")
Back to Journals » OncoTargets and Therapy » Volume 10
The activity, safety, and evolving role of brigatinib in patients with ALK-rearranged non-small cell lung cancers
Authors Sabari JK, Santini FC , Schram AM, Bergagnini I, Chen R, Mrad C, Lai WV, Arbour KC, Drilon A
Received 25 January 2017
Accepted for publication 15 March 2017
Published 6 April 2017 Volume 2017:10 Pages 1983—1992
DOI https://doi.org/10.2147/OTT.S109295
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Joshua K Sabari,1 Fernando C Santini,1,2 Alison M Schram,2 Isabella Bergagnini,1 Ruqin Chen,1 Chebli Mrad,1 W Victoria Lai,1 Kathryn C Arbour,1 Alexander Drilon2,3
1Thoracic Oncology Service, Division of Solid Tumor Oncology, Department of Medicine, Memorial Sloan Kettering Cancer Center, 2Developmetal Therapeutics, Division of Solid Tumor Oncology, Department of Medicine, Memorial Sloan Kettering Cancer Center, 3Thoracic Oncology Service, Division of Solid Tumor Oncology, Department of Medicine, Memorial Sloan Kettering Cancer Center, Weill Cornell Medical College, New York, NY, USA
Abstract: Brigatinib (AP26113) is a dimethylphosphine oxide group-containing tyrosine kinase inhibitor (TKI) constructed around a bisanilinopyrimidine scaffold with potent activity against the anaplastic lymphoma kinase (ALK) and several other targets. Despite the activity of first- and second-generation ALK inhibitors in advanced ALK-rearranged lung cancers, the development of acquired resistance represents an ongoing challenge. Later generation ALK inhibitors such as brigatinib are important potential tools in the management of patients with acquired resistance characterized by continued dependency on ALK. Brigatinib is active in vitro against many ALK kinase domain mutations that may mediate acquired resistance to other ALK TKIs, with reported activity (IC50 <50 nM) against ALK C1156Y, I1171S/T, V1180L, L1196M, L1152R/P, E1210K, and G1269A. In patients with ALK-rearranged lung cancers who receive brigatinib after crizotinib, substantial and durable responses and intracranial disease control can be achieved based on early-phase clinical trial data. The drug is also being explored in TKI-naïve patients. From a safety perspective, early pulmonary toxicity has been observed, prompting the decision to pursue lead-in dosing for the drug. Early data point to ALK G1202R and ALK E1210K as potential mechanisms of clinical resistance to brigatinib.
Keywords: NSCLC, ALK, acquired resistance, tyrosine kinase inhibitor, brigatinib, crizotinib, ceritinib, alectinib, lorlatinib
Introduction
Lung cancer remains the leading cause of cancer-related mortality in the USA.1 Targeting genomic alterations has led to a paradigm shift in the treatment of patients with lung cancer. The majority of patients harboring fusions involving the ALK gene, most commonly EML4–ALK (Figure 1), respond to ALK tyrosine kinase inhibition with the Food and Drug Administration-approved agents crizotinib, ceritinib, and alectinib;2 however, patients universally develop acquired resistance to targeted therapy, and a better understanding of the molecular mechanisms underlying resistance and how to sequence these therapeutics is needed.3
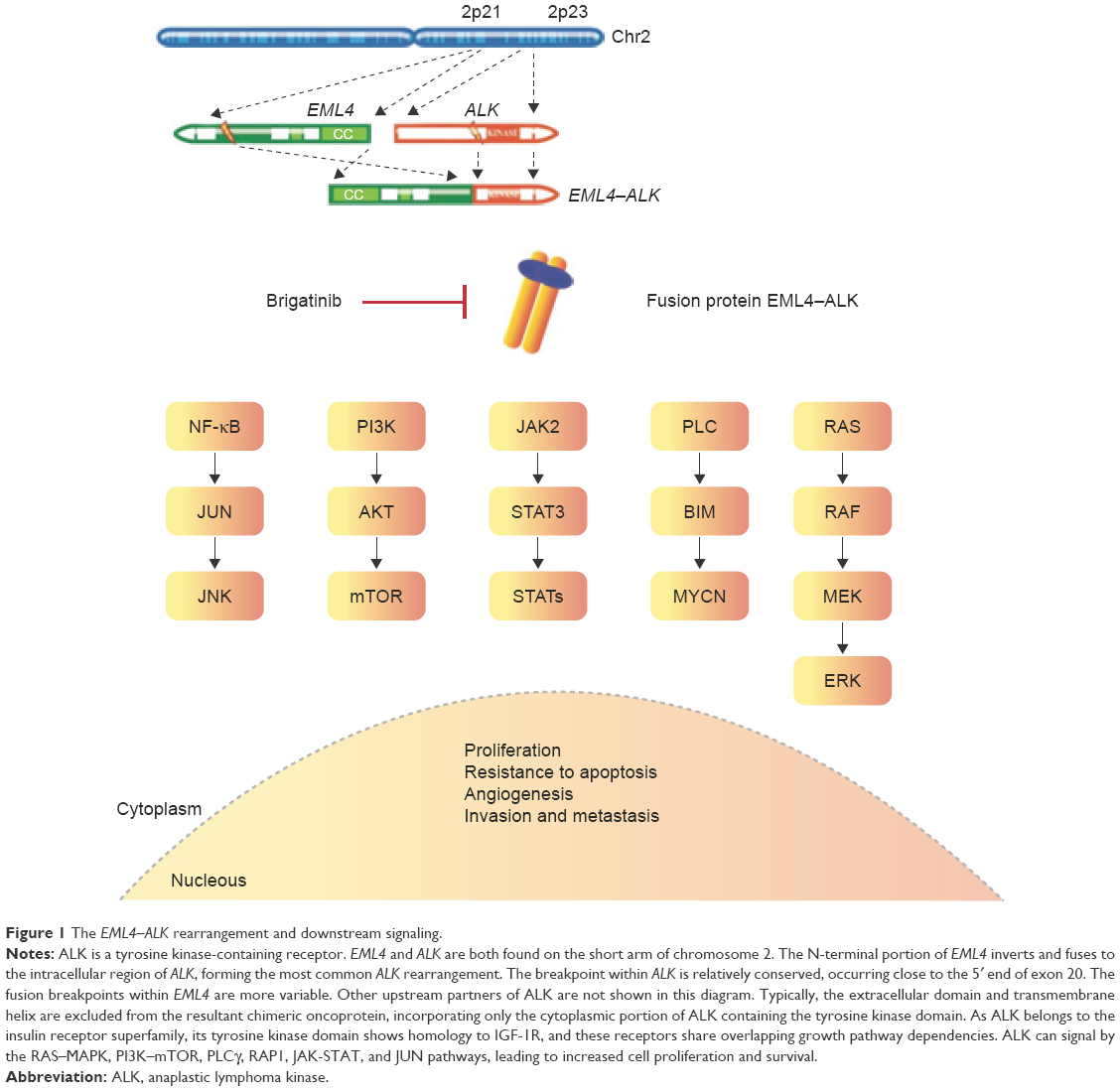 | Figure 1 The EML4–ALK rearrangement and downstream signaling. |
In the setting of acquired resistance to crizotinib, ~30% of patients present with ALK-dependent mechanisms including ALK amplification and ALK mutation. The proportion of patients that develop ALK mutations in the setting of acquired resistance increases after treatment with the second-generation tyrosine kinase inhibitors (TKIs), alectinib and ceritinib. The ALK mutations most commonly found after TKI exposure include: L1196M for crizotinib, G1202R and compound ALK mutations after ceritinib, and G1202R after alectinib.4,5 Beyond ALK-dependent mechanisms of acquired resistance, the activation of alternative pathway-mediated survival signals (bypass pathways involving epidermal growth factor receptor [EGFR], KIT, insulin growth factor 1 receptor [IGF-1R], hepatocyte growth factor receptor MET/HGFR, and Kirsten rat sarcoma) has been observed. Other mechanisms that may be responsible for disease progression include suboptimal central nervous system penetration, epithelial to mesenchymal transition, and microenvironment dynamics. Furthermore, different mechanisms may coexist in the same patient.6
Brigatinib (AP26113) is an orally administered ALK TKI that has broad-spectrum preclinical activity against a variety of ALK mutations that potentially mediate resistance to other ALK TKIs.7 This review will focus on the development of brigatinib, including its pharmacology, safety, and efficacy.
Preclinical data
Drug discovery and pharmacology
The high sequence homology of ALK with other members of the insulin receptor superfamily poses a significant challenge to the design of ALK-selective inhibitors. In 2009, Shakespeare et al reported the identification of a series of compounds that inhibit ALK both in vitro and in vivo, while maintaining relative selectivity over the homologous (IGF-1R) and insulin receptor kinase.8 A compound from this series, AP26113, later named brigatinib, inhibited the kinase activity of ALK, IGF-1R, and the insulin receptor kinase with IC50 values of 0.58, 38, and 262 nmol/L, respectively. For control cell lines that did not express ALK, the IC50 for proliferation was >1,000 nmol/L. Daily oral administration of brigatinib to mice bearing subcutaneous xenografts of ALK-rearranged anaplastic large cell lymphoma (ALCL) Karpas-299 cells elicited a dose-dependent antitumor effect, with near-complete tumor regression observed at the highest doses of 50 and 100 mg/kg. In rats, doses that achieved plasma levels that were effective in the Karpas-299 mouse model had no effect on either insulin or glucose levels, supporting its relative selectivity for ALK.8
In contrast to crizotinib that is constructed around an aminopyridine scaffold, brigatinib is constructed around a bisanilinopyrimidine scaffold. This scaffold occupies the adenosine triphosphate-binding site of ALK in a U-shaped conformation.9,10 Two aniline groups are attached to the C2 and C4 positions of the pyrimidine core (shown in red in Figure 2), and a chlorine atom is attached to C5. In addition, there is a methoxy substituent and an extended solubilization group on the aniline at C2. The former binds to a pocket located under the ALK L1198 hinge residue, and the latter fills part of the ribose-binding pocket and interacts with additional residues. The chlorine atom at C5 interacts with the ALK L1196 gatekeeper residue.
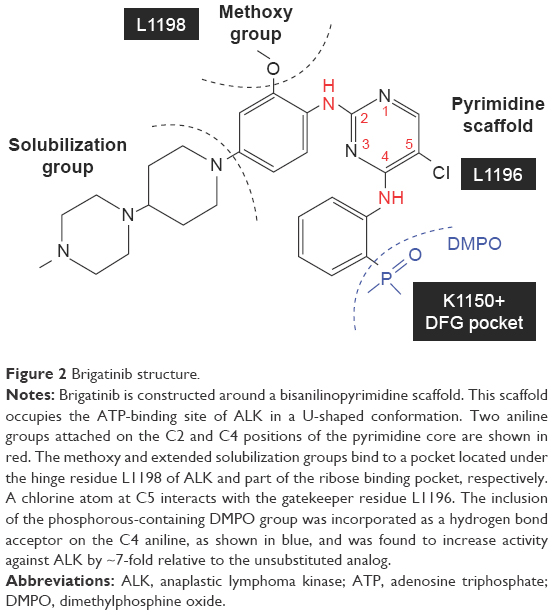 | Figure 2 Brigatinib structure. |
Also unique to the development of brigatinib is the inclusion of a phosphorous-containing dimethylphosphine oxide (DMPO) group. The DMPO group was incorporated as a hydrogen bond acceptor on the aniline at C4 (shown in blue in Figure 2) and increased activity against ALK by ~7-fold relative to the unsubstituted analog.10 This moiety interacts with a small pre-DFG (aspartic acid, phenylalanine, and glycine) pocket and with the ALK K1150 catalytic lysine.10 The DMPO structural feature of brigatinib is critical for balancing ALK potency and selectivity over the highly homologous IGF-1R and insulin receptor kinases. These features impart several important properties, including increased aqueous solubility, decreased lipophilicity, and reduced protein binding.10
In vitro kinase activity
To assess the kinase selectivity profile of brigatinib beyond ALK, IGF-1R, and the insulin receptor, the drug was screened against a panel of 289 kinases and mutants and isoforms (in vitro HotSpotSM kinase assay). Brigatinib inhibited eleven additional native or mutant kinases with IC50 <10 nmol/L. Among these kinases were ROS1 and FLT3 (IC50 values of 1.9 and 2.1 nmol/L, respectively). The drug exhibited more modest activity against T790M-mutant EGFR and, as previously mentioned, IGF-1R and the insulin receptor (IC50 29–160 nmol/L).
The activity of brigatinib was also examined in cellular assays. Consistent with the kinase assay data, brigatinib inhibited ALK and ROS1 with similar potencies (IC50S of 14 and 18 nmol/L, respectively). Compared to ALK, brigatinib inhibited FLT3, mutant variants of FLT3, T790M-mutant EGFR, and IGF-1R with ~11-fold to 35-fold lower potency. Brigatinib did not show significant activity toward the insulin receptor, wild-type EGFR, or MET (IC50 >1,000 nmol/L).10
In 2012, Rivera et al studied brigatinib’s activity as a reversible inhibitor of activated EGFR (exon 19 deletion and L858R-mutant) as well as T790M-mutant EGFR in non-small cell lung cancer (NSCLC). Ba/F3 cell lines were used to characterize the activity of the drug against wild-type or activated EGFR (exon 19 del E746_A750) with or without T790M. In Ba/F3 cell lines expressing EGFR with an exon 19 deletion, brigatinib inhibited EGFR phosphorylation and viability with IC50s of 75 and 114 nmol/L, respectively. In Ba/F3 cells expressing EGFRT790M, the drug inhibited EGFR phosphorylation and viability with IC50s of 15 and 281 nM, respectively. Brigatinib also exhibited in vivo potency against HCC827 (NSCLC) cells expressing EGFR exon 19 deletions or the T790M mutation in a xenograft model. Brigatinib did not inhibit wild-type EGFR phosphorylation in an NSCLC cell line (H358) or in engineered Ba/F3 cells (IC50 >3,000 nmol/L). Effective doses in mice against activated and T790M-mutant EGFR are similar to doses active against crizotinib-resistant ALK variants such as L1196M.11
In 2013, Squillace et al showed that brigatinib effectively inhibited the viability of Ba/F3 cells expressing CD74-ROS1 (IC50 18 nmol/L), FIG-ROS1 (IC50 31 nmol/L), SDC4-ROS1 (IC50 16 nmol/L), and EZR-ROS1 (IC50 41 nmol/L). Ba/F3 cells driven by the ROS1 L2026M gatekeeper-mutant forms of CD74-ROS1 and FIG-ROS1 were developed to test the drug’s activity. The inhibitory capacity of brigatinib was unaffected by the ROS1 L2026M gatekeeper mutation. In contrast, crizotinib potencies were reduced ~4-fold.12
Preclinical activity against ALK mutations
ALK kinase domain mutations have been identified as a major mechanism for the development of ALK TKI resistance. An in vitro mutagenesis screen in Ba/F3 cells expressing native EML4–ALK was performed by Zhang et al,13 with cells grown in plates containing various concentrations of brigatinib, crizotinib, ceritinib, and alectinib. DNA was extracted from the resistant cells and the ALK kinase region was sequenced. Treatment with 500 nmol/L brigatinib was sufficient to suppress the emergence of any mutant, whereas higher concentrations of all of the other TKIs (1,000 nmol/L) were needed. The same group generated a panel of Ba/F3 cell lines containing native EML4-ALK, or 17 variants of ALK mutations that were either previously associated with clinical resistance or identified in the aforementioned mutagenesis screen. Brigatinib was a potent inhibitor of native EML4–ALK (IC50 14 nmol/L), with crizotinib, ceritinib, and alectinib having IC50 of 107, 37, and 25 nmol/L, respectively. Brigatinib was active (IC50s <200 nmol/L) against all resistant mutations tested. These include F1174C/V, I1171N, and G1202R mutations, which have been reported in patients with progression of disease on ceritinib and alectinib. Brigatinib inhibited five mutants with ≥3-fold greater potency than ceritinib (G1269A, C1156Y, L1152R/P, and L1198F) and inhibited six mutants with ≥3-fold greater potency than alectinib (L1152R, I1171N, V1180L, L1196M, G1202R, and G1269A). Furthermore, brigatinib demonstrated greater activity than crizotinib for all mutants, except L1198F. Brigatinib also had the greatest selectivity over parental cells and exhibited >17-fold selectivity for all mutants, with G1202R being the most resistant in relative terms (IC50 184 nmol/L). These results suggest that brigatinib is a highly potent ALK inhibitor, with substantial in vitro activity against select ALK secondary resistance mutations when compared to crizotinib, ceritinib, and alectinib.
Additional in vitro and in vivo activity
In 2010, Rivera et al first reported the preclinical efficacy and activity of brigatinib in comparison with crizotinib, establishing brigatinib as a potent ALK inhibitor with a broad therapeutic index.14 In a panel of seven ALK-rearranged ALCL and NSCLC cell lines, the concentration of brigatinib that inhibited ALK phosphorylation by 50% (GI50) ranged from 4 to 31 nmol/L. It demonstrated 100-fold selectivity for ALK-rearranged versus ALK wild-type cell lines.
To study the in vivo efficacy of brigatinib as compared to crizotinib, severe combined immunodeficiency beige mice were implanted subcutaneously with either ALK-rearranged ALCL Karpas-299 or ALK-rearranged NSCLC H2228 cells and dosed orally with brigatinib (10, 25, or 50 mg/kg once daily) or crizotinib (25, 50, or 100 mg/kg once daily) for 14 days (Karpas-299) or 21 days (H2228). Tumor regression was observed at significantly lower doses of brigatinib compared to crizotinib: brigatinib was dosed at 25 mg/kg (ALCL) and 10 mg/kg (NSCLC), in comparison with crizotinib dosed at 100 mg/kg. In a pharmacokinetic/pharmacodynamic study in the ALCL model, inhibition of ALK phosphorylation after administration of 100 mg/kg of crizotinib was intermediate between that observed after administration of 10 or 25 mg/kg of brigatinib. Results from the analysis of plasma levels of each drug showed that brigatinib had equivalent efficacy to crizotinib at 4- to 10-fold lower levels of exposure (AUC and 24-hour trough plasma levels). Brigatinib had 10-fold greater potency and a 10-fold broader therapeutic index than crizotinib. Early animal studies showed favorable features of brigatinib, including good oral bioavailability across multiple animal species, moderate in vitro plasma binding (mouse, rat, monkey, and human plasma), as well as minimal inhibition of the major cytochrome P450 isoforms. These data supported further clinical evaluation of brigatinib in patients with ALK-rearranged tumors.14
Clinical data
Phase I/II trial data
A single-arm, open-label, Phase I/II trial of brigatinib was recently reported.15 The Phase I dose escalation was performed in patients with advanced malignancies, with the primary objective of establishing the recommended Phase II dose. The Phase I escalation dosing of the drug ranged from 30 to 300 mg daily. Dose-limiting toxicities were seen at 240 and 300 mg daily, and the recommended Phase II dose was determined to be 180 mg daily. Of note, given the concern for early pulmonary toxicity (occurring within the first 7 days of therapy, discussed more extensively later in this review) at 180 mg, two additional regimens were assessed: 90 mg once daily and 180 mg once daily with a 7-day lead-in at 90 mg daily. The primary objective of the Phase II study was to determine the efficacy with the primary endpoint of best objective response to therapy. The Phase II component of the study was divided into five histologically and molecularly defined cohorts where three oral once-daily regimens were assessed: 90, 180, and 180 mg with a 7-day lead-in at 90 mg daily.
The five cohorts enrolled in the expansion Phase II and their respective objective response rates (ORRs) were as follows: cohort 1, ALK-inhibitor-naïve, ALK-rearranged NSCLC patients with an ORR of 100% (4/4); cohort 2 – prior crizotinib-treated, ALK-rearranged NSCLC patients with an ORR of 74% (31/42); cohort 3 – EGFRT790M NSCLC patients with resistance to one prior EGFR TKI with an ORR of 0% (0/1); cohort 4 – patients with other cancers with ROS1, EGFR alterations with an ORR of 17% (3/18); and cohort 5 – crizotinib-naïve or crizotinib-treated, ALK-rearranged NSCLC with active, measurable, intracranial metastases with an ORR of 50% (3/6).
Among patients who had previously received crizotinib (Phases I and II combined), 72% (51/71) had an objective response (Table 1). The median progression-free survival (PFS) in crizotinib-pretreated patients was 13.2 months (9.1–18.7).15 Among crizotinib-naïve patients (four patients in Phase I and four patients in Phase II), 100% (8/8) achieved confirmed objective responses, including three complete responses (CRs).15 The median duration of treatment for patients with ALK-rearranged NSCLC was 15.4 months (7.1–20.9).
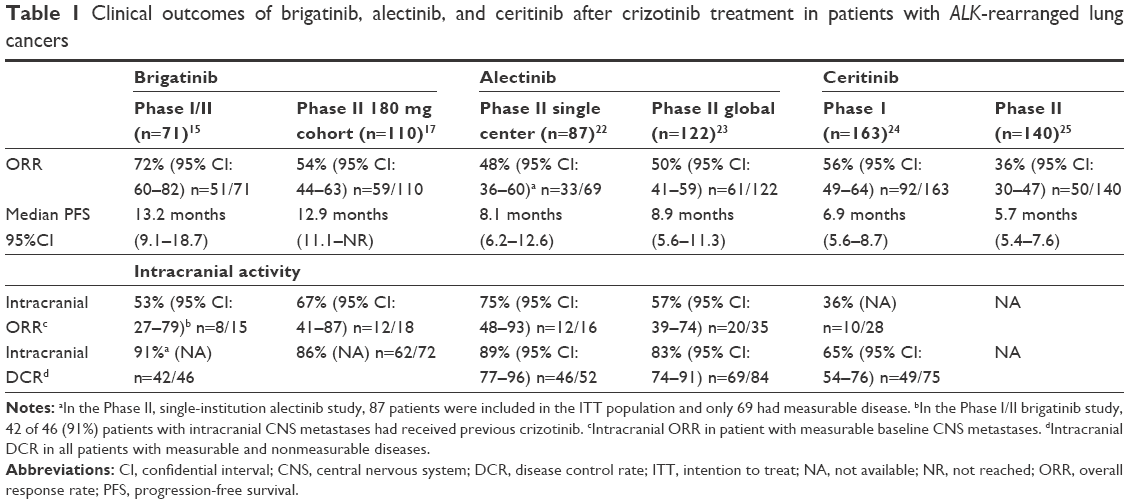 | Table 1 Clinical outcomes of brigatinib, alectinib, and ceritinib after crizotinib treatment in patients with ALK-rearranged lung cancers |
In the same Phase I/II trial, 50 patients with ALK-rearranged NSCLC (50/79; 63%) had brain metastases at baseline. Of the 50 patients with brain metastases, 23 had previously untreated brain metastases (23/50; 46%). In a post hoc analysis, 46/50 (92%) of patients with brain metastases had magnetic resonance imaging (MRI) of the brain 1 month prior to beginning therapy with brigatinib and at least one follow-up MRI brain to allow for the assessment of intracranial response, with a median follow-up of 17.5 months.
Of the 46 patients with baseline brain metastases who had MRI 1 month prior to starting treatment, 33% (15/46) had measurable central nervous system (CNS) metastases with an objective intracranial response rate of 53% (8/15; Table 1). Also, 67% (31/46) had nonmeasurable intracranial metastases with an objective intracranial response rate of 35% (11/31). The median intracranial PFS for all 46 evaluable patients (measurable and nonmeasurable intracranial disease) was 15.6 months (13.0–not reached).15 Recently presented updated response data showed an 18.4-month median intracranial PFS in ALK-rearranged NSCLC patients with CNS metastases.16
Ongoing clinical trials
The ALK in Lung Cancer Trial of brigatinib (ALTA, ClinicalTrials.gov number NCT02094573) is a randomized, Phase II, open-label, multicenter, international study designed to evaluate the efficacy and safety of two different dosing regimens of brigatinib (90 mg daily and 180 mg daily with a 7-day lead-in of 90 mg) in patients with ALK-rearranged, locally advanced or metastatic NSCLC, who have previously been treated with crizotinib. Patients were stratified by the presence of brain metastases at baseline and the best response to prior crizotinib, and were randomized 1:1 to receive oral brigatinib at 90 mg daily (arm A) or 90 mg daily for 7 days followed by 180 mg daily (arm B). The primary endpoint was investigator-assessed confirmed ORR. The secondary endpoints included PFS and independent review committee-assessed confirmed ORR and CNS response/PFS. The randomized, Phase II design was not intended for statistical comparisons between arms; however, post hoc comparisons performed on progression free survival (PFS) and overall survival (OS) were used to support dose selection.
An interim analysis of this study was presented in 2016 by Kim et al.24 A total of 112 patients were randomized to arm A and 110 patients to arm B. Approximately 70% of the patients in both groups had brain metastases. As of February 29, 2016, the investigator-assessed ORR in arm A was 45% (95% confidence interval (CI): 34–56) including one confirmed CR. The ORR in arm B was 54% (95% CI: 43–65) including four confirmed CRs (Table 1). Responses included a confirmed partial response in arm B in a patient with G1202R mutation at baseline. The median PFS in arm A was 9.2 months and in arm B was 12.9 months. Among patients with measurable, active brain metastases at baseline (defined as lesions with no prior radiotherapy or those with progression after radiotherapy), the intracranial ORR was 36% at 90 mg and 67% at 180 mg.17 This study recently completed accrual with an estimated primary completion date of November 2017.
ALTA-1L (ClinicalTrials.gov number NCT02737501) is an ongoing Phase III head-to-head trial of brigatinib versus crizotinib in the first-line setting with an estimated primary completion date of April 2019. Also planned and not yet open for enrollment is a Phase II study (ClinicalTrials.gov number NCT02706626) of brigatinib in patients with progression of disease on the second-generation ALK TKIs, ceritinib and alectinib. The results of this trial may help elucidate a population of patients in which brigatinib could become a rational second- or third-line treatment option.
Activity by mutation status and acquired resistance to brigatinib
Using next-generation sequencing (NGS) of tumor tissue collected from the Phase I/II and ALTA trials, Gettinger et al18 characterized the activity of brigatinib in patients who progressed on crizotinib according to ALK mutation status in the context of concurrent ALK fusion. Tumor samples were analyzed using the FoundationOne NGS platform. A total of 32 samples collected after progression on crizotinib and prior to brigatinib treatment were analyzed and nine patients with secondary ALK mutations were found (F1174L, S1206F, G1269A, L1196M, G1202R, F1245V). Confirmed ORR in patients with known secondary ALK kinase domain mutations was 67% (6/9).
In the same study by Gettinger et al, a total of six samples were evaluated by NGS from patients after progression on brigatinib and five had detectable secondary mutations in the ALK kinase domain. Three patients had complex mutation patterns with compound mutations and one BRAF G469A driver mutation in conjunction with an ALK G1202R mutation.18 A similar analysis was performed by Gainor et al, where 103 repeat biopsies from ALK-rearranged NSCLC patients progressing on a variety of ALK inhibitors were evaluated (Table 2).19 Six patients underwent biopsies following progression on brigatinib. Five of six patients previously received crizotinib. None of these patients had received another second-generation ALK inhibitor. The median duration of treatment was 20.2 months (12.1–44.4). None of the patients had intrinsic resistance to brigatinib. ALK resistance mutations were seen in 71% of brigatinib-resistant specimens. The most common ALK resistance mutation was G1202R, which was observed in three specimens (one with CNS progression). The other patients had ALK E1210K, D1203N, or S1206Y/C mutations.
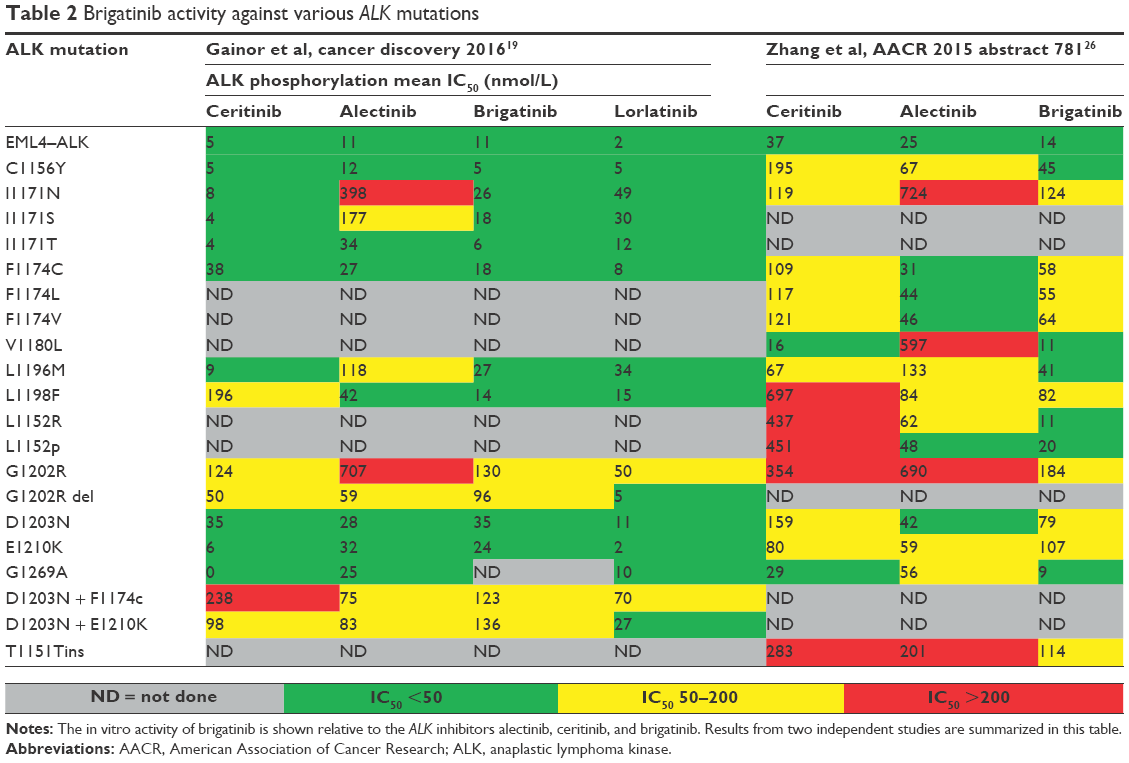 | Table 2 Brigatinib activity against various ALK mutations |
Safety profile
With regard to the Phase I/II trial previously discussed, patients received 30–300 mg of brigatinib in the initial dose-escalation stage. Dose-limiting toxicities observed during the dose-escalation phase included grade 3 increased alanine aminotransferase at 240 mg daily and grade 4 dyspnea at 300 mg daily. The decision was made to restart dose escalation at 90 mg daily and was escalated to 240 mg, including twice-daily regimens. The maximum tolerated dose was not formally established as specified in the protocol, and the study authors initially chose to move the 180 mg daily dose forward as the recommended Phase II dose based on the absence of dose-limiting toxicities, preliminary activity, and pharmacokinetic profiles.15 The most common treatment-emergent adverse events included nausea (53%), fatigue (43%), diarrhea (41%), headache and cough (39%), predominantly grades 1 and 2. The most common grade 3–4 treatment-emergent adverse events across all doses (Phases I and II) were increased lipase (9%), dyspnea (6%), and hypertension (5%). Serious treatment-emergent adverse events were dyspnea (7%), pneumonia (7%), and hypoxia (5%).
A subset of pulmonary events, including dyspnea and hypoxia, were observed to occur within 7 days of treatment initiation or reinitiation following a prolonged period of dose interruption. Most events occurred within 2 days of dosing and were managed with dose interruption and empiric treatment with steroids and antibiotics. Rates of these events were lower with lower starting doses. Fourteen percent of events occurred in patients starting on 180 mg daily dosing and only 2% in patients starting with 90 mg daily dosing.
In an effort to better understand the early pulmonary toxicity, two other dosing regimens were explored at 90 mg daily and 180 mg daily, with a 7-day lead-in at 90 mg daily. Among 32 patients treated with a loading dose of 90 mg daily for 7 days followed by 180 mg daily, no such events were reported after dose escalation. Sixteen patients (12%, 16/137) died during treatment or within 31 days of the last dose of brigatinib. Eight patients (6%, 6/137) died from progression of disease. Of the eight remaining patients, three were judged by the investigator as at least possibly related to treatment.15 Treatment-emergent adverse events of brigatinib and other ALK TKIs are reviewed in Table 3.
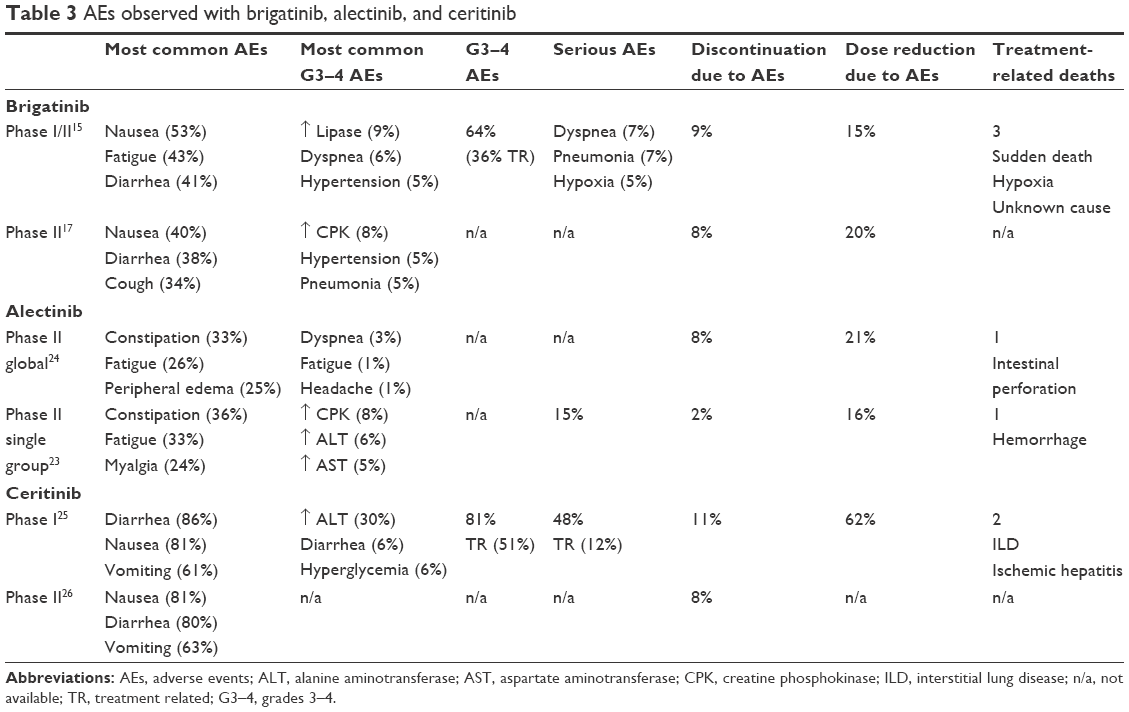 | Table 3 AEs observed with brigatinib, alectinib, and ceritinib |
Discussion
Brigatinib is a next-generation ALK inhibitor with broad activity against several acquired resistance mutations that can emerge after treatment with other ALK TKIs. The drug has clear activity in patients with ALK-rearranged lung cancers previously treated with crizotinib, with response rates of 54%–72%, intracranial response rates of 53%–67%, and a median PFS of between 12.9 and 13.2 months based on Phase I and II trial data. Brigatinib received breakthrough designation therapy from the US Food and Drug Administration based on this data, and was granted orphan designation for the treatment of ALK-rearranged NSCLCs.
Considering the current therapeutic landscape for ALK-rearranged lung cancers where crizotinib is approved as frontline therapy, and alectinib and ceritinib are approved post crizotinib, the prospect of brigatinib receiving approval post crizotinib raises the question as to which of the three TKIs to choose in the second-line setting. From an activity perspective, brigatinib seems to have the broadest in vitro coverage against potential mechanisms of resistance to therapy mediated by the acquisition of an ALK mutation and would not be an unreasonable choice over alectinib and ceritinib. Furthermore, recognizing the substantial limitations of cross-trial comparisons, the median PFS achieved with brigatinib is numerically increased in comparison with the activity achieved with alectinib and ceritinib post crizotinib. From a safety perspective, ceritinib has the highest incidence of clinically significant drug-related adverse events of the three drugs and is viewed by many practitioners as the least preferable TKI from a safety standpoint. In contrast, alectinib is perceived, in general, as a tolerable therapy, and the more widespread adoption of brigatinib will hinge on physicians’ level of comfort with the drug’s profile of pulmonary toxicity and the recommended dosing of 90 mg initially, followed by 180 mg thereafter.
The targeted therapy space for ALK-rearranged lung cancers continues to evolve rapidly, however, and the role of brigatinib is likely to be impacted by a number of factors. Recently presented data from the J-ALEX study in Japan of alectinib versus crizotinib in ALK inhibitor-naïve, ALK-rearranged NSCLC patients showed improved PFS with alectinib compared to crizotinib.20 Based on the J-ALEX data and further confirmation from the ALEX study, there is a possibility that alectinib will be approved in the frontline setting, effectively replacing crizotinib as the first-line standard of care. This would open up room for brigatinib as a potential second-line therapy in patients with progression of disease on alectinib; however, ALK G1202R mutations are predicted to emerge in a substantial proportion of tumors post alectinib, and other drugs with possibly improved ALK G1202R coverage, such as lorlatinib, are currently in development. A planned trial of brigatinib in patients with ALK-rearranged NSCLC who have previously been treated with second-generation ALK inhibitors (NCT02706626) may help elucidate the role of brigatinib in the post-alectinib setting.
In the first-line setting, we hypothesize that brigatinib may, similar to alectinib, offer increased duration of disease control compared to crizotinib, secondary to delaying the onset of resistance and possibly improved intracranial disease control. A pivotal Phase III study (ALTA-1L) is currently underway (NCT02737501) to investigate the activity of the drug in treatment-naïve patients.
We envision a molecularly driven approach to the treatment of ALK-rearranged NSCLC patients, where patients’ tissue samples are sequenced at baseline and at the time of progression on ALK TKIs in an effort to select the next best therapy based on ALK secondary acquired resistance mutations or other genomic alterations that emerge. The availability of multiple drugs will allow the clinician to sequence these therapies in an effort to prolong disease control, assuming that tumors remain dependent on signaling through ALK. Shaw et al recently reported the resensitization to crizotinib by lorlatinib ALK resistance mutation L1198F.21 This exciting finding reveals the complexity of determining proper drug sequencing, with the ability to potentially revisit a previously used ALK inhibitor should the molecular profile suggest activity. As this space continues to evolve rapidly, the treatment landscape for ALK-rearranged NSCLC will continue to change. The biology will continue to predict response to therapy and this will guide the development and sequencing of brigatinib and other ALK TKIs.
Conclusion
Brigatinib is a potent ALK inhibitor with activity against select ALK TKI-induced ALK secondary resistance mutations. It is active in patients with ALK-rearranged lung cancers who previously received crizotinib, and good intracranial disease control has been reported. Early pulmonary toxicity has been observed, prompting the choice to dose the drug at a 90 mg 7-day lead-in, followed by 180 mg daily. A randomized Phase III study of brigatinib versus crizotinib in ALK inhibitor-naïve, ALK-rearranged lung cancer patients is currently underway. Further clinical trials, utilizing both cell free DNA plasma-based and tissue-based biomarkers, are needed to better understand resistance mechanisms and to help correctly sequence the multiple ALK-directed therapies that are currently at our disposal.
Acknowledgments
Joshua K Sabari and Fernando C Santini are the first coauthors. Joshua K Sabari and Alison M Schram are supported by the National Institue of Health and National Cancer Institute T32 CA009207. Alexander Drilon is supported by the National Institute of Health and National Cancer Institute P30 CA008748.
Disclosure
Alexander Drilon has received honoraria from Exelixis, Ignyta, and Loxo Oncology and financial support through a grant from Foundation Medicine. The other authors report no conflicts of interest in this work.
References
American Cancer Society: Cancer Facts and Figures 2016. Atlanta: GACS. | ||
Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non–small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. | ||
Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–1482. | ||
Tchekmedyian N, Ali SM, Miller VA, Haura EB. Acquired ALK L1152R mutation confers resistance to ceritinib and predicts response to alectinib. J Thorac Oncol. 2016;11:e87–e88. | ||
Katayama R, Lovly CM, Shaw AT. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: a paradigm for precision cancer medicine. Clin Cancer Res. 2015;21(10):2227–2235. | ||
Isozaki H, Takigawa N, Kiura K. Mechanisms of aacquired resistance to ALK inhibitors and the rationale for treating ALK-positive lung cancer. Cancers (Basel). 2015;7(2):763–783. | ||
Zhang S, Anjum R, Squillace R, et al. The potent ALK inhibitor brigatinib (AP26113) overcomes mechanisms of resistance to first- and second-generation ALK inhibitors in preclinical models. Clin Cancer Res. 2016;22(22):5527–5538. | ||
Shakespeare W, Fantin V, Wang F, et al. Abstract #3738: discovery of potent and selective orally active inhibitors of anaplastic lymphoma kinase (ALK). Cancer Res. 2009;69:3738–3738. | ||
Huang Q, Johnson TW, Bailey S, et al. Design of potent and selective inhibitors to overcome clinical anaplastic lymphoma kinase mutations resistant to crizotinib. J Med Chem. 2014;57(4):1170–1187. | ||
Huang W, Liu S, Zou D, et al. Discovery of brigatinib (AP26113), a phosphine oxide-containing, potent, orally active inhibitor of anaplastic lymphoma kinase. J Med Chem. 2016;59(10):4948–4964. | ||
Rivera VM, Wang F, Anjum R, et al. Abstract 1794: AP26113 is a dual ALK/EGFR inhibitor: characterization against EGFR T790M in cell and mouse models of NSCLC. Cancer Res. 2014;72:1794–1794. | ||
Squillace RM, Anjum R, Miller D, et al. Abstract 5655: AP26113 possesses pan-inhibitory activity versus crizotinib-resistant ALK mutants and oncogenic ROS1 fusions. Cancer Res. 2014;73(8):5655–5655. | ||
Zhang S, Wang F, Keats J, et al. Abstract LB-298: AP26113, a potent ALK inhibitor, overcomes mutations in EML4-ALK that confer resistance to PF-02341066 (PF1066). Cancer Res. 2014;70(8):LB-298–LB-298. | ||
Rivera VM, Anjum R, Wang F, et al. Abstract 3623: efficacy and pharmacodynamic analysis of AP26113, a potent and selective orally active inhibitor of Anaplastic Lymphoma Kinase (ALK). Cancer Res. 2010;70(8):3623–3623. | ||
Gettinger SN, Bazhenova LA, Langer CJ, et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: a single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016;17(12):1683–1696. | ||
D Ross Camidge, Tiseo M, Myung-Ju Ahn, et al. Brigatinib in Crizotinib-Refractory ALK+NSCLC: central Assessment and Updates from ALTA, a Pivotal Randomized Phase 2 Trial: World Conference on Lung Cancer 2016 Poster 4046; 2016. | ||
Dong-Wan Kim MT, D Ross Camidge. Brigatinib (BRG) in patients (pts) with crizotinib (CRZ)-refractory ALK+ non-small cell lung cancer (NSCLC): first report of efficacy and safety from a pivotal randomized phase (ph) 2 trial (ALTA). Abstract 9007 ASCO 2016. J Clin Oncol. 2016;34(Suppl; abstr 9007). | ||
Gettinger SN, Zhang S, Hodgson JG, et al. Activity of brigatinib (BRG) in crizotinib (CRZ) resistant patients (pts) according to ALK mutation status. ASCO 2016 Abstract 9060. J Clin Oncol. 2016;34(Suppl; abstr 9060). | ||
Gainor JF, Dardaei L, Yoda S, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016;6(10):1118–1133. | ||
Nokihara H, Hida T, Kondo M, et al. Alectinib (ALC) versus crizotinib (CRZ) in ALK-inhibitor naive ALK-positive non-small cell lung cancer (ALK+ NSCLC): primary results from the J-ALEX study. Abstract 9008 ASCO 2016. J Clin Oncol. 2016;34(Suppl; abstr 9008). | ||
Shaw AT, Friboulet L, Leshchiner I, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med. 2016;374(1):54–61. | ||
Shaw AT, Gandhi L, Gadgeel S, et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: a single-group, multicentre, phase 2 trial. Lancet Oncol. 2016;17(2):234–242. | ||
Ou SH, Ahn JS, De Petris, et al. Alectinib in crizotinib-refractory ALK-rearranged non–small-cell lung cancer: a phase II global study. J Clin Oncol. 2016;34(7):661–668. | ||
Kim DW, Mehra R, Tan DS, et al. Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol. 2016;17:452–463. | ||
Mok T, Spiegel D, Felip E, et al. ASCEND-2: a single-arm, open-label, multicenter phase II study of ceritinib in adult patients (pts) with ALK-rearranged (ALK+) non-small cell lung cancer (NSCLC) previously treated with chemotherapy and crizotinib (CRZ). Abstract 8059 ASC0 Annual Meeting 2015. J Clin Oncol. 2015;33(Suppl; abstr 8059). | ||
Zhang S, Nadworny S, Scott D, et al. Abstract 781: the potent ALK inhibitor AP26113 can overcome mechanisms of resistance to first- and second-generation ALK TKIs in preclinical models. Cancer Res. 2015;75(15):781–781. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.