")
Back to Journals » Cancer Management and Research » Volume 9
The applications of liquid biopsy in resistance surveillance of anaplastic lymphoma kinase inhibitor
Authors Chen Y, Guo W, Fan J , Chen Y, Zhang X, Chen X, Luo P
Received 8 September 2017
Accepted for publication 26 October 2017
Published 7 December 2017 Volume 2017:9 Pages 801—811
DOI https://doi.org/10.2147/CMAR.S151235
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Video abstract presented by Jiaqi Ye on behalf of Chen et al
Views: 292
Yating Chen,1,* Wenjie Guo,1,* Junsheng Fan,2,* Yuqing Chen,1 Xiaoli Zhang,1 Xin Chen,1 Peng Luo1
1Department of Respiratory, Zhujiang Hospital of Southern Medical University, Guangzhou, China; 2Department of Respiratory, Shanghai Tenth People’s Hospital, Tongji University, Shanghai, China
*These authors contributed equally to this work
Abstract: With the clinical promotion of precision medicine and individualized medical care, molecular targeted medicine has been used to treat non-small cell lung cancer (NSCLC) patients and proved to be significantly effective. Anaplastic lymphoma kinase (ALK) inhibitor is one of the most important specific therapeutic agents for patients with ALK-positive NSCLC. It can extend the survival of patients. However, resistance to the ALK inhibitor inevitably develops in the application process. So, the real-time resistance surveillance is particularly important, and liquid biopsy is one of the most potential inspection methods. Circulating tumor cells, circulating free tumor DNA and exosome in body fluid are used as the main detection biomarkers to reflect the occurrence of resistance in real time through sequencing or counting and then to guide the follow-up treatment.
Keywords: NSCLC, ALK, resistance, liquid biopsy, CTCs, ctDNA, exosome
Introduction
As a malignant disease, lung cancer ranks first in incidence and mortality of cancer, and has so for a long period of time. It is predicted that there still will be more than 220,000 new cases and more than 160,000 deaths in the US in 2017.1 Non-small cell lung cancer (NSCLC) is the most common type of lung cancer, accounting for about 80%–90%.2–4 However, more than 50% of newly diagnosed patients have been in advanced stage or have gained metastases, and thus they have lost the opportunity for surgical treatment. Radiotherapy or chemotherapy is not ideal for them.5 Radiotherapy and chemotherapy are typical traditional tumor treatments but they lack cell killing specificity. This results in these treatments to not only kill tumor cells but also damage normal cells.. Therefore, they will produce a variety of toxic side effects on all systems in the body, and some patients cannot tolerate treatment. Though chemotherapy is still the main treatment for advanced NSCLC patients, clinical data suggest that the development of chemotherapy has encountered the situation of bottleneck.6 It cannot further extend remission and survival. Radiotherapy could only serve as a palliative reduction therapy.7 Furthermore, though radiotherapy and specific chemotherapy could be received by outpatients, compared with oral medication, they are considerably inconvenient, since patients who receive the treatment need to visit the hospital frequently. Due to the shortcomings of radiotherapy and chemotherapy, targeted therapy has gradually been paid more attention to and has gradually become a first-line treatment program.
With genetic and precision medicine developing in recent years, it has been found that the development of lung cancer is closely related to tumor-driven gene. Anaplastic lymphoma kinase (ALK) fusion gene and epidermal growth factor receptor (EGFR) are the NSCLC-driven genes that are currently being recognized.8–12 ALK fusion gene occurs in 3%–7% of patients with NSCLC, which includes echinoderm microtubule-associated protein-like 4-ALK (EML4-ALK), TRK-fused gene-ALK, nucleophosmin-ALK and kinesin family member 5B-ALK.13–18 ALK inhibitor (ALKi) is an effective targeted medicine for patients with ALK mutations, which can significantly prolong their survival. Crizotinib is a first-generation ALKi, and it has become a recommendation for the treatment of ALK-positive NSCLC patients in the guidelines of Europe and the US, and has been used as a first-line therapy for patients in advanced stage.19,20 Crizotinib can not only significantly prolong the progression-free survival (PFS), but also increase the objective response rate (ORR). The clinical trial of crizotinib (PROFILE 1014) indicated that the median PFS was 10.9 months, while the ORR was 74% and 1-year survival rate was 84%, demonstrating better efficacy than chemotherapy.21 But before long, it was found that about 30% of patients who used crizotinib as initial treatment have primary resistance to it, some patients developed secondary resistance within 1–2 years and about 40% developed metastasis in the central nervous system.22
With the emergence of crizotinib resistance, the second- and third-generation ALKi were gradually introduced and approved for clinical application. It was observed that the ORR and the median PFS of crizotinib-resistant patients increased to 50% and 8.9 months, respectively, after being treated with alectinib, a second-generation ALKi.23 The third-generation ALKi lorlatinib had a strong inhibitory effect on known resistant mutations and had a good therapeutic effect on ALK-driven brain metastasis.24 The ongoing I/II trial shows that the ORR of lorlatinib is 26%, indicating that the timely changing of treatment program can still bring about a positive effect when the original treatment escapes.25 But the direct application of the latest and most effective ALKi is not equal to permanent efficacy and benefit. A study has showed that a patient with lorlatinib escape had a new tumor gene mutation that caused his re-sensitization to crizotinib.26 All of these show that it is of clinical significance to guide the treatment at the molecular level after resistance. The real-time detection technology that can be applied to cancer patients is needed urgently in clinical practice. At present, the regular surveillance of lung cancer during treatment mainly depends on the pathological diagnosis after puncture biopsy and imaging examination. But pathological biopsy is an invasive operation that cannot be carried out in all patients, and the compliance of the patients becomes worse during repeated operation. Furthermore, the error resulting from tumor heterogeneity cannot be overcome now, while the imaging examination is often delayed.
As early as the 19th century, Paget had put forward the seed-soil theory.27 It was thought that “seeds” would fall off from the primary tumor with the appropriate distal organs as “soil” to form metastases, which was the most original definition of what is called the circulating tumor cell (CTC) now. Subsequently, the researchers also found other tumor-derived biomarkers, such as circulating tumor DNA (ctDNA), exosome, etc. These biomarkers carry information about the source of the organization and flow freely in various body fluids.28 Liquid biopsy distinguishes itself as a new technique for separation and analysis of the tumor-derived biomarkers in body fluid, which obtain the coding information of tumor. It is an ideal technique for ALK-positive NSCLC resistance surveillance for its advantages such as simplicity, minimally invasive, real time and repeatability.
The mechanisms of ALKI resistance
Despite the constant upgrading of ALKi and its enhancing efficacy, the resistance still cannot be overcome. ALKi resistance can be divided into primary resistance and secondary resistance.
Primary resistance
Primary resistance refers to the ineffective medical treatment in the early stage.29 About 25% of patients treated with crizotinib have primary resistance.30 Although the mechanism of primary resistance is not fully understood yet, it is speculated that it is closely related to ALK variations. ALK variations include the fusion of ALK with other partner genes encoding protein other than EML4 and structural variations of ALK itself.31 Experiments showed that there is a significant difference in sensitivity to crizotinib among tyrosine kinase receptors coded by different ALK fusion genes or different subtypes of same ALK fusion gene.32 In addition, tumor heterogeneity can also lead to primary resistance. Studies have found that 5% -8% of cancer cells in ALK-positive NSCLC patients contain EGFR mutation, which causes the failure of ALKi. Another cause may be false-positive genotype. ALK tumor-driven mutation can be monitored by a variety of techniques, and ALK fluorescence in situ hybridization (FISH) testing is considered the current gold standard procedure.33–35 It is believed that false-positive results could be led by artificial cutting gene fragment error, cell morphological differences, probe abnormalities or pathological misunderstanding in the detection process.36 Even VENTANA ALK (D5F3) CDx assay with higher specificity and sensitivity in detecting ALK-positive NSCLC, which is recently approved by the US Food and Drug Administration (FDA), can also cause false-positive results.37 Thus, in a few cases, ALKi primary resistance may be caused by technical factors rather than a real existence of biological resistance. Finally, although ALK FISH could recognize true ALK translocation, these patients did not have functional rearrangements, which is also a pseudo-positive resistance that results from pseudo-primary genotype.
Secondary resistance
Secondary resistance, also known as acquired resistance, refers to the recurrence of tumor progression that the original ALKi-sensitive patients suffer after complete remission or partial remission after more than 6 months with medical treatment.38 Secondary resistance can be divided into dominant and non-dominant. Dominant secondary resistance mainly refers to ALK kinase domain mutation (29%) and increase of ALK gene copy number (9%), which accounts for one-third of crizotinib resistance by increasing the activity of tyrosine kinases.39–41 Other types of resistance are non-dominant, including ALK signal bypass activation, tumor heterogeneity, etc. In fact, two or more resistance patterns could be generated in the same patient. A large part of the resistance mechanism still remains unknown.
Mutation in the target ALK gene
When the first mutation emerges in the ALK fusion gene, it induces the production of tumor; when secondary mutation comes up, it means the emergence of resistance. Most of the mutations currently found in the target ALK gene are mainly point mutation. C1156Y and L1196M were the first discovered mutant types, followed by L1152R, G1202R, G1269A, F1174L, 1151Tins, S1206Y, I1171T, D1203N, V1180L, etc.32,42,43 Crizotinib is an ATP-competitive selective inhibitor of the ALK and MET tyrosine kinases, which inhibits their tyrosine phosphorylation.44 With low level of tyrosine kinase phosphorylation, aberrant downstream pathways of ALK mutations will be inhibited. In general, these aberrant downstream pathways can lead to apoptosis insufficiency, excessive cell proliferation and the development of NSCLC.45 Ultimately, crizotinib reaches the pharmacological effect of restraining the development of tumor. As the secondary mutation emerges, the protein composition of tyrosine kinase changes and its activity increases, which leads to the decrease of the inhibitory effect. But the point mutations do not destroy the effect of ALK signaling pathway, and so the tumor can still rely on the pathway for survival and development. Now the most common mutation of ALK kinase domain in the crizotinib resistance is L1196M, leading to replacement of leucine by methionine in the kinase region. It helps in the formation of the protein active conformation and increases the activity of protein kinase, interfering with the inhibitory effect of the targeted treatment on kinase activity, and finally leading to treatment failure.46 Studies have shown that each ALKi will specifically correspond to a group of resistant mutant types. G1202R is one of the specific resistance mutation types that responses to the second generation of ALKi.47
Abnormal copy number of fusion gene
ALK fusion gene copy number abnormalities include copy number gain (CNG) and gene deletion. The resistance mechanism of abnormal ALK gene copy number is similar to the gene mutation, increasing the activity of kinase. The CNG means that the average number of the rearranged genes in lung cells are more than triple. Katayama et al analyzed the genes in crizotinib-resistant patients and found that ALK fusion gene copy number increased remarkably, and confirmed that CNG is to blame for the resistance in vitro experiments.41 Doebele et al happened to discover a special sample of resistance that may be caused by deletion of the ALK fusion gene.46 It was after exclusion of common causes of resistance such as gene mutations, CNG, signal bypass activation and after multiple biopsies, that researchers thought that the resistance was indeed caused by genetic deficiency. But the view that ALK fusion gene deficiency is the mechanism of ALKi resistance has not been fully verified.
Emergence of bypass tracks
Signal bypass activation refers to the activation of other carcinogenic drivers. When ALKi inhibits the level of tyrosine kinase phosphorylation and blocks the downstream signaling pathways, tumor cells can reduce its dependence on ALK and downstream signals by activating other signaling pathways or the ALK signaling pathway in downstream that is not inhibited, resulting in the failure of ALKi. It results in 20% of the cases of crizotinib resistance.49 The key signaling transduction pathways in downstream of ALK include Ras/MEK/ERK, PI3K/AKT and JAK3-STAT3.50,51 Takezawa et al found that ALKi could inhibit the amplification of EML4-ALK positive tumor cells and induce apoptosis by inhibiting the transduction pathways of ERK and STAT3, while the pathway of PI3K/AKT was not affected or less affected and finally causes resistance.52 In addition, tumors can continue to develop by activating other tumor driven genes. The most common driven genes include EGFR and KRAS mutations, EGFR phosphorylation and c-KIT amplification.39,48 Activation of a variety of different carcinogenic drivers can occur simultaneously in the tumor. In an in vitro experiment, the expression of EGFR mRNA in cells with resistance was upregulated after applying crizotinib to ALK-positive cell lines, indicating that the continual progression of tumor could be owing to the activation of EGFR and persistent activation of downstream signal transduction pathway independent from ALK.53
Tumor heterogeneity
There still lacks a thorough understanding about resistance on account of tumor heterogeneity. ALK-positive solid tumor could be a mixture of a majority of ALK-positive tumor cells and other tumor cells induced by different tumor-driven genes. The growth and amplification of predominant ALK-positive tumor cells would be inhibited and the original minority of tumor cells driven by other tumor genes was transformed into a dominant cell line after the application of ALKi. It is also possible that there are multiple dominant and non-dominant mechanisms of resistance in the tumor, for which ALKi would be helpless. The American Society of Clinical Oncology meeting reported a study on the distribution of driven genes in NSCLC patients in 2014. The study included 35 cases of patients with multiple pulmonary nodules, and it showed that the inconsistency rate of mutation-driven gene of different nodules reached 68.6%.54 But the current mainstream surveillance techniques, such as pathological biopsy and imaging examination, still cannot overcome the detection error of tumor heterogeneity.
Others
In addition, there are some rare types of non-dominant resistance. Researchers had found an abdominal nodule transformed from small cell lung cancer (SCLC) in a patient with crizotinib treatment failure.55 In another case, SCLC-transformed issue was detected in the lung tissue of alectinib-resistant patients.56 They both indicated the potential of SCLC transformation to be a novel resistance mechanism. Moreover, ALKi resistance is probable to be achieved by epithelial-mesenchymal transition, autophagy, etc.57,58 The current discovered mechanisms are only a small part; many other mechanisms of resistance still need further study.
Liquid biopsy
Due to the insurmountable shortcomings of imaging and pathological biopsy methods, and the necessity for real-time resistance surveillance in the treatment of ALK-positive NSCLC patients, the liquid biopsy technology that can achieve minimally invasive and real-time surveillance has attracted more and more attention and expectation. The most common biomarkers of liquid biopsy are CTCs, ctDNA and exosome. They are permissible for separation, purification and analysis of tumor DNA, mRNA, miRNA and other genetic materials. Since biomarkers can be derived from different parts of the tumor or different cancer foci, liquid biopsy can overcome the interference of tumor heterogeneity and reflect the overall information of tumor. Therefore, liquid biopsy is the most potential and ideal detection method for resistance surveillance at present. The research progress of liquid biopsy in resistance surveillance is shown in Figure 1.
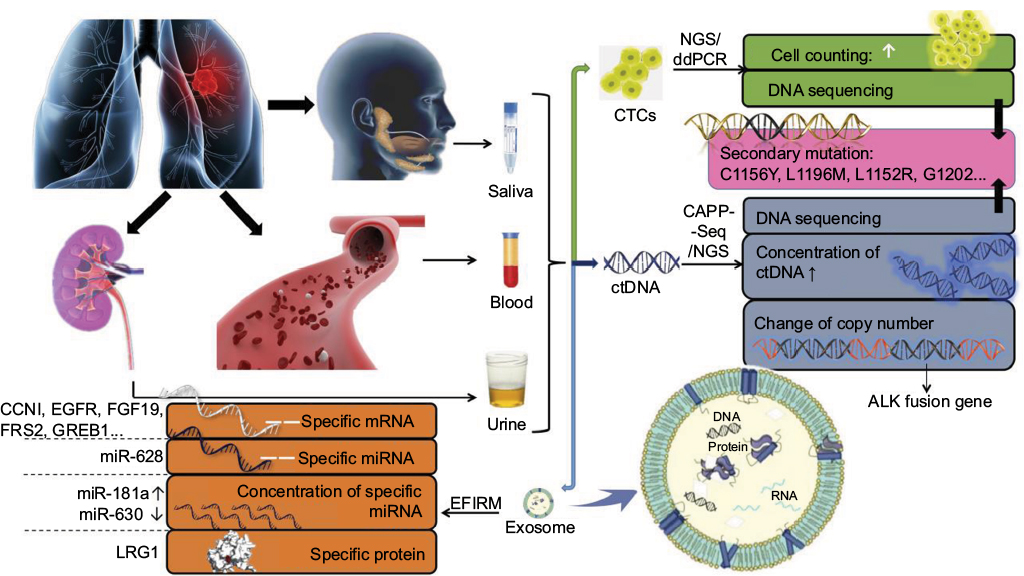 | Figure 1 The research progress of liquid biopsy in resistance surveillance. Notes: Biomarkers of liquid biopsy can be extracted from saliva, peripheral blood, urine and other body fluid. CTCs, ctDNA and exosome are main detection biomarkers. CTC counting and DNA sequencing of CTC could be detected by NGS and ddPCR for resistance surveillance. With CAPP-Seq and NGS, ctDNA detection is helpful to find out secondary mutations, concentration increase of ctDNA and change of ALK gene copy number, which suggest tumor resistance. Exosome is an extracellular vesicle containing specific mRNA, miRNA and protein that would provide clue to resistance. EFIRM helps to release the contents of exosome and then detect them. Abbreviations: CTC, circulating tumor cell; ctDNA, circulating tumor DNA; NGS, next-generation sequencing; ddPCR, digital droplet polymerase chain reaction; CAPP-Seq, cancer personalized profiling by deep sequencing; EFIRM, electric field-induced release and measurement technology. |
CTC
CTC refers to the tumor cells that fall off from primary lesions or metastases of tumor and then enter into the blood circulation.59 But the detection rate of CTC is low; only one CTC can be detected in an average of 105–107 monocytes in the peripheral blood of patients with advanced tumors.60 Genetic materials, such as DNA, RNA and protein, are contained in CTCs. Digital droplet polymerase chain reaction combined with next-generation sequencing (NGS) is currently used to detect and sequence genes in CTCs, which help to obtain the information about the source of the organization.61 CTC can also be detected by CellSearch, which is approved by FDA.62 When secondary resistance occurs or other carcinogenic drivers are activated, theoretically, resistance can be indicated by the percentage of mutated CTCs and the mutations of gene, mRNA and protein. The half-life of CTC in blood is only a few hours. Therefore, it can be used as a real-time detection target.63,64
The mutation of L1196M was found by enrichment and gene sequencing of the CTCs in peripheral blood of patients with crizotinib secondary resistance, indicating that the resistance mutation can be found at an early stage through the gene sequencing on mutated CTCs.65 Thus, it would be beneficial to conduct a large sample of gene sequencing to obtain the specific resistant mutation spectrum of different ALKi, which may be helpful for the diagnosis of resistance. In another study, researchers tried to obtain the quantitative change of EML4-ALK+ CTC detected in serum of patients with adenocarcinoma as a prompt of resistance.66 At first, three CTCs were detected before the treatment with crizotinib, one of which was EML4-ALK + CTC. After treatment, no EML4-ALK+ CTC was found, while two EML4-ALK + CTC recurred in the further progress of tumor. The reappearance of EML4-ALK + CTC indicated that the patients acquired crizotinib resistance. As there was no gene detection on CTC for surveillance, researchers give priority to the dominant resistance when the reasons for resistant are unknown. So, ceritinib was used for treatment, but EML4-ALK + CTC and tumor progression were still detectable subsequently. At last, it was considered as non-dominant resistance, and had chemotherapy as adjuvant therapy. Although the method of CTCs counting for resistance surveillance is less accurate than gene sequencing, it is simpler. Coupled with the clinical and imaging performance, the resistance and its mechanism can be roughly determined. TKI of other target genes is often used in combination for treatment when the non-dominant resistance occurs, especially when combined with other tumor-driven genes or in the presence of signal bypass activation. A study based on Asians found that 18.6% of ALK-positive patients had EGFR mutations, while 3.9% of EGFR mutation patients had ALK fusion.67 The combination of ALKi and EGFRi can achieve a better inhibitory effect than the use of each of the specific inhibitors alone. In addition, using the treatment-resistant ALKi, or a combination of heat shock protein inhibitors (HSP90) or chemotherapy with the treatment-resistant ALKi has a positive significance for patients.21,68
Despite the low concentration of CTCs in peripheral blood and a lack of detection technology with high sensitivity and specificity, CTC is ideal for detection and analysis, for its capability of overcoming the disturbance of tumor heterogeneity because it is derived from different parts of the solid tumor or different cancer foci. Moreover, CTC has a full set of tumor genetic information that can be used to study tumor phenotype and cellular function. Due to the presence of resistance, a series of biopsies is usually required to detect the progression of the tumor after the use of ALKi. Though it is possible to perform multiple biopsy tests on the patients technically, it will be followed by operational risk and poor tolerability of the frail patients. Now DNA in CTCs is considered to be a more ideal detection target for re-biopsy.69–71
ctDNA
Circulating free DNA (cfDNA) is defined as the tissue-specific DNA fragment that is released into the blood.72 CtDNA refers to the cfDNA that is secreted by tumor cells or released after apoptosis and necrosis.73 The level of ctDNA in tumor patients is positively correlated with tumor progression, and the half-life is generally about 2 hours, which can better reflect tumor burden and the effectiveness of the current treatment in real time than imaging examination.74,75 Secondly, ctDNA sequencing can be performed by the technique of NGS and cancer personalized profiling by deep sequencing (CAPP-Seq), with which the tumor resistance or the change of gene after progression can be found.76,77 It is also possible for it to reflect the dominant tumor cell population at different points in time, regardless of the heterogeneity.78
In a study, a tracing blood test was performed on two advanced NSCLC patients with crizotinib treatment, and found that the ctDNA in serum and the allele frequency of ALK rearrangements in ctDNA significantly increased after 10 months of medication, suggesting that the treatment failed.79 Three resistance mutations, L1152R, I1171T and L1196M, could be detected in the ctDNA of one of the patients, indicating that the change of nucleic acid sequence and blood concentration of ctDNA can be sensitive to detect the resistance. At this point, the second generation of ALKi should be put into use. The experimental data showed that the ORR of the second-generation ALKi, ceritinib and alectinib, after the treatment with crizotinib failed, were 56% and 50% respectively.22,80 Especially, alectinib had a significant effect on secondary resistance caused by L1196R.36 The other patient with central nervous system metastasis who had not used crizotinib in the same study sequentially used the second-generation ALKi ceritinib and alectinib, and had progress of metastases 1 year later.81 The ALK G1202R mutation was detected in ctDNAs. The clinical manifestations and the results of ctDNA detection pointed out the occurrence of resistance, and then the patient switched to the third-generation ALKi lorlatinib. The clinical symptoms caused by metastases improved, and the concentration of ctDNA of G1202R in peripheral blood also decreased, showing the significance of ctDNA in resistance surveillance and guiding follow-up therapy. It is particularly important to detect ctDNA mutation after using the second-generation ALKi resistance. The second-generation ALKi can be used for patients who had or did not have the resistant mutation of the first-generation ALKi. While the presence of new secondary resistance mutation in ALK fusion gene, such as G1202R, is an essential indication of lorlatinib, the third-generation ALKi, otherwise, lorlatinib will not work.82
Because ctDNA is freely available in the body fluids with low concentration, it is easily decomposed by enzymes or phagocytosed by cells. There still lacks effective separation and analysis technique at present. The current detection technique is prone to false-negative or false-positive results. Moreover, ctDNA is dissociated from the extracellular region, resulting in the detection result of nucleic acid not being co-located with protein, nor can the function of cells be studied. CtDNA is now the most commonly used biomarker in all reports of ALK-positive NSCLC resistance surveillance. But the testing standard and evaluation criteria are different in different laboratories. Therefore, it is necessary to provide a unified industry measurement standard before putting it into clinical use. But overall, ctDNA is indeed an ideal biomarker. It has the characteristics of minimally invasive, strong specificity and can overcome the tumor heterogeneity and reflect the integral information of the tumor. It is more sensitive to the detection of the mutation of the tumor and permissible for recognizing known resistance mutations. All of these traits meet the requirements of a repeatable biopsy technique that is urgently needed in the resistance surveillance process.83–85
Exosome
Exosome is an extracellular vesicle with a diameter of 30–120 nm, the surface of which is made up of lipid bilayer vesicles, containing DNA, mRNA, miRNA, protein and other genetic materials.86,87 In physiological and pathological conditions, exosome can be secreted by different types of cells.88 It carries different components closely based on its parental-derived cells. The specific components can be referred in the “exosome content database” (www.exocarta.org).89 The current exosome content database contains approximately 1,639 mRNAs, 764 miRNAs, 4,563 proteins and 194 lipids. The peripheral blood of tumor patients has higher concentration of exosome than that in normal ones, and it carries tumor-specific molecular substances such as DNA and gene mutations.90 Although changes in protein-coding genes are key targets for elucidating tumor-specific changes, recent studies have shown that miRNAs have a greater advantage in detecting convenience and demonstrating tumor specificity. A study about ovarian cancer found that 8 miRNA-specific diagnostic markers could be detected simultaneously in tumor tissue and exosome of patients.91 The miRNAs of exosome were introduced into the exosomes through a special sorting mechanism, implying that the exosome miRNA spectrum was highly similar to the miRNA spectrum of the parental cancer cells.92–94 Its information is more tumor-specific and better than global mRNA spectrum, which is used to define the types of cancer and other subtypes of RNA.95,96 Although there is no report illustrating the use of exosome in resistance surveillance currently, similar to tumor DNA, changes in copy number of exosome miRNAs and nucleic acid sequences are considered to be available for resistance surveillance.
Though the specific research of exosome miRNAs in the field of resistance surveillance is deficient temporarily, we can find its potential and future development according to the appliance of tumor miRNA. MiRNA is a small non-coding RNA, consisting of about 18–25 nucleotides and being responsible for negative regulatory gene expression after transcription. Specific miRNA is able to indicate its binding effect on proto-oncogenes or tumor suppressor genes.97,98 Studies have found that the expression of tumor miRNAs is associated with tumor biology. When the expression of certain miRNAs increases, it may lead to downregulation of tumor suppressor genes, while the decrease in expression of other miRNAs can lead to an increase in the expression of oncogenes.99,100 Both of them can lead to tumor progression and resistance. The sensitivity of NSCLC to cisplatin is associated with the upregulation of miR-181a, while the upregulation of miR-630 is related to resistance.101 However, there is still a lack of research on the correlation between miRNA expression and ALKi resistance.
The abnormal expression of miRNA is a common feature of dysplasia and cancer. The expression profile of miRNA is associated with the progression of disease and prognosis.102,103 A total of 12 special miRNAs were found to be particularly present in the exosomes of patients with NSCLC, indicating that the current technique is capable of supporting miRNAs’ detection that were produced after resistance.104 MiR-628, another marker in crizotinib-resistant cell line, shows an overexpression, indicating the significance of miRNA in resistance surveillance.105
As with CTCs and ctDNA, exosomes all have important implications in resistance testing, and it may be more valuable than the former two. The concentration of exosomes in peripheral blood is higher than that of CTCs, and it can be detected in urine, saliva, cerebrospinal fluid, semen, milk, pleural effusion, ascites and other body fluids in addition to serum or plasma.106 The exosome contents are protected by the vesicle membrane. They can be prevented from being destroyed by enzymes in the body fluids and become more complete and stable. The surveillance of platelet endocytosis of tumor-derived RNA is even earlier than imaging to detect tumor progression.107
Other body liquid
Currently, liquid biopsy used in clinical and laboratory applications primarily uses peripheral blood to enrich and analyze the biomarkers. In fact, studies have proved that urine, saliva and pleural effusion are available for noninvasive detection.
Urine
A total of 150 patients with positive EGFR gene mutation (that is, L858R or L861Q positive) and EGFR-tyrosine kinase inhibitor (TKI) treatment accepted a long-term surveillance.108 The data showed that the level of cfDNA in plasma and urine was significantly decreased in early treatment. With the application of TKI, resistant individuals gradually appeared. In all, 53% of the treatment-resistant patients had double growth of the cfDNA in plasma and urine. The secondary mutation EGFR T790M can be detected in some patients. Both liquid biopsy and pathologic biopsy were conducted to detect patients with resistance in this study. When urine was used as the detection sample of liquid biopsy, the detection results of liquid biopsy and pathologic biopsy overlapped by 88%, while the detection results of both biopsy techniques were exactly the same when using plasma samples for liquid biopsy. It indicated that urine could also serve as a liquid biopsy material, and urine cfDNA could be used as an indicator of tumor progression. In addition, comparing exosome protein in urine and lung cancer tissue of the normal control group with that of NSCLC patients, it was found that leucine-rich α-2-glycoprotein (LRG1) was highly expressed in urinary exosome and lung tissues of NSCLC patients, showing that the exosome LRG1 in urine may be a candidate biomarker for noninvasive diagnosis of NSCLC.109
Saliva
With the development of real-time quantitative PCR detection system, the five biomarkers (CCNI, EGFR, FGF19, FRS2 and GREB1) of salivary exosome mRNA are effective in distinguishing lung cancer patients from normal controls.110 These salivary mRNA biomarkers have the ability to detect lung cancer. Electric field-induced release and measurement technology (EFIRM) is also a popular technique for the analysis of exosomes currently. It can effectively cleave exosomes; release proteins, RNA and DNA; and detect tumor biomarkers.111 At present, this technique has been proved to succeed in detecting tumor-driven mutations in saliva of NSCLC patients.112 The difference between saliva detection and plasma detection is not significant.113 It is expected to be used in diagnosis and treatment guidance.
Pleural effusion
EGFR mutation has been detected in the CTCs derived from pleural effusion, indicating that biomarkers from pleural effusion can offer a clue to the existence or progression of tumor.114 Pleural effusion may be more specific than plasma, urine and saliva for its proximity to cancer foci. However, pleural effusion sampling, like tissue biopsy, is an invasive operation, which restricts its development.
The value of other body fluids on the diagnosis of NSCLC has been fully supported according to the current experimental results, of which there are a great many studies on EGFR-TKI resistance surveillance. More studies and experiments are expected to confirm their viability in ALKi resistance surveillance.
Discussion and conclusion
This paper focused on the use of liquid biopsy on ALKi resistance surveillance. Actually, liquid biopsy is applicable to the surveillance of all lung cancer patients. Although targeted therapy is superior to chemotherapy in PFS and tolerance, targeted therapy has not surpassed chemotherapy in terms of overall survival.115 Patients with genetic mutations are a minority after all, and chemotherapy and radiotherapy are still the treatment of most patients with lung cancer.116 Liquid biopsy can predict tumor progression, which can also guide the adjustment of chemotherapy program or increase the radiotherapy dose. Certainly, the best indication for liquid biopsy is targeted therapy, which is mainly for NSCLC patients. The existing clinical data point out that the molecular targeted medicine has a significant effect on NSCLC patients with gene mutation. Although it is not possible to solve the problem of resistance up to now, liquid biopsy plays an important role in warning resistance as well as its related mutation in early stage and guiding the alteration of treatment program, which has a positive effect on relieving the disease and prolonging the survival. With the further study of NSCLC, more and more specific tumor-driven genes and the corresponding targeted medicine are bound to be found. Liquid biopsy will keep promoting the development of targeted therapies.
Liquid biopsy is superior to traditional pathologic biopsy and imaging for its unique noninvasive nature, convenience and repeatability. However, many obstacles need to be overcome before it becomes a practical method to monitor treatment resistance. With small size and low concentration, there is still a lack of specific and sensitive separation and analysis techniques for the biomarkers used in the experimental study. The results of these studies from different laboratories also lack unified interpretation for resistance evaluation. It is prone to be false negative or false positive when using FISH, in situ hybridization (ICH) and other detection technology in disease surveillance, and their ratio of signal-to-noise tends to be low when detecting the early disease. The application of exosomes in resistance surveillance is still to be developed.
To overcome the obstacles of using liquid biopsy in clinical research and clinical practice in ALK-translocated patients, researchers came up with several solutions. Setting ALK-CNG as an example, an enrichment technique is supposed to be performed before detecting ALK-CNG, such as isolation by size of epithelial tumor cells, which is more effectively in CTC recovery than CellSearch.117 Then, Pailler et al developed filter-adapted FISH to increase cell recovery as well as to detect mutations in ALK-positive CTCs.118 For ctDNA, FISH is generally available in pathology laboratories for gene sequencing, but it is often used to identify copy number alternation of known gene.119 A capture-based NGS ctDNA detection method called CAPP-Seq was developed with ultrasensitivity for even unknown genes and all major classes of mutations in large portions of the genome, while NGS were developed with ultrasensitive for specific genomic positions.120 EFIRM is a newly developed technique that combines the rapid extracellular vesicle lysis procedure and detection and capture of molecular content from the EV with high sensitivity, which reduces the content degradation caused by exposure.121 Though it is impossible to totally overcome false-negative and false-positive results in theory, it is advised to apply two kinds of detection techniques simultaneously to improve accuracy. For example, a combination of ICH assay and FISH is usually used in clinical research. ICH is usually recommended as a primary assay because of its greater sensitivity and specificity, while FISH is reserved for use only in ICH equivocal cases.119,122 To date, the absence of unified interpretation for resistance evaluation results from the standpoints varying from one researcher to another and different detection techniques used in studies. Only by developing an acknowledged detection technique and stimulating a great deal of experimental and clinical data will the unified interpretation gradually be brought about.
With the promotion of precision medicine in NSCLC and the continued development of ALKis, more accurate auxiliary detection technology is the focus of liquid biopsy. Meanwhile, more laboratory evidence is needed to demonstrate the practicability of liquid biopsy in resistance surveillance. Real-time liquid biopsy is the trend in the application of resistance surveillance of targeted NSCLC therapy in ALK-positive patients. It will effectively guide patients with ALKi resistance to change therapy in time, prolong their lives and avoid the toxic side effects caused by ineffective treatment.
Acknowledgment
This work was supported by Nature Science Foundation of Guangdong Province, People’s Republic of China (2016A030313620).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Chunhacha P, Chanvorachote P. Roles of caveolin-1 on anoikis resistance in non small cell lung cancer. Int J Physiol Pathophysiol Pharmacol. 2012;4(3):149–155. | ||
Vázquez S, Casal J, Afonso Afonso FJ, et al. EGFR testing and clinical management of advanced NSCLC: a Galician Lung Cancer Group study (GGCP 048-10). Cancer Manag Res. 2016;8:11–20. | ||
Guo WJ, Liu SH, Zhang XL, et al. The coexpression of multi-immune inhibitory receptors on T lymphocytes in primary non-small-cell lung cancer. Drug Des Devel Ther. In press 2017. | ||
Hamilton G, Rath B. Detection of circulating tumor cells in non-small cell lung cancer. J Thorac Dis. 2016;8:1024–1028. | ||
Scagliotti GV, De Marinis F, Rinaldi M, et al; Italian Lung Cancer Project. Phase III randomized trial comparing three platinum-based doublets in advanced non-small-cell lung cancer. J Clin Oncol. 2002;20(21):4285–4291. | ||
Nestle U, Nieder C, Walter K, et al. A palliative accelerated irradiation regimen for advanced non-small-cell lung cancer vs. conventionally fractionated 60 GY: results of a randomized equivalence study. Int J Radiat Oncol Biol Phys. 2000;48(1):95–103. | ||
Yochum ZA, Socinski MA, Burns TF. Paradoxical functions of ZEB1 in EGFR-mutant lung cancer: tumor suppressor and driver of therapeutic resistance. J Thorac Dis. 2016;8(11):E1528–E1531. | ||
Valdespino V, Valdespino PM. Potential of epigenetic therapies in the management of solid tumors. Cancer Manag Res. 2015;7:241–251. | ||
Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4(6):662–673. | ||
Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol. 2016;9:34. | ||
Wang S, Song Y, Yan F, Liu D. Mechanisms of resistance to third-generation EGFR tyrosine kinase inhibitors. Front Med. 2016;10(4):383–388. | ||
Iragavarapu C, Mustafa M, Akinleye A,. Novel ALK inhibitors in clinical use and development. J Hematol Oncol. 2015;8:17. | ||
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. | ||
Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–4253. | ||
Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15(9):3143–3149. | ||
Schaefer ES, Baik C. Proactive management strategies for potential gastrointestinal adverse reactions with ceritinib in patients with advanced ALK-positive non-small-cell lung cancer. Cancer Manag Res. 2016;8:33–38. | ||
Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–1203. | ||
Ettinger DS, Akerley W, Borghaei H, et al; National comprehensive cancer network. Non-small cell lung cancer. J Natl Compr Canc Netw. 2012;10:1236–1271. | ||
Joshi M, Rizvi SM, Belani CP. Afatinib for the treatment of metastatic non-small cell lung cancer. Cancer Manag Res. 2015;7:75–82. | ||
Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. | ||
Ou SH, Jänne PA, Bartlett CH, et al. Clinical benefit of continuing ALK inhibition with crizotinib beyond initial disease progression in patients with advanced ALK-positive NSCLC. Ann Oncol. 2014;25(2):415–422. | ||
Ou SH, Ahn JS, De Petris L, et al. Alectinib in crizotinib-refractory ALK-rearranged non-small-cell lung cancer: a phase II global study. J Clin Oncol. 2016;34(7):661–668. | ||
Zou HY, Friboulet L, Kodack DP, et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell. 2015;28(1):70–81. | ||
Solomon BJ, Bauer TM, Felip E, et al. Safety and efficacy of lorlatinib (PF-06463922) from the dose-escalation component of a study in patients with advanced ALK+ or ROS1+ non-small cell lung cancer (NSCLC). J Clin Oncol. 2016;34:9009. | ||
Shaw AT, Friboulet L, Leshchiner I, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med. 2016;374(1):54–61. | ||
Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8(2):98–101. | ||
Rolfo C, Castiglia M, Hong D, et al. Liquid biopsies in lung cancer: the new ambrosia of researchers. Biochim Biophys Acta. 2014;1846(2):539–546. | ||
Quintás-Cardama A, Kantarjian HM, Cortes JE. Mechanisms of primary and secondary resistance to imatinib in chronic myeloid leukemia. Cancer Control. 2009;16(2):122–131. | ||
Mok T, Kim DW, Wu YL, et al. First-line crizotinib versus pemetrexed-cisplatin or pemetrexed-carboplatin in patients (pts) with advanced ALK-positive non-squamous non-small cell lung cancer (NSCLC): results of a phase III study (PROFILE 1014). J Clin Oncol. 2014;32:8002. | ||
Heuckmann JM, Balke-Want H, Malchers F, et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin Cancer Res. 2012;18(17):4682–4690. | ||
Heuckmann JM, Hölzel M, Sos ML, et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin Cancer Res. 2011;17(23):7394–7401. | ||
Shaw AT, Solomon B, Kenudson MM. Crizotinib and testing for ALK. J Natl Compr Canc Netw. 2011;9(12):1335–1341. | ||
Kaneda H, Yoshida T, Okamoto I. Molecularly targeted approaches herald a new era of non-small-cell lung cancer treatment. Cancer Manag Res. 2013;5:91–101. | ||
Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol. 2013;8(7):823–859. | ||
Camidge DR, Kono SA, Flacco A, et al. Optimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatment. Clin Cancer Res. 2010;16(22):5581–5590. | ||
Conde E, Hernandez S, Prieto M, Martinez R, Lopez-Rios F. Profile of Ventana ALK (D5F3) companion diagnostic assay for non-small-cell lung carcinomas. Expert Rev Mol Diagn. 2016;16(6):707–713. | ||
Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28(2):357–360. | ||
Wu J, Savooji J, Liu D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J Hematol Oncol. 2016;9:19. | ||
Toyokawa G, Seto T. Updated evidence on the mechanisms of resistance to ALK inhibitors and strategies to overcome such resistance: clinical and preclinical data. Oncol Res Treat. 2015;38(6):291–298. | ||
Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4(120):120ra17. | ||
Choi YL, Soda M, Yamashita Y, et al; ALK Lung Cancer Study Group. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–1739. | ||
Sasaki T, Okuda K, Zheng W, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70(24):10038–10043. | ||
Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. | ||
Croegaert K, Kolesar JM. Role of anaplastic lymphoma kinase inhibition in the treatment of non-small-cell lung cancer. Am J Health Syst Pharm. 2015;72(17):1456–1462. | ||
Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–1482. | ||
Gainor JF, Dardaei L, Yoda S, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016;6(10):1118–1133. | ||
Peters S, Adjei AA, Gridelli C, Reck M, Kerr K, Felip E; ESMO Guidelines Working Group. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(Suppl 7):vii56–vii64. | ||
Boland JM, Jang JS, Li J, et al. MET and EGFR mutations identified in ALK-rearranged pulmonary adenocarcinoma: molecular analysis of 25 ALK-positive cases. J Thorac Oncol. 2013;8(5):574–581. | ||
Mossé YP, Wood A, Maris JM. Inhibition of ALK signaling for cancer therapy. Clin Cancer Res. 2009;15(18):5609–5614. | ||
Chiarle R, Simmons WJ, Cai H, et al. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005;11(6):623–629. | ||
Takezawa K, Okamoto I, Nishio K, Jänne PA, Nakagawa K. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res. 2011;17(8):2140–2148. | ||
Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108(18):7535–7540. | ||
Ren S, Wu C, Li X, Zhao C, Hou L, Zhou C. Incidence of inconsistent driver mutations between multiple lung ground-glass nodules in patients with non-small cell lung cancer. J Clin Oncol. 2014;32(15 Suppl):11068. | ||
Cha YJ, Cho BC, Kim HR, Lee HJ, Shim HS. A case of ALK-rearranged adenocarcinoma with small cell carcinoma-like transformation and resistance to crizotinib. J Thorac Oncol. 2016;11(5):e55–e58. | ||
Takegawa N, Hayashi H, Iizuka N, et al. Transformation of ALK rearrangement-positive adenocarcinoma to small-cell lung cancer in association with acquired resistance to alectinib. Ann Oncol. 2016;27(5):953–955. | ||
Ignatius Ou SH, Azada M, Hsiang DJ, et al. Next-generation sequencing reveals a Novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J Thorac Oncol. 2014;9(4):549–553. | ||
You L, Shou J, Deng D, et al. Crizotinib induces autophagy through inhibition of the STAT3 pathway in multiple lung cancer cell lines. Oncotarget. 2015;6(37):40268–40282. | ||
Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127(4):679–695. | ||
Allan AL, Keeney M. Circulating tumor cell analysis: technical and statistical considerations for application to the clinic. J Oncol. 2010;2010:426218. | ||
Toss A, Mu Z, Fernandez S, Cristofanilli M. CTC enumeration and characterization: moving toward personalized medicine. Ann Transl Med. 2014;2(11):108. | ||
Tamminga M, Groen HH, Hiltermann TJ. Investigating CTCs in NSCLC-a reaction to the study of Jia-Wei Wan: a preliminary study on the relationship between circulating tumor cells count and clinical features in patients with non-small cell lung cancer. J Thorac Dis. 2016;8(6):1032–1036. | ||
Meng S, Tripathy D, Frenkel EP, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10(24):8152–8162. | ||
Leong SM, Tan KM, Chua HW, et al. Sampling circulating tumor cells for clinical benefits: how frequent? J Hematol Oncol. 2015;8:75. | ||
Zhang Z, Shiratsuchi H, Palanisamy N, Nagrath S, Ramnath N. Expanded circulating tumor cells from a patient with ALK-positive lung cancer present with EML4-ALK rearrangement along with resistance mutation and enable drug sensitivity testing: a case study. J Thorac Oncol. 2017;12(2):397–402. | ||
Aieta M, Facchinetti A, De Faveri S, et al. Monitoring and characterization of circulating tumor cells (CTCs) in a patient with EML4-ALK-positive non-small cell lung cancer (NSCLC). Clin Lung Cancer. 2016;17(5):e173–e177. | ||
Yang JJ, Zhang XC, Su J, et al. Lung cancers with concomitant EGFR mutations and ALK rearrangements: diverse responses to EGFR-TKI and crizotinib in relation to diverse receptors phosphorylation. Clin Cancer Res. 2014;20(5):1383–1392. | ||
Normant E, Paez G, West KA, et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene. 2011;30(22):2581–2586. | ||
Gainor JF, Shaw AT. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J Clin Oncol. 2013;31(31):3987–3996. | ||
Kuang Y, Rogers A, Yeap BY, et al. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin Cancer Res. 2009;15(8):2630–2636. | ||
Sun W, Yuan X, Tian Y, et al. Non-invasive approaches to monitor EGFR-TKI treatment in non-small-cell lung cancer. J Hematol Oncol. 2015;8:95. | ||
Shaw JA, Stebbing J. Circulating free DNA in the management of breast cancer. Ann Transl Med. 2014;2(1):3. | ||
Jahr S, Hentze H, Englisch S, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61(4):1659–1665. | ||
Diehl F, Schmidt K, Choti MA. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985–990. | ||
Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–554. | ||
Jenkins S, Yang JC, Ramalingam SS, et al. Plasma ctDNA analysis for detection of the EGFR T790M mutation in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2017;12(7):1061–1070. | ||
Chabon JJ, Simmons AD, Lovejoy AF, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. | ||
Escriu C, Field JK. Circulating tumour DNA and resistance mechanisms during EGFR inhibitor therapy in lung cancer. J Thorac Dis. 2016;8(9):2357–2359. | ||
Wang Y, Tian PW, Wang WY, et al. Noninvasive genotyping and monitoring of anaplastic lymphoma kinase (ALK) rearranged non-small cell lung cancer by capture-based next-generation sequencing. Oncotarget. 2016;7(40):65208–65217. | ||
Cooper MR, Chim H, Chan H, Durand C. Ceritinib: a new tyrosine kinase inhibitor for non-small-cell lung cancer. Ann Pharmacother. 2015;49(1):107–112. | ||
Ou SI, Lee TK, Young L, et al. Dual occurrence of ALK G1202R solvent front mutation and small cell lung cancer transformation as resistance mechanisms to second generation ALK inhibitors without prior exposure to crizotinib. Pitfall of solely relying on liquid re-biopsy? Lung Cancer. 2017;106:110–114. | ||
Gridelli C, Peters S, Sgambato A, Casaluce F, Adjei AA, Ciardiello F. ALK inhibitors in the treatment of advanced NSCLC. Cancer Treat Rev. 2014;40(2):300–306. | ||
Punnoose EA, Atwal S, Liu W, et al. Evaluation of circulating tumor cells and circulating tumor DNA in non-small cell lung cancer: association with clinical endpoints in a phase II clinical trial of pertuzumab and erlotinib. Clin Cancer Res. 2012;18(8):2391–2401. | ||
Geva S, Roisman LC, Peled N. Liquid biopsy in the practice of neo-oncology. J Thorac Dis. 2016;8(10):E1279–E1281. | ||
Zhou Q, Yang JJ, Chen ZH, et al. Serial cfDNA assessment of response and resistance to EGFR-TKI for patients with EGFR-L858R mutant lung cancer from a prospective clinical trial. J Hematol Oncol. 2016;9(1):86. | ||
Rodríguez M, Silva J, López-Alfonso A, et al. Different exosome cargo from plasma/bronchoalveolar lavage in non-small-cell lung cancer. Genes Chromosomes Cancer. 2014;53(9):713–724. | ||
Taverna S, Giallombardo M, Gil-Bazo I, et al. Exosomes isolation and characterization in serum is feasible in non-small cell lung cancer patients: critical analysis of evidence and potential role in clinical practice. Oncotarget. 2016;7(19):28748–28760. | ||
Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–579. | ||
Mathivanan S, Fahner CJ, Reid GE, Simpson RJ. ExoCarta 2012: database of exosomal proteins, RNA and lipids. Nucleic Acids Res. 2012;40(Database issue):D1241–D1244. | ||
Demory Beckler M, Higginbotham JN, Franklin JL, et al. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol Cell Proteomics. 2013;12(2):343–355. | ||
Taylor DD, Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol. 2008;110(1):13–21. | ||
Kosaka N, Takeshita F, Yoshioka Y, et al. Exosomal tumor-suppressive microRNAs as novel cancer therapy: “exocure” is another choice for cancer treatment. Adv Drug Deliv Rev. 2013;65(3):376–382. | ||
Jaiswal R, Gong J, Sambasivam S, et al. Microparticle-associated nucleic acids mediate trait dominance in cancer. FASEB J. 2012;26(1):420–429. | ||
Pigati L, Yaddanapudi SC, Iyengar R, et al. Selective release of microRNA species from normal and malignant mammary epithelial cells. PLoS One. 2010;5(10):e13515. | ||
Chuang JC, Jones PA. Epigenetics and microRNAs. Pediatr Res. 2007;61(5 Pt 2):24R–29R. | ||
Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–838. | ||
Anglicheau D, Muthukumar T, Suthanthiran M. MicroRNAs: small RNAs with big effects. Transplantation. 2010;90(2):105–112. | ||
Shyu AB, Wilkinson MF, van Hoof A. Messenger RNA regulation: to translate or to degrade. EMBO J. 2008;27(3):471–481. | ||
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–866. | ||
Garzon R, Calin GA, Croce CM. MicroRNAs in cancer. Annu Rev Med. 2009;60:167–179. | ||
Galluzzi L, Morselli E, Vitale I, et al. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res. 2010;70(5):1793–1803. | ||
Dacic S, Kelly L, Shuai Y, Nikiforova MN. miRNA expression profiling of lung adenocarcinomas: correlation with mutational status. Mod Pathol. 2010;23(12):1577–1582. | ||
Wu X, Xiao H. miRNAs modulate the drug response of tumor cells. Sci China C Life Sci. 2009;52(9):797–801. | ||
Yanaihara N, Caplen N, Bowman E, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9(3):189–198. | ||
Enfield KS, Stewart GL, Pikor LA, et al. MicroRNA gene dosage alterations and drug response in lung cancer. J Biomed Biotechnol. 2011;2011:474632. | ||
Vlassov AV, Magdaleno S, Setterquist R, Conrad R. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta. 2012;1820(7):940–948. | ||
Nilsson RJ, Karachaliou N, Berenguer J, et al. Rearranged EML4-ALK fusion transcripts sequester in circulating blood platelets and enable blood-based crizotinib response monitoring in non-small-cell lung cancer. Oncotarget. 2016;7(1):1066–1075. | ||
Chen S, Zhao J, Cui L, Liu Y. Urinary circulating DNA detection for dynamic tracking of EGFR mutations for NSCLC patients treated with EGFR-TKIs. Clin Transl Oncol. 2017;19(3):332–340. | ||
Li Y, Zhang Y, Qiu F, Qiu Z. Proteomic identification of exosomal LRG1: a potential urinary biomarker for detecting NSCLC. Electrophoresis. 2011;32(15):1976–1983. | ||
Zhang L, Xiao H, Zhou H, et al. Development of transcriptomic biomarker signature in human saliva to detect lung cancer. Cell Mol Life Sci. 2012;69(19):3341–3350. | ||
Wei F, Yang J, Wong DT. Detection of exosomal biomarker by electric field-induced release and measurement (EFIRM). Biosens Bioelectron. 2013;44:115–121. | ||
Wei F, Lin CC, Joon A, et al. Noninvasive saliva-based EGFR gene mutation detection in patients with lung cancer. Am J Respir Crit Care Med. 2014;190(10):1117–1126. | ||
Pu D, Liang H, Wei F, et al. Evaluation of a novel saliva-based epidermal growth factor receptor mutation detection for lung cancer: a pilot study. Thorac Cancer. 2016;7(4):428–436. | ||
Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2(1):e17. | ||
Melosky B. Treatment algorithms for patients with metastatic non-small cell, non-squamous lung cancer. Front Oncol. 2014;4:256. | ||
Ye T, Pan Y, Wang R, et al. Analysis of the molecular and clinicopathologic features of surgically resected lung adenocarcinoma in patients under 40 years old. J Thorac Dis. 2014;6(10):1396–1402. | ||
Farace F, Massard C, Vimond N, et al. A direct comparison of CellSearch and ISET for circulating tumour-cell detection in patients with metastatic carcinomas. Br J Cancer. 2011;105(6):847–853. | ||
Pailler E, Oulhen M, Borget I, et al. Circulating tumor cells with aberrant ALK copy number predict progression-free survival during crizotinib treatment in ALK-rearranged non-small cell lung cancer patients. Cancer Res. 2017;77(9):2222–2230. | ||
Zito Marino F, Rocco G, Morabito A, et al. A new look at the ALK gene in cancer: copy number gain and amplification. Expert Rev Anticancer Ther. 2016;16(5):493–502. | ||
Bratman SV, Newman AM, Alizadeh AA, Diehn M. Potential clinical utility of ultrasensitive circulating tumor DNA detection with CAPP-Seq. Expert Rev Mol Diagn. 2015;15(6):715–719. | ||
Wang C, Wang A, Wei F, Wong DTW, Tu M. Electric field-induced disruption and releasing viable content from extracellular vesicles. Methods Mol Biol. 2017;1660:367–376. | ||
Reck M, Popat S, Reinmuth N, De Ruysscher D, Kerr KM, Peters S; ESMO Guidelines Working Group. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;(25 Suppl 3):iii27–iii39. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.