")
Back to Journals » Clinical Interventions in Aging » Volume 17
The Association of Plasma Amyloid-β and Cognitive Decline in Cognitively Unimpaired Population
Authors Wang J, Gao L, Liu J, Dang L, Wei S, Hu N, Gao Y, Peng W, Shang S, Huo K, Wang J, Qu Q
Received 12 January 2022
Accepted for publication 11 April 2022
Published 20 April 2022 Volume 2022:17 Pages 555—565
DOI https://doi.org/10.2147/CIA.S357994
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Maddalena Illario
Jin Wang,1,* Ling Gao,1,* Jie Liu,1 Liangjun Dang,1 Shan Wei,1 Ningwei Hu,1 Yao Gao,1 Wei Peng,1 Suhang Shang,1 Kang Huo,1 Jingyi Wang,2 Qiumin Qu1,3
1Department of Neurology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, People’s Republic of China; 2Huyi Hospital of Traditional Chinese Medicine, Xi’an, People’s Republic of China; 3Center for Brain Science, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qiumin Qu, Department of Neurology, The First Affiliated Hospital of Xi’an Jiaotong University, 277 West Yanta Road, Xi’an, 710061, People’s Republic of China, Tel/Fax +86 29 8532 4083, Email [email protected]
Purpose: This study investigates the relationship between baseline plasma Aβ and cognitive decline during follow-up in cognitively unimpaired population.
Materials and Methods: Cognitively unimpaired population was selected from people who lived in the suburbs of Xi’an, China. The levels of plasma Aβ1-42 and Aβ1-40 were tested using commercial enzyme-linked immunosorbent assay (ELISA). The mini-mental state examination (MMSE) and neuropsychological battery were used to assess cognition. Two years later, MMSE was tested again, and significant cognitive decline was defined as a decrease in MMSE scores ≥ 5 points. Logistic regression analysis was performed to analyze the relationship between baseline plasma Aβ and cognitive change during the two-year follow-up.
Results: A total of 1144 participants completed the study, among whom 59 subjects (5.2%) presented significant cognitive decline. The high plasma Aβ1-42 level group had more significant cognitive decline (P = 0.023). Multivariable logistic regression analysis showed that significant cognitive decline was associated with the high levels of baseline plasma Aβ1-42 (OR = 1.043, 95% CI: 1.005– 1.083, P = 0.026). However, significant cognitive decline was not associated with baseline plasma Aβ1-40 levels and Aβ1-42 /Aβ1-40 ratio.
Conclusion: Population with high level of baseline plasma Aβ1-42 manifested significant cognitive decline over 2 years; however, further investigation on the dynamics of plasma Aβ and long-term follow-up are needed.
Keywords: Alzheimer’s disease, cognitive decline, plasma amyloid β, cohort study, plasma biomarker
Introduction
Alzheimer’s disease (AD) is a progressive and irreversible brain degenerative disease with progressive cognitive decline, mental and behavioral symptoms, and decreased ability of daily living as the main clinical manifestations. As the aging of population, the incidence of the disease tends to increase dramatically.1 The pathological hallmarks of AD include senile plaques composed of amyloid-β (Aβ) deposition, and neurofibrillary tangles composed of tau phosphorylation.2 Although amyloid hypothesis, which is the predominant framework for research in AD, has been the source of considerable controversy,3,4 many studies support that Aβ likely is the key initiator of a complex pathogenic cascade that causes AD.5,6
An imbalance of Aβ generation and clearance may be the main cause of Aβ aggregation in the brain. Aβ are generated from the cleavage of amyloid precursor protein (APP) by sequential β and γ secretase.7 Aβ can be cleared from the central nervous system (CNS) to the blood via transporters through the blood-brain barrier (BBB), while reverse transport of peripheral Aβ across the BBB into the brain does occur.8,9 There is a complex dynamic equilibrium between amyloid burden in the brain and plasma Aβ,10 and plasma Aβ levels can reflect Aβ deposition in the brain to a certain extent.11–15
Our previous studies have shown that the plasma Aβ levels in people with suspected cognitive impairment were higher than those in cognitively normal people and people with cognitive impairment.16 Elevated plasma Aβ may be associated with cognitive impairment. However, in prospective studies, the evidence for whether plasma Aβ levels can predict cognitive decline in people with normal cognition has been inconsistent.17,18 Therefore, in the present study, we investigated the relationship between baseline plasma Aβ levels and cognitive decline during follow-up in cognitively unimpaired population.
Materials and Methods
Study Cohort
This was a prospective cohort study. All the participants came from the rural cognitive impairment cohort established in Qubao village, Huyi district of Xi’an city, from October 2014 to March 2015. The inclusion criteria of the participants in this study were as follows: (1) a member of the permanent population of Qubao Village, Huyi District, Xi’an City, with residence time ≥3 years; (2) aged ≥40 years old, that is, those born before December 31, 1973; (3) identified as having normal cognitive function during the baseline test (see details in Cognitive assessment section); (4) voluntarily participated in this study and signed an informed consent; (5) cooperated to complete the questionnaire survey and scale assessment; (6) had blood samples collected and had completed measurements of plasma Aβ and APOE genotypes. The exclusion criteria were as follows: (1) individuals who suffered from diseases that can cause cognitive impairment including central nervous system infection (such as AIDS, syphilis, etc.), traumatic dementia, epilepsy, Parkinson’s disease, optic neuromyelitis, intellectual disability, other physical and chemical factors (such as drug poisoning, alcohol poisoning, carbon monoxide poisoning, etc.), severe physical diseases (such as hepatic encephalopathy, pulmonary encephalopathy, etc.), subdural hematoma, brain tumor, or endocrine system diseases (such as thyroid disease, parathyroid disease) and cognitive impairment caused by lack of vitamins or other reasons; (2) individuals diagnosed with psychiatric disorders, based on the “Diagnosis and Statistical Manual of Mental Diseases (Fourth Edition)” (DSM-IV-TR) criteria, including schizophrenia, bipolar disorder, severe depression or delirium; (3) individuals with unstable or severe heart, lung, liver, kidney, hematopoietic system diseases; and (4) individuals with data considered an outliers, that is, plasma Aβ levels exceeding 3 times the standard deviation.
Ethics Approval and Informed Consent
The study protocol conformed to the ethical guidelines of the Declaration of Helsinki and was approved by the Medical Ethics Committee of the First Affiliated Hospital of Xi’an Jiaotong University. Written informed consent was obtained from all participants.
Data Collection
In 2014, we collected baseline data for this population through face-to-face questionnaires, cognitive function assessment, and laboratory tests. The detailed experimental procedures have been described in previous articles.16 A total of 1155 cognitively unimpaired subjects were included at baseline.
To assess the changes of cognitive function, all subjects who finished baseline investigation were been followed-up. Two years later, all subjects accepted a face-to-face questionnaire, cognitive function assessment, and laboratory tests again. The procedure was same as baseline investigation. If the subjects died or did not complete two follow-up visits, it was considered lost. In the follow-up in 2016, 11 persons were lost. Finally, 1144 participants were included in the analysis. The study protocol and the selection of subjects are shown in Figure 1.
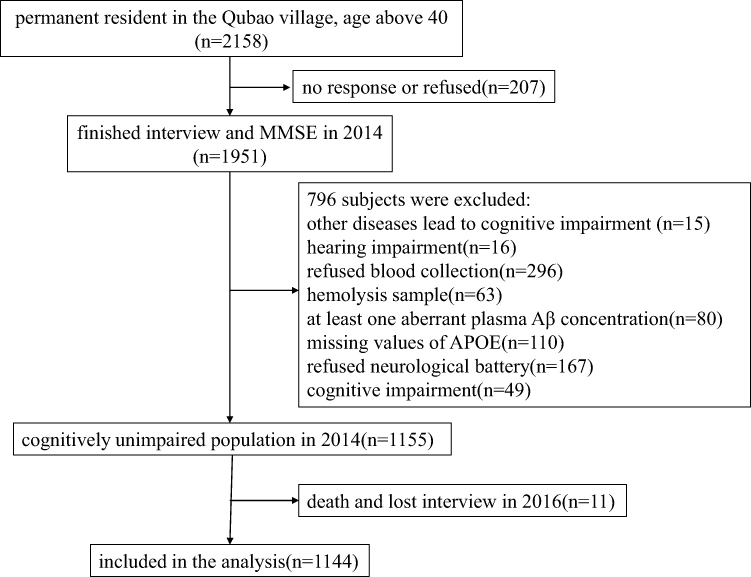 |
Figure 1 The enrollment flow chart of this study. |
Cognitive Assessment
The diagnosis of cognitive impairment followed the three-step protocol as described in previous articles.19 First, all participants underwent the Mini-Mental State Examination (MMSE) to assess the global cognitive functions. Cutoff values for the MMSE were ≤17 for illiterate subjects, ≤20 for subjects whose education time was less than 6 years, and ≤24 for subjects whose education time was more than 6 years.20,21 Second, individuals whose MMSE score was below the cutoff value underwent a neuropsychological battery. Clock Drawing Task22 and Trail Making Test were used to assess the executive function, Fuld Object Memory Evaluation23 was for memory assessment, Rapid Verbal Retrieval24 was for language function, Block Design Test25 was for visuospatial function, Digit Span Test26 was for attention evaluation. Third, a neurologist gave the subjects who underwent both the MMSE and the neuropsychological battery a diagnosis based on their clinical manifestations and outcomes of the neuropsychological tests. The diagnosis of mild cognitive impairment (MCI), dementia and its subtypes was determined in accordance with international criteria.27–30 Two years later, the three-step protocol was also used to assess cognitive function.
The investigators were neurologists and graduate students. Strict training was carried out before the survey, and a unified questionnaire and standardized survey language were used during the survey. All investigators passed the consistency evaluation after the training (kappa = 0.76~1). The same interviewers participated in the two surveys.
Definition of Significant Cognitive Decline
Some studies have shown that MMSE scores are reduced by 2–4 points in 1.5 years31 or that a difference of 3 or 4 points in MMSE scores reliably reflect cognitive decline.32 Therefore, to avoid uncertainty, a decline in MMSE score ≥521 was defined as significant cognitive decline, while a decline in MMSE score <5 was defined as non-significant cognitive decline in our study.
Plasma Aβ Measurement
Fasting cubital venous blood (3 mL) was collected from all subjects between 8 and 10 am, placed in an ethylene diamine tetraacetic acid (EDTA) anticoagulant purple-top tube, centrifuged at 3000 g for 10 min at room temperature (20°C), and had the supernatant plasma extracted and aliquoted. Aliquots of plasma were stored at –80°C pending biochemical analyses. Double-antibody sandwich enzyme-linked immunoassay (ELISA) was used to determine baseline plasma Aβ concentrations. The kit was purchased from Shanghai Yuanye Biotechnology Co., Ltd. All samples were measured in duplicate on an RT-6000 analyzer (Rayto Co. Shenzhen, China) at 450 nm, and the operation was strictly in accordance with the instructions. The concentration was calculated based on the standard curve, and the average was taken as the sample concentration.
Baseline plasma Aβ concentrations were classified into four subtypes according to its quartile. The lowest quartile was Quartile 1 group, the highest quartile was Quartile 4 group.
APOE Genotyping
APOE genotyping was detected using polymerase chain reaction (PCR) and Sanger sequencing. A blood genomic deoxyribonucleic acid (DNA) extraction kit (Tiangen Co. Beijing, China) was used to extract genomic DNA from blood samples. A polymerase chain reaction thermocycler was used to amplify 244 base pairs of the APOE gene fragment that included two polymorphic sites at amino acid residues 112 and 158. Sanger sequencing (Sangon Co. Shanghai, China) was used to detect the sequence of the polymerase chain reaction products. Chromas2 peak map software was used to analyze the sequenced DNA sequence and peak map, and determine the APOE genotype. E2/2, E2/3, and E3/3 genotypes were defined as APOEε4(-), and E2/4, E3/4, and E4/4 genotypes were defined as APOEε4(+).
Covariates
Demographic data (age, sex and level of education), lifestyle information (smoking, drinking), and comorbidities (hypertension, diabetes mellitus) were collected through face-to-face interviews. Laboratory test parameters including blood lipid levels including triglyceride (TG), total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C) were examined in the clinical laboratory of the First Affiliated Hospital of Xi’an Jiaotong University. TG ≤1.70 mmol/L, TC ≤5.18 mmol/L, LDL-C ≤3.37 mmol/L, HDL-C ≥1.04 mmol/L were the criteria for normal values. Smoking was defined as ten cigarettes daily for at least six months, with no distinction made between current and past smokers. Drinking was defined as an alcoholic beverage at least once a week. Hypertension and diabetes mellitus were diagnosed in accordance with relevant guidelines.33,34
Statistical Analysis
Continuous variables with approximately normal distribution were described as mean±SD. Median (25% percentile, 75% percentile) was used for skewed continuous variables, and frequencies and percentages were administrated to describe categorical variables. For univariate analyses, we chose χ2 tests, t-tests, ANOVA, and non-parametric tests according to different types of variables. Then we explored the relationship between baseline plasma Aβ and significant cognitive decline with univariate logistic regression model, in which plasma Aβ was fitted as a restricted cubic spline (smooth curve).
In the multivariate logistic regression models, covariates were chosen from the significant variables (p < 0.20) in the univariate analysis as well as covariates reported to be related to cognition in previous studies. In the pre-analysis process, multiple groups of models were established by adding covariates one by one. The analysis found that the correlation between baseline plasma Aβ levels and significant cognitive decline was consistent in several models. At last, we established a logistic regression model with significant cognitive decline (yes or no) as the dependent variable, plasma Aβ parameters (Aβ1-40, Aβ1-42 and Aβ1-42/Aβ1-40) and covariates (age, sex, education, hypertension, and APOEε4) as independent variables. We also conducted a sensitivity analysis with the excluded cases due to the outliers of plasma Aβ in the logistic regression model to determine whether the findings were similar. SPSS version 18.0 (SPSS Inc., IBM, Chicago) was used to perform statistical analyses. GraphPad Prism version 5.0 (GraphPad Software, Inc., San Diego) was used to draw all the graphs. A p value of less than 0.05 was considered significant.
Results
Demographics and Clinical Characteristics of the Population
Of the 1144 participants aged 40–85 years (mean 55.10 ± 9.63 years), 462 (40.4%) subjects were male; 681 (59.5%) subjects had high school or above education; the prevalence of hypertension and diabetes mellitus were 27.1% (310/1144) and 9.0% (103/1144), respectively; and 15.1% (173/1144) of subjects were APOE ε4 carriers. After the 2-year follow-up, 59 subjects (5.2%) presented cognitive decline (defined as a decrease in MMSE scores ≥5 points).
Compared with non-significant cognitive decline group, the participants with significant cognitive decline were older (P<0.001), had lower education level (P = 0.001), higher levels of plasma Aβ1-42 (P =0.016), and higher prevalence of hypertension (P = 0.014), while gender, fasting blood lipids, diabetes mellitus, APOE ε4 allele, plasma Aβ1-40 levels, the Aβ1-42/Aβ1-40 ratio and baseline MMSE had no significant difference between two groups (Table 1).
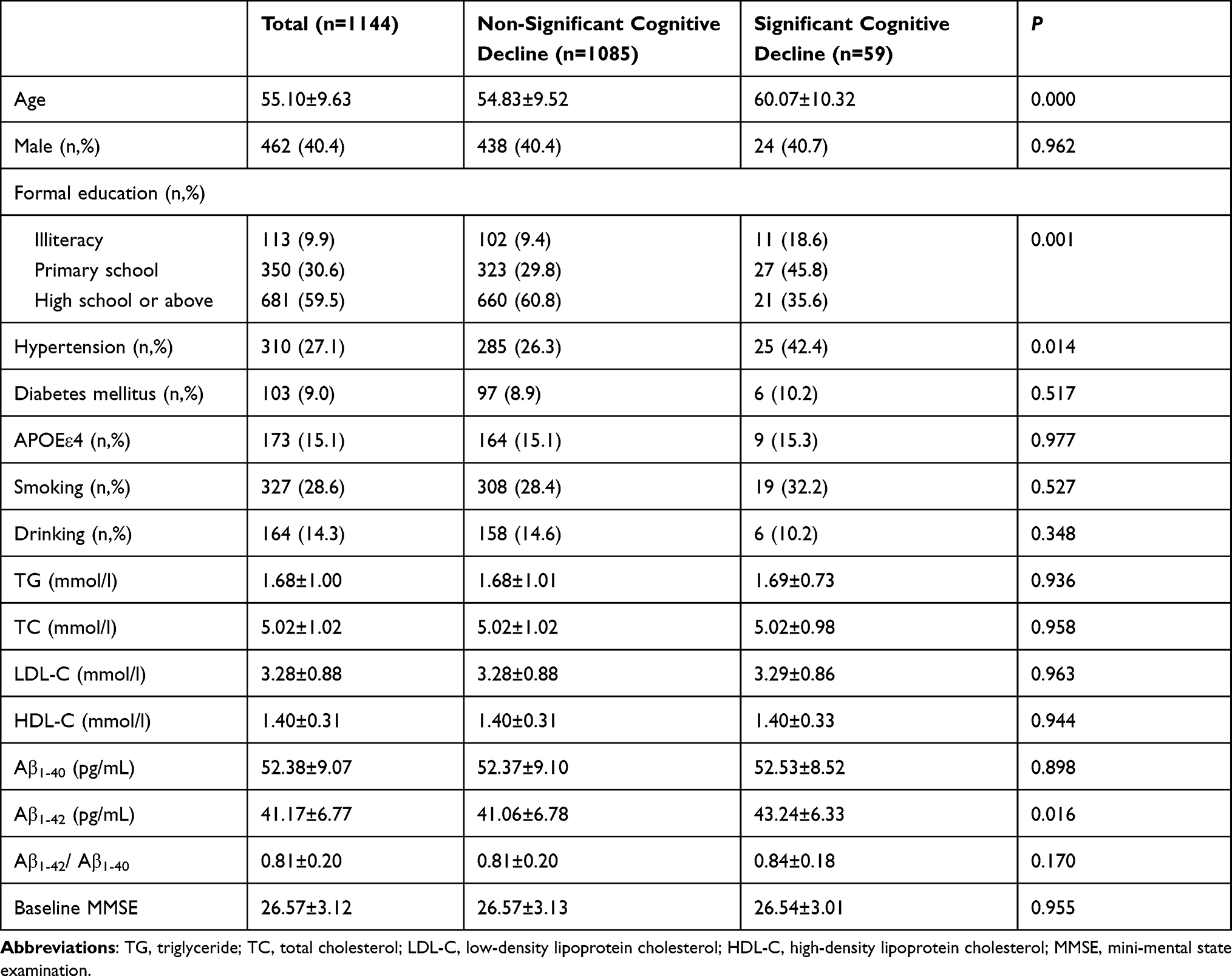 |
Table 1 Demographics and Clinical Characteristics of the Population |
Association Between Baseline Plasma Aβ and Significant Cognitive Decline
As shown in Figure 2, when baseline plasma Aβ was fitted as a restricted cubic spline, the univariate logistic regression analysis showed that baseline plasma Aβ1-42 had a linear relationship to significant cognitive decline; however, plasma Aβ1-40 and the ratio of Aβ1-42/Aβ1-40 had no obvious relationship to significant cognitive decline.
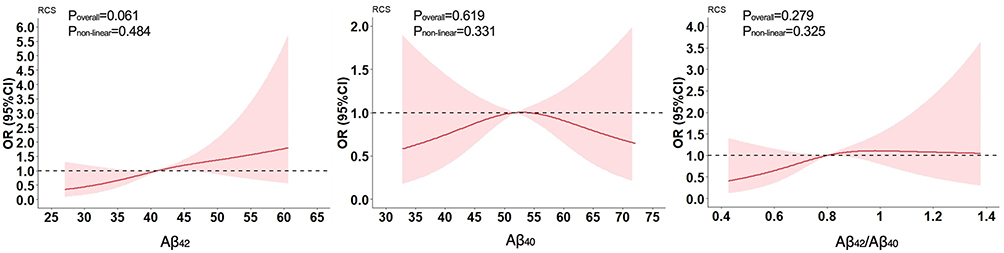 |
Figure 2 Association between baseline plasma Aβ and significant cognitive decline. |
Univariate Analysis of Baseline Plasma Aβ42 Levels
After 2-year follow-up, the Quartile 4 (highest plasma Aβ1-42 levels) group had more cognitive decline than that in other groups (P = 0.023), while age, sex, education level, hypertension, diabetes mellitus, blood lipid levels, APOE ε4 allele, smoking, drinking and baseline MMSE scores had no significant difference among different plasma Aβ1-42 level groups (Table 2).
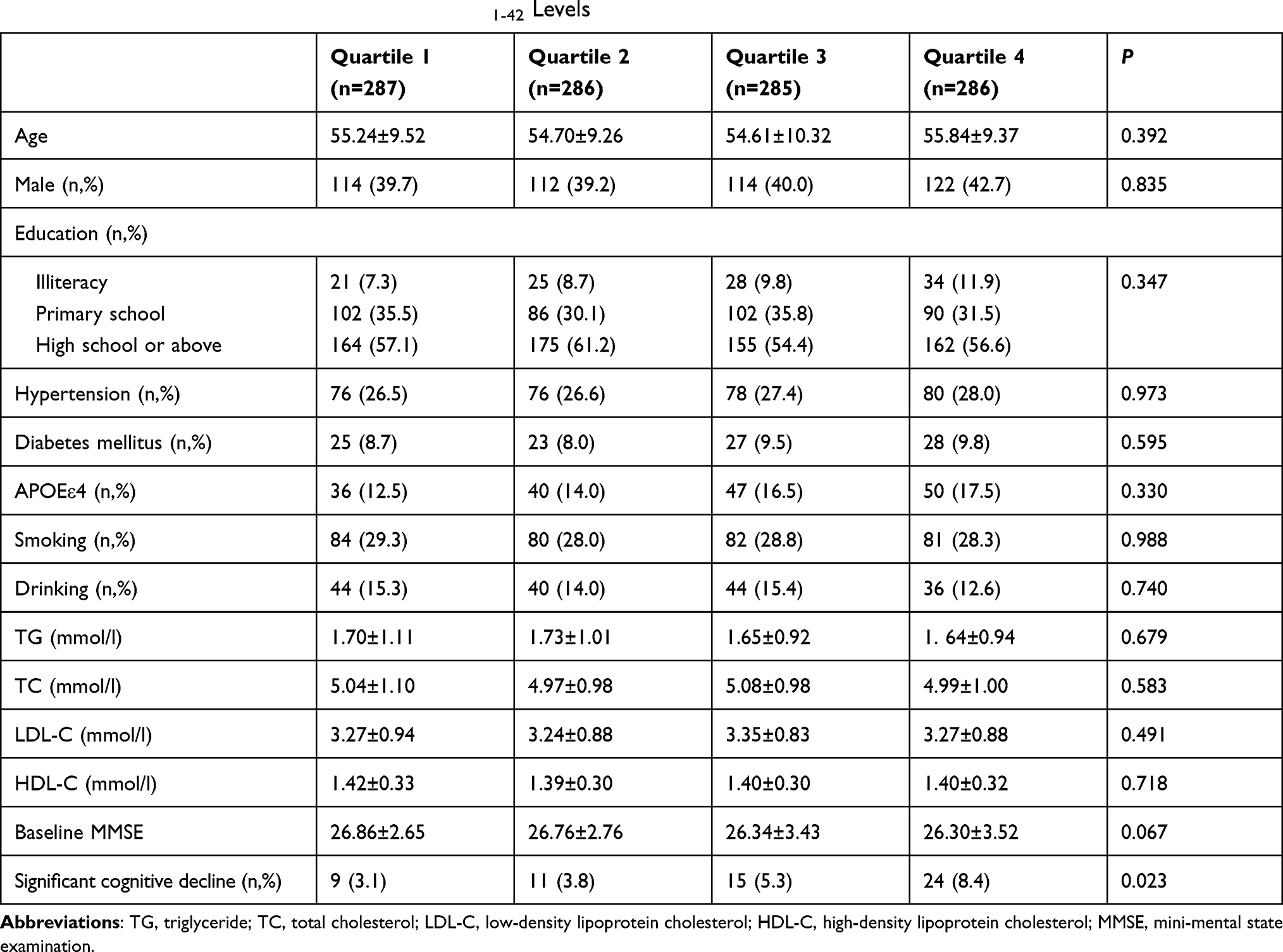 |
Table 2 Univariate Analysis of Baseline Plasma Aβ1-42 Levels |
Multivariate Logistic Regression Analysis of Significant Cognitive Decline and Baseline Plasma Aβ
Multivariate logistic regression analysis was used to analyze correlations between baseline plasma Aβ levels and significant cognitive decline. We established a logistic regression model with significant cognitive decline (yes or no) as the dependent variable, plasma Aβ parameters (Aβ1-40, Aβ1-42 and Aβ1-42/Aβ1-40) and covariates (age, sex, education, hypertension, and APOEε4) as independent variables.
When the baseline plasma Aβ concentration was a continuous variable, multivariate logistic regression analysis showed that significant cognitive decline was significantly related to high levels of baseline plasma Aβ1-42 (OR = 1.043, 95% CI: 1.005–1.083, P = 0.026). However, significant cognitive decline was not associated with baseline plasma Aβ1-40 levels or Aβ1-42 /Aβ1-40 ratio (Table 3).
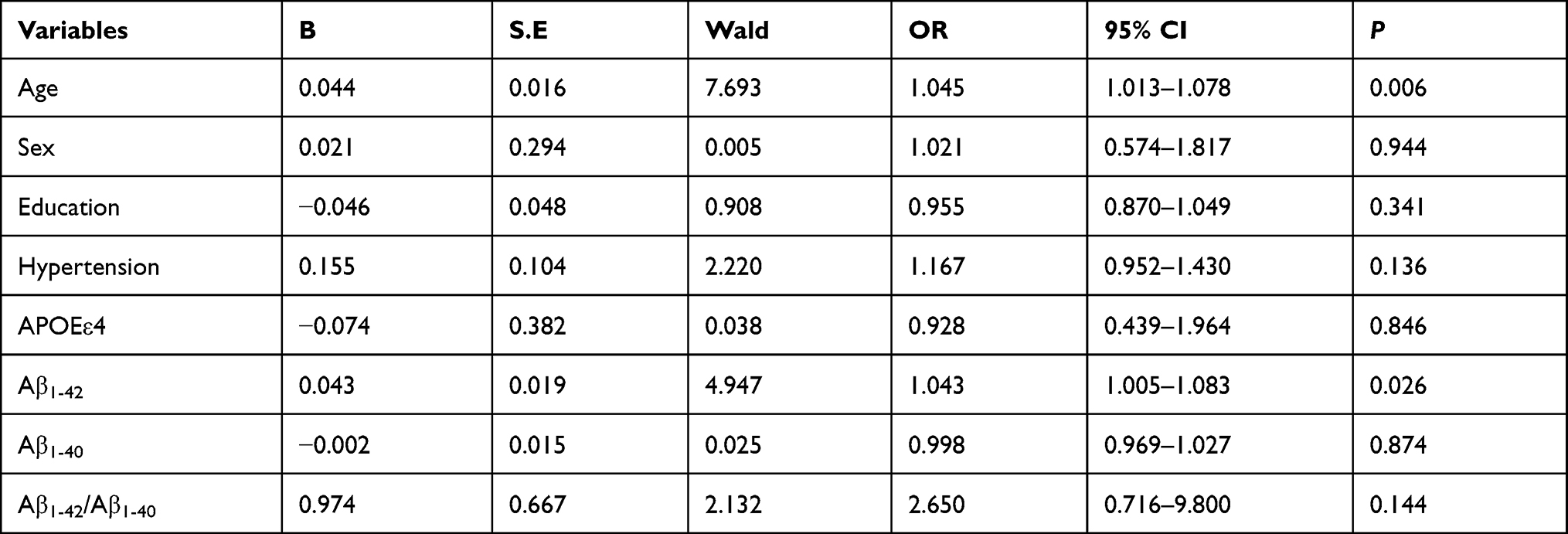 |
Table 3 Multivariate Logistic Regression Analysis of Significant Cognitive Decline and Baseline Plasma Aβ |
When the baseline plasma Aβ concentrations were classified into four groups according to its quartile and Quartile 1 (the lowest level of the plasma Aβ) group was the reference, multivariate logistic regression analysis also showed that Quartile 4 (the highest level of plasma Aβ1-42) group were obviously associated with significant cognitive decline (OR = 2.721, 95% CI: 1.232–6.009, P = 0.013). However, the levels of plasma Aβ1-40 and the Aβ1-42 /Aβ1-40 ratio were not associated with significant cognitive decline (Table 4).
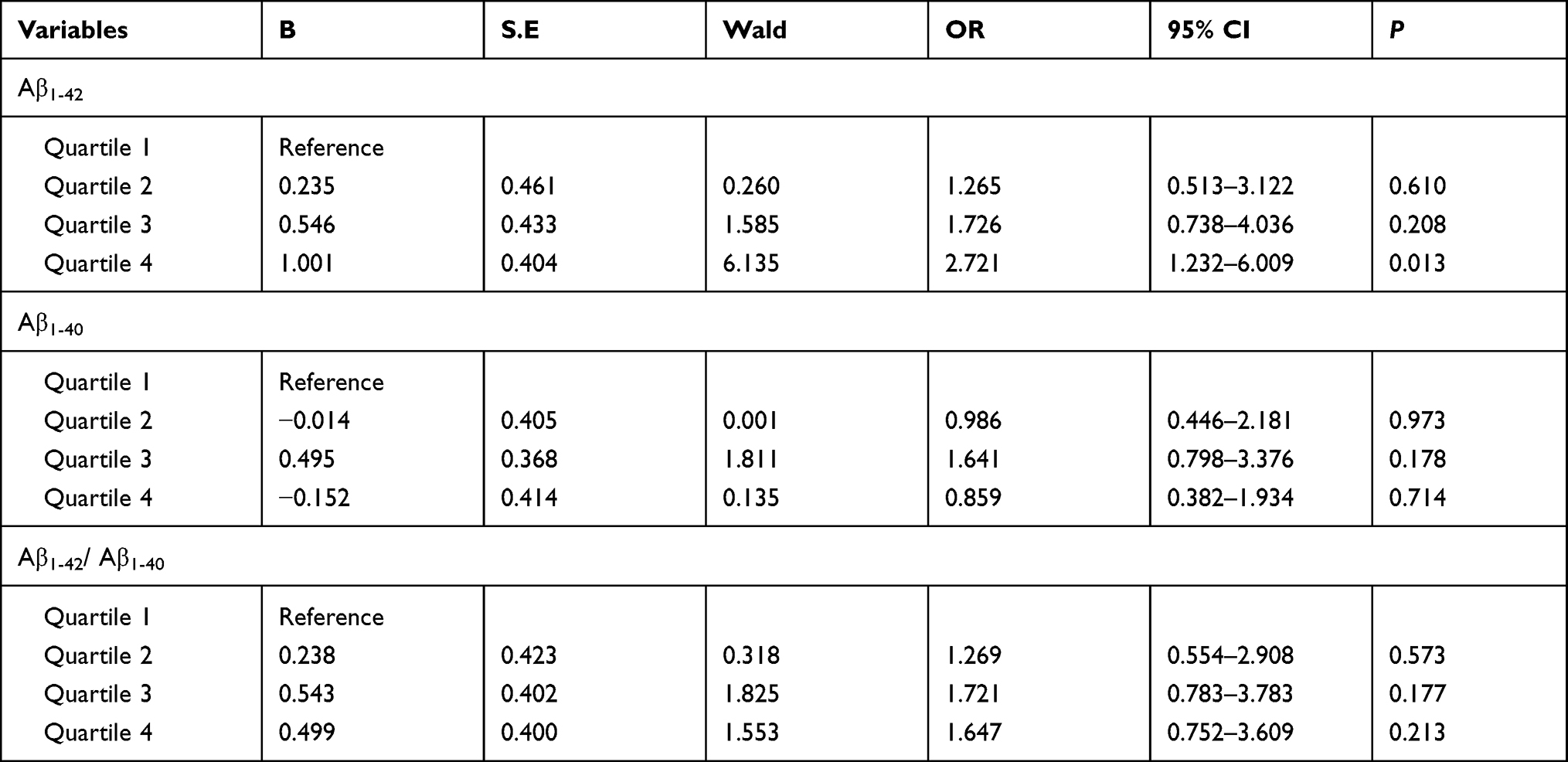 |
Table 4 Multivariate Logistic Regression Analysis of Significant Cognitive Decline and Different Levels of Baseline Plasma Aβ |
Sensitivity Analysis
In the present study, 80 cases were excluded due to the outliers of plasma Aβ. We have conducted the sensitivity analysis with these cases in the logistic regression models. Similar findings were obtained when the analyses were performed in the entire study population (Supplementary Table 1), suggesting a solid correlation between plasma Aβ1-42 and significant cognitive decline in our population.
Discussion
In the present study, we investigated the relationship between baseline plasma Aβ levels and cognitive decline during a 2-year follow-up in a cognitively unimpaired population. Subjects with high baseline plasma Aβ1-42 levels manifested cognitive decline over the 2-year period. However, the levels of plasma Aβ1-40 and the Aβ1-42 /Aβ1-40 ratio were not associated with cognitive decline.
The results regarding the relationships between plasma Aβ levels and cognitive decline are conflicting. A meta-analysis analyzed data showed that plasma Aβ1-42 and Aβ1-40 levels were not associated with AD.35 In a cohort of 1125 elderly persons without dementia with 4.6 years of follow-up, higher plasma Aβ1-42 levels at the onset of the study were associated with a threefold increased risk of AD. However, conversion to AD was accompanied by a significant decline in plasma Aβ1-42 levels and a decreased Aβ1-42/Aβ1-40 ratio.36 The Rotterdam study including 1756 persons showed that higher plasma Aβ40 levels were associated with an increased risk of dementia with 8.6 years of follow-up, while plasma Aβ1-42 levels had no relationship with the risk of dementia.17 On the contrary, the results from the Framingham Heart Study including 2189 cognitively normal participants older than 60 years of age with 10-year follow-up showed that increased baseline Aβ1-42 levels indicated a lower risk of both dementia and AD.18 Another study analyzed plasma Aβ levels in 458 individuals from the Rotterdam Study with 14.8 years of follow-up demonstrated that both higher plasma Aβ1-42 and Aβ1-38 levels were associated with a decreased risk of AD, which was consistent with the Framingham Heart Study.37 Recently, most studies have shown that decreased plasma Aβ1-42 levels38,39 or plasma Aβ1-42/Aβ1-40 ratio40–44 indicates a higher risk of dementia or cognitive decline. In our previous study, we reported that the relationships between baseline plasma Aβ1-40 level and cognitive decline was not linear over the 2-year period.45
Several reasons contributed to the conflicting relationships between plasma Aβ levels and cognitive decline: First, previous studies selected elderly population as study participants, while both middle-aged and elderly people were included in the present study. Researchers had found that levels of plasma Aβ were influenced by many factors, including age,46 therefore the differences in inclusion criteria might be one reason for the inconsistent results. Second, definition of cognitive decline differed greatly between studies, with some defined incidence AD or MCI as major outcome. In our study, the drop of MMSE score ≥5 was defined as significant cognitive decline, which may also affect the relationship between plasma Aβ and cognitive impairment. Third, the follow-up time varied greatly between different studies, fluctuating from 2 to 14 years. With longer follow-up, we believed it would be easier to detect the relationship between plasma Aβ and cognitive impairment. Fourth, methods to detect plasma Aβ might also influence the outcome. Apart from ELISA, xMAP technology, immunoprecipitation-mass spectrometry (IP-MS) assays, Elecsys immunoassay and Simoa-based assay were sensitive to detect plasma Aβ. The accuracy of these methods was different, which would inevitably affect the relationship between plasma Aβ and cognitive impairment. Hence, differences in inclusion criteria, definition of cognitive decline, follow-up time and the method of plasma Aβ detection would all contribute to the conflicting results among these studies.
The population in our study came from cluster sampling of the community population cohort, selecting middle-aged and elderly people as the research subjects. Two years after the completion of the baseline survey, a face-to-face questionnaire survey and cognitive assessment were conducted again. The survey methods and the composition of the interviewers were identical. The criteria for significant cognitive decline was defined as a decline in MMSE scores ≥5 to avoid uncertainty in our study, which make sure the patients who met the criteria of cognitive decline may have a reliably cognitive impairment. The results showed that the MMSE scores of people with high baseline plasma Aβ1-42 levels significantly dropped. Correcting the confounding factors of cognitive function in the multivariate logistic regression analysis, cognitive function significantly declined in those with high baseline plasma Aβ1-42 levels, which supported the findings that high plasma Aβ levels were associated with cognitive decline.
We excluded those with secondary cognitive impairment caused by stroke, epilepsy, hypothyroidism, and Parkinson’s disease during the baseline and follow-up surveys. Therefore, changes in cognitive function were more likely to be related to AD-like pathological changes in the brain. Brain is considered to be the main source of Aβ, and Aβ is deposited in senile plaques in AD patients. Aβ is in dynamic balance in the cerebrospinal fluid and plasma. Increases in the production of Aβ in the brain have been related to increases in the concentration of Aβ in the plasma. Studies have shown that high levels of plasma Aβ may reflect reduced peripheral Aβ clearance or increased Aβ production in the brain, which has been related to an increased risk of AD.47 The reduction in plasma Aβ can promote the outflow of Aβ from the brain to the blood circulation through the “peripheral pool” effect,48 which reduces the deposition of Aβ in the brain and reduces the risk of AD and cognitive decline. Aβ1-42 is the main component of senile plaques, and it more easily accumulates in the brain than Aβ1-40 and is more closely related to the pathological process of AD.49 This study also found that plasma Aβ1-42 levels were related to cognitive decline. The results are consistent with previous studies. The mechanism may be that high levels of plasma Aβ in the cognitively unimpaired population indicated a disturbance in Aβ clearance; therefore, such people are prone to cognitive decline over time.
However, the study also had some limitations. The follow-up time of this study was only 2 years, and the plasma Aβ concentrations was measured only at baseline, the plasma concentrations did not reflect long-term changes in plasma Aβ levels. Therefore, further long-term follow-up of the study population and dynamic determination of plasma Aβ changes may have more important significance for clarifying the relationship between plasma Aβ levels and cognitive function changes.
Conclusion
In summary, our study found that people with higher baseline plasma Aβ1-42 levels had significant cognitive decline through a 2-year follow-up, suggesting that early high plasma Aβ1-42 levels may be a predictive factor of cognitive deterioration.
Abbreviations
Aβ, amyloid-β; AD, Alzheimer’s disease; APOE, apolipoprotein E; APP, amyloid precursor protein; BBB, blood-brain barrier; CNS, central nervous system; DNA, deoxyribonucleic acid; EDTA, ethylene diamine tetraacetic acid; ELISA, enzyme-linked immunosorbent assay; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; MCI, mild cognitive impairment; MMSE, mini-mental state examination; PCR, polymerase chain reaction; TC, total cholesterol; TG, triglyceride.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
The authors thank the participants and the team members of the study for their participation and support given.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The study was supported by the First Affiliated Hospital of Xi’an Jiaotong University Foundation (Grant Numbers: 2019QN-22) and the Nature Science Foundation of China (No.81771168) and the Key Research & Development Programs of Shaanxi Province (No. 2018ZDXM-SF-052 and No.2019SF-227) and Clinical research award of the First Affiliated Hospital of Xi’an Jiaotong University (No. XJTU1AF-CRF-2018-004).
Disclosure
The authors have no conflict of interest to report.
References
1. Jia L, Quan M, Fu Y, et al. Dementia in China: epidemiology, clinical management, and research advances. Lancet Neurol. 2020;19(1):81–92. doi:10.1016/S1474-4422(19)30290-X
2. Busche MA, Hyman BT. Synergy between amyloid-beta and tau in Alzheimer’s disease. Nat Neurosci. 2020;23(10):1183–1193. doi:10.1038/s41593-020-0687-6
3. Frisoni GB, Altomare D, Thal DR, et al. The probabilistic model of Alzheimer disease: the amyloid hypothesis revised. Nat Rev Neurosci. 2022;23(1):53–66. doi:10.1038/s41583-021-00533-w
4. Kametani F, Hasegawa M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front Neurosci. 2018;12:25. doi:10.3389/fnins.2018.00025
5. Magalingam KB, Radhakrishnan A, Ping NS, Haleagrahara N. Current concepts of neurodegenerative mechanisms in Alzheimer’s disease. Biomed Res Int. 2018;2018:3740461. doi:10.1155/2018/3740461
6. Paroni G, Bisceglia P, Seripa D. Understanding the amyloid hypothesis in Alzheimer’s disease. J Alzheimers Dis. 2019;68(2):493–510. doi:10.3233/JAD-180802
7. Tiwari MK, Kepp KP. beta-amyloid pathogenesis: chemical properties versus cellular levels. Alzheimers Dement. 2016;12(2):184–194. doi:10.1016/j.jalz.2015.06.1895
8. Roberts KF, Elbert DL, Kasten TP, et al. Amyloid-beta efflux from the central nervous system into the plasma. Ann Neurol. 2014;76(6):837–844. doi:10.1002/ana.24270
9. Deane R, Du Yan S, Submamaryan RK, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9(7):907–913. doi:10.1038/nm890
10. Giedraitis V, Sundelof J, Irizarry MC, et al. The normal equilibrium between CSF and plasma amyloid beta levels is disrupted in Alzheimer’s disease. Neurosci Lett. 2007;427(3):127–131. doi:10.1016/j.neulet.2007.09.023
11. Ovod V, Ramsey KN, Mawuenyega KG, et al. Amyloid beta concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 2017;13(8):841–849. doi:10.1016/j.jalz.2017.06.2266
12. Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature. 2018;554(7691):249–254. doi:10.1038/nature25456
13. Schindler SE, Bollinger JG, Ovod V, et al. High-precision plasma beta-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93(17):e1647–e1659. doi:10.1212/WNL.0000000000008081
14. Vergallo A, Megret L, Lista S, et al. Plasma amyloid beta 40/42 ratio predicts cerebral amyloidosis in cognitively normal individuals at risk for Alzheimer’s disease. Alzheimers Dement. 2019;15(6):764–775. doi:10.1016/j.jalz.2019.03.009
15. Perez-Grijalba V, Romero J, Pesini P, et al. Plasma Abeta42/40 ratio detects early stages of Alzheimer’s disease and correlates with CSF and neuroimaging biomarkers in the AB255 Study. J Prev Alzheimers Dis. 2019;6(1):34–41. doi:10.14283/jpad.2018.41
16. Wang J, Qiao F, Shang S, et al. Elevation of plasma amyloid-beta level is more significant in early stage of cognitive impairment: a Population-Based Cross-Sectional Study. J Alzheimers Dis. 2018;64(1):61–69. doi:10.3233/JAD-180140
17. van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM. Plasma Abeta(1-40) and Abeta(1-42) and the risk of dementia: a prospective case-cohort study. Lancet Neurol. 2006;5(8):655–660. doi:10.1016/S1474-4422(06)70501-4
18. Chouraki V, Beiser A, Younkin L, et al. Plasma amyloid-beta and risk of Alzheimer’s disease in the Framingham Heart Study. Alzheimers Dement. 2015;11(3):249–257 e241. doi:10.1016/j.jalz.2014.07.001
19. Yuan J, Zhang Z, Wen H, et al. Incidence of dementia and subtypes: a cohort study in four regions in China. Alzheimers Dement. 2016;12(3):262–271. doi:10.1016/j.jalz.2015.02.011
20. Katzman R, Zhang MY, Ouang Ya Q, et al. A Chinese version of the Mini-Mental State Examination; impact of illiteracy in a Shanghai dementia survey. J Clin Epidemiol. 1988;41(10):971–978. doi:10.1016/0895-4356(88)90034-0
21. Bohm M, Schumacher H, Leong D, et al. Systolic blood pressure variation and mean heart rate is associated with cognitive dysfunction in patients with high cardiovascular risk. Hypertension. 2015;65(3):651–661. doi:10.1161/HYPERTENSION-AHA114.04568
22. Shulman KI. Clock-drawing: is it the ideal cognitive screening test? Int J Geriatr Psychiatry. 2000;15(6):548–561. doi:10.1002/1099-1166(200006)15:6<548::aid-gps242>3.0.co;2-u
23. Fuld PA, Masur DM, Blau AD, Crystal H, Aronson MK. Object-memory evaluation for prospective detection of dementia in normal functioning elderly: predictive and normative data. J Clin Exp Neuropsychol. 1990;12(4):520–528. doi:10.1080/01688639008400998
24. Henry JD, Crawford JR, Phillips LH. Verbal fluency performance in dementia of the Alzheimer’s type: a meta-analysis. Neuropsychologia. 2004;42(9):112–1222. doi:10.1016/j.neuropsychologia.2004.02.001
25. Johanson AM, Gustafson L, Risberg J. Behavioral observations during performance of the WAIS Block Design Test related to abnormalities of regional cerebral blood flow in organic dementia. J Clin Exp Neuropsychol. 1986;8(3):201–209. doi:10.1080/01688638608401312
26. Simard M, van Reekum R. Memory assessment in studies of cognition- enhancing drugs for Alzheimer’s disease. Drugs Aging. 1999;14(3):197–230. doi:10.2165/00002512-199914030-00004
27. First MB. Diagnostic and statistical manual of mental disorders, 5th edition, and clinical utility. J Nerv Ment Dis. 2013;201(9):727–729. doi:10.1097/NMD.0b013e3182a2168a
28. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of department of health and human services task force on Alzheimer’s disease. Neurology. 1984;34(7):939–944. doi:10.1212/wnl.347.939
29. Roman GC, Tatemichi TK, Erkinjuntti T, et al. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology. 1993;43(2):250–260. doi:10.1212/wnl.43.2.250
30. Vega JN, Newhouse PA. Mild cognitive impairment: diagnosis, longitudinal course, and emerging treatments. Curr Psychiatry Rep. 2014;16(10):490. doi:10.1007/s11920-014-0490-8
31. Hensel A, Angermeyer MC, Riedel-Heller SG. Measuring cognitive change in older adults: reliable change indices for the Mini-Mental State Examination. J Neurol Neurosurg Psychiatry. 2007;78(12):1298–1303. doi:10.1136/jnnp.2006.109074
32. Anderson C, Teo K, Gao P, et al. Renin-angiotensin system blockade and cognitive function in patients at high risk of cardiovascular disease: analysis of data from the ONTARGET and TRANSCEND studies. Lancet Neurol. 2011;10(1):43–53. doi:10.1016/S1474-4422(10)70250-7
33. Lay-Flurrie SL, Sheppard JP, Stevens RJ, et al. Impact of changes to national hypertension guidelines on hypertension management and outcomes in the United Kingdom. Hypertension. 2020;75(2):356–364. doi:10.1161/HYPERTENSIONAHA.119.13926
34. Chatterjee S, Khunti K, Davies MJ. Type 2 diabetes. Lancet. 2017;389(10085):2239–2251. doi:10.1016/S0140-6736(17)30058-2
35. Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 2016;15(7):673–684. doi:10.1016/S1474-4422(16)00070-3
36. Schupf N, Tang MX, Fukuyama H, et al. Peripheral Abeta subspecies as risk biomarkers of Alzheimer’s disease. Proc Natl Acad Sci USA. 2008;105(37):14052–14057. doi:10.1073/pnas.0805902105
37. Hilal S, Wolters FJ, Verbeek MM, et al. Plasma amyloid-beta levels, cerebral atrophy and risk of dementia: a population-based study. Alzheimers Res Ther. 2018;10(1):63. doi:10.1186/s13195-018-0395-6
38. de Wolf F, Ghanbari M, Licher S, et al. Plasma tau, neurofilament light chain and amyloid-beta levels and risk of dementia; a population-based cohort study. Brain. 2020;143(4):1220–1232. doi:10.1093/brain/awaa054
39. Iulita MF, Ganesh A, Pentz R, et al. Identification and preliminary validation of a plasma profile associated with cognitive decline in Dementia and at-risk individuals: a retrospective cohort analysis. J Alzheimers Dis. 2019;67(1):327–341. doi:10.3233/JAD-180970
40. Giudici KV, de Souto Barreto P, Guyonnet S, et al. Assessment of plasma Amyloid-β 42/40 and cognitive decline among community-dwelling older adults. JAMA Netw Open. 2020;3(12):e2028634. doi:10.1001/jamanetworkopen.2020.28634
41. Shahpasand-Kroner H, Klafki HW, Bauer C, et al. A two-step immunoassay for the simultaneous assessment of Abeta38, Abeta40 and Abeta42 in human blood plasma supports the Abeta42/Abeta40 ratio as a promising biomarker candidate of Alzheimer’s disease. Alzheimers Res Ther. 2018;10(1):121. doi:10.1186/s13195-018-0448-x
42. Verberk IMW, Hendriksen HMA, van Harten AC, et al. Plasma amyloid is associated with the rate of cognitive decline in cognitively normal elderly: the SCIENCe project. Neurobiol Aging. 2020;89:99–107. doi:10.1016/j.neurobiolaging.2020.01.007
43. Verberk IMW, Slot RE, Verfaillie SCJ, et al. Plasma amyloid as prescreener for the earliest Alzheimer pathological changes. Ann Neurol. 2018;84(5):648–658. doi:10.1002/ana.25334
44. Cullen NC, Leuzy A, Janelidze S, et al. Plasma biomarkers of Alzheimer’s disease improve prediction of cognitive decline in cognitively unimpaired elderly populations. Nat Commun. 2021;12(1):3555. doi:10.1038/s41467-021-23746-0
45. Gao F, Shang S, Chen C, et al. Non-linear relationship between plasma amyloid-β 40 level and cognitive decline in a cognitively normal population. Front Aging Neurosci. 2020;12:557005. doi:10.3389/fnagi.2020.557005
46. Lopez OL, Kuller LH, Mehta PD, et al. Plasma amyloid levels and the risk of AD in normal subjects in the Cardiovascular Health Study. Neurology. 2008;70(19):1664–1671. doi:10.1212/01.wnl.0000306696.82017.66
47. Locascio JJ, Fukumoto H, Yap L, et al. Plasma amyloid beta-protein and C-reactive protein in relation to the rate of progression of Alzheimer disease. Arch Neurol. 2008;65(6):776–785. doi:10.1001/archneur.65.6.776
48. Sagare A, Deane R, Bell RD, et al. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. 2007;13(9):1029–1031. doi:10.1038/nm1635
49. Younkin SG. Evidence that A beta 42 is the real culprit in Alzheimer’s disease. Ann Neurol. 1995;37(3):287–288. doi:10.1002/ana.410370303
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.