")
Back to Journals » Cancer Management and Research » Volume 13
The Antitumor Activity of CAR-T-PD1 Cells Enhanced by HPV16mE7-Pulsed and SOCS1-Silenced DCs in Cervical Cancer Models
Authors Zheng J, Huang J, Ma W, Yang W, Hu B
Received 24 May 2021
Accepted for publication 21 July 2021
Published 4 August 2021 Volume 2021:13 Pages 6045—6053
DOI https://doi.org/10.2147/CMAR.S321402
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kenan Onel
Jingwei Zheng,1,* Jingsong Huang,2,* Wei Ma,3 Wenqiang Yang,3 Bicheng Hu3
1Clinical Medical College of Jilin University, Changchun, 130012, People’s Republic of China; 2Department of Transfusion, Xiang’an Hospital of Xiamen University, School of Medicine, Xiamen University, Xiamen, 361101, People’s Republic of China; 3The Central Laboratory, Wuhan No.1 Hospital, the Hospital of Traditional Chinese and Western Medicine Affiliated to Hubei University of Chinese Medicine, Wuhan, 430022, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Bicheng Hu
The Central Laboratory, Wuhan No.1 Hospital, the Hospital of Traditional Chinese and Western Medicine Affiliated to Hubei University of Chinese Medicine, Wuhan, 430022, Hubei, People’s Republic of China
Tel/Fax +86 27-85332367
Email [email protected]
Background: Genetically T cells modified with cancer-specific chimeric antigen receptors (CARs) showed great promise in mediate tumor regression, especially in patients with advanced leukemia. However, the therapeutic effect against solid tumors is not as prominent as anticipated to exhibit potent antitumor efficacy. The underlying mechanism maybe attributed to the inhibitory co-stimulatory pathways such as (PD1/PDL1), which provide tumor cells an escape mechanism from immunosurveillance. Therefore, by exchanging the transmembrane and cytoplasmic tail of PD1 with positive costimulatory molecules, such as CD28 and 4– 1BB signaling domains (PD1-CD28-4-1BB, PD1-CAR), the T cell-negative co-stimulatory PD1/PDL1 signal pathway was thus converted into a positive one. This study aimed to investigate whether the genetically modified CAR-T-PD1 cells activated by SOCS1 silenced DCs have enhanced anti-neoplastic potential in vitro/in vivo.
Methods: In order to enhance the antigenicity and reduce transformation activity, a modified HPV16 E7 (HPV16mE7) was employed to load on dendritic cells (DCs) with SOCS1 silenced to improve its antitumor efficiency and targeting ability against cervical cancer. The CAR-T-PD1 cells activated by the generated DCs were transfused into murine models bearing tumor of CaSki cells that expressing PDL1 and HPV16 E6/E7 for in vitro/in vivo antitumor activity assay.
Results: The data showed that DC-activated CAR-T-PD1 cells significantly increased the secretion of IL-2, IFN-γ and TNF-α, whilst enhanced cytotoxic activity, suppressed tumor growth and prolong the survival time compared with the controls.
Conclusion: These results indicated that the genetically engineered T cells activated by DCs had improved antitumor efficiency and targeting ability. Furthermore, it was suggested that it may have important implications for the improvement of T cell immunotherapy against cervical cancer.
Keywords: PD1/PDL1, CAR-T, dendritic cells, suppressor of cytokine signaling 1, cervical cancer
Introduction
High-risk human papillomavirus (hr-HPV) persistent infection has been recognized as the oncogenesis cause of cervical cancer which ranks the second most common cause of cancer-related deaths among women worldwide.1 Due to lack of effective therapies, cervical cancer mortality remains very high. Fortunately, adoptive cell immunotherapy constitutes a feasible strategy against cervical cancer. Chimeric antigen receptor (CAR) T cells present potent tumoricidal activity, especially in advanced leukemia.2 However, the therapeutic efficacy of CAR-T cells against solid tumors is not as efficient as anticipated in clinical application. It is mainly attributed to the inhibitory co-stimulatory pathway such as CTLA-4 and PD1, which provide a mechanism for tumor cells to evade immune surveillance.1,3 As they can not only irritate T cell dysfunction and exhaustion but also induce apoptosis by impairing the proliferation, cytotoxicity, and production of cytokines (especially TNF-α and IFN-γ) of activated T cells.4,5 Notably, PD1 was found up-regulated on the surface of the activated T lymphocytes and its ligand PDL1 was expressed in most human cancers such as myeloma, renal, ovarian, pancreatic, gastric and breast cancer, which is considered as the sign of poor clinical prognosis.6
Due to its profound immunosuppressive effect, PD1/PDL1 has become the focus of recent substantial studies, aiming at neutralizing its detrimental effects on T cell antitumor efficacy by blocking the interaction between PD1 and PDL1 through anti-PD1/PDL1 antibodies. The PD1/PDL1 blockade can restore T cell functions and enhance the antitumor efficacy of adoptively transferred T cells in vitro and in vivo. It demonstrated unprecedented rates of durable clinical responses with an activity ranged from 10% to 45% in unselected populations affected by advanced solid tumors.7–9
Being contrary to PD1, several co-stimulatory molecules such as CD28 and 4–1BB have proven central to T cell persistence and function through synergizing with the TCR to activate T cells, which resulted in an increase of cytokines production needed for clonal expansion and differentiation, and improved antitumor activity and survival.10,11 In addition, the lack of positive co-stimulators in the tumor microenvironment can further dampen T cell functions. Therefore, by constructing chimeric activated receptors composed of the extracellular domain of PD1, CD28 transmembrane and intracellular domains, and 4–1BB intracellular co-stimulatory domain (PD1-CAR), the T cell negative co-stimulatory PD1/PDL1 signaling pathway was thus converted to a positive one.12
As the most professional antigen-presenting cells (APCs), DCs were considered the center of the immune system because of their ability to regulate both immunity and immune tolerance. Antigen-loaded mature DCs could induce antigen-specific T cells to differentiate into effectors in contrary to immature DCs that induce immune tolerance by generating suppressor T cells or T cell deletion.13 In this study, DCs were loaded with modified HPV16 E7 (HPV16mE7) with reduced transformatory activity and enhanced antigenicity to produce antigen-specific T cells against cervical cancer,14,15 as HPV16 was considered the “high risk” (HR) virus most commonly in oncogenesis and development of over 50% cervical cancer.1
Suppressor of cytokine signaling 1 (SOCS1), an inducible negative inhibitor of the JAK/STAT signaling pathway, exerts potent immunosuppressive effect on DCs maturation by blocking the activation of constitutive immune response.14,15 Silencing SOCS1 in DCs promotes the maturation of DCs and triggers an allogeneic T cell expansion.16,17 Our previous study showed that DCs increased the expressions of CD1a/CD80/CD83 as well as cytokines production with SOCS1 knockdown.14,15
In this study, the HPV16mE7 pulsed DCs with silenced SOCS1 were co-cultured with CAR-T-PD1 cells to improve targeting ability and tumoricidal efficacy against cervical cancer. The generated DC-CAR-T-PD1 cells exhibited enhanced cytokine production, cytotoxic activity, suppressed tumor growth, and prolonged survival time in murine models.
Materials and Methods
Ethics Statement
This study was performed in accordance with Declaration of Helsinki and approved by the Conduct of Human Ethics Committee of Wuhan No. 1 Hospital. All experiments were carried out in strict accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the China National Institutes of Health, and the animal experiments were approved by the Committee on the Ethics Animal Experiment of Wuhan No.1 Hospital, the Hospital of Traditional Chinese and Western Medicine Affiliated to Hubei University of Chinese Medicine (approval No. 2017004). Each patient was informed with written consents.
Cell Lines and Reagents
HEK293, HCC38 and CaSki cell lines were purchased from American Type Culture Collection (ATCC). Cells were maintained in RPMI-1640 or DMEM culture media (Gibco, Life Technologies, USA) supplemented with 10% (v/v) fetal bovine serum (FBS) (HyClone Laboratories, USA). Normal human AB serum was acquired from Gibco, Life Technologies (USA). Anti-CD3 antibody, rhIL-2, rhTNF-γ, rhGM-CSF, rhIL-4 and TNF-α were purchased from Peprotech Inc. Lymphocyte separation medium Ficoll was purchased from GE Healthcare (USA). Mouse anti-human monoclonal antibodies of CD3 (FITC labeled) and CD8 (PE labeled) were purchased from eBioscience Co, Ltd. Anti-human PD1 and PDL1 monoclonal antibodies were purchased from BioLegend (San Diego, CA). Mouse anti-human monoclonal antibodies of CD80-PE, CD83-PE, CD86-PE, CD1a-FITC, and CD40-FITC were all purchased from Santa Cruz Co, Ltd. Female BALB/c nude mice (8-week-old, weight 16–22g) raised under SPF circumstance were acquired from the Guangzhou Traditional Chinese Medicine University. All animal studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animal in accordance with the research protocols approved by Wuhan No.1 Hospital Animal Facility and Use Committee. PBMCs were generated from healthy donors with informed consent.
Adeno Sh-SOCS1 Plasmid Construction
The shRNA-SOCS1 and its mutant form shRNA-mSOCS1 were constructed according to http://www.bioon.com.cn. Primers used are as follows:
shRNASOCS1 (F): 5′-GATCC CTA CCT GAG TTC CTT CCC CT TCAAGAG AG GGG AAG GAA CTC AGG TAGTTTTTT G-3′ (BamHI and EcoRI);
shRNA-SOCS1 (R): 5′-AATTC AAAAAACTA CCT GAG TTC CTT CCC CT CTCTTGA AG GGG AAG GAA CTC AGG TAG G-3′;
shRNA-mSOCS1 (F): 5′-GATCC ACT ATC TAA GTT ACT ACC CCT TCAAGAG AGG GGT AGT AAC TTA GAT AGT TTTTTTG-3′;
shRNA-mSOCS1 (R): 5′-AATTC AAAAAAACT ATC TAA GTT ACT ACC CCT CTCTTGA AGG GGT AGT AAC TTA GAT AGT G-3′.
The shRNA-SOCS1 and shRNA-mSOCS1 were cloned into the plasmid vector RNAi-SOCS1-pShuttle (BD Clontech) and then inserted into the replication-deficient pAdeno-X vector (BD Clontech). The recombinant adenovirus plasmids were generated according to manufacturer’s instructions and verified by PCR and sequencing and titrated using Adeno-X Rapid Titer kits (BD Bioscience).
Preparation of DCs and CAR-T Cells
The PBMCs were prepared from human 100mL peripheral blood by using Ficoll-Paque. The obtained cells were subsequently transferred into culture flasks and cultured for 2h in RPMI-1640 medium supplemented with 10% autologous serum. Using RosetteSep CD8+ Human T cell Enrichment Cocktail (Stem cell Technologies), CD8+ T cells were isolated from nonadherent cells and stimulated for 48h in the presence of 100ng/mL OKT3 (eBioscience, San Diego, CA) and rhIL-2 (100U/mL) before transduction. Lentivirus containing PD1-CAR was produced by Umibio (Shanghai) Co. Ltd.18 The activated T cells were transduced at a multiplicity of infection (MOI) of 5 in the presence of 10μg/mL protamine sulphate. The transduced T cells were expanded in RPMI-1640 medium supplemented with 10% FCS and rhIL-2 (50U/mL). The remaining adherent cells were cultured in RPMI-1640 medium containing 40ng/mL rhGM-CSF and 40ng/mL rhIL-4. After 7 days of culture, the transducted adherent cells were exposed to Ad-shSOCS1 at 5 MOI for 8h, washed with PBS and pulsed with HPVm16E7 protein at 25µg/mL in fresh culture medium for 6h. HPVm16E7 were gifted by Dr. Zheng Yi (Graduate School at Shenzhen, Tsinghua University), and the silencing of SOCS1 was verified with western-blotting analysis (Figure 1). Then, mature DCs were developed after 24h stimulation with 10ng/mL TNF-α. The supernatant was replaced with fresh medium every 3 days. All cultures were incubated at 37°C in 5% humidified CO2.
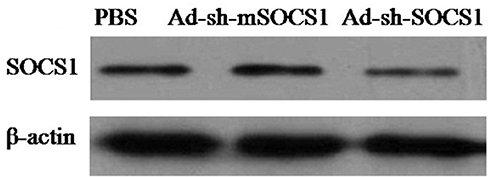 |
Figure 1 Western blot analysis of SOCS1 silencing. SOCS1 and β-actin proteins were identified using anti-SOCS1 polyclonal and anti-β-actin monoclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA), respectively. |
Surface Marker Staining
HCC38 and CaSki cells were co-cultured with CD8+ T cells at a ratio of 10:1 for 24h. The PD-L1 expression levels of HCC38 and CaSki cells were analyzed by FACS. The CD1a, CD80, CD83, CD86 and HLA-DR expression levels of DCs were also examined. Cells were stained in a FACS buffer containing PBS and 0.5% BSA.
Cytokine Release Assay
The CAR-T-PD1 cells were co-cultured with matured DCs at a ratio of 10:1 in CAR-T medium for 3 days to generate DC-CAR-T-PD1 cells, and then co-cultured with HCC38 (45% PDL1+) and CaSki cells (22% PDL1+) at a ratio of 10:1 for 24h. The supernatants were analyzed for the secretion levels of IL-2, interferon γ (IFN-γ) and tumor necrosis factor α (TNF-α) using commercially available ELISA kits (R&D Systems, Minneapolis, UK). The cytokines secreted in the culture supernatant were measured using ELISA.
In vitro Cytotoxicity Analysis
The transduced lymphocytes activated by matured DCs were co-cultured with target CaSki and HCC38 at E:T ratios of 10:1, 30:1, and 90:1 at 37°C in 96-well plates for 24h. Then, the cytotoxicity was determined by the CCK8 assay (Dojindo Molecular Technologies, Inc., Kumamoto, Japan). The groups comprising specific mixture of cell types were the experimental groups, whereas the control groups contained only one cell type of the CaSki, HCC38, CAR-T-PD1, DC-CAR-T-PD1 or cultivating solution. The CCK8 assay was performed in triplicate to evaluate cell viability, and the optical density (OD) was read at 450 nm. The cytotoxic activity was calculated as follows:
Cytotoxic activity (%)= [OD(effector and target cells)-OD(control)/OD(target cells) –OD(control)] x 100.
In vivo Antitumor Assay
By abdominal subcutaneous injection of 1×106 CaSki cells into 6-week-old female BALB/c nude mice and the xenografted tumor mouse models were established. On day 9, all mice had palpable tumors were assigned to 3 groups as follows: T lymphocytes (served as control), CAR-T-PD1, and DC-CAR-T-PD1(10 mice in each group). Mice were treated with a total of 1.0×107 lymphocytes, initiated intravenously into tail vein for 2 times and 5×106 cells were administered each time on days 10 and 13. The tumor volume was calculated by the following formula: (major axis x minor axis2) x 0.52.
Statistical Analysis
All data were expressed as means ± standard deviation (SD). The statistical significance of group differences was analyzed using the Student’s t test. P-values <0.05 were considered statistically significant (StatXact4 software, Cytel Corporation, Cambridge, MA). The mouse survival rates were analyzed using the Kaplan-Meier method (Log rank test).
Results
Cell Phenotype Analysis
The PDL1 expression levels of CaSki cells and HCC38 cells were increased 26.3% and 11.5%, respectively, after challenged by CD8+ T lymphocytes (Figure 2). Compared with control, the CD1a, CD80, CD83, CD86 and HLA-DR expression levels were significantly increased by 2–4 folds during DCs maturation in SOCS1 silenced group (Table 1). The CD1a, CD80, and CD83 expression levels were higher in SOCS1 silenced group than those in TNF-α group. These results suggested that CD8+T cells stimulated tumor cells to up-regulate PDL1 expression.
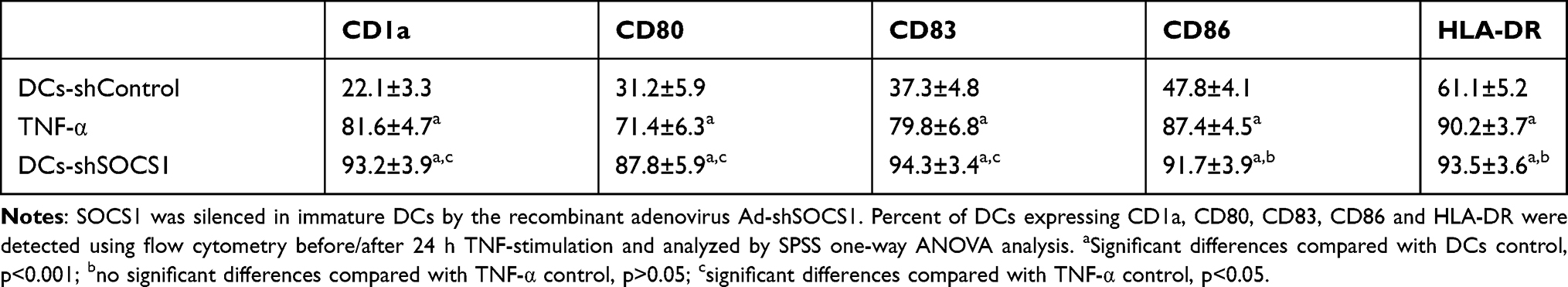 |
Table 1 Percentage of DCs Expressing CD1a, CD80, CD83, CD86, and HLA-DR |
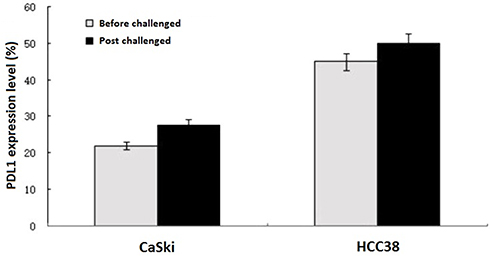 |
Figure 2 PDL1 expression levels. Relative PDL1 expression levels of CaSki and HCC38 cells after challenged by CD8+ T lymphocytes. The PD-L1 expression levels of CaSki cells and HCC38 cells increased 26.3% and 11.5% respectively after challenged by CD8+ T lymphocytes. |
Cytokine Release Assay
After stimulated by CaSki and HCC38, CAR-T-PD1 and DC-CAR-T-PD1 cells significantly increased the secretion of IL-2, IFN-γ and TNF-α in comparison to the control T cells (P<0.05). The secretion levels of IL-2, IFN-γ and TNF-α in DC-CAR-T-PD1 showed 20–40% higher than those of CAR-T-PD1 cells. Moreover, after being stimulated by HCC38 cells, CAR-T-PD1 and DC-CAR-T-PD1 cells demonstrated 22–33% higher secretion levels of IL-2, IFN-γ and TNF-α with respect to CaSki cells (Figure 3), indicating the interaction between PD1-PDL1 induced CAR-T-PD1 cells activation and secretion of cytokines such as IL-2, IFN-γ and TNF-α.
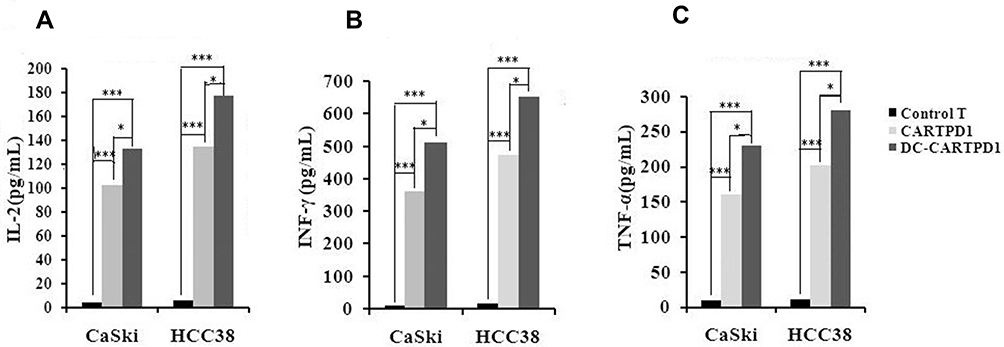 |
Figure 3 Cytokine release assay. (A) IL-2 secretion levels of T lymphocytes, CARTPD1 and DC-CARTPD1 cells after stimulated by CaSki or HCC38 cells. (B) INF-γ secretion levels of T lymphocytes, CARTPD1 and DC-CARTPD1 cells after stimulated by CaSki or HCC38 cells. (C) TNF-α secretion levels of T lymphocytes, CARTPD1 and DC-CARTPD1 cells after stimulated by CaSki or HCC38 cells. T lymphocytes compared with CARTPD1 and DC-CARTPD1 cells (P<0.001); CARTPD1 cells versus DC-CARTPD1 cells (P<0.05). ***Indicated extremely significant difference (P <0.001); *Indicated significant difference (P <0.05). |
Cytotoxicity Assay
The transduced lymphocytes activated by matured DCs were co-cultured with target CaSki and HCC38 cells at E:T ratios of 10:1, 30:1, and 90:1 for 24 hours. Then, the cytotoxicity was determined using the CCK8 assay kit in vitro. It revealed that DC-CAR-T-PD1 lymphocytes exhibited the highest cytotoxic activity against CaSki and HCC38 cells. Meanwhile with the increase of the ratio of E:T cells, the cytotoxicity effect of DC-CAR-T-PD1 lymphocytes was significantly enhanced. Compared with the CaSki group (Figure 4A), DC-CAR-T-PD1 lymphocytes had a stronger effect in HCC38 group (Figure 4B), possibly because HCC38 cells expressed more PDL1.
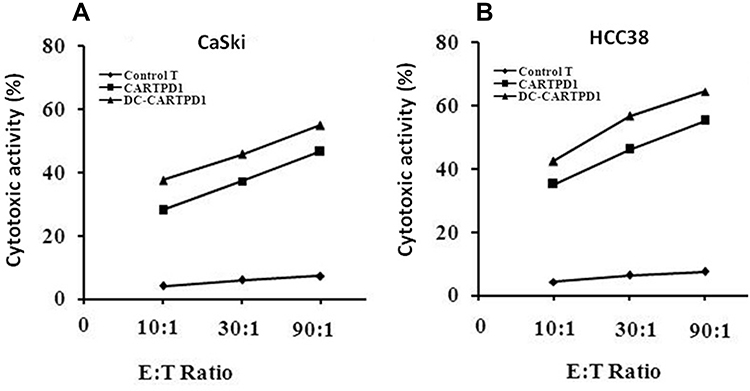 |
Figure 4 Cytotoxicity assay. The cytotoxic activity of T lymphocytes, CARTPD1 and DC-CARTPD1 cells against CaSki and HCC38 cells. Meanwhile with the increased of the ratio of E:T cells, the cytotoxicity effect of DC-CAR-T-PD1 lymphocytes was significantly enhanced. Compared with the CaSki group (A), DC-CAR-T-PD1 lymphocytes had a stronger effect in HCC38 group (B), possibly because HCC38 cells expressed more PDL1. T lymphocytes compared with CARTPD1 and DC-CARTPD1 cells (P<0.001); CARTPD1 cells versus DC-CARTPD1 cells (P<0.01). |
In vivo Antitumor Activity Analysis
The therapeutic effect of the DC-CAR-T-PD1 lymphocytes was investigated in established xenograft tumor mouse models. When the mice had palpable tumors on day 9, the therapeutic treatments were initiated with different lymphocytes. As shown in Figure 5A, CAR-T-PD1 and DC-CAR-T-PD1 lymphocytes significantly regressed the tumor growth compared with T lymphocyte controls, while DC-CAR-T-PD1 lymphocytes presented enhanced antineoplastic activity in respect to CAR-T-PD1 lymphocytes (p<0.01). Although the therapeutic CAR-T-PD1 and DC-CAR-T-PD1 cells could not eradicate the tumor completely, the survival time was significantly prolonged and reached more than 60 days in 40% of the DC-CAR-T-PD1 cells treated group and in 20% of the CAR-T-PD1 cells treated mice, with respect to T lymphocytes control group in which 90% of the mice were dead in 30 days (Figure 5B). These results demonstrated the therapeutic potential of DC-CAR-T-PD1 cells for HPV16 E7-positive tumors in vivo.
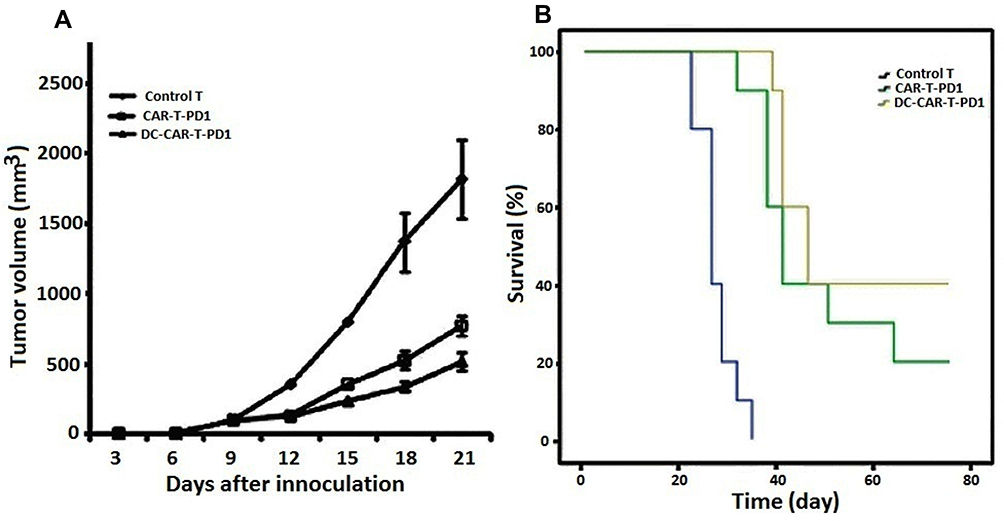 |
Figure 5 In vivo antitumor activity analysis. (A) The therapeutic effect CARTPD1 and DC-CARTPD1 lymphocytes against mice bearing CaSki tumors was investigated. CARTPD1 and DC-CARTPD1 lymphocytes significantly regressed the tumor volume compared with T cell controls in which progressive tumor growth was observed (P<0.001); CARTPD1 cells versus DC-CARTPD1 cells (P<0.01). (B) The survival rates were analyzed using the Kaplan–Meier method (Log rank test). T cell control versus CARTPD1 cells or DC-CARTPD1 cells (P<0.001); CARTPD1 cells versus DC-CARTPD1 cells (P<0.05). |
Discussion
Chimeric antigen receptors (CARs) could orient the activity of immune cells towards specific molecular targets expressed on the surface of tumor cells. When exposed to specific target antigen, cytokine secretion of genetically engineered T lymphocytes expressing CARs was increased and showed specific antitumor activity.18,19 Moreover, different CARs have been generated so far against a wide range of solid tumors and haematological malignancies.20–22 CARs containing early co-stimulator CD28 and late co-stimulator OX40 or 4–1BB can substantially improve effector T-cell functions and promote T effector memory cell differentiation by up-regulating Bcl-XL. The co-stimulation mediated by CD28 and 4–1BB suggested some complementary effects.23–26 Recent studies have shown that at lower doses, 4–1BB- CAR-T cells retained their cytotoxic and cytokine secretion functions longer than CD28-CAR-T cells. The prolonged function of 4–1BB- CAR-T cells correlated with improved survival.27
CAR-CD19 modified T cells against lymphoid leukemia achieved great success in clinic. But, therapeutic efficacy of other CAR-T cells against solid tumors was not as efficient as anticipated. The reason was assumed that tumors using immunomodulatory checkpoints such as PDL1 and CTLA-4 could evade immune surveillance, hampering activated tumor-specific T cell activities, and rendering them functionally exhausted. Tumors frequently co-opt immunomodulatory signaling pathways to evade immune recognition and elimination, as the mechanism of maintaining tolerance in normal immune physiology, the PD1/PDL1 interaction contributes to clone-depletion and effector lymphocytes exhaustion.28 Up-regulation of constitutive PD1 expression was also observed in infiltrating T lymphocytes (TIL) in solid tumors, suggesting that blocking PD1 may have therapeutic potential in patients.29
Different strategies had developed to reduce the negative effects of PD1/PDL1 signaling on CAR-T in the tumor microenvironment. There were combination of CAR-T with PD1 or PDL1 antibodies, silencing or knockout of PD1 with siRNA or CRISPR/Cas9, and expression of anti-PDL1 antibodies with oncolytic viruses, etc.27,30–34 This study aimed to investigate whether the genetically modified CAR-T-PD1 cells activated by SOCS1 silenced DCs have enhanced anti-neoplastic potential in vitro/in vivo. By exploiting the chimeric costimulator redirecting molecules such as CD28 and 4–1BB, the negative costimulatory signal of PDL1 was converted into a positive one composed of PD1-CD28-4-1BB (CAR-PD1). And the CAR-T-PD1 cells presented enhanced cytokine secretion of IL-2, TNF-α and IFN-γ, improved the antitumor activity in vitro/in vivo.12,18 It was reported that PDL1 was expressed in 19% and PDL2 by 29% of 115 cervical tumors.35 Our results showed that PDL1 was expressed in 24% of CaSki cells, while the CaSki cells were challenged by T lymphocytes, the PDL1 expression level was increased to 30%. In order to further improve the targeting ability of CAR-T-PD1 cells against cervical cancer, the engineered CAR-T-PD1 cells were co-cultured with DCs which could induce antigen-specific immune responses.
E6 and E7 oncoproteins encoded by HPV16 are crucial for the viral life cycle in the transformation and maintenance of malignant phenotype and there are commonly immunotherapeutic targets against cervical cancer. In this study, the modified HPV16 E7 (HPV16mE7) with reduced transformation activity and enhanced antigenicity was utilized to load on DCs to generate specific cytotoxicity against cervical tumors. Moreover, when SOCS1, an inducible negative feedback inhibitor of the JAK/STAT signal pathway was silenced in DCs,14,15 enhanced secretions of pro-inflammatory cytokines such as IL-12 and INF-γ, allogeneic T-cell expansion, and DC maturation were observed.36
CAR-T-PD1 and DC-CAR-T-PD1 cells significantly increased cytokines secretion of IL-2, TNF-α and INF-γ in respect to T cells. Compared with CaSki cells, HCC38 stimulated CAR-T-PD1 and DC-CAR-T-PD1 cells showed higher cytokine secretion, indicating PD1-CAR shifted the negative co-stimulatory signal to a positive one, here HCC38 as antigen presenting cells. Our results demonstrated that the expression of PDL1 on the surface of HCC38 cells was higher than that of CaSki cells, and in vitro, CAR-T-PD1 and DC-CAR-T-PD1 showed higher cytotoxicity to HCC38 cells than CaSki cells. The interaction of DCs and CAR-T promoted the secretion of cytokines, and SOCS1 silencing enhanced the related functions of DCs.
Furthermore, the cytokine levels as well as the cytolytic activity were further increased by the co-cultivation with HPVm16E7-pulsed and SOCS1-silenced DCs. Moreover, the in vivo antineoplastic activities of CAR-T-PD1 and DC-CAR-T-PD1 cells were also enhanced in the xenograft mice models of CaSki cells.
Conclusion
Cervical cancer patients were usually accompanied with serious cellular immune deficiency and improving the cellular immune function is critical in clinical therapy. The genetically engineered T cells activated by DCs had improved antitumor efficiency and targeting ability. A combination of comprehensive immunotherapy applications such as genetic engineered lymphocytes, PDL1/PD1 blockage and antigen activated DCs might be a promising approach against cervical cancer.
Acknowledgments
This work was supported by Natural Science Foundation of Hubei Province (2016CFB474) and the Clinical Medical Research Project of Wuhan Health and Family Planning Commission (WX13A01) to Bicheng Hu; Medical and Health guidance project of Xiamen Municipal Science and Technology Bureau (3502Z20199052) to Jingsong Huang.
Disclosure
The authors declared that they have no conflicts of interest for this work.
References
1. Galliverti G, Wullschleger S, Tichet M, et al. Myeloid cells orchestrate systemic immunosuppression, impairing the efficacy of immunotherapy against HPV+ cancers. Cancer Immunol Res. 2020;8(1):131–145. doi:10.1158/2326-6066.CIR-19-0315
2. Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. doi:10.1126/scitranslmed.3002842
3. Fife BT, Pauken KE, Eagar TN, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009;10(11):1185–1192. doi:10.1038/ni.1790
4. Wang W, Lau R, Yu D, Zhu W, Korman A, Weber J. PD1 blockade reverses the suppression of melanoma antigen-specific CTL by CD4+ CD25(Hi) regulatory T cells. Int Immunol. 2009;21(9):1065–1077. doi:10.1093/intimm/dxp072
5. Robertson SA, Care AS, Moldenhauer LM. Regulatory T cells in embryo implantation and the immune response to pregnancy. J Clin Invest. 2018;128(10):4224–4235. doi:10.1172/JCI122182
6. Alsaab HO, Sau S, Alzhrani R, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. doi:10.3389/fphar.2017.00561
7. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PDL1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi:10.1038/nature14011
8. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–668. doi:10.1038/s41577-20-0306-5
9. Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558–562. doi:10.1038/nature13904
10. Bour-Jordan H, Esensten JH, Martinez-Llordella M, et al. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol Rev. 2011;241(1):180–205. doi:10.1111/j.1600-065X.2011.01011.x
11. Ware CF. The TNF receptor super family in immune regulation. Immunol Rev. 2011;244(1):5–8. doi:10.1111/j.1600-065X.2011.01065.x
12. Ellis GI, Sheppard NC, Riley JL. Genetic engineering of T cells for immunotherapy. Nat Rev Genet. 2021;22(7):427–447. doi:10.1038/s41576-021-00329-9
13. Duewell P, Steger A, Lohr H, et al. RIG-I-like helicases induce immunogenic cell death of pancreatic cancer cells and sensitize tumors toward killing by CD8+ T cells. Cell Death Differ. 2014;21(12):1825–1837. doi:10.1038/cdd.2014.96
14. Zhu YQ, Zheng Y, Mei L, et al. Enhanced immunotherapeutic effect of modified HPV16 E7-pulsed dendritic cell vaccine by an adeno-shRNA-SOCS1 virus. Int J Oncol. 2013;43(4):1151–1159. doi:10.3892/ijo.2013.2027
15. Zheng Y, Hu B, Xie S, et al. Dendritic cells infected by Ad-sh-SOCS1 enhance cytokine-induced killer (CIK) cell immunotherapeutic efficacy in cervical cancer models. Cytotherapy. 2017;19(5):617–628. doi:10.1016/j.jcyt.2017.01.008
16. Yoshimura A, Ito M, Chikuma S, Akanuma T, Nakatsukasa H. Negative regulation of cytokine signaling in immunity. Cold Spring HarbPerspect Biol. 2018;10(7):a028571. doi:10.1101/cshperspect.a028571
17. Hanada T, Yoshida H, Kato S, et al. Suppressor of cytokine signaling-1 is essential for suppressing dendritic cell activation and systemic autoimmunity. Immunity. 2003;19(3):437–450. doi:10.1016/S1074-7613(03)00240-1
18. Tang X, Li Q, Zhu Y, et al. The advantages of PD1 activating chimeric receptor (PD1-ACR) engineered lymphocytes for PDL1(+) cancer therapy. Am J Transl Res. 2015;7(3):460–473. eCollection 2015
19. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi:10.1056/NEJMoa1103849
20. DeRenzo C, Gottschalk S. Genetically modified T-cell therapy for osteosarcoma: into the roaring 2020s. Adv Exp Med Biol. 2020;1257:109–131. doi:10.1007/978-3-030-43032-0_10
21. Holstein SA, Lunning MA. CAR T-cell therapy in hematologic malignancies: a voyage in progress. Clin Pharmacol Ther. 2020;107(1):112–122. doi:10.1002/cpt.1674
22. Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. 2019;36(5):471–482. doi:10.1016/j.ccell.2019.09.006
23. Vinay DS, Kwon BS. 4-1BB (CD137), an inducible costimulatory receptor, as a specific target for cancer therapy. BMB Rep. 2014;47(3):122–129. doi:10.5483/bmbrep.2014.47.3.283
24. Chacon JA, Pilon-Thomas S, Sarnaik AA, Radvanyi LG. Continuous 4-1BB co-stimulatory signals for the optimal expansion of tumor-infiltrating lymphocytes for adoptive T-cell therapy. Oncoimmunology. 2013;2(9):e25581. doi:10.4161/onci.25581
25. Voo KS, Bover L, Harline ML, et al. Antibodies targeting human OX40 expand effector T cells and block inducible and natural regulatory T cell function. J Immunol. 2013;191(7):3641–3650. doi:10.4049/jimmunol.1202752
26. Habibagahi M, Razmkhah M, Niri NM, Hosseini A, Ghaderi A, Jaberipour M. Combined 4-1BB and CD28 costimulation could unleash lymphocytes from immunosuppression induced by adipose derived stem cell soluble products. Immunol Invest. 2013;42(4):307–323. doi:10.3109/08820139.2013.764315
27. Cherkassky L, Morello A, Villena-Vargas J, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126(8):3130–3144. doi:10.1172/JCI83092
28. Reynolds J, Sando GS, Marsh OB, et al. Stimulation of the PD-1/PDL-1 T-cell co-inhibitory pathway is effective in treatment of experimental autoimmune glomerulonephritis. Nephrol Dial Transplant. 2012;27(4):1343–1350. doi:10.1093/ndt/gfr529
29. Eikawa S, Mizukami S, Udono H. Monitoring multifunctionality of immune-exhausted CD8 T cells in cancer patients. Methods Mol Biol. 2014;1142:11–17. doi:10.1007/978-1-4939-0404-4_2
30. Zhang Y, Zhang X, Cheng C, et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med. 2017;11(4):554–562. doi:10.1007/s11684-017-0543-6
31. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23(9):2255–2266. doi:10.1158/1078-0432.CCR-16-1300
32. Tanoue K, Shaw AR, Watanabe N, et al. Armed oncolytic adenovirus–expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. 2017;77(8):2024–2051. doi:10.1158/0008-5472.CAN-16-1577
33. Choi BD, Yu X, Castano AP, et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J ImmunoTher Cancer. 2019;7(1):304. doi:10.1186/s40425-019-0806-7
34. Rafiq S, Yeku OO, Jackson HJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. 2018;36(9):847–856. doi:10.1038/nbt.4195
35. Karim R, Jordanova ES, Piersma SJ, et al. Tumor-expressed B7-H1 and B7-DC in relation to PD-1+ T-cell infiltration and survival of patients with cervical carcinoma. Clin Cancer Res. 2009;15(20):6341–6347. doi:10.1158/1078-0432.CCR-09-1652
36. Hashimoto M, Ayada T, Kinjyo I, et al. Silencing of SOCS1 in macrophages suppresses tumor development by enhancing antitumor inflammation. Cancer Sci. 2009;100(4):730–736. doi:10.1111/j.1349-7006.2009.01098.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.