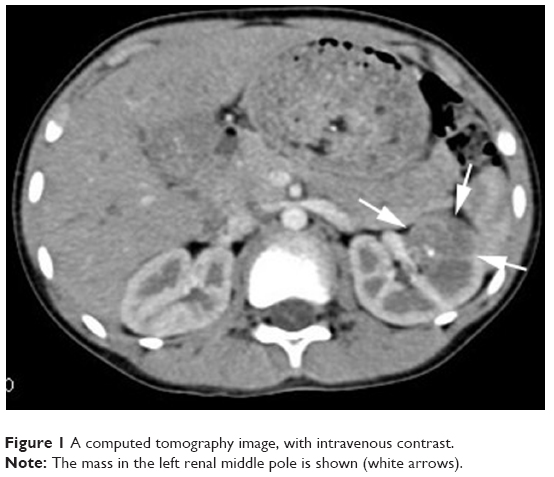 | Figure 1 A computed tomography image, with intravenous contrast. |
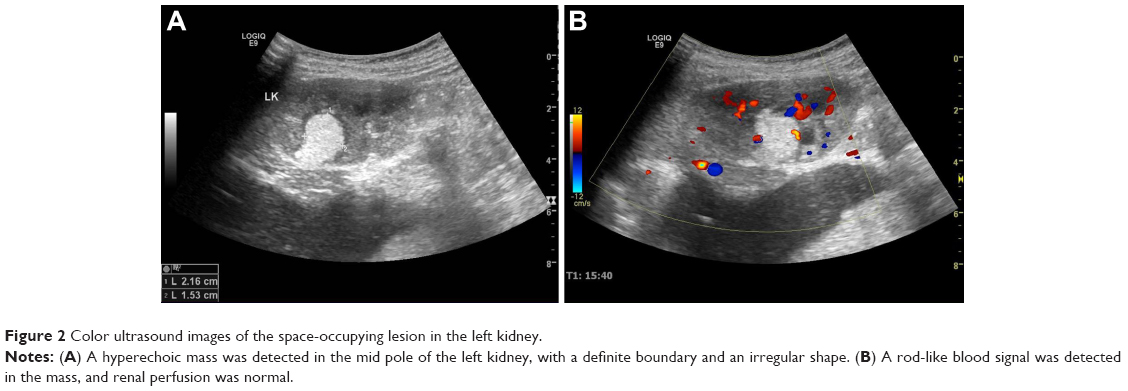 | Figure 2 Color ultrasound images of the space-occupying lesion in the left kidney. |
The patient underwent resection of the tumor on 22 October 2012. In the process of the resection, the tumor was seen to be located in the mid pole of the left kidney. The papillary tumor invaded the renal parenchyma and pelvis, but the kidney capsule and ureter were tumor free. Further, lymphadenectasis was not found surrounding the renal pedicle.
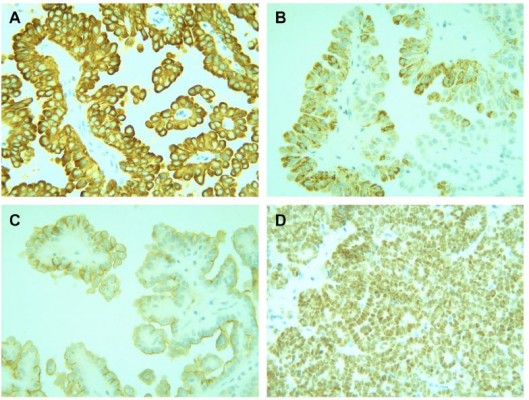
Gross examination showed a grayish-white irregular 2.5×2.5×2 cm sized mass in the left kidney, with a definite boundary. The surface of the mass was partly adhered to the kidney capsule. The cut surface showed that the tumor had spread from the renal cortex to the medulla and finally to the renal pelvis, with formation of several cysts. Some large cysts included grayish yellow papillary excrescences (Figure 3). Under a microscope, the tumor consisted of a papillary RCC and WT. The papillary RCC was located in the cyst, and the cancer cells were arranged in a papillary shape. The thin fibrovascular cores could be observed in these papillae, and some of the tumor tissue invaded the cyst wall (Figure 4A–C). The majority of the WT tissue was epithelium, and well-differentiated tiny tubes coexisted with poorly differentiated original tubular structures (Figure 4D and E). Loose connective tissues (Figure 4A) and immature mesenchymal tissues were discovered between the WT and papillary RCC (Figure 4F). The surgical margins were clear. As shown by immunohistochemical staining, the papillary RCC region was positive for cytokeratin 7 (CK7), alpha-methylacyl coenzyme A racemase, and cluster of differentiation 10 but negative for Wilms tumor gene 1 (WT1). The immunoprofile of WT region was contrary to the former (Figure 5). The pathological diagnosis was an epithelial WT coexisting with a papillary RCC in the left kidney.
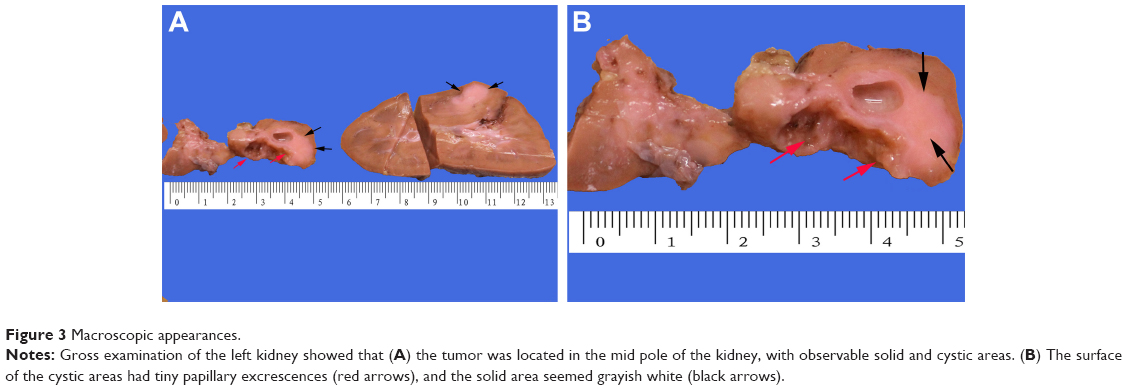 | Figure 3 Macroscopic appearances. |
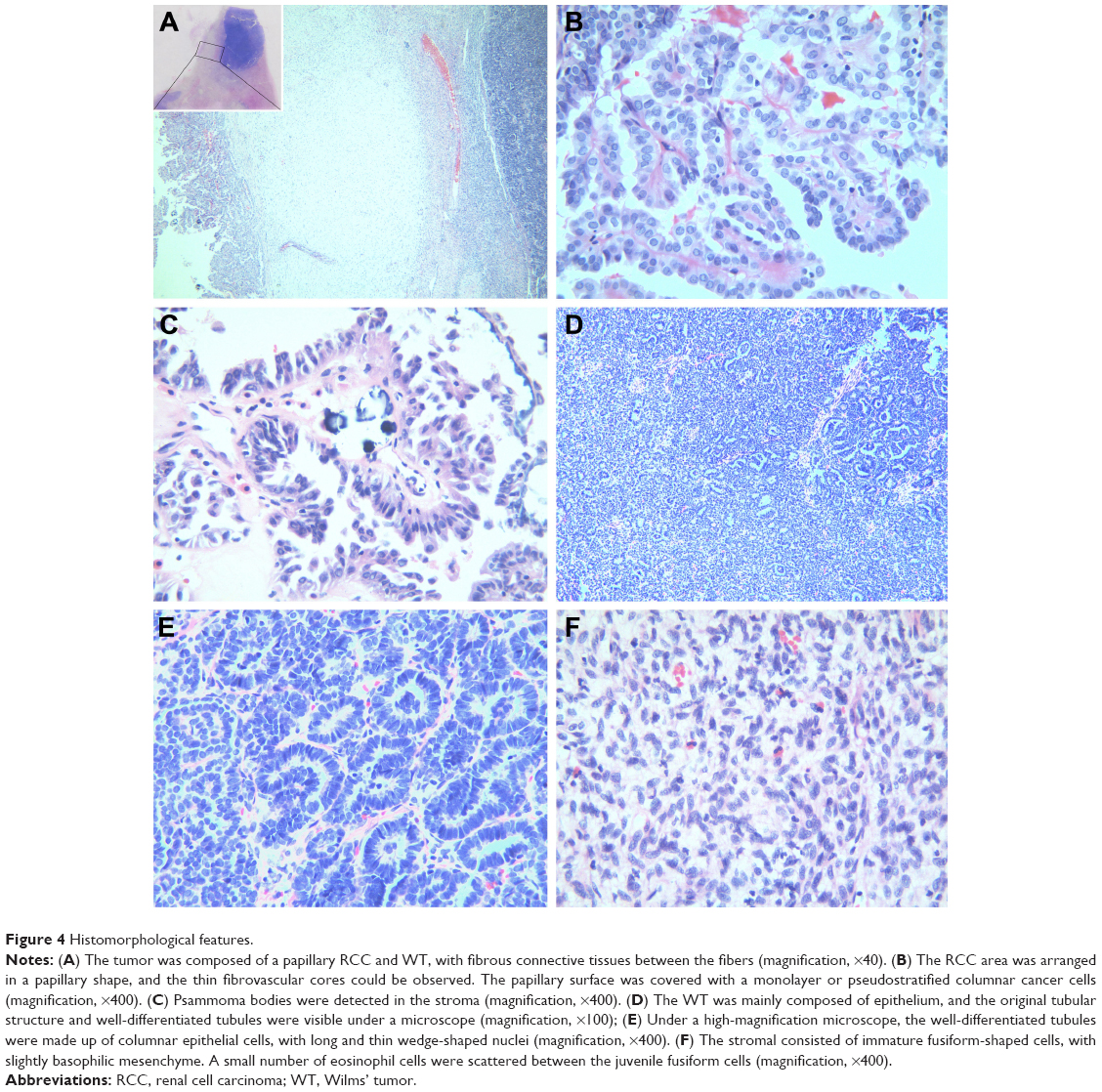 | Figure 4 Histomorphological features. |
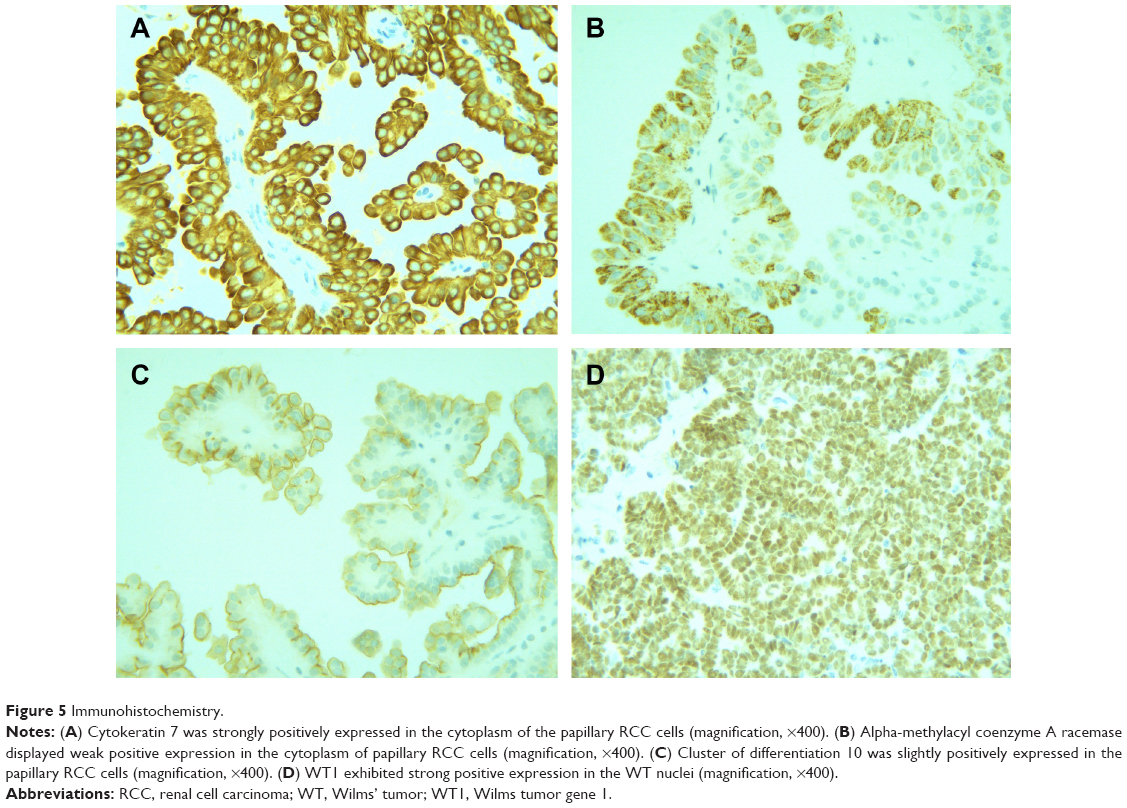 | Figure 5 Immunohistochemistry. |
According to the criteria of the National Wilms’ Tumor Study, the WT was classified as Stage I. Based on the pathological grading, the RCC was classified as Grade 2, and the TNM staging was T1aN0M0. Therefore, the treatment provided was in accordance with that for a Stage I WT (ie, radical nephrectomy of the left kidney, with complete resection of the tumor). After the operation, the patient recovered quickly. One month after the operation, the patient received vincristine plus dactinomycin (EE-4A regimen) as chemotherapy and underwent regular CT and B-mode ultrasound examinations. At the 69-month follow-up, there was no recurrence or metastasis.
This study was approved by the medical ethics committee of the First Affiliated Hospital of Guangxi Medical University. Written informed consent has been provided by the legal guardian of the patient to have the case details and any accompanying images published.
Discussion
The coexistence of a WT and an RCC is an extremely rare medical situation, with only nine similar cases published in the English literature (Table 1). These cases comprised five boys and four girls aged 1–14 years, with the average age being 4.8 years and median age being 3 years.6–9 The age of the patient in the present study with a WT coexisting with an RCC was 5 years and 10 months, which was consistent with the age of onset reported in the literature.
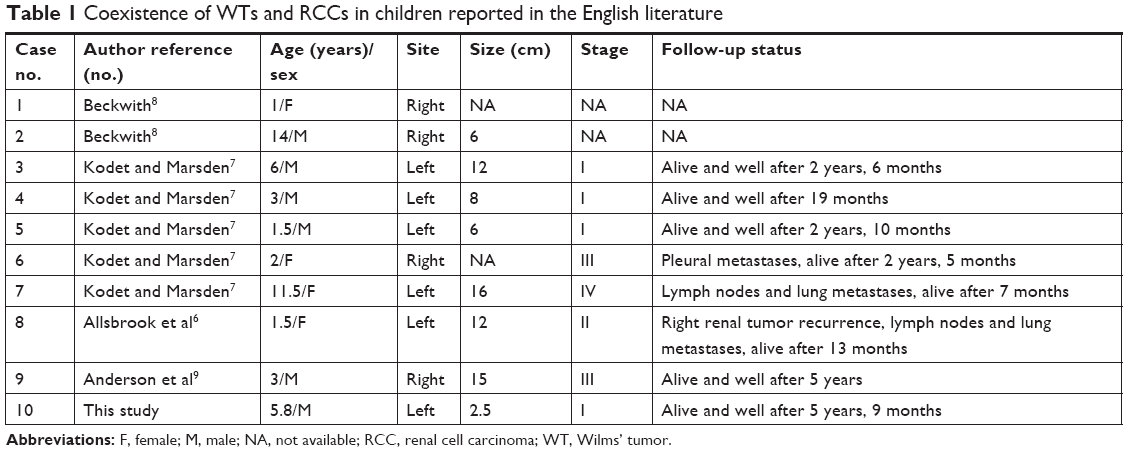 | Table 1 Coexistence of WTs and RCCs in children reported in the English literature |
The clinical features of a WT coexisting with an RCC are not specific and can be manifested as hematuria, fever, and an abdominal mass.9 Medical imaging aids the preoperative diagnosis, staging, and postoperative monitoring of renal tumors in children.3,10–12 In the present case, the patient had recurring hematuria and weight loss. A CT examination revealed a mass with an uneven density and scattered calcifications, which could aid the diagnosis of RCCs.13,14 Both color ultrasound and ECT showed increased vascularity and intense blood signals, indicating the possibility of a malignant tumor.
Prior to surgery, it is difficult to diagnose the coexistence of WTs and RCCs in children, and the diagnosis mainly depends on histology and immunohistochemical staining. A WT is composed of blastema, epithelium, and stromal. Pathologically, a WT is categorized into two types: a favorable histological type and an unfavorable histological type.15–17 In the present case, the WT largely consisted of epithelium, with little mesenchyme and no blastema. The gross examination showed few differences as compared with observations of other tumors, including pediatric RCCs, which are generally characterized by a small size, solid mass, necrosis, hemorrhages, and cystic changes.1 It also revealed several papillary excrescences in some large cysts. These were later confirmed as structures of the papillary RCC in the microscopic analysis. Occasionally, papillary RCCs exhibit similarities to epithelial WTs with papillary structures.5 In the present case, positive immunohistochemical staining of CK7 served as a marker of the papillary RCC. The area of the papillary RCC showed positive expression of CK7 and negative expression of WT1, which validated the diagnosis.
The existence of a biological relationship between RCCs and WTs has not been confirmed. However, since the first report of WT, many studies have discovered RCCs secondary to WTs.18–20 In a review article, the authors found that WT patients had a genetic predisposition to tumor formation, whereas others revealed that secondary RCCs were associated with radiotherapy and chemotherapy.18 In the present case, the WT and RCC were of a similar size, and the two tumors were separated by fibrous tissues, with no direct connection or transitions.
Due to their infrequency and short follow-up periods, the prognosis in cases of the coexistence of WTs and RCCs remains unclear. In patients with WTs, the overall survival rate was over 90%, and factors associated with a poor prognosis included an anaplastic type, more advanced stage, and loss of heterozygosity (LOH) for chromosomes 1p and 16q.15,21 The treatment for WTs is multidisciplinary and involves surgery and chemotherapy, which is individualized based on risk stratification.15 Biological markers associated with a poor WT prognosis include LOH at 1p/16q, LOH at 11q, LOH at 4p and 14q, 1q gain, MYCN amplification, and TP53 and WT1 mutations.21,22 The prognosis of RCCs in children is poorer than that in WT cases, with an overall survival rate of 64%. The tumor stage plays a vital role in the overall survival rate.1 In patients with RCCs insensitive to chemotherapy and radiotherapy, the appropriate treatment is radical nephrectomy, with complete resection of the tumor.23 In a previous study, the researchers suggested that the treatment applied in cases of WTs could also be applied in cases of WTs coexisting with RCCs, regardless of the size of each tumor.9 In the present case, the WT was the favorable histological type, and it was classified as Stage I according to the National Wilms’ Tumor Study criteria. The RCC was categorized as histological Grade 2, and the TNM stage was T1aN0M0. The patient was treated with radical nephrectomy, with complete resection of the tumors, in combination with chemotherapy and postsurgery. Regular medical imaging and the findings at a 69-month follow-up showed no recurrence of the tumors.
Conclusion
The coexistence of a WT and an RCC is a rare occurrence in children, and it is difficult to diagnose the coexistence of these tumors before surgery. The diagnosis of a WT coexisting with an RCC may be determined by histopathology and immunohistochemistry. No standard treatment is available owing to the infrequency of these renal neoplasms. The treatment for a WT seems appropriate in cases of a WT coexisting with an RCC.
Acknowledgment
The authors thank the Department of Radiology and Pediatric Surgery, the First Affiliated Hospital of Guangxi Medical University, for providing the imaging data and clinical information.
Disclosure
The authors report no conflicts of interest in this work.
References
George M, Perez-Rosello JM, Yikilmaz A, Lee EY. Pediatric urinary system neoplasms: an overview and update. Radiol Clin North Am. 2017;55(4):767–784. | ||
Walz AL, Ooms A, Gadd S, et al. Recurrent DGCR8, Drosha, and six homeodomain mutations in favorable histology Wilms tumors. Cancer Cell. 2015;27(2):286–297. | ||
Chung EM, Graeber AR, Conran RM. Renal tumors of childhood: radiologic-pathologic correlation part 1. The 1st decade: from the radiologic pathology archives. Radiographics. 2016;36(2):499–522. | ||
Pastore G, Znaor A, Spreafico F, Graf N, Pritchard-Jones K, Steliarova-Foucher E. Malignant renal tumours incidence and survival in European children (1978–1997): report from the automated childhood cancer information system project. Eur J Cancer. 2006;42(13):2103–2114. | ||
Cajaiba MM, Dyer LM, Geller JI, et al. The classification of pediatric and young adult renal cell carcinomas registered on the Children’s Oncology Group (COG) protocol AREN03B2 after focused genetic testing. Cancer. 2018;124(16):3381–3389. | ||
Allsbrook WC, Boswell WC, Takahashi H, et al. Recurrent renal cell carcinoma arising in Wilms’ tumor. Cancer. 1991;67(3):690–695. | ||
Kodet R, Marsden HB. Papillary Wilms’ tumour with carcinoma-like foci and renal cell carcinoma in childhood. Histopathology. 1985;9(10):1091–1102. | ||
Beckwith JB. Wilms’ tumor and other renal tumors of childhood: a selective review from the National Wilms’ Tumor Study Pathology Center. Hum Pathol. 1983;14(6):481–492. | ||
Anderson BW, Halverstadt DB, Culkin DJ, Johnson SF, Parkhurst JB, Kropp BP. Wilms’ tumor with renal cell carcinoma overgrowth in a 3-year-old child. Urology. 1998;52(1):128–130. | ||
McDonald K, Duffy P, Chowdhury T, McHugh K. Added value of abdominal cross-sectional imaging (CT or MRI) in staging of Wilms’ tumours. Clin Radiol. 2013;68(1):16–20. | ||
Downey RT, Dillman JR, Ladino-Torres MF, McHugh JB, Ehrlich PF, Strouse PJ. CT and MRI appearances and radiologic staging of pediatric renal cell carcinoma. Pediatr Radiol. 2012;42(4):410–417. | ||
Servaes S, Khanna G, Naranjo A, et al. Comparison of diagnostic performance of CT and MRI for abdominal staging of pediatric renal tumors: a report from the Children’s Oncology Group. Pediatr Radiol. 2015;45(2):166–172. | ||
Abdellah A, Selma K, Elamin M, et al. Renal cell carcinoma in children: case report and literature review. Pan Afr Med J. 2015;20:84. | ||
Young EE, Brown CT, Merguerian PA, Akhavan A. Pediatric and adolescent renal cell carcinoma. Urol Oncol. 2016;34(1):42–49. | ||
Davidoff AM. Wilms tumor. Adv Pediatr. 2012;59(1):247–267. | ||
Jurić I, Pogorelić Z, Kuzmić-Prusac I, et al. Expression and prognostic value of the Ki-67 in Wilms’ tumor: experience with 48 cases. Pediatr Surg Int. 2010;26(5):487–493. | ||
Vujanić GM, Sandstedt B. The pathology of Wilms’ tumour (nephroblastoma): The International Society of Paediatric Oncology Approach. J Clin Pathol. 2010;63(2):102–109. | ||
Rich BS, McEvoy MP, La Quaglia MP. A case of renal cell carcinoma after successful treatment of Wilms tumor. J Pediatr Surg. 2010;45(9):1883–1886. | ||
Lazarus J, Moolman C. Renal cell carcinoma as second malignancy in patient with previous Wilms tumor. Urology. 2009;74(3):598–600. | ||
Kraushaar G, Wiebe S. Renal cell carcinoma as a second malignant neoplasm in a patient with non-syndromic hemihypertrophy and previous Wilms tumor. Pediatr Radiol. 2005;35(12):1208–1211. | ||
Grundy PE, Breslow NE, Li S, et al. Loss of heterozygosity for chromosomes 1P and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms tumor Study Group. J Clin Oncol. 2005;23(29):7312–7321. | ||
Phelps H, Kaviany S, Borinstein S, Lovvorn H. Biological drivers of Wilms tumor prognosis and treatment. Children. 2018;5(11):145. | ||
Syed JS, Nguyen KA, Wu CQ, et al. Distinguishing pediatric and adolescent renal cell carcinoma from other renal malignancies. Pediatr Blood Cancer. 2017;64(5):e26315. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.