")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
The co-delivery of a low-dose P-glycoprotein inhibitor with doxorubicin sterically stabilized liposomes against breast cancer with low P-glycoprotein expression
Authors Gao W, Lin Z, Chen M, Yang X, Cui Z, Zhang X, Yuan L, Zhang Q
Received 16 October 2013
Accepted for publication 6 December 2013
Published 21 July 2014 Volume 2014:9(1) Pages 3425—3437
DOI https://doi.org/10.2147/IJN.S56070
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Wei Gao,1 Zhiqiang Lin,1 Meiwan Chen,2 Xiucong Yang,1 Zheng Cui,1 Xiaofei Zhang,1 Lan Yuan,3 Qiang Zhang1
1State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing, 2State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macau, 3Medical and Healthy Analytical Center, Peking University, Beijing, People's Republic of China
Introduction: P-glycoprotein (P-gp) inhibitors are usually used to treat tumors that overexpress P-gps. However, most common types of breast cancers, such as Luminal A, are low-P-gp expressing, at least during the initial phases of treatment. Therefore, it would be interesting to know if P-gp inhibitors are still useful in treating low-P-gp-expressing tumors.
Methods: In the study reported here, the human breast-cancer cell line MCF-7, chosen as a model of Luminal A, was found to be low-P-gp expressing. We designed a novel doxorubicin (DOX) sterically stabilized liposome system co-loaded with the low-dose P-gp inhibitor cyclosporine A (CsA) (DOX/CsA/SSL).
Results: The co-delivery system showed good size uniformity, high encapsulation efficiency, and a desirable release profile. The cell-uptake and cytotoxicity studies demonstrated that CsA could significantly enhance the intracellular accumulation and toxicity of free DOX and the liposomal DOX in MCF-7 cells. The confocal microscopy and in vivo imaging study confirmed the intracellular and in vivo targeting effect of DOX/CsA/SSL, respectively. Finally, the in vivo study proved that DOX/CsA/SSL could achieve significantly better antitumor effect against MCF-7 tumor than controls, without inducing obvious systemic toxicity.
Conclusion: This study demonstrated that the co-delivery of a low-dose P-gp inhibitor and liposomal DOX could improve the therapy of low-P-gp-expressing cancer, which is of significance in clinical tumor therapy.
Keywords: liposomes, low-P-gp-expressing tumor, antitumor activity, cyclosporine A, targeted delivery
Introduction
Tumor therapy is currently far from satisfactory in clinical practice.1 A major factor in this problem is the expression of adenosine triphosphate-binding cassette transporters, mainly P-glycoproteins (P-gps), on tumor cell membranes.2 When nanomedicines deliver drugs to tumor sites, some parts of the drugs are expelled by the P-gps expressed on cell membranes.2,3 This inevitably lowers the therapeutic efficiency of the treatment, and cancer cells soon develop resistance to a variety of drugs.4 Thus, the inhibition of P-gp function is an effective strategy to enhance the intracellular concentration and therapeutic efficacy of nanomedicines.5 Nowadays, some P-gp inhibitors have attracted researchers’ attention, and many small-molecule P-gp inhibitors have been investigated in combination with chemotherapeutics in clinical application.6
The long-term use of chemotherapy drugs increases the expression of P-gps in tumor cells, and induces multidrug resistance (MDR).7 Extensive studies are focusing on using P-gp inhibitors to reverse MDR in cancers that overexpress P-gps.6 However, large doses of the P-gp inhibitor and chemotherapy drugs are required in the treatment of MDR, thus inducing severe side effects in vivo.6 However, most common type of cancers, such as breast cancer Luminal A, are not P-gp negative, but express low levels of P-gps, at least during the initial phase of treatment.8,9 Thus, we were interested in finding out whether a low level of P-gps influences the antitumor efficiency of chemotherapy drugs or nanoparticles loaded with chemotherapy drug, and if P-gp inhibitors are still useful in treating tumors that express P-gps at a low level when combined with chemotherapy drugs.
Cyclosporine A (CsA), a well-known P-gp inhibitor, shows significant effects in terms of decreasing the efflux of doxorubicin (DOX) in tumor cells.10 The co-administration of CsA and chemotherapeutic agents has elicited promising clinical results in acute myeloid leukemia.11 However, CsA cannot be used long-term because of its immunosuppressive effects as well as severe nephrotoxicity at high doses.12
Liposomal DOX (Doxil®; Janssen Pharmaceuticals, Inc., Titusville, NJ, USA), the first nanomedicine approved by the US Food and Drug Administration,13 has been used to treat cancer for more than two decades. Liposomes can prolong the circulation time of DOX in vivo, accumulate relatively more of the drug at the tumor site specifically, are internalized into the tumor cells effectively, and penetrate deeply into the tumor parenchyma through the enhanced permeability and retention (EPR) effect. In long chemotherapy terms, the therapetic efficacy of liposomal DOX does not improve much comparing to the efficacy of free DOX.1
In this study, we sought to improve the antitumor efficiency of the liposomal drug in low-P-gp-expressing tumors by simultaneously using a P-gp inhibitor. To this end, we prepared a novel DOX liposome system co-loaded with a low-dose of CsA (DOX/CsA/sterically stabilized liposome system [SSL]). MCF-7 was chosen as a cell model, representing the most common type of breast cancer. First, we evaluated the effect of CsA on cell uptake and the cytotoxicity of free DOX and liposomal DOX. The in vitro intracellular targeting effect of DOX/CsA/SSL was tested by confocal microscopy, and the in vivo tumor target effect was observed by in vivo imaging. Finally, we investigated the anticancer effect of DOX/CsA/SSL in vivo, as well as the systemic toxicity.
Materials and methods
Materials and animals
DOX was purchased from Hisun Pharmaceutical Co, Ltd (Zhejiang, People’s Republic of China) as doxorubicin hydrochloride. CsA was obtained from Taizhou Pharmaceutical Co, Ltd (Zhejiang, People’s Republic of China). DSPE-PEG (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-n-[methoxy(polyethylene glycol)-2000] was provided by the NOF Corporation (Tokyo, Japan). Cholesterol (Chol) and Sephadex® G-50 were obtained from Pharmacia Biotech Inc. (Piscataway, NJ, USA). Lipoid E 80 (PC) was purchased from Lipoid GmbH (Ludwigshafen, Germany). Sulforhodamine B (SRB) and Tris base were purchased from Sigma-Aldrich (St Louis, MO, USA). Fluorescein isothiocyanate (FITC) mouse antihuman P-gp 17F9 and FITC mouse IgG2bκ isotype control were purchased from BD Biosciences (San Diego, CA, USA), while 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR) and Hoechst 33258 were obtained from Molecular Probes Inc. (Eugene, OR, USA). All other reagents used were of analytical grade.
The human breast-cancer cell line MCF-7 and HeLa cervical cancer cell line were obtained from the Institute of Basic Medical Science (Beijing, People’s Republic of China). The DOX-resistant breast-cancer cell line MCF-7/ADR was kindly supplied by the Institute of Hematology and Blood Diseases Hospital (Chinese Academy of Medical Sciences and Peking Union Medical College, Tianjin, People’s Republic of China). The cell-culture medium Roswell Park Memorial Institute (RPMI)-1640, fetal bovine serum, and antibiotics (penicillin 100 U/mL and streptomycin 100 mg/mL) were obtained from MacGene Biotech Co, Ltd (Beijing, People’s Republic of China).
Healthy female BALB/c nude mice, approximately 6 weeks old and weighing 18–20 g, were supplied by the Peking University Health Science Center (Beijing, People’s Republic of China). The mice were kept in specific pathogen free (SPF) conditions for 1 week before use. All animal experiments conducted in this study complied with the Principles of Care and Use of Laboratory Animals, prepared by the Peking University Institutional Animal Care and Use Committee.
Preparation of liposomes
DOX-loaded liposomes
Liposomes were prepared by the hydration of dried lipid films as previously reported.14 DOX was loaded in the liposomes using the ammonium sulfate gradient method.15 The lipid components were PC:Chol:DSPE-PEG2000 (24.0:1.4:5.4, weight/weight [w/w]). Briefly, the lipid components were weighed and dissolved in chloroform. The mixture was evaporated under vacuum in a rotary evaporator until a thin lipid film formed. Ammonium-sulfate solution (123 mM) was then added. The mixture was sonicated using a bath-type sonicator to obtain empty liposomes. The liposomes were then eluted with phosphate-buffered saline (PBS; 120 mM) using a Sephadex G-50 column. DOX (DOX:PC =1:24, w/w) was added and incubated for 20 minutes at 37°C with continuous shaking. The liposomes were eluted with PBS using a Sephadex G-50 column to remove the free DOX. The DOX liposomes (DOX/SSL) were stored at 4°C.
DOX and CsA co-loaded liposomes
DOX/CsA/SSL was prepared using the method described above, but with the addition of CsA in the lipid components. The formulation was PC:Chol:DSPE-PEG2000:CsA (24.0:1.4:5.4:1, w/w). Free CsA was removed in the elution step using a Sephadex G-50 column.
DiR-loaded liposomes
For the in vivo imaging study, DiR-loaded liposomes (DiR/SSL) and DiR and CsA co-loaded liposomes (DiR/CsA/SSL) were prepared using the method just described, but with the addition of the near-infrared fluorescent probe DiR to the lipid components at the desired amount. PBS was used as the hydration medium. The obtained liposomes were eluted with PBS to remove the free DiR.
Characterization of liposomes
Size distribution and zeta potential
The size, polydispersity, and zeta potential of the obtained liposomes were measured by means of dynamic light scattering using a Malvern Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). The zeta potential of the liposomes was determined by electrophoretic light scattering using a Malvern Zetasizer Nano ZS (Malvern Instruments, Malvern UK). The morphology of the liposomes was identified by transmission electron microscope (TEM) using a negative staining method.
Encapsulation efficiency
The prepared liposomes were solubilized in methanol (liposomes:methanol =1:9, volume/volume [v/v]). The DOX concentration was determined by ultraviolet (UV)–visible spectrophotometry at 480 nm. The CsA concentration was determined using an Agilent 1100 Series HPLC System (Agilent Technologies, Inc., Santa Clara, CA, USA) with a UV detector (LC-10AT, Shimadzu Corp, Kyoto, Japan). An octadecylsilyl column (Agilent Zorbax® SB-C18; Agilent Technologies, Inc.; 4.6 × 250.0 mm, 5 μm) was used for the analysis. The mobile phase was water (0.1% trifluoroacetic acid) and acetonitrile (30:70, v/v). The UV detection wavelength was set at 220 nm, the column temperature was 70°C, and the flow rate was 1.0 mL/minute. The encapsulation efficiency (%EE) was calculated according to Equation 1. The data were obtained using triplicate liposome preparations.
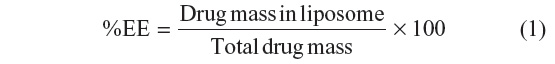
Drug release
The in vitro DOX and CsA release from liposomes was measured with a dialysis method. DOX/CsA/SSL was dissolved in PBS supplemented with 0.05% sodium dodecyl sulfate. The mixture was placed in a dialysis bag (MW cut off 12,000–14,000), which was sealed at both ends with clips. The dialysis bag was sunk into a beaker with 50 mL PBS. This was then was incubated at 37°C with continuous shaking at a speed of 100 times per minute for 24 hours. At 0.5, 1, 2, 4, 6, 8, 12, 24 hours, samples were withdrawn and replaced with an equal volume of medium. The CsA content in the released medium was determined by high-performance liquid chromatography (HPLC), as described earlier. The DOX content was also determined by HPLC using the LC-10AT UV detector, the analysis was performed using the octadecylsilyl column. The mobile phase consisted of 500 mL of water containing 1.44 g sodium dodecyl sulfate and 0.68 mL phosphoric acid (H3PO4), methanol, and acetonitrile (40:5:50, v/v). The detection wavelength was set at 233 nm, the flow rate was 1.0 mL/minute, and the column temperature was 25°C.
P-gp expression
The expression of P-gps in MCF-7, MCF-7/ADR, and HeLa cells was studied by flow cytometry using an FITC-labeled 17F9 monoclonal P-gp antibody (BD Biosciences). The cells (circa 1 × 106) were harvested, washed with PBS, and fixed by adding 1 mL 4% (v/v) of paraformaldehyde solution for 15 minutes at room temperature. FITC mouse antihuman P-gp 17F9 monoclonal antibody and its isotype control were then added and the cells were incubated for 90 minutes at 4°C. After this, the cells were washed with PBS and examined by flow cytometry using the BD FACScan™ System (Becton Dickinson, San Jose, CA, USA). The P-gp expression level was determined by the ratio of the mean fluorescence intensity (MFI) value to the isotype MFI value.16,17
Cellular-uptake studies
The cellular-uptake studies of various DOX formulations were performed by flow cytometry and confocal microscopy observation. For flow-cytometric analysis, MCF-7 and MCF-7/ADR cells were cultured on 12-well plates at 37°C for 24 hours. When cells reached approximately 80% confluence, the medium was removed and the cells washed with PBS. The DOX formulations (DOX concentration 35 μg/mL) diluted with serum-free media were subsequently added and incubated for 3 hours. The cells were then harvested for flow-cytometric analysis using the FACScan. The excitation and emission wavelengths of DOX were 488 and 560 nm, respectively. Ten thousand gated events were collected and analyzed with the FCS Express V3 software.
For confocal microscopy analysis, MCF-7 cells were seeded on glass coverslips in 24-well plates for 24 hours until total adhesion. Various DOX formulations were added and incubated for 3 hours as just described. The cells were then washed with cold PBS and fixed with 4% (v/v) paraformaldehyde solution for 20 minutes at room temperature. The cell nuclei were then stained with Hoechst 33258 for 15 minutes. Confocal microscopy analysis was performed using a laser-scanning confocal microscope (TCS SP5; Leica, Solms, Germany). The excitation and emission wavelengths of DOX were 480 and 555–590 nm, respectively, while the excitation and emission wavelengths of Hoechst 33258 were 405 and 425–465 nm, respectively.
Cytotoxicity assay
The cytotoxicity of various DOX formulations to the MCF-7 cell line was tested by SRB colorimetric assay. Briefly, MCF-7 cells (2,000–3,000 cells/well) were incubated in 96-well plates at 37°C overnight. Various concentrations of free DOX, CsA plus DOX, DOX/SSL, and DOX/CsA/SSL were added and the plates were incubated at 37°C for another 24 hours. Following this, the cells were fixed by cold trichloroacetic acid, then washed and dried in the air. SRB dye (0.4%) was applied to each well and allowed to stain for 15 minutes at room temperature. The excess dye was washed off with 1% acetic acid and the bound dye was dissolved in 10 mM Tris-base solution and measured at 540 nm using a the Thermo Scientific Multiskan® FC Microplate Photometer (Shanghai, People’s Republic of China). Drug inhibition curves were generated and the drug concentration inhibiting the cell growth by 50% (half-maximal inhibitory concentration [IC50]) was obtained from semi-logarithmic dose–response plots.
Observation of intercellular delivery of DOX/CsA/SSL
FITC is a typical fluorescence probe widely applied in labeling peptides.18 In our study, CsA was first conjugated to ethylenediamine (data not shown), and then reacted with FITC to obtain FITC-CsA following the manufacturer’s (BD Biosciences) protocol. DOX and FITC-CsA co-loaded liposome (DOX/FITC-CsA/SSL) was prepared as DOX/CsA/SSL. MCF-7 cells were seeded on a glass-bottomed cell-culture dish for 24 hours until total adhesion. DOX/CsA/SSL was then added. At preset time points, the living cells were washed and observed by confocal microscopy. The excitation wavelengths of DOX, FITC, and Hoechst 33258 were 480, 488, and 405 nm, respectively, while the detection wavelengths were 555–590, 510–540, and 425–465 nm, respectively.
In vivo imaging
In vivo fluorescence imaging was performed using an in vivo image system (FX Pro; Kodak, Rochester, NY, USA). The MCF-7-cell-bearing female BALB/c nude mice models were established by subcutaneous inoculation of MCF-7 cells in the right flanks of nude mice. The imaging experiment was performed when the tumor volume reached circa 150 mm3. The mice were randomly assigned to three groups (three animals per group) and injected via the tail vein with free DiR, DiR/SSL, or DiR/CsA/SSL according to which group they were in. At predetermined time intervals, the mice were anesthetized and fluorescence images captured by the in vivo FX Pro imaging system. The images were analyzed using Carestream Molecular Imaging software (v 5.0.7.23; Carestream Health Inc., Rochester, NY, USA). The relative DiR tumor distributions were quantified by the sum intensity of DiR in tumors divided by the sum intensity of DiR in whole bodies.
In vivo antitumor efficacy
The antitumor efficacy study was performed on MCF-7-tumor cell-bearing mice as described. Treatments were initiated when the average tumor volume reached 50–100 mm3. The mice were randomly assigned into four groups (five animals per group), with one group each treated with 5% glucose (control), DOX solution (DOX 2 mg/kg), DOX/SSL (DOX 2 mg/kg), and DOX/CsA/SSL (DOX 2 mg/kg). The treatments were injected via the tail vein every 2 days a total of five times. Tumor volume (mm3) and mice weights were measured and recorded every 2 days. Tumor volume was measured and calculated as [(major axis) × (minor axis)2]/2. Mice were sacrificed on the eleventh day, and the tumors were excised and weighed. Hearts and kidneys were also excised to make hematoxylin and eosin staining sections for evaluation of cardiotoxicity and nephrotoxicity after drug treatments.
Statistical analysis
The experiments in this study were all performed at least three times. Quantitative data are expressed as the mean ± standard deviation. Differences between groups were analyzed by two-tailed Student’s t-test for pairs. Statistical significance was set for a P-value, with cases lower than 0.05 considered statistically significant and those lower than 0.01 highly significant.
Results and discussion
Characterization of liposome systems
We designed a novel liposome co-loaded with CsA and DOX (DOX/CsA/SSL). The liposomes were prepared by a thin-film hydration method. As shown in the schematic illustration, Figure 1A, the lipophilic compound CsA was located in the bilayer of the liposomes, while the hydrophilic drug DOX was loaded in the aqueous phase of the liposomes, in the form of aggregated and gelatinous anthracycline sulfate salt. The average diameter of the DOX/CsA/SSL liposomes was about 99 nm with good uniformity (polydispersity index =0.245) determined by Malvern Zetasizer Nano ZS (Figure 1B). Such a size might be optimal for tumor targeting by the EPR effect.19 The typical TEM image in Figure 1C shows the morphology of the DOX/CsA/SSL. The particles observed by TEM were of good uniformity and their diameter was smaller than 100 nm. The DOX/CsA/SSL was electrically neutral due to the lipid materials used. In addition, DOX-loaded liposomes (DOX/SSL) were prepared with the same process. As shown in Table 1, the characteristics of the DOX/SSL were similar to those of DOX/CsA/SSL. DiR, a near-infrared fluorescent dye commonly used in in vivo imaging, was used to label the liposomes. Both the size and zeta potential of the DiR-loaded liposomes exhibited no significant difference from those of the DOX/CsA/SSL (data not shown). The %EE of the DOX and CsA in the DOX/CsA/SSL was 88% and 75%, respectively. The various liposomes were stored at 4°C. There were no significant changes in size, potential, or %EE for at least 4 weeks.
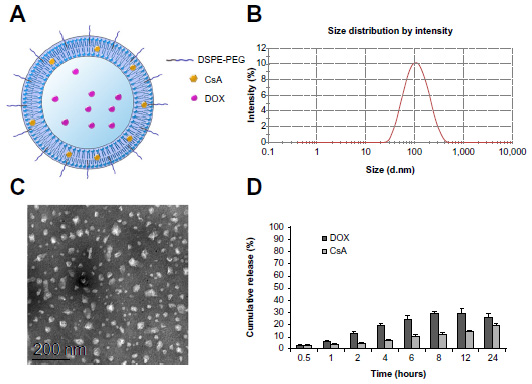 | Figure 1 Characterization of the sterically stabilized liposomes co-loaded with cyclosporine A (CsA) and doxorubicin (DOX) (DOX/CsA/SSL). (A) Schematic illustration. (B) Representative particle-size distribution. (C) Typical transmission electron microscope image. Scale bar is 200 nm. (D) In vitro release profiles of both DOX and CsA from DOX/CsA/SSL in phosphate-buffered saline with 0.05% sodium dodecyl sulfate. |
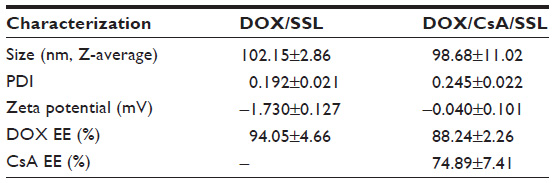 | Table 1 Characterization of doxorubicin (DOX)-loaded liposomes (n=3) |
Figure 1D shows the in vitro release profiles of DOX and CsA from the DOX/CsA/SSL. Both drugs were released slowly from the liposomes, without burst effect, and the release of CsA and DOX was nearly simultaneous. The total cumulative release of DOX and CsA was approximately 30% and 20%, respectively, indicating that most of the drugs were maintained in the liposomes for 24 hours. This result proved the stability of DOX and CsA in the liposomes. The stability of liposome in circulation is also an important characteristic because this stability is the prerequisite of the long circulation and the EPR effects. The instability of liposomes in plasma may cause the release of the encapsulated drug. Previous reports have proven that modifying polyethylene glycol (PEG) on the surface of liposomes can significantly reduce the interference of serum proteins and increase liposome stability in blood circulation.20 In our study, PEG was added to the liposome formulation (17% of the total weight before the addition of DOX). This amount of PEG could cover the surface of the liposomes, increasing the stability of the DOX liposomes in circulation.21 Further, both CsA and DOX are substrates of P-gps and can be transported by P-gps. CsA inhibits the efflux of DOX by competitively binding to P-gps.22 CsA could only enhance the accumulation of DOX when simultaneously located in the tumor cells with DOX. Thus, the maintenance of the CsA and DOX in the liposomes and the similar release properties of the two drugs would guarantee that both drugs would arrive and be released simultaneously at the tumor site.
P-gp expression in MCF-7 cell line
The human breast-cancer cell line MCF-7 was used as the low-P-gp-expressing cell line in our study. The flow-cytometry study was performed to assess the P-gp expression of MCF-7, MCF-7/ADR, and HeLa cells. Figure 2A–C show the histograms of MCF-7, MCF-7/ADR, and HeLa, respectively, after incubation with the P-gp antibody (17F9). The level of P-gp expression was determined by the ratio of the MFI value of each sample to the isotype MFI value (Figure 2D).16,17 The MFI of MCF-7 was significantly higher than its isotype (18.11 versus [vs] 9.06), indicating that most of the cells expressed P-gps.16,17 It should be noted that the curve was partially overlaid with the isotype control, and the ratio of the MFI value to the isotype was 2.0, indicating a relatively low expression level on MCF-7 cells.23 Multidrug-resistant MCF-7/ADR cells were used as a positive control,24 and the ratio of the MFI to isotype for these was 7.0, much higher than for the MCF-7 cells. HeLa was a P-gp-negative cell line, and there was no significant difference between its MFI value and that of its isotype (3.75 vs 3.36). The P-gp expression results demonstrated that MCF-7 could be used as a cell model of low P-gp expression. In many studies, MCF-7 has been used as a negative control of MCF-7/ADR to study MDR,25 but in the study reported here, it was found that MCF-7 only expressed P-gps at a low level. It seems that this level of P-gp expression in MCF-7 cannot be ignored, even though this cell line is reported to be sensitive to chemotherapy.26
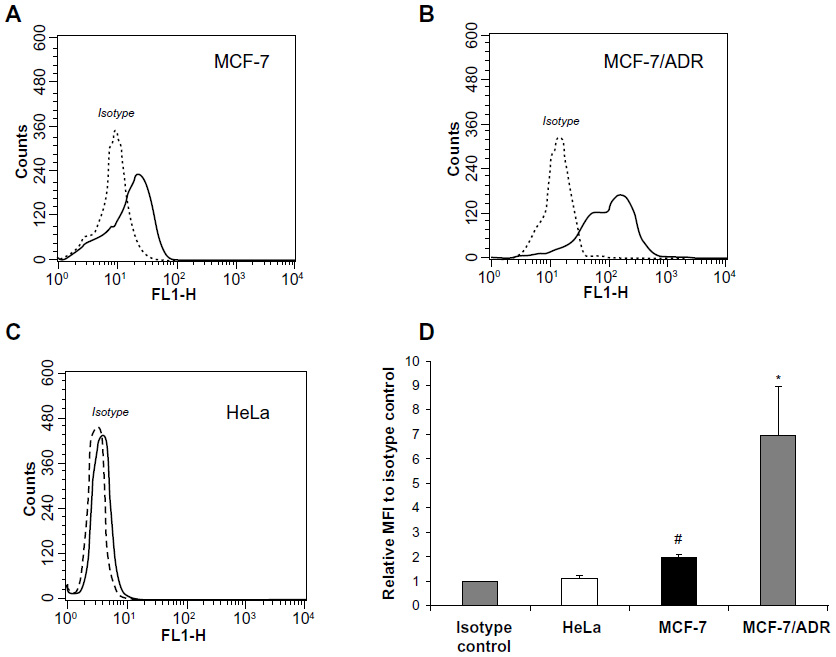 | Figure 2 P-glycoprotein (P-gp) expression. Representative overlay histograms of antihuman P-gp 17F9 monoclonal antibody binding in (A) MCF-7, (B) MCF-7/ADR, and (C) HeLa cells with isotype control (dashed line) and fluorescein isothiocyanate-labeled 17F9 (solid line). (D) P-gp expression level. Relative mean fluorescence intensity (MFI) to isotype control was calculated by the MFI of each cell line divided its isotype control MFI. |
Effect of CsA on cell uptake of free DOX and liposomal DOX
First, we studied the effect of free CsA on the internalization of free DOX in MCF-7 and MCF-7/ADR cells by flow cytometry. As seen in Figure 3, the DOX level in the MCF-7/ADR cell line was lower than that in MCF-7, due to the different levels of P-gp expression. After the addition of free CsA, the level of DOX in MCF-7/ADR substantially increased by 1.7-fold. This result is identical with previous reports that CsA can reverse MDR by inhibiting the cytotoxic drug efflux by P-gps.27,28 However, the effect of CsA in MCF-7 has been barely considered in previously studies as far as we know. We found that CsA could also profoundly increase the DOX level in MCF-7 (1.3-fold), thus indicating the effect of P-gps on chemotherapy. In other words, it was demonstrated that the outward transport of DOX by the low-P-gp-expressing MCF-7 cells should be considered.
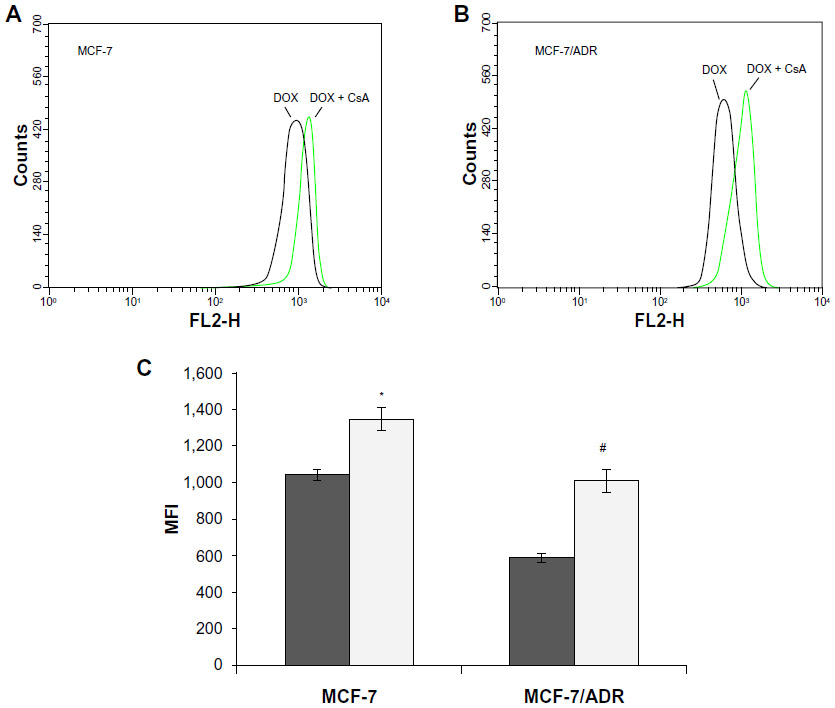 | Figure 3 Flow-cytometry studies on cell uptake of free doxorubicin (DOX). Flow-cytometry curves of (A) MCF-7 and (B) MCF-7/ADR cells. (C) Fluorescence intensity graph of intracellular DOX in MCF-7 cells and MCF-7/ADR cells after being incubated with DOX and DOX plus cyclosporine A (CsA) for 3 hours at 37°C.a |
Second, the effect of CsA on the cell uptake of liposomal DOX was investigated by flow cytometry and confocal microscopy. In the experiments, CsA was either added as free drug (DOX/SSL + CsA) or loaded in the liposome system (DOX/CsA/SSL). As shown in Figure 4A and B, the uptake of DOX in the DOX/CsA/SSL and DOX/ SSL + CsA groups was significantly higher than that in the DOX/SSL group, indicating that CsA could increase the uptake of liposomal DOX. The confocal images (Figure 4C) also show that almost all of the cells in the DOX/CsA/SSL and DOX/SSL + CsA groups exhibited an increment of fluorescence signal, which was due to the low expression of P-gps on most of the MCF-7 cells. Moreover, there was a significant increase in the uptake of DOX in the DOX/CsA/SSL group compared with the DOX/SSL + CsA group, which was probably due to the simultaneous cell uptake of DOX and CsA in the form of liposomes in the DOX/CsA/SSL group. In the DOX/CsA/SSL group, DOX and CsA were loaded in the same liposomes. The liposomes simultaneously delivered both drugs into the cells. When DOX and CsA enters the cells at the same time, the CsA could competitively bind to P-gps, reducing the efflux of DOX. If CsA enters the cells before DOX, it could be effluxed directly by P-gps. As a result, DOX and CsA co-loaded in liposomes could maximize the P-gp inhibition effect of CsA.
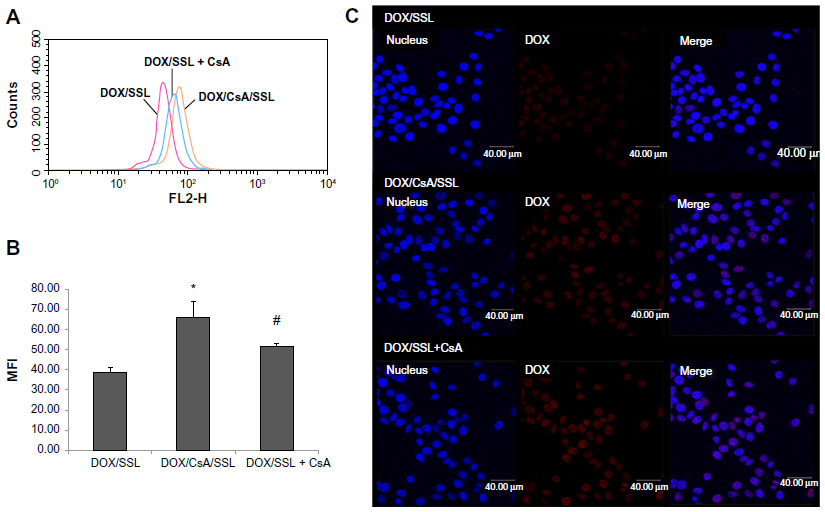 | Figure 4 Cell-uptake studies of liposomal doxorubicin (DOX). (A) Flow-cytometry curves of MCF-7 cells and (B) quantification of intracellular DOX after incubation with DOX/SSL, DOX/CsA/SSL, and DOX/SSL plus cyclosporine A (CsA) for 3 hours at 37°C. (C) Confocal microscopy images of MCF-7 incubated with DOX/SSL, DOX/CsA/SSL, and DOX/SSL plus CsA for 3 hours at 37°C, respectively.** |
Effect of CsA on the cytotoxicity of free DOX and liposomal DOX
The effect of CsA on the cytotoxicity of free DOX was further tested in MCF-7 cells. Figure 5A shows the inhibition rates of DOX when 2 or 10 μg/mL CsA was added. The corresponding IC50 values are listed in Table 2. It was found that adding CsA increased the cytotoxicity of free DOX significantly and the effect of CsA was concentration-dependent. This phenomenon indicated that the cytotoxicity of free DOX was affected by P-gp-mediated efflux in low-P-gp-expressing cell lines.
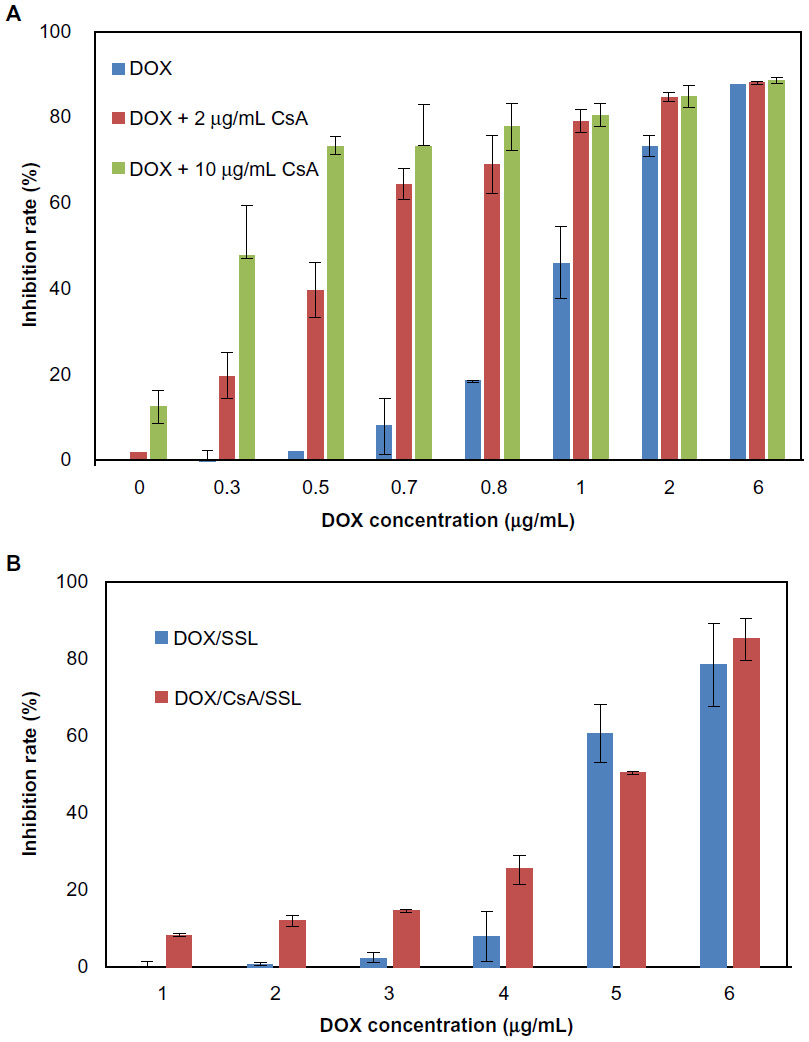 | Figure 5 In vitro cytotoxicity assay. (A) The inhibition ratio of doxorubicin (DOX), DOX plus 2 μg/mL cyclosporine A (CsA), DOX plus 10 μg/mL CsA against MCF-7 cells for 24 hours. (B) Inhibition ratio of DOX/SSL and DOX/CsA/SSL against MCF-7 cells for 24 hours. |
 | Table 2 Cytotoxicity of various doxorubicin (DOX) formulations against MCF-7 cells (n=6) |
The cytotoxicity of the DOX/SSL and DOX/CsA/SSL was also tested in MCF-7 cells. Figure 5B and Table 2 show the inhibition rates and IC50 values, respectively, of the DOX/CsA/SSL and DOX/SSL. The cytotoxicity of the DOX/CsA/SSL was found to be significantly higher than that of DOX/SSL, indicating the advantage of the DOX/CsA/SSL in terms of antitumor efficacy in MCF-7 cells. This was in accord with the findings of the cell-uptake study.
Conclusively, the low expression of P-gps on MCF-7 cells influenced the intracellular accumulation of DOX. CsA could largely increase the cell uptake and cytotoxicity of DOX and liposomal DOX. The result indicates that P-gp inhibitors in combination with cytotoxic drugs might be favorable in the treatment of MCF-7 tumors, as well as other low-P-gp-expressing tumors. Liposomal drugs, such as liposomal DOX, have become favorable alternatives to conventional small-molecular drugs in cancer therapy. As such, we further evaluated the targeting effect and in vivo antitumor efficacy of our CsA and DOX co-loaded liposomes.
Intracellular targeted delivery of DOX/CsA/SSL
To find out about the intracellular delivery characteristics of both drugs, we observed the internalization process of DOX/CsA/SSL using a confocal microscope. As shown in Figure 6, the red color represents DOX, the green CsA, and the yellow in the merge group represents the co-localization of DOX and CsA in liposomes. Liposomes carrying both drugs were first adsorbed on the surface of cells (yellow color). After 210 minutes, the liposomes successfully delivered the two drugs to their therapeutic target organelles: DOX to the nucleus, and CsA to the cell membrane and cytoplasm. The results prove that the DOX/CsA/SSL can achieve intercellular targeted delivery of both drugs at the same time, which is important to achieve the synergetic effect of the two drugs.
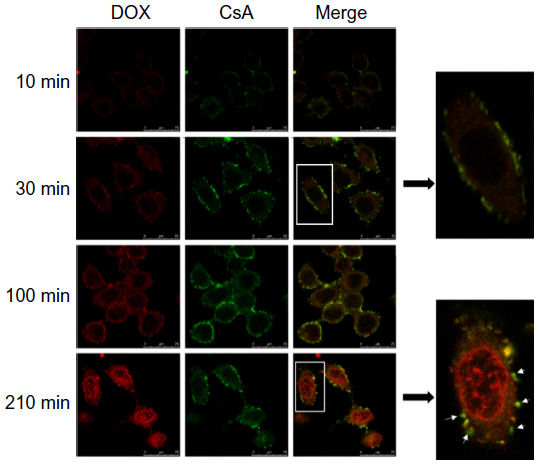 | Figure 6 The intracellular delivery of DOX/FITC-CsA/SSL. MCF-7 cells were incubated with DOX/FITC-CsA/SSL at 37°C. At preset time points, the living cells were washed and observed by confocal microscopy. |
In vivo imaging assay
The biodistribution of the DOX liposomes was evaluated by in vivo fluorescence imaging. Figure 7A shows representative images of mice bearing MCF-7 tumors (white arrows) 48 hours after being administered free DiR, DiR/SSL, or DiR/CsA/SSL. The free DiR was mainly distributed in liver during the whole test. No signal was detected in tumor tissues, indicating that free DiR had no specificity to tumor tissues. On the contrary, both liposomal DiR groups exhibited strong signals in tumors from 1 to 24 hours. In particular, when the signal in the liver gradually decreased after 8 hours, the signal in the tumor continued to increase over time until 48 hours.
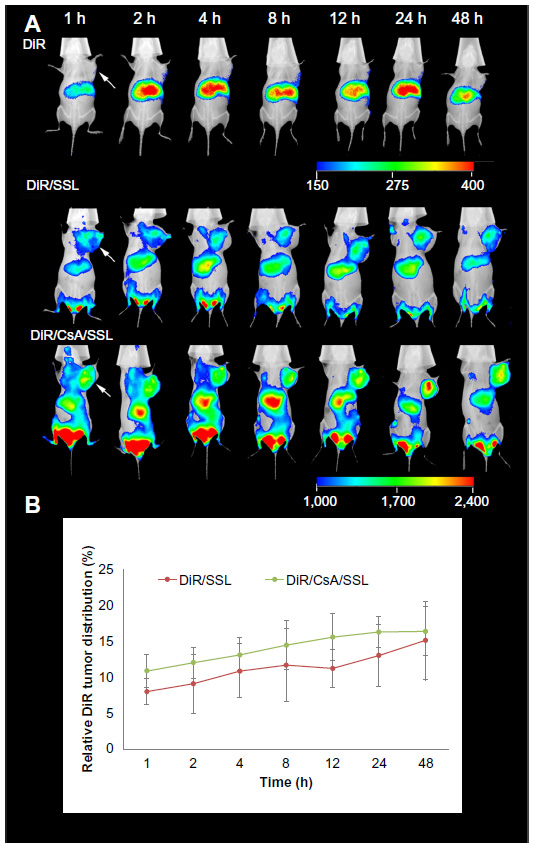 | Figure 7 In vivo fluorescence images. (A) Representative images of MCF-7-tumor-bearing mice administered free 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR), DiR/SSL, and DiR/CsA/SSL via the tail vein at various time points after dosing. (B) Semi-quantity analysis of relative DiR tumor distribution. |
Semi-quantitative analysis was performed to further quantify the targeting efficiency of DiR/SSL and DiR/CsA/SSL. Figure 7B shows the relative DiR tumor distribution, which was calculated as the sum intensity of the tumor fluorescence signal divided by the whole-body signal. The DiR tumor distribution of the two liposome formulations increased with time. Besides, the tumor distribution of DiR/CsA/SSL was slightly higher than that of DiR/SSL. This better tumor distribution of DiR/CsA/SSL might be attributed to the composition of the liposome membrane.29 The load of hydrophobic CsA in the liposome membrane might increase the lipophilicity of the liposomes and improve the affinity of the liposomes to the cell membrane.29
To summarize, the liposomes prepared in our study could achieve targeted delivery of DOX and CsA in vivo, guaranteeing the accumulation of both drugs in tumor tissue and achieving the synergetic antitumor effect. Moreover, such specific accumulation of liposomes in tumor tissue might reduce the toxicity of both CsA and DOX by reducing the distribution of these drugs to normal tissues as well as reducing the therapeutic dose required.
In vivo antitumor efficacy of DOX/CsA/SSL
In this study, nude mice bearing MCF-7 tumors were intravenously administered 5% glucose (control), free DOX, DOX/SSL, and DOX/CsA/SSL. Figure 8A displays the tumor volume of each group during the 10-day treatment. In all mice in the DOX group, there was strong suppression of tumor growth compared with in the control group. The most significant inhibition rate was observed in the DOX/CsA/SSL group compared with the other DOX groups (P<0.01 vs DOX/SSL; P<0.05 vs DOX). The tumor weights were obtained at the end of the test (Day 11), and, as shown in Figure 8B, the results were consistent with the tumor volumes at the end of the test. The DOX/CsA/SSL group exhibited the smallest tumor weights among the four groups (P<0.05 vs DOX/SSL). As shown in Figure 8C, no significant loss of body weight was observed in any of the three DOX formulation groups compared with the control group, indicating the low toxicity of DOX at efficacious doses. As shown in Figure 8D, there was no obvious toxicity to cardiac muscle cells or renal cells observed in any of the groups, except for some minor vacuolar degeneration. In conclusion, the addition of CsA to the DOX liposome formulation increased the antitumor efficacy of DOX liposomes in MCF-7-tumor-bearing mice with no apparent toxicity.
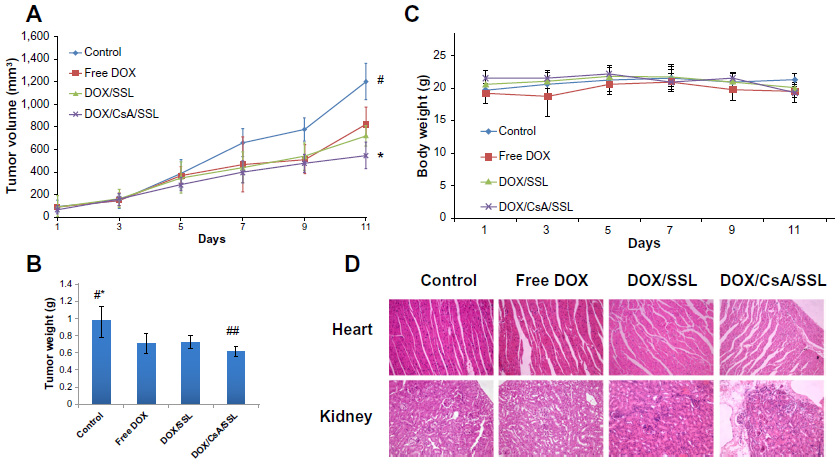 | Figure 8 In vivo antitumor efficacy. Mice were given 5% glucose (control), free doxorubicin (DOX), DOX/SSL, or DOX/CsA/SSL of 2 mg/kg DOX via the tail vein every 2 days a total of five times. Tumor volume and body weight were measured each day following the day of administration. (A) Tumor volumes of MCF-7-tumor-bearing nude mice after treatment with various DOX formulations. (B) Tumor weights at the end of test. (C) Body weights of mice during the efficacy test. (D) Representative light microscopy pictures (×200) of heart and kidney tissue sections stained with hematoxylin and eosin. |
In the treatment of MDR tumors, a large dose of CsA is required, which can induce severe nephrotoxicity and lead to the failure of clinical trials.6 The CsA dose in the treatment of MDR tumors is usually 50–200 mg/kg orally30,31 or 10–25 mg/kg intraperitoneally or intravenously.32,33 However, the dosage of CsA used in our study was about 2.0 mg/kg, which is much lower than that usually used in MDR tumors. Low-dose CsA significantly increased the antitumor efficiency in MCF-7 tumors, without inducing obvious nephrotoxicity. This is because the low expression of P-gps in MCF-7 tumors requires a lower dose of inhibitors, and the targeting effect of the liposomes also increased the drug distribution in tumors.
Figure 9 illustrates the possible advantages of using DOX/CsA/SSL in the treatment of low-P-gp-expressing tumors. In the initial phase of drug treatment, many tumors express low levels of P-gps.8,9 Compared with DOX/SSL, DOX/CsA/SSL significantly increased the cell accumulation of DOX and improved the therapeutic efficiency of liposomal DOX in these low-P-gp-expressing tumors. Moreover, the long-term use of liposomal DOX could gradually increase the level of P-gps on cells.4 This might eventually induce MDR, leading to poor prognosis. When DOX/CsA/SSL is administrated before MDR, the CsA in the liposomes might avoid drug efflux from those cells with relatively high levels of P-gps and suppress the progression of MDR in the long term. Altogether, nanoparticles co-loaded with a P-gp inhibitor and anticancer drug might be a new strategy to improve the antitumor efficiency of chemotherapy in the treatment of various low-P-gp-expressing tumors.
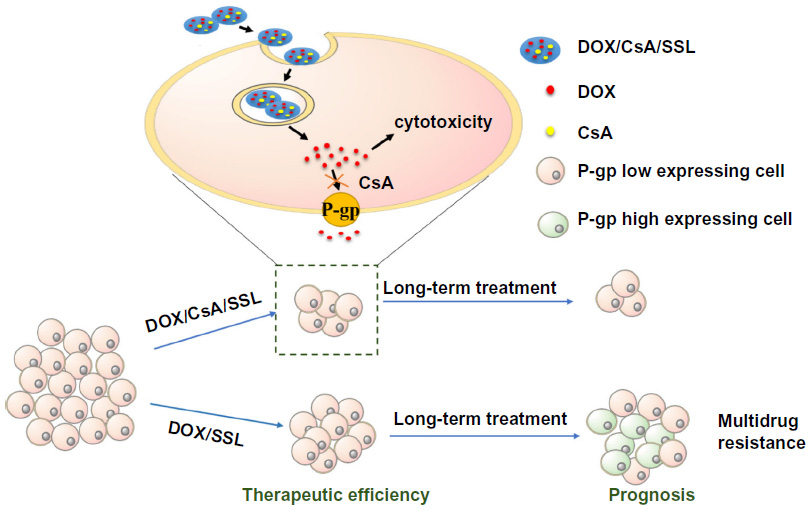 | Figure 9 Schematic illustration of the possible advantages of using DOX/CsA/SSL in the treatment of a low-P-glycoprotein (P-gp)-expressing tumor. |
Conclusion
In this study, we designed a novel DOX liposome system incorporating the low-dose P-gp inhibitor CsA (DOX/CsA/SSL). The DOX/CsA/SSL was successfully prepared and had good size uniformity, a high %EE, and a desirable release profile. Unlike many other studies using CsA to reverse MDR, in this study, it was discovered that CsA could significantly increase the cell uptake and cytotoxicity of free and liposomal DOX in low-P-gp-expressing MCF-7 cells. Intracellular and in vivo targeted delivery of both drugs via the liposomes was achieved. Moreover, incorporating low-dose CsA in the DOX liposomes significantly enhanced the therapeutic efficiency in MCF-7-tumor-bearing mice, without inducing apparent systematic toxicity. Generally, we demonstrated, for the first time, as far as we are aware, that the low expression of P-gps on tumors might affect the efficacy of chemotherapy. The co-delivery of low-dose P-gp inhibitors with liposomal drugs might be an effective approach to improve therapy for a wide range of cancers with low P-gp levels.
Acknowledgments
This study was supported by the National Science Foundation of China (No 81130059), the National Research Fund for Fundamental Key Project (No 2009CB930300), and the Innovation Team of the Ministry of Education (No BMU20110263).
Disclosure
The authors report no conflicts of interest in this work.
References
Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3(1):16–20. | |
Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006; 5(3):219–234. | |
Chavanpatil MD, Patil Y, Panyam J. Susceptibility of nanoparticle-encapsulated paclitaxel to P-glycoprotein-mediated drug efflux. Int J Pharm. 2006;320(1–2):150–156. | |
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. | |
Soma CE, Dubernet C, Bentolila D, Benita S, Couvreur P. Reversion of multidrug resistance by co-encapsulation of doxorubicin and cyclosporin A in polyalkylcyanoacrylate nanoparticles. Biomaterials. 2000;21(1):1–7. | |
Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003;10(2):159–165. | |
Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2(1):48–58. | |
Cordon-Cardo C, O’Brien JP, Boccia J, Casals D, Bertino JR, Melamed MR. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J Histochem Cytochem. 1990;38(9):1277–1287. | |
Goldstein LJ, Galski H, Fojo A, et al. Expression of a multidrug resistance gene in human cancers. J Natl Cancer Inst. 1989;81(2):116–124. | |
Watanabe T, Tsuge H, Oh-Hara T, Naito M, Tsuruo T. Comparative study on reversal efficacy of SDZ PSC 833, cyclosporin A and verapamil on multidrug resistance in vitro and in vivo. Acta Oncol. 1995;34(2):235–241. | |
List AF, Kopecky KJ, Willman CL, et al. Benefit of cyclosporine modulation of drug resistance in patients with poor-risk acute myeloid leukemia: a Southwest Oncology Group study. Blood. 2001;98(12):3212–3220. | |
Varma MV, Ashokraj Y, Dey CS, Panchagnula R. P-glycoprotein inhibitors and their screening: a perspective from bioavailability enhancement. Pharmacol Res. 2003;48(4):347–359. | |
Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | |
Wang Z, Yu Y, Dai W, et al. The use of a tumor metastasis targeting peptide to deliver doxorubicin-containing liposomes to highly metastatic cancer. Biomaterials. 2012;33(33):8451–8460. | |
Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151(2):201–215. | |
Leslie G. Estimating population size. In: Flow Cytometry – A Basic Guide. Institute of Medical Biology, University of Southern Denmark, Odense M 2006:28–29. Available from: http://www.flowcytometri.dk/literature/Leslie-FCBasic.pdf. Accessed December 14, 2013. | |
Givan AL. Information: harnessing the data. In: Flow Cytometry: First Principles. 2nd ed. New York, NY: Wiley; 2001:41–57. | |
Sasaki Y, Minamizawa M, Ambo A, Sugawara S, Ogawa Y, Nitta K. Cell-penetrating peptide-conjugated XIAP-inhibitory cyclic hexapeptides enter into Jurkat cells and inhibit cell proliferation. FEBS J. 2008;275(23):6011–6021. | |
Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71(3):409–419. | |
Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1(3):297–315. | |
Dadashzadeh S, Mirahmadi N, Babaei MH, Vali AM. Peritoneal retention of liposomes: Effects of lipid composition, PEG coating and liposome charge. J Control Release. 2010;148(2):177–186. | |
Sharom FJ. The P-glycoprotein multidrug transporter. Essays Biochem. 2011;50(1):161–178. | |
Gong JZ, Lagoo AS, Peters D, Horvatinovich J, Benz P, Buckley PJ. Value of CD23 determination by flow cytometry in differentiating mantle cell lymphoma from chronic lymphocytic leukemia/small lymphocytic lymphoma. Am J Clin Pathol. 2001;116(6):893–897. | |
Mealey KL, Barhoumi R, Burghardt RC, Safe S, Kochevar DT. Doxycycline induces expression of P glycoprotein in MCF-7 breast carcinoma cells. Antimicrob Agents Chemother. 2002;46(3):755–761. | |
Fu LW, Zhang YM, Liang YJ, Yang XP, Pan QC. The multidrug resistance of tumour cells was reversed by tetrandrine in vitro and in xenografts derived from human breast adenocarcinoma MCF-7/adr cells. European Journal of Cancer. 2002;38(3):418–426. | |
Lee YJ, Galoforo SS, Berns CM, et al. Effect of ionizing radiation on AP-1 binding activity and basic fibroblast growth factor gene expression in drug-sensitive human breast carcinoma MCF-7 and multidrug-resistant MCF-7/ADR cells. The Journal of biological chemistry. Dec 1 1995;270(48):28790–28796. | |
Toffoli G, Corona G, Sorio R, Bertola A, Boiocchi M. Reversal activity of cyclosporin A and its metabolites M1, M17 and M21 in multidrug-resistant cells. Int J Cancer. 1997;71(5):900–906. | |
Sonneveld P, Durie BG, Lokhorst HM, et al. Modulation of multidrug-resistant multiple myeloma by cyclosporin. The Leukaemia Group of the EORTC and the HOVON. Lancet. Aug 1 1992;340(8814):255–259. | |
Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145(3):182–195. | |
Watanabe T, Naito M, Kokubu N, Tsuruo T. Regression of established tumors expressing P-glycoprotein by combinations of adriamycin, cyclosporin derivatives, and MRK-16 antibodies. J Natl Cancer Inst. 1997;89(7):512–518. | |
Boesch D, Gavériaux C, Jachez B, Pourtier-Manzanedo A, Bollinger P, Loor F. In vivo circumvention of P-glycoprotein-mediated multidrug resistance of tumor cells with SDZ PSC 833. Cancer Res. 1991;51(16):4226–4233. | |
Binkhathlan Z, Shayeganpour A, Brocks DR, Lavasanifar A. Encapsulation of P-glycoprotein inhibitors by polymeric micelles can reduce their pharmacokinetic interactions with doxorubicin. Eur J Pharm Biopharm. 2012;81(1):142–148. | |
Colombo T, Zucchetti M, D’Incalci M. Cyclosporin A markedly changes the distribution of doxorubicin in mice and rats. J Pharmacol Exp Ther. 1994;269(1):22–27. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.