")
Back to Journals » Journal of Inflammation Research » Volume 14
The β3/5 Integrin-MMP9 Axis Regulates Pulmonary Inflammatory Response and Endothelial Leakage in Acute Lung Injury
Authors Tong Y , Bao C, Xu YQ, Tao L, Zhou Y, Zhuang L, Meng Y, Zhang H, Xue J, Wang W, Zhang L, Pan Q, Shao Z, Hu T, Guo Q, Xue Q, Lu H, Luo Y
Received 4 August 2021
Accepted for publication 23 September 2021
Published 5 October 2021 Volume 2021:14 Pages 5079—5094
DOI https://doi.org/10.2147/JIR.S331939
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Yao Tong,* Chengrong Bao,* Yi-Qiong Xu,* Lei Tao,* Yao Zhou, Lei Zhuang, Ying Meng, Hui Zhang, Jingjing Xue, Weijun Wang, Lele Zhang, Qingbo Pan, Zhenzhen Shao, Tianran Hu, Qian Guo, Qingsheng Xue, Han Lu, Yan Luo
Department of Anesthesiology, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yan Luo; Han Lu Email [email protected]; [email protected]
Background: Acute lung injury (ALI) is a severe respiratory disease with high rates of morbidity and mortality. Many mediators regarding endogenous or exogenous are involved in the pathophysiology of ALI. Here, we have uncovered the involvement of integrins and matrix metalloproteinases, as critical determinants of excessive inflammation and endothelial permeability, in the regulation of ALI.
Methods: Inflammatory cytokines were measured by quantitative real-time PCR for mRNA levels and ELISA for secretion levels. Endothelial permeability assay was detected by the passage of rhodamine B isothiocyanate-dextran. Mice lung permeability was assayed by Evans blue albumin (EBA). Western blot was used for protein level measurements. The intracellular reactive oxygen species (ROS) were evaluated using a cell-permeable probe, DCFH-DA. Intratracheal injection of lipopolysaccharide (LPS) into mice was conducted to establish the lung injury model.
Results: Exogenous MMP-9 significantly aggravated the inflammatory response and permeability in mouse pulmonary microvascular endothelial cells (PMVECs) treated by LPS, whereas knockdown of MMP-9 exhibited the opposite phenotypes. Knockdown of integrin β 3 or β 5 in LPS-treated PMVECs significantly downregulated MMP-9 expression and decreased inflammatory response and permeability in the presence or absence of exogenous MMP-9. Additionally, the interaction of MMP-9 and integrin β 5 was impaired by a ROS scavenger, which further decreased the pro-inflammatory cytokines production and endothelial leakage in PMVECs subjected to co-treatment (LPS with exogenous MMP-9). In vivo studies, exogenous MMP-9 treatment or knockdown β 3 integrin significantly decreased survival in ALI mice. Notably, knockdown of β 5 integrin alone had no remarkable effect on survival, but which combined with anti-MMP-9 treatment significantly improved the survival by ameliorating excessive lung inflammation and permeability in ALI mice.
Conclusion: These findings support the β 3/5 integrin-MMP-9 axis as an endogenous signal that could play a pivotal role in regulating inflammatory response and alveolar-capillary permeability in ALI.
Keywords: acute lung injury, endothelial cells, integrin, MMP-9
Introduction
Acute lung injury (ALI) is a lethal pulmonary disease, characterized by increased vascular permeability, intrapulmonary retention of neutrophils, and synthesis of proinflammatory cytokines.1 Lung endothelial cells (ECs) injury plays an important role in the dysfunction of alveolar-capillary barrier, which is a characteristic feature of ALI.2,3 ECs were activated in the setting of inflammation, and resulted in increasing pro-inflammatory cytokines release, recruitment of circulating leukocytes adhesion to ECs, and deteriorated ECs permeability.4,5 Lipopolysaccharide (LPS), also named endotoxin, has a pro-inflammatory effect and involves in the pathogenesis of Gram-negative bacterial infection, which stimulates innate immunity and triggers biochemical and cellular responses that lead to inflammation and toxicity.6,7 LPS is a primary pathogenic mediator of ECs activation, which activates the classic NF-κB pathway, to induce the expression of several endogenous molecules, such as high-mobility group box 1 (HMGB1), integrins, and matrix metalloproteinases.8–10 This study was focused on integrins and matrix metalloproteinases in ECs and ALI.
Integrins are a family of cell surface glycoproteins, which include 24 receptors, each involving a single α subunit and a single β subunit. Integrins are expressed in all cell types and serve as a family of cell-cell and cell-extracellular matrix (ECM) binding transmembrane receptors capable of signal transduction.11 Studies have indicated many integrin receptors play pivotal roles in the process of inflammatory responses.9,12 We and others have demonstrated critical roles for three integrins (αvβ3, αvβ5, and αvβ6) in regulating lung inflammation and permeability.13–15 These results have identified these three integrins as potential therapeutic targets in ALI. Integrins are transmembrane adhesion receptors essential for cell communication with the environment and in particular with the ECM, which can be processed by several enzymes. Matrix metalloproteinases (MMPs) are one of the largest members involved in this process.
MMPs, a family of zinc-dependent extracellular enzymes secreted into the extracellular space and have important roles in many physiological and pathological processes, involving tissue repair, tumor invasion, and acute lung injury.16–18 Matrix metalloproteinase-9 (MMP-9 or gelatinase B) belongs to a family of MMPs that are released by inflammatory and noninflammatory cells, such as neutrophils and endothelial cells.19,20 Previous studies have focused on the role of MMP-9 in various LPS-induced acute inflammatory models, and MMP-9 can act as a regulator to promote the secretion of other cytokines by leukocytes.21,22 However, the precise mechanism for MMP-9 in the regulation of ECs and ALI remains unclear. In the current study, we performed to uncover the regulatory effect of β3/5 integrin and MMP-9 on mouse pulmonary microvascular endothelial cells (PMVECs) in vitro, as well as the LPS induced murine lung injury models in vivo.
Materials and Methods
Reagents
Antibodies against MMP-9 (ab228402), V5 (ab27671), Integrin β3 (ab119992), ICAM-1 (ab222736), VCAM-1 (ab134047), TRAF-6 (ab33915) and β-actin (ab8227) were purchased from Abcam. Anti- Integrin β5 (#3629), and NF-κB proteins (NF-κB Pathway Sampler Kit #9936) were purchased from Cell Signaling Technology. Pierce™ Protein A/G Plus Agarose was obtained from Invitrogen. LPS, rhodamine B isothiocyanate-dextran, and p-aminophenylmercuric acetate (APMA) were purchased from Sigma-Aldrich. Murine recombinant MMP-9 (R&D, 909-MM), MMP-9 ELISA kits (MMPT90), IL-6 ELISA kits (M6000B), TNF-α ELISA kits (DY410), CXCL-1 ELISA kits (MKC00B), and MPO ELISA kits (DY3667) were from R&D Systems. MMP-9 neutralizing monoclonal antibody (IM09L) was from Sigma-Aldrich. NF-κB inhibitor, PDTC, and ROS scavenger, N-acetyl-L-cysteine (Nac), were obtained from MedChem Express (Shanghai, China). LipoJet™ reagent and GeneMute siRNA transfection reagent were purchased from SignaGen (Ijamsville, MD, USA).
Animals and ALI Models
Male wild-type mice (C57BL/6; 8–12 weeks) were housed and cared in the specific pathogen-free animal care facility at Shanghai Jiao Tong University following the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals as well as the guidelines of Shanghai Ruijin Hospital (approval number: SYXK 2018-0027). Mice in this study were randomly assigned to indicated groups by an investigator who was blinded to the group allocation. Acute lung injury (ALI) models were performed by intratracheal administration of LPS (2 mg per kg body weight) for 24h. Besides, C57/BL6 mice were intravenously administered of lentivirus vectors or lenti-Itgb3/5 (5 × 107 plaque-forming units per mouse) for 5 days prior to intratracheal administration of LPS.
Cell Culture and Transfection
Mouse pulmonary microvascular endothelial cells (PMVECs) and HEK-293T cells were obtained from Procell and cultured in DMEM with 10% fetal bovine serum. Penicillin (50 μg/mL) and streptomycin (50 μg/mL) were added to cell cultures. PMVECs were authenticated and tested for mycoplasma contamination before conducting experiments. LipoJet™ reagent was used to overexpress Itgb3/5 in PMVECs, and 24 or 48hr later, cells were treated with stimulators. The coding sequence of mouse Itgb3 or Itgb5 with C-terminal V5 epitope was synthesized and cloned into pCMV between HindIII and BamHI sites. For target genes knockdown, scramble siRNA, MMP-9 siRNA, Itgb3 siRNA, Itgb5 siRNA were used to transfect PMVECs for 48 h by GeneMute siRNA transfection reagent. Cell lysates were collected in Lysis Buffer (150 mM NaCl, 50 mM Tris, 1.0 mM EDTA, 2 mM dithiothreitol, 0.025% sodium azide, and 1 mM phenylmethylsulfonyl fluoride), and prepared by brief sonication at 4°C.
Western Blot Analysis
The lysates of cells or lung tissues were extracted in ice-cold lysis buffer RIPA with Protease and Phosphatase Inhibitor Cocktail (Invitrogen). Lysates were prepared by brief sonication at 4°C. Insoluble cellular debris was precipitated via centrifugation at 12,000g for 15 min at 4°C. Lysate supernatant was normalized for protein concentrations, which were measured with use of a Bio-Rad Protein Assay Kit. Protein was eluted by dilution in denaturing loading buffer to a final 1X mixture and then incubated at 90°C for five minutes. Equal amounts of proteins were resolved by SDS-PAGE and then electroblotted onto polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA, USA). These membranes were blocked with 5% skim milk for 1 h at room temperature and subsequently incubated with a primary antibody overnight at 4 °C. The membranes were then washed three times at 10 min intervals with PBST. After the addition of a secondary antibody for 1 h at room temperature, membranes were screened by the Odyssey Infrared Imaging System (LI-COR) and quantified through the Image J software.
RNA Extraction, Reverse Transcription PCR, Quantitative PCR
The total RNA was isolated from PMVECs cells using TRIzol reagent (Life Technologies, Grand Island, NY) and 1μg total RNA was reverse-transcribed to cDNA using the first-strand cDNA synthesis kit (Takara) according to standard protocols. The target genes were amplified and quantified in a LightCycler 480 Instrument II machine (Roche Life Science) using SYBR Green PCR mixture (KAPA Biosystems). The data were normalized to GAPDH and analyzed with the 2−ΔΔCq method. Primers for qPCR in this study are presented in Supplementary Table 1.
Intracellular ROS Detection
A cell-permeable probe, DCFH-DA was used to measure intracellular ROS levels. PMVECs were seeded in 6-well plates and treated as indicated before incubation with DCFH-DA for 30 min at 37 °C. Cells were washed with PBS and then measured using flow cytometry.
Immunoprecipitation Assay
PMVECs were seeded in 10-cm dishes and treated with LPS with or without Nac before collection. Cell lysates were extracted in ice-cold IP buffer (0.25% Triton-X-100 in 1x PBS, pH 7.6, 0.025% sodium azide, and 1 mM phenylmethylsulfonyl fluoride) and prepared by brief sonication at 4°C. Lysate supernatant was collected by centrifugation at 12,000g for 15 min at 4°C and normalized for protein concentration. Supernatants were incubated with the indicated antibody for three hours at 25 °C. The immunoprecipitated protein was captured with 25 μL resuspended volume of Protein A/G PLUS-Agarose for 1 h before three rounds of washing with IP buffer (1 mL). Protein was eluted by dilution in denaturing loading buffer and incubation at 88°C for five minutes. Eluted samples were resolved by SDS-PAGE and subjected to immunoblotting.
Endothelial Permeability Assay
PMVECs monolayers were seeded in transwell filters (0.4-μm pore size; Corning) and treated as indicated prior to measurement by the passage of rhodamine B isothiocyanate-dextran (average MW ~70,000; Sigma) as described.23 Briefly, the top well was placed with rhodamine-dextran at 20 mg/mL, and the fluorescence in the bottom well was screened by detecting 40 μL medium aliquots in a time course using a SpectraMax microplate reader at 544 nm excitation and 590 nm emission.
Alveolar-Capillary Permeability by Evans Blue Albumin (EBA)
Lung alveolar-capillary permeability was estimated using EBA based on our previous description.10 Mice were anesthetized and injected vena jugularis externa with EBA (20 mg/kg). At 30 min after EBA injection, mice were killed, and lung tissue was harvested for EBA measurement.
NF-κB Promotor Assay
Cignal NF-κB Reporter luciferase plasmids were co-transfected with empty, Itgb5, control siRNA, or Itgb5 siRNA for 24 to 48 hours before vehicle, LPS with or without rMMP-9 treatment for an additional 6 to 18 hours. Cells were then collected and assayed for firefly and Renilla luciferase activity. NF-kB promoter activity was normalized by firefly and Renilla luciferase activity ratio.
H&E Staining and Lung Injury Scoring
Hematoxylin&eosin (H&E) staining and assessment of histological lung damage were performed as described previously.24 Briefly, the left lungs from mice were dehydrated and embedded in paraffin. All lung fields were performed to H&E for the measurement of inflammatory cells infiltration and pulmonary morphology and determined at ×20 magnification for each sample. Lung histology was scored by an expert clinical panel blinded to the group allocation.
Lentivirus Construction
To generate lentivirus encoding Itgb3/5, pLVX-Itgb3/5 plasmid was co-transfected with Lenti-X HTX packaging plasmids (Clontech) into 293FT cells following the manufacturer’s instructions. Seventy-two hours later, virus was collected and concentrated using Lenti-X concentrator.
Statistical Analysis
All data in this study were expressed as means ± SEM of independent experiments. Group comparisons were performed using t-test or one-way analysis of variance (ANOVA) with Tukey’s post hoc test. Survival curve was measured by Mantel–Cox test. P<0.05 (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001) was regarded as statistically significant. All statistical analyses were carried out using the GraphPad Prism 8.0 program.
Results
MMP-9 Regulates Inflammation and Permeability in LPS-Induced PMVECs
As MMP-9 was regarded as an inflammatory mediator, we determined to explore its role in PMVECs treated with or without LPS. We found exogenous recombinant MMP-9 (rMMP-9) significantly enhanced the mRNA levels of IL-6, CXCL-1, ICAM-1 and VCAM-1 in PMVECs treated by LPS, whereas rMMP-9 treated alone did not notably affect these genes expression (Figure 1A–D). The protein levels also showed a similar pattern, with rMMP-9/LPS co-treatment increasing TRAF6, ICAM-1, and VCAM-1 protein amounts among treatment groups (Figure 1E). In addition, the NF-κB signaling inhibitor, PDTC (pyrrolidinedithiocarbamate), drastically decreased MMP-9 protein amounts in LPS-treated PMVECs (Figure 1F).
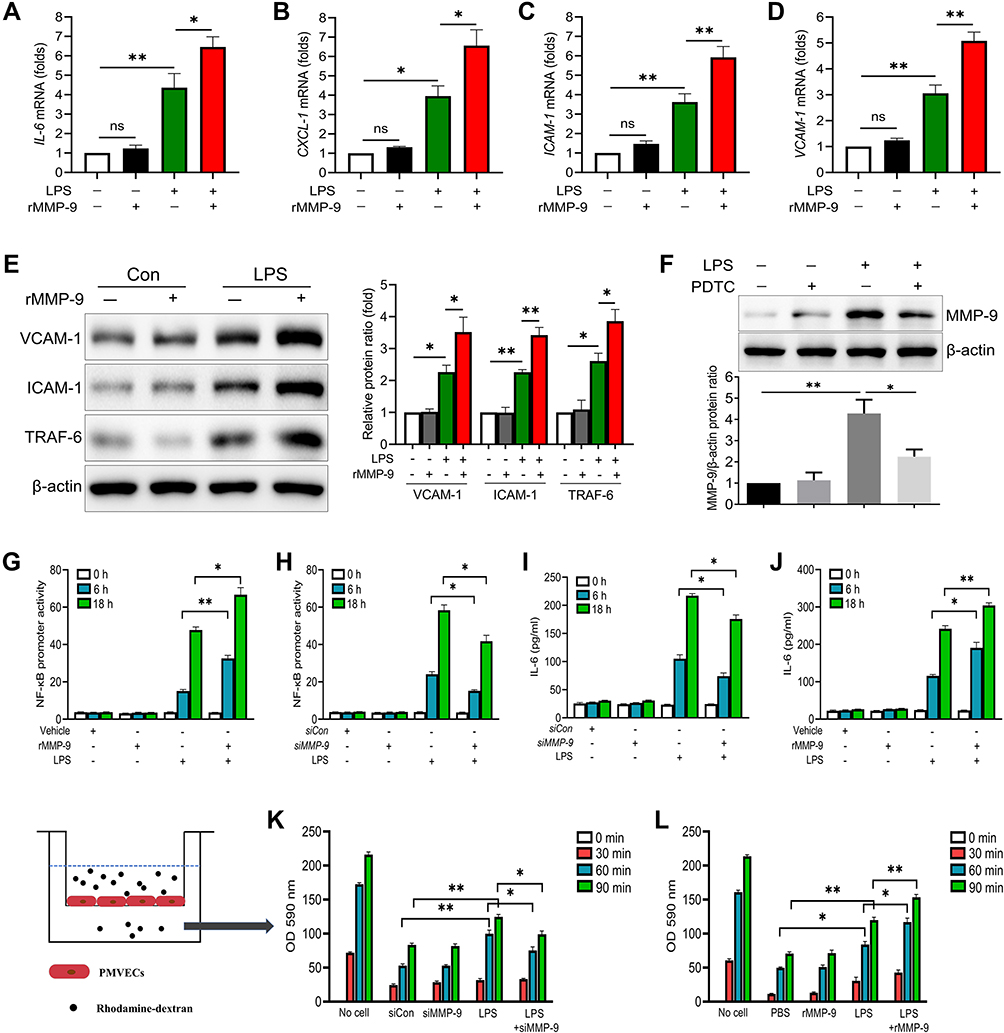 |
Figure 1 MMP-9 regulates inflammation and permeability in LPS-treated cells. (A–D) PMVECs were treated with LPS (1μg/mL) with or without rMMP-9 (10 ng/mL) for 4 h. The mRNA levels of IL-6, CXCL-1, ICAM-1, and VCAM-1 were detected. (E) The protein levels of VCAM-1, ICAM-1 and TRAF-6 were detected by Western blot analysis in PMVECs exposed to LPS with or without rMMP-9 treatment. (F) MMP-9 protein levels were detected by immunoblotting in PMVECs exposed to LPS with or without PDTC treatment. (G) 293T cells were transfected with Cignal NF-kB dual luciferase reporter plasmids for 48 h, and then cells were treated with LPS (1μg/mL) with or without rMMP-9 (10 ng/mL) for an additional 6 h or 18 h. (H) 293T cells were co-transfected with NF-kB dual luciferase reporter plasmids along with control siRNA, or MMP-9 siRNA for 48 h, and then cells were treated with LPS (1μg/mL) for an additional 6 h or 18 h. Cells were then collected and assayed for luciferase activity to evaluate NF-kB promoter activity. (I) PMVECs were transfected with control siRNA, or MMP-9 siRNA for 48 h, and then cells were treated with LPS (1μg/mL) in a time course. Supernatant IL-6 secretion was measured by ELISA. (J) PMVECs were treated by APMA (APMA was used to active rMMP9 in vitro, and served as a control of active rMMP9) or active rMMP9 (10 ng/mL) 1 h before LPS challenged at indicated times, and then supernatant IL-6 was detected. (K and L) The permeability of treated PMVEC monolayers grown on 0.4 mm filters was determined by the appearance of rhodamine-dextran, which was added to the top well at the beginning of the experiment and detected the absorbance at 590 nm in the bottom well during a 90 min time course. *P<0.05, **P<0.01. All the results are from at least three independent experiments; Data represent means ±SEM. |
To further explore whether MMP-9 would affect NF-κB signaling, exogenous rMMP-9 was used to treat 293T cells and showed a significant increase in LPS (100 ng/mL) mediated NF-κB promoter activity (Figure 1G), whereas MMP-9 knockdown drastically reduced NF-κB promoter activity (Figure 1H). The expression of endogenous MMP-9 in protein and mRNA levels was efficiently inhibited by siRNAs (Figure S1A and B). Based on the NF-κB promoter activity assay, we found siRNA to MMP-9 treated cells showed a significant decrease in LPS mediated increases in phospho-NF-κB proteins, including p-IKK, p-P65, and p-IκBα at different time points (Figure S1C). Exogenous rMMP-9 did not affect NF-κB signaling, but in the presence of LPS, rMMP-9 significantly increased phospho-NF-κB proteins in PMVECs (Figure S1D). Next, mouse pulmonary microvascular endothelial cells (PMVECs) were transfected with siRNA (or scrambled control) to MMP-9 and exposed to LPS (1 μg/mL) at indicated time points. siRNA to MMP-9 treated cells showed a significant decrease in LPS mediated increases in IL-6 levels (Figure 1I). Exogenous rMMP-9, by itself, did not affect IL-6 secretion, but in the presence of LPS, rMMP-9 increased expression of IL-6 (Figure 1J). Further, the permeability assay was showed by detecting the traversing of rhodamine-dextran probes via PMVECs monolayers growing on 0.4-μm filters. Similar to the above-mentioned results, MMP-9 knockdown significantly reduced PMVECs leakage (Figure 1K), whereas treatment of rMMP-9 exhibited a significant increase in LPS mediated permeability (Figure 1L).
We next used PDTC to explore the role of MMP-9 in the inflammation and permeability of PMVECs. We found PDTC drastically diminished the effect of MMP-9 on promoting inflammatory cytokine production and rhodamine penetration in LPS-treated cells (Figure S1E and F). Collectively, these results suggest that MMP-9 modulates inflammatory response and permeability partially via NF-κB signaling in PMVECs.
MMP-9 Potentiates Lung Injury and Lowers Survival in LPS-Induced ALI Mice
To determine whether MMP-9 contributed to the pathology of lung injury, we used a clinically relevant model for pneumonia and severe lung injury initiated by intratracheal administration of LPS. Mice were treated with vehicle (PBS), LPS (2mg/kg, i.t.), recombinant MMP-9 (rMMP-9, 50μg/kg, i.v.), or their combination, and we collected lung tissues and Bronchoalveolar lavage fluid (BALF) 18 h later. LPS increased BALF protein amounts, inflammatory cytokines IL-6 and TNF-α, cell counts, neutrophils, and the MPO levels. In contrast, rMMP-9 treatment alone had only a modest effect on the same parameters. However, co-treatment of rMMP-9 with LPS dramatically increased the amount of BALF protein, cell counts, inflammatory cytokines and MPO (Figure 2A–F) compared to LPS alone. The EBA permeability and lung histology also displayed a similar pattern, with rMMP-9/LPS co-treatment increasing EBA uptake and the extent of lung damage most dramatically among treatment groups (Figure 2G–I). Additionally, we used a lethal dose of LPS (20mg/kg) to detect survival. Mice were divided into three groups: Control group (rMMP-9, i.v.), ALI group (LPS, i.t.), and Co-treatment group (rMMP-9 with LPS), and seven days survival were recorded. Mice in ALI group had a significantly lower survival rate than in the control group, whereas co-treatment group further reduced the survival (Figure 2J).
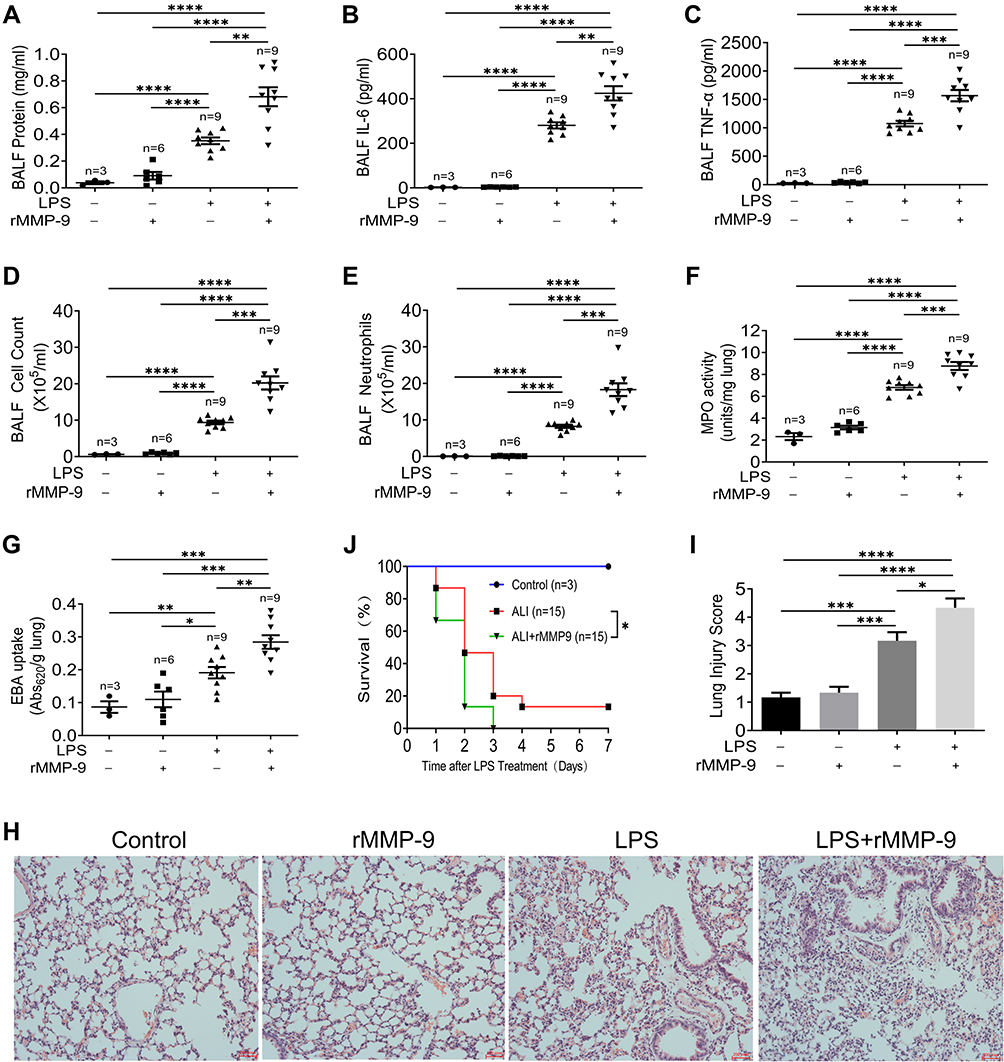 |
Figure 2 MMP-9 worsens LPS-induced lung injury in vivo. Mice were treated with LPS (2mg/kg, i.t.), recombinant MMP-9 (rMMP-9, 50μg/kg, i.v.), or their combination. 18 h later, bronchoalveolar lavage fluid (BALF) and lung tissues were collected. The protein concentration (A), IL-6 (B), TNF-α (C), cell counts (D), neutrophils (E) in Bronchoalveolar lavage fluid (BALF), and MPO activity (F) from the lung of mice were measured. (G) EBA uptake and (H) Hematoxylin and eosin (H&E)–stained lung sections (×200 magnification) with the injury score (I) were detected. Mice were treated by a lethal dose of LPS (20mg/kg, i.t.) with or without rMMP-9 (50μg/kg, i.v.), and then survival rates (J) were investigated. Data were analyzed by the Mantel-Cox test (survival). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Data represent means ±SEM. |
Next, we determined to explore whether MMP-9 neutralizing antibody ameliorates lung injury in the LPS-induced mouse model. Mice were co-treated with LPS (2 mg/kg, i.t.) and either IgG (control) or a neutralizing antibody against MMP-9 (3 mg/kg, i.v.). We discovered that BALF cell counts, neutrophils, MPO levels, inflammatory cytokines IL-6 and TNF-α were all significantly reduced in mice treated with anti-MMP-9 compared to IgG-treated control mice (Figure S2A–E). However, blocking MMP-9 treatment had no significant effect on BALF protein amounts (Figure S2F), EBA uptake (Figure S2G), and the survival rate of ALI mice (Figure S2H). Taken together, these results suggest that MMP-9 aggravates LPS-induced lung injury.
Integrin β3/5 Dampens MMP-9 Expression in LPS-Induced PMVECs
Many integrin receptors involve in the process of lung injury, and three (Integrin β3/5/6) of these integrins are related to vascular endothelial damage.25 We determined whether Integrin β3/5/6 would regulate any MMPs expressions in LPS-induced PMVECs and uncovered that Integrin β3/5 knockdown (siItgb3/5) significantly downregulated MMP-9 transcription in LPS treated PMVECs (Figure 3A–D). In line with the gene expression, MMP-9 protein levels were drastically decreased in PMVECs transfected with siItgb3/5 (Figure 3E and F). Conversely, we over-expressed Itgb3/5 in PMVECs and led to a dose-dependent increase of MMP-9 protein amounts (Figure 3G and H). MMP-9 secretion was also detected in cell supernatant by ELISA. Over-expression of Itgb3/5 in PMVECs resulted in a significant increase in LPS mediated MMP-9 release (Figure 3I), whereas Itgb3/5 knockdown drastically reduced MMP-9 secretion (Figure 3J).
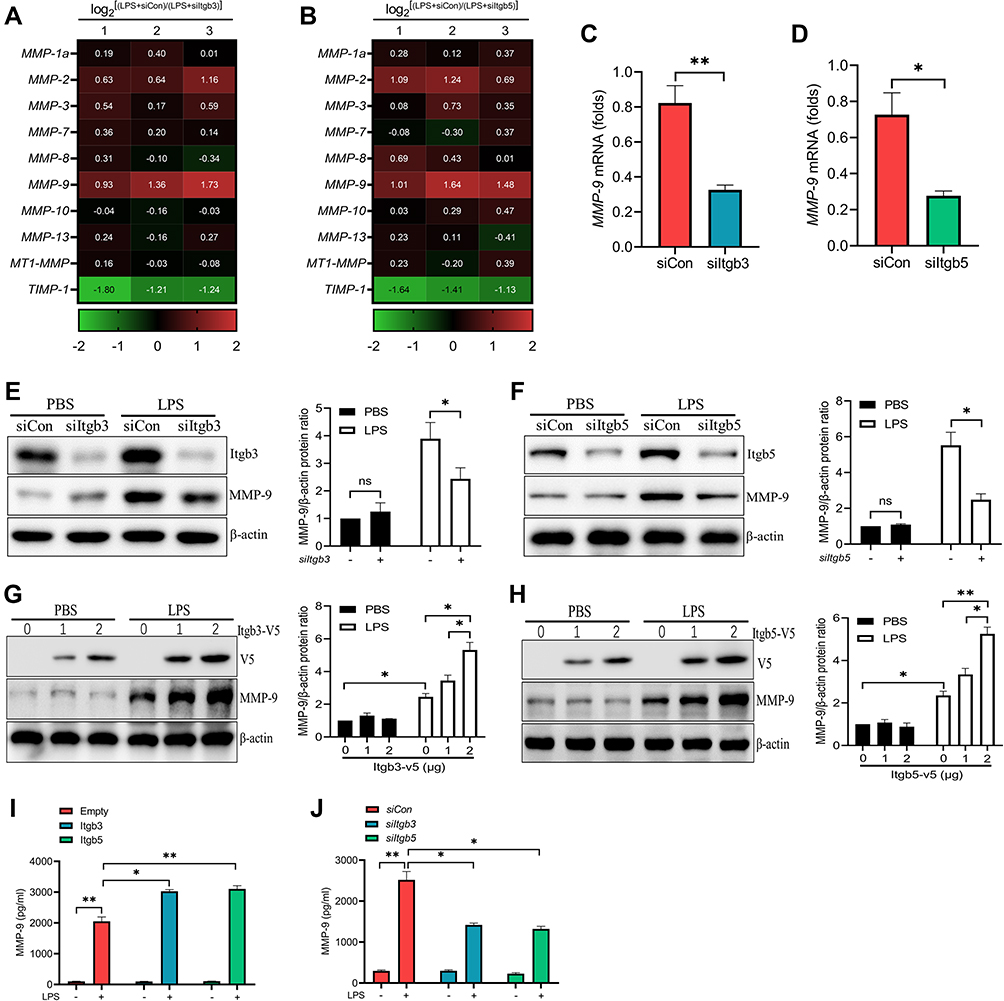 |
Figure 3 Integrin β3/5 regulates MMP-9 expression in LPS-treated PMVECs. (A and B) Heat map of downregulated (green) and upregulated (red) mRNAs from PMVECs transfected with Itgb3 siRNA (siItgb3) or Itgb5 siRNA (siItgb5) for 48 h, and then subjected to 18 h of LPS (1μg/mL) treatment. Numbers 1, 2, and 3 refer to three independent experimental analyses of log2[(LPS+siCon)/(LPS+siItgb3/5)]. (C and D) MMP-9 gene expressions were measured in PMVECs transfected with siItgb3/5 for 48 h prior to 18 h of LPS (1μg/mL) treatment. (E and F) Western blot analysis of MMP-9 protein levels from PMVECs transfected with control siRNA (siCon) or siItgb3/5 for 48 h prior to 18 h of PBS or LPS (1μg/mL) treatment. (G and H) Western blot analysis of MMP-9 protein levels from PMVECs transfected with Itgb3/5 plasmid in a dose course and then exposed to LPS (1μg/mL). ELISA analysis of MMP-9 secretion from PMVECs transfected with Itgb3/5 plasmid (I) or siItgb3/5 (J) and then exposed to PBS or LPS (1μg/mL). *P<0.05, **P<0.01. All the results are from at least three independent experiments; Data represent means ±SEM. |
Integrin β3 Mitigates Permeability and Protects Survival in vitro and in vivo
Integrin β3 has been verified in modulating the NF-κB signaling pathway.26 We first detected whether integrin β3 would regulate NF-κB signaling in LPS/MMP-9 co-treated PMVECs. Over-expressed integrin β3 can give rise to a significant increase in phospho-NF-κB proteins, including p-IKK, p-P65, and p-IκBα (Figure 4A), whereas integrin β3 knockdown significantly decreased phospho-NF-κB proteins (Figure 4B) in PMVECs caused by LPS and rMMP-9 co-treatment at different times. We also observed increased IL-6 and CXCL-1 in the supernatant of LPS/MMP-9 co-treated PMVECs compared to LPS treated alone at 18h (Figure 4C and E). PMVECs were transfected with siItgb3 and we found IL-6 and CXCL-1 secretions were drastically decreased in LPS/MMP-9 co-treatment compared to LPS treated alone (Figure 4D and F). The permeability assay revealed that over-expressed itgb3 did not affect permeation improvement (Figure 4G), whereas itgb3 knockdown enhances the rhodamine penetration (Figure 4H).
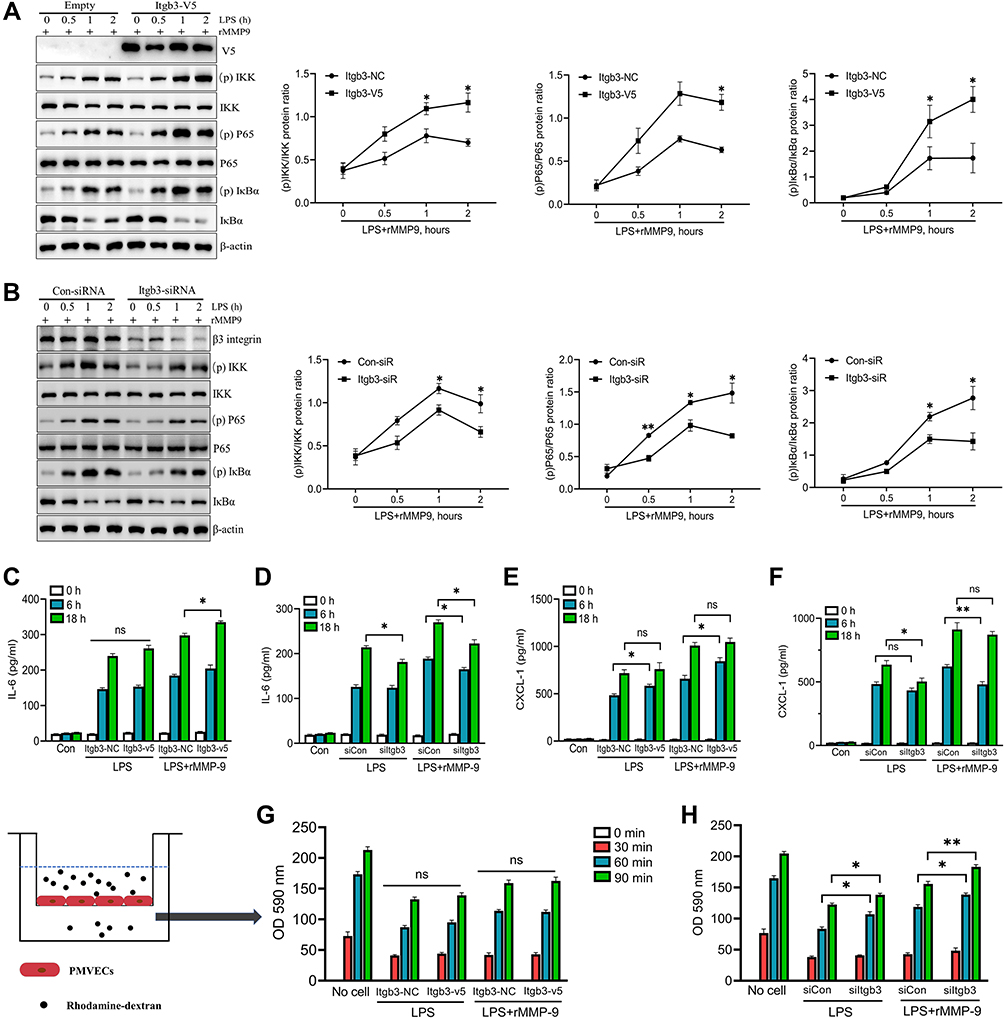 |
Figure 4 Integrin β3 regulates inflammatory response and permeability in vitro. PMVECs were transfected with empty or Itgb3 plasmid for 48 h, and then cells were exposed to rMMP-9 (10 ng/mL) for 1h, followed by treatment with LPS (1μg/mL) at indicated times. (A) NF-κB signaling proteins (p-IKK, IKKβ, p-P65, P65, IκBα, and p-IκBα) were measured by Western blot. Supernatant IL-6 (C) and CXCL-1 (E) secretions were measured by ELISA. (G) The permeability of treated PMVEC monolayers grown on 0.4 mm filters was determined by the appearance of rhodamine-dextran. PMVECs were transfected with control siRNA, or Itgb3 siRNA for 48 h, and then cells were exposed to rMMP-9 for 1h, following by treatment with LPS at indicated times. (B) NF-κB signaling proteins were measured by Western blot. Supernatant IL-6 (D) and CXCL-1 (F) secretions were measured by ELISA. (H) The permeability of treated PMVECs was determined by the appearance of rhodamine-dextran. *P<0.05, **P<0.01. All the results are from at least three independent experiments; Data represent means ±SEM. |
Next, we infected mice intravenously with empty lentivirus or lentivirus encoding Itgb3 for 5 days and subsequently challenged them with LPS (2mg/kg, i.t.) with or without rMMP-9 (50 μg/kg, i.v.). Ectopic expression of Itgb3 in the lungs was confirmed by immunochemistry staining (Figure S3A). Itgb3 gene transfer significantly aggravated lung inflammation caused by LPS and rMMP-9 co-treatment as shown by increased BALF cell counts, neutrophils, and cytokine release (Figure S3B–D). However, there was no significant difference in the lung EBA permeability and BALF protein concentrations (Figure S3E and F). To determine the role of anti-integrin β3 in LPS/MMP-9-induced lung injury, we injected lentivirus intravenously encoding control shRNA or Itgb3 shRNA and then suffered from LPS with or without rMMP-9. Itgb3 knockdown significantly alleviated lung inflammation, but enhanced lung permeability as shown by decreased BALF cell counts, neutrophils, cytokine release (Figure S3G–I), and increased EBA uptake and BALF protein concentrations (Figure S3J and K). The silencing efficiency of Itgb3 in lungs was detected by Western blot (Figure S3L). We also observed the survival in mice intravenously with lentivirus encoding Itgb3 shRNA, and subsequently challenged them with LPS (2mg/kg, i.t.) with or without rMMP-9 (50 μg/kg, i.v.). Itgb3 knockdown drastically decreased the survival in LPS/MMP-9 co-treatment group compared to LPS treated group (Figure S3M). Taken together, these results suggest that inhibition of integrin β3 could mitigate lung inflammation, but have the unwanted side effect of increasing lung endothelial permeability and lead to high mortality.
β5 Integrin/MMP-9 Axis Modulates Lung Injury in vitro and in vivo
We and others have uncovered that integrin β5 contributes to NF-κB-dependent pathological process.15,27 Thus, we explored whether integrin β5 would regulate NF-κB signaling in LPS/MMP-9 treated PMVECs. Over-expressed integrin β5 (itgb5) resulted in a significant increase in phospho-NF-κB proteins, including p-IKK, p-P65, and p-IκBα (Figure 5A), whereas integrin β5 knockdown significantly decreased phospho-NF-κB proteins (Figure 5B) in PMVECs caused by LPS and rMMP-9 co-treatment at different times. We also found itgb5 overexpression dramatically increased NF-κB promoter activity (Figure 5C) and IL-6 secretion (Figure 5D). Conversely, itgb5 knockdown significantly reduced NF-κB promoter activity (Figure 5E) and IL-6 production (Figure 5F) in LPS or LPS/MMP9 co-treatment models. Because vascular endothelial cell injury is associated with reactive oxygen species (ROS) production, we next investigated whether integrin β5 modulates ROS levels. We showed that LPS significantly enhanced the ROS production regardless of integrin β5 expression in LPS-treated PMVECs (Figure 5G). Notably, the ROS scavenger was able to inhibit the interaction of MMP-9 and integrin β5 (Figure 5H) and alleviate the secretion of the pro-inflammatory cytokine (Figure S4A and B) and endothelial leakage (Figure S4C) in PMVECs subjected to co-treatment (LPS with rMMP-9). Moreover, itgb5 overexpression significantly increased the permeability in PMVECs as shown by enhanced rhodamine penetration (Figure 5I), and Itgb5 knockdown exhibited the opposite phenotype (Figure 5J).
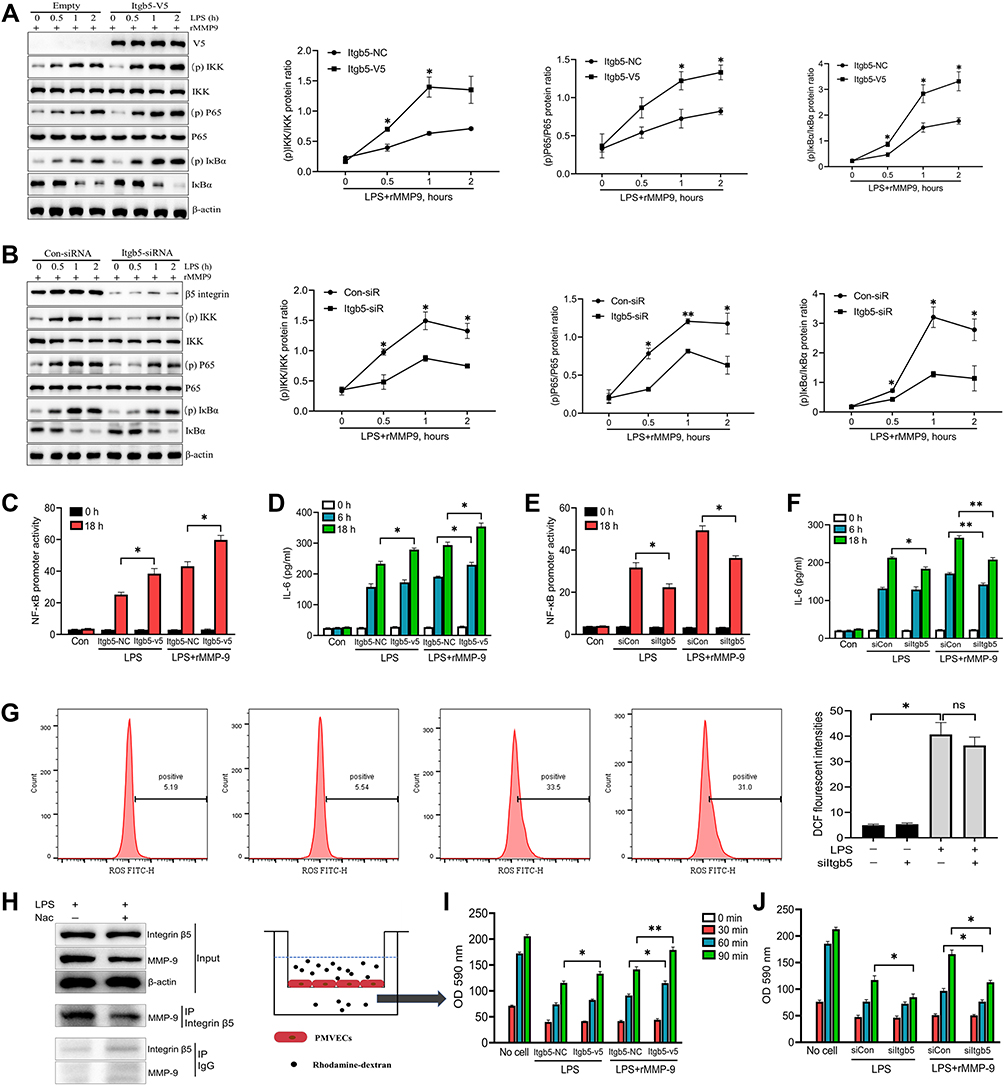 |
Figure 5 β5 Integrin/MMP-9 axis modulates inflammatory response and permeability in vitro. PMVECs were transfected with empty or Itgb5 plasmid, or control siRNA or Itgb5 siRNA for 48 h, and then cells were exposed to rMMP-9 (10 ng/mL) for 1h, followed by treatment with LPS (1 μg/mL) in a time course. (A and B) Immunoblot analysis of NF-κB signaling protein abundances (p-IKK, IKKβ, p-P65, P65, IκBα, and p-IκBα) was measured. (D and F) Supernatant IL-6 secretion was measured by ELISA. (I and J) The permeability of treated PMVECs was determined by the appearance of rhodamine-dextran. (C and E) 293T cells were co-transfected with NF-kB dual luciferase reporter plasmids along with empty or Itgb5 plasmid, or control siRNA or Itgb5 siRNA for 48 h, and then cells were treated with LPS (1 μg/mL) for an additional 6 h or 18 h. Cells were then collected and assayed for luciferase activity to evaluate NF-kB promoter activity. (G) PMVECs were transfected with control siRNA or Itgb5 siRNA for 48 h, and then cells were exposed to LPS for 18 h. The intercellular ROS levels were detected by flow cytometry. (H) PMVECs were treated with or without ROS scavenger, Nac (10 mM) for 1 h, prior to LPS (1 μg/mL) administration for 18 h, and immunoblot analysis of PMVECs and immunoprecipitation (IP) of Integrin β5. *P<0.05, **P<0.01. All the results are from at least three independent experiments; Data represent means ±SEM. |
We next sought to investigate the role of β5 integrin/MMP-9 axis during acute lung injury in vivo. Integrin β5 and MMP-9 protein amounts were detected in lung homogenates after intratracheal LPS and found that both of them were increased compared to control mice (Figure 6A). Further, we measured Itgb5 gene expression via RT-qPCR from BALF cells of ALI mice, and found a significant correlation to MMP-9 secretion from the same BALF (Figure 6B). We also detected enhanced MMP-9 in the BALF of LPS-treated mice compared to controls (Figure S5A). To explore the role of β5 integrin in LPS/MMP-9 induced lung injury, we infected mice intravenously with empty lentivirus or lentivirus encoding Itgb5 for 5 days prior to intratracheal LPS with or without intravenous rMMP-9 challenge. Itgb5 gene transfer significantly aggravated lung inflammation and permeability caused by LPS/MMP-9 co-treatment as shown by increased EBA uptake, BALF protein concentrations, cell counts, neutrophils, lung histology (Figure 6C–F, K), and cytokines release (Figure S5B and C). Additionally, we administered lentivirus intravenously encoding control shRNA or itgb5 shRNA and then exposed to LPS with or without MMP-9. Itgb5 knockdown significantly alleviated lung inflammation and injury caused by LPS and rMMP-9 co-treatment as shown by decreased EBA uptake, BALF protein concentrations, cell counts, neutrophils, (Figure 6G–J), and cytokines release (Figure S5D and E). Intriguingly, histological analysis showed that Itgb5 knockdown had no significant mitigating effect on the lung damage induced by LPS, but combined with anti-MMP9 treatment dramatically decreased the degree of lung injury (Figure S5F). Meanwhile, the survival rate revealed that Itgb5 knockdown combined with anti-MMP9 treatment significantly increased the survival in a lethal dose of LPS (20mg/kg) induced ALI mice, but not Itgb5 knockdown treated alone (Figure 6L). Ectopic expression of Itgb5 in the lungs was confirmed by immunochemistry staining (Figure S5G). The silencing efficiency of Itgb5 in the lungs was detected by Western blot (Figure S5H). These results suggest that the β5 integrin/MMP-9 axis modulates acute lung injury in vivo.
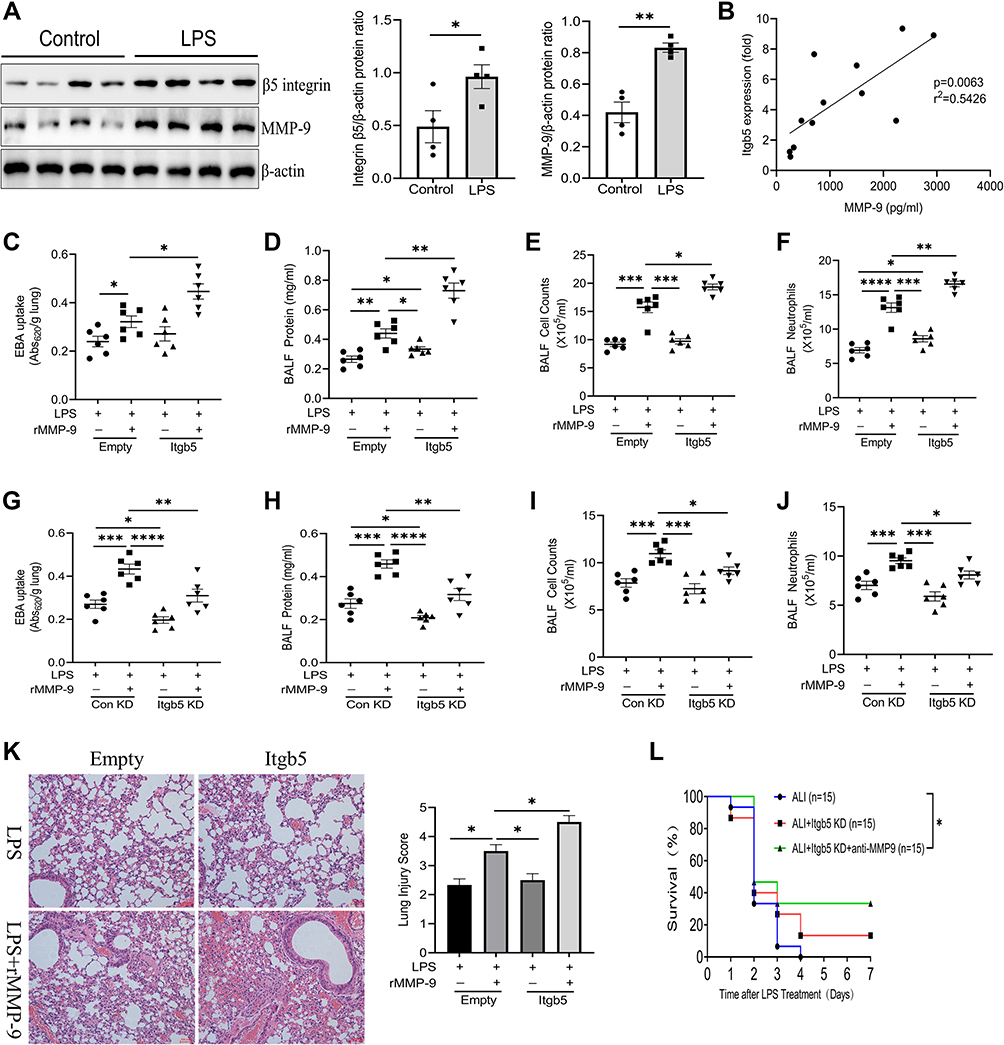 |
Figure 6 β5 Integrin/MMP-9 axis regulates acute lung injury in vivo. (A) Immunoblot analysis of β5 Integrin and MMP-9 from mice intratracheally treated with control or LPS for 18 h. Data and means ± SEM of 4 mice per group. (B) Linear regression of Itgb5 gene expression normalized to control mice vs BALF MMP-9 in ALI subjects (n=12). (C) EBA uptake, (D) protein concentrations, (E) total cell counts, and (F) neutrophils from BAL fluid of mice intratracheally treated with Lenti-Empty or Lenti-Itgb5 and then treated with LPS and PBS or recombinant MMP-9, as indicated. Data and means ± SEM pooled of 3 mice per group are from 2 independent experiments. (G) EBA uptake, (H) protein concentrations, (I) total cell counts, and (J) neutrophils from BAL fluid of mice intratracheally treated with Lenti-control shRNA or Lenti-Itgb5 shRNA and then treated with LPS and PBS or recombinant MMP-9, as indicated. Data and means ± SEM pooled of 3 mice per group are from 2 independent experiments. (K) Histological analysis of lung samples from mice treated as indicated. Images are representative of all independent experiments. (L) Survival rates of mice treated as indicated. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
Discussion
Matrix metalloproteinases (MMPs) are a family of proteolytic enzymes, including gelatinases, collagenases, stromelysins, matrilysins, and membrane-type MMPs.28 MMP-9 belongs to one of MMPs and has a critical role in the pathogenesis of inflammatory diseases with systemic or local destruction, such as sepsis, rheumatoid arthritis, and lung injury.29–31 Acute lung injury (ALI) is characterized by lesions of both lung endothelial and alveolar epithelial cells and finally leads to acute hypoxic respiratory insufficiency. Studies have reported the correlation between alveolar epithelial cells and MMP-9.32,33 Nonetheless, the role of MMP-9 is somewhat unclear in lung endothelial cells. MMP-9 can be secreted by many cell types, including neutrophils, macrophages, lung epithelial cells, and fibroblasts. Here, we showed pulmonary endothelial cells (ECs) also release MMP-9 in the setting of LPS stimulation. The activation of MMP-9 expression in ECs is concomitant with increases in pro-inflammatory cytokine production. This finding is in line with our previous study proposing that MMP-9 is released by immune cells and aggravates inflammatory response in macrophages exposed to LPS.10 MMP-9 degrades extracellular matrix (ECM), and leads to subsequent activation of major proangiogenic factors, including vascular endothelial growth factor (VEGF) and intercellular adhesion molecule 1 (ICAM-1).34,35 Excessive VEGF or ICAM-1 expression is associated with vascular endothelial cell injury. In this regard, we noted enhancement of endothelial permeability caused by rMMP-9 in a simple model of LPS-treated PMVECs, whereas rMMP-9 itself did not affect the permeability. Studies have found MMP-9 levels can be regulated by transcription factor NF-κB in vascular smooth muscle cells and macrophages.36,37 We found PDTC, as a NF-κB inhibitor, drastically reduced MMP-9 levels and abrogated the effect of rMMP-9 on promoting pro-inflammatory cytokine production and permeability in LPS-treated PMVECs.
MMP-9, as an important regulator in lung injury, has been reported. Nonetheless, its certain role in ALI is not well established. Albaiceta et al and Yoon et al have indicated that MMP-9 has a protective effect on lung injury caused by ventilator or ozone.38,39 In contrast, Warner et al and Kim et al have found MMP-9 acts as a negative regulator in the lung by which blocking MMP-9 can against lung injury.40,41 In this study, we found endogenous MMP-9 aggravates lung injury induced by LPS, and anti-MMP-9 inhibits excessive inflammatory response in ALI mice. Relevant to these findings, many other studies have demonstrated that MMP-9 may be injurious in ALI caused by cardiopulmonary bypass, pancreatitis, and endotoxin.42–44 However, despite the protective effect of MMP-9 deletion on lung inflammatory injury, we did not see any benefit in regards to the lung edema from ALI mice. This may be a reflection of the fact that there was no significant improvement in the survival rate of ALI mice treated with MMP-9 neutralizing antibody in this model. MMP-9 expression can be regulated by transcription factors, such as NF-κB and AP-1.37,45 Besides, studies have implicated the importance of integrins in regulating the expression of MMP-9.46,47 By using a model of LPS-treated PMVECs, we demonstrated that blocking integrin β3 or integrin β5 could reduce MMP-9 expression at the gene and protein levels.
Integrins are a family of transmembrane, heterodimeric receptors that act a pivotal part in cell adhesion to the ECM and initiate “outside-in” signal transduction events that regulate cell functions, including gene expression, cell migration, and invasion.48,49 Redondo-Muñoz et al verified α4β1 integrin as a novel MMP-9 cell surface docking complex in chronic leukemic.50 In the current study, we found extracellular MMP-9 also could bind to β5 integrin receptor, and knockdown of β5 integrin ameliorates the role of rMMP-9 in promoting inflammatory cytokine production and permeability in LPS-treated PMVECs. These findings could explain exogenous MMP9 aggravates the inflammatory response and permeability partially through β5 integrin in PMVECs exposed to LPS. Integrin β5 is a closely related receptor to ECM components by showing its interaction with MMP-9 in vitro. Additionally, we further found this interaction was dampened by a ROS scavenger, which also drastically decreased the pro-inflammatory cytokines secretion and endothelial permeability in PMVECs with co-treatment (LPS with rMMP-9) in this study. LPS induced excessive ROS production independent of β5 integrin, whereas the excessive ROS promoted the interaction between β5 integrin with MMP-9, suggesting the ROS seems to affect the role of β5 integrin/MMP-9 axis in acute lung injury.
Studies have documented several integrins involve in the process of acute lung injury by using either specific blocking antibodies or gene knockout mice, and identified critical roles for two integrins (αvβ3 and αvβ5) in regulating alveolar endothelial permeability.25,51 Nonetheless, integrin β3 appears to be a protective regulator in the setting of lung injury, as we found blocking integrin β3 aggravates lung edema and even decreased the survival in mice subjected to LPS with or without rMMP-9. Our findings are in line with the studies that noted the agonistic effects of β3-blocking on induced permeability in vitro and lung edema in vivo,25,52 but the certain mechanism is not distinct and will need further clarification. Endothelial cells widely express the integrin αvβ3, which has overlapping ligand specificity with αvβ5. Su et al have confirmed that targeting integrin β5 can potently inhibit lung vascular permeability and pulmonary endothelial barrier during acute lung injury.51 We reported that the increase in permeability was inhibited by transfection with integrin β5 siRNA in PMVECs exposed to LPS with or without rMMP-9. Our study showed the integrin β3 and β5 have completely opposite roles in cell leakage. We wonder whether they mediate different and antagonistic effects on the actin stress fibers and cortical actin, which are regarded as critical mediators in endothelial permeability. Further functional study will be conducted to prove the reciprocal phenotype.
We next characterized integrin β5 as an injurious modulator in vivo by showing its gene transfer or knockdown also has a similar phenotype in cytokine release and albumin extravasation into the lung from ALI mice treated with or without exogenous MMP-9. Intriguingly, the survival of ALI mice was significantly increased by combined blocking integrin β5 and MMP-9, whereas blocking integrin β5, by itself, did not show any dramatic improvement in the survival of mice suffered from LPS. Based on our findings, combination therapy with drugs or biomaterials that specifically targeting integrin β5 and MMP-9 could be a potential method to antagonize ALI.
Collectively, our data imply that MMP-9 functions as an inflammatory accelerator protein that promotes lung innate immune response and albumin extravasation during acute lung injury (ALI). Inhibition of the β3 integrin on endothelial cells can potently aggravate ALI, whereas β5 integrin has the opposite effect of decreasing susceptibility to ALI. Besides, we found that β3/5 integrin negatively mediated the expression of MMP-9, which could interact with β5 integrin involves in the regulation of endothelial function. This study demonstrated a novel insight into the mechanism by which the β3/5 Integrin-MMP-9 axis plays a critical role in lung inflammatory response and permeability, and might be a potential therapeutic target in ALI (Figure 7).
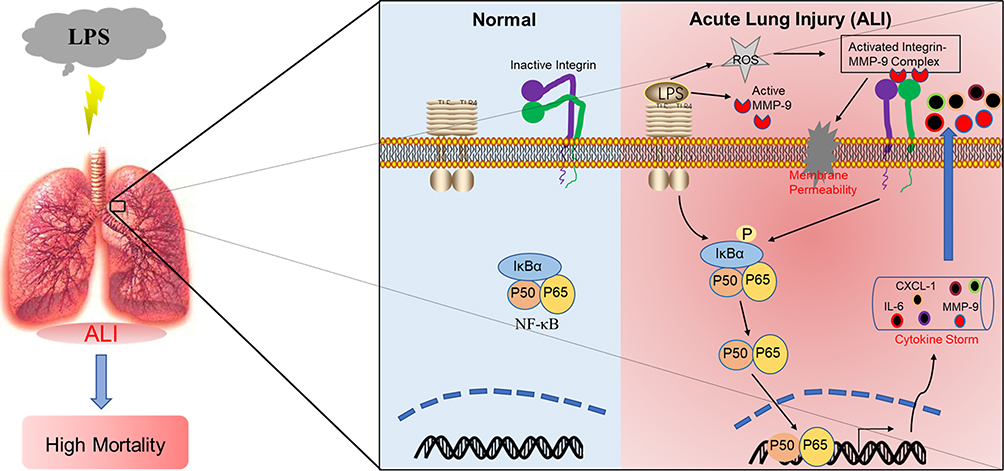 |
Figure 7 Scheme showing that β5 integrin/MMP-9 axis-mediated acute lung injury by promoting pulmonary inflammatory disorders and endothelial permeability. |
Funding
This study was supported by the National Natural Science Foundation of China (No. 81871101 to Luo Y, No. 81771138 to Lu H, No. 81900073 to Tong Y), and Shanghai Sailing Program (No.19YF1430700 to Tong Y).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bernard GR. Acute respiratory distress syndrome: a historical perspective. Am J Respir Crit Care Med. 2005;172:798–806. doi:10.1164/rccm.200504-663OE
2. Huang LS, Hong Z, Wu W, et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity. 2020;52:475–86.e5. doi:10.1016/j.immuni.2020.02.002
3. Cen M, Ouyang W, Zhang W, et al. MitoQ protects against hyperpermeability of endothelium barrier in acute lung injury via a Nrf2-dependent mechanism. Redox Biol. 2021;41:101936. doi:10.1016/j.redox.2021.101936
4. Li L, Wei J, Mallampalli RK, et al. TRIM21 mitigates human lung microvascular endothelial cells’ inflammatory responses to LPS. Am J Respir Cell Mol Biol. 2019;61:776–785. doi:10.1165/rcmb.2018-0366OC
5. Bhattacharya J, Matthay MA. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu Rev Physiol. 2013;75:593–615. doi:10.1146/annurev-physiol-030212-183756
6. Nova Z, Skovierova H, Calkovska A. Alveolar-capillary membrane-related pulmonary cells as a target in endotoxin-induced acute lung injury. Int J Mol Sci. 2019;20:831. doi:10.3390/ijms20040831
7. Tong Y, Lear TB, Evankovich J, et al. The RNFT2/IL-3Rα axis regulates IL-3 signaling and innate immunity. JCI Insight. 2020;5. doi:10.1172/jci.insight.133652
8. Abdulmahdi W, Patel D, Rabadi MM, et al. HMGB1 redox during sepsis. Redox Biol. 2017;13:600–607. doi:10.1016/j.redox.2017.08.001
9. Ley K, Laudanna C, Cybulsky MI, et al. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi:10.1038/nri2156
10. Tong Y, Yu Z, Chen Z, et al. The HIV protease inhibitor Saquinavir attenuates sepsis-induced acute lung injury and promotes M2 macrophage polarization via targeting matrix metalloproteinase-9. Cell Death Dis. 2021;12:67. doi:10.1038/s41419-020-03320-0
11. De Franceschi N, Hamidi H, Alanko J, et al. Integrin traffic - the update. J Cell Sci. 2015;128:839–852. doi:10.1242/jcs.161653
12. Perera PY, Mayadas TN, Takeuchi O, et al. CD11b/CD18 acts in concert with CD14 and Toll- like receptor (TLR) 4 to elicit full lipopolysaccharide and taxol-inducible gene expression. J Immunol. 2001;166:574–581. doi:10.4049/jimmunol.166.1.574
13. Sheppard D. Roles of alphav integrins in vascular biology and pulmonary pathology. Curr Opin Cell Biol. 2004;16:552–557. doi:10.1016/j.ceb.2004.06.017
14. Ganter MT, Roux J, Miyazawa B, et al. Interleukin-1beta causes acute lung injury via alphavbeta5 and alphavbeta6 integrin-dependent mechanisms. Circ Res. 2008;102:804–812. doi:10.1161/CIRCRESAHA.107.161067
15. Ding X, Tong Y, Jin S, et al. Mechanical ventilation enhances extrapulmonary sepsis-induced lung injury: role of WISP1-αvβ5 integrin pathway in TLR4-mediated inflammation and injury. Crit Care. 2018;22:302. doi:10.1186/s13054-018-2237-0
16. Rohani MG, Parks WC. Matrix remodeling by MMPs during wound repair. Matrix Biol. 2015;44–46:113–121. doi:10.1016/j.matbio.2015.03.002
17. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi:10.1016/j.cell.2010.03.015
18. Davey A, McAuley DF, O’Kane CM. Matrix metalloproteinases in acute lung injury: mediators of injury and drivers of repair. Eur Respir J. 2011;38:959–970. doi:10.1183/09031936.00032111
19. Vandooren J, Van den Steen PE, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit Rev Biochem Mol Biol. 2013;48:222–272. doi:10.3109/10409238.2013.770819
20. Adya R, Tan BK, Punn A, et al. Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: novel insights into visfatin-induced angiogenesis. Cardiovasc Res. 2008;78:356–365. doi:10.1093/cvr/cvm111
21. Chang CC, Tien CH, Lee EJ, et al. Melatonin inhibits matrix metalloproteinase-9 (MMP-9) activation in the lipopolysaccharide (LPS)-stimulated RAW 264.7 and BV2 cells and a mouse model of meningitis. J Pineal Res. 2012;53:188–197. doi:10.1111/j.1600-079X.2012.00986.x
22. Opdenakker G, Van den Steen PE, Dubois B, et al. Gelatinase B functions as regulator and effector in leukocyte biology. J Leukoc Biol. 2001;69:851–859. doi:10.1189/jlb.69.6.851
23. Zhou W, Fong MY, Min Y, et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell. 2014;25:501–515. doi:10.1016/j.ccr.2014.03.007
24. D’Alessio FR, Tsushima K, Aggarwal NR, et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest. 2009;119:2898–2913. doi:10.1172/JCI36498
25. Sheppard D. Modulation of acute lung injury by integrins. Proc Am Thorac Soc. 2012;9:126–129. doi:10.1513/pats.201112-052AW
26. Hong SK, Lee H, Kwon OS, et al. Large-scale pharmacogenomics based drug discovery for ITGB3 dependent chemoresistance in mesenchymal lung cancer. Mol Cancer. 2018;17:175. doi:10.1186/s12943-018-0924-8
27. Ballana E, Pauls E, Clotet B, et al. β 5 integrin is the major contributor to the α v integrin-mediated blockade of HIV-1 replication. J Immunol. 2011;186:464–470. doi:10.4049/jimmunol.1002693
28. Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–573. doi:10.1016/j.cardiores.2005.12.002
29. Gäddnäs FP, Sutinen MM, Koskela M, et al. Matrix-metalloproteinase-2, −8 and −9 in serum and skin blister fluid in patients with severe sepsis. Crit Care. 2010;14:R49. doi:10.1186/cc8938
30. Shi Y, Wu Q, Xuan W, et al. Transcription factor SOX5 promotes the migration and invasion of fibroblast-like synoviocytes in part by regulating MMP-9 expression in collagen-induced arthritis. Front Immunol. 2018;9:749. doi:10.3389/fimmu.2018.00749
31. Villalta PC, Rocic P, Townsley MI. Role of MMP2 and MMP9 in TRPV4-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;307:L652–9. doi:10.1152/ajplung.00113.2014
32. Buisson AC, Zahm JM, Polette M, et al. Gelatinase B is involved in the in vitro wound repair of human respiratory epithelium. J Cell Physiol. 1996;166:413–426. doi:10.1002/(SICI)1097-4652(199602)166:2<413::AID-JCP20>3.0.CO;2-A
33. Shyamsundar M, McAuley DF, Ingram RJ, et al. Keratinocyte growth factor promotes epithelial survival and resolution in a human model of lung injury. Am J Respir Crit Care Med. 2014;189:1520–1529. doi:10.1164/rccm.201310-1892OC
34. Bekes EM, Schweighofer B, Kupriyanova TA, et al. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol. 2011;179:1455–1470. doi:10.1016/j.ajpath.2011.05.031
35. Fiore E, Fusco C, Romero P, et al. Matrix metalloproteinase 9 (MMP-9/gelatinase B) proteolytically cleaves ICAM-1 and participates in tumor cell resistance to natural killer cell-mediated cytotoxicity. Oncogene. 2002;21:5213–5223. doi:10.1038/sj.onc.1205684
36. Bond M, Chase AJ, Baker AH, et al. Inhibition of transcription factor NF-kappaB reduces matrix metalloproteinase-1, −3 and −9 production by vascular smooth muscle cells. Cardiovasc Res. 2001;50:556–565. doi:10.1016/S0008-6363(01)00220-6
37. Lim EJ, Lee SH, Lee JG, et al. Toll-like receptor 9 dependent activation of MAPK and NF-kB is required for the CpG ODN-induced matrix metalloproteinase-9 expression. Exp Mol Med. 2007;39:239–245. doi:10.1038/emm.2007.27
38. Albaiceta GM, Gutiérrez-Fernández A, Parra D, et al. Lack of matrix metalloproteinase-9 worsens ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;294:L535–L543. doi:10.1152/ajplung.00334.2007
39. Yoon HK, Cho HY, Kleeberger SR. Protective role of matrix metalloproteinase-9 in ozone-induced airway inflammation. Environ Health Perspect. 2007;115:1557–1563. doi:10.1289/ehp.10289
40. Warner RL, Beltran L, Younkin EM, et al. Role of stromelysin 1 and gelatinase B in experimental acute lung injury. Am J Respir Cell Mol Biol. 2001;24:537–544. doi:10.1165/ajrcmb.24.5.4160
41. Kim JH, Suk MH, Yoon DW, et al. Inhibition of matrix metalloproteinase-9 prevents neutrophilic inflammation in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;291:L580–L587. doi:10.1152/ajplung.00270.2005
42. Wang C, Li D, Qian Y, et al. Increased matrix metalloproteinase-9 activity and mRNA expression in lung injury following cardiopulmonary bypass. Lab Invest. 2012;92:910–916. doi:10.1038/labinvest.2012.50
43. Yang D, Song Y, Wang X, et al. Deletion of peroxiredoxin 6 potentiates lipopolysaccharide-induced acute lung injury in mice. Crit Care Med. 2011;39:756–764. doi:10.1097/CCM.0b013e318206befd
44. Keck T, Balcom JH, Fernández-del Castillo C, et al. Matrix metalloproteinase-9 promotes neutrophil migration and alveolar capillary leakage in pancreatitis-associated lung injury in the rat. Gastroenterology. 2002;122:188–201. doi:10.1053/gast.2002.30348
45. Moon SK, Cha BY, Kim CH. ERK1/2 mediates TNF-alpha-induced matrix metalloproteinase- 9 expression in human vascular smooth muscle cells via the regulation of NF-kappaB and AP-1: involvement of the ras dependent pathway. J Cell Physiol. 2004;198:417–427. doi:10.1002/jcp.10435
46. Iyer V, Pumiglia K, DiPersio CM. Alpha3beta1 integrin regulates MMP-9 mRNA stability in immortalized keratinocytes: a novel mechanism of integrin-mediated MMP gene expression. J Cell Sci. 2005;118:1185–1195. doi:10.1242/jcs.01708
47. Redondo-Muñoz J, Escobar-Díaz E, Samaniego R, et al. MMP-9 in B-cell chronic lymphocytic leukemia is up-regulated by alpha4beta1 integrin or CXCR4 engagement via distinct signaling pathways, localizes to podosomes, and is involved in cell invasion and migration. Blood. 2006;108:3143–3151. doi:10.1182/blood-2006-03-007294
48. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi:10.1016/S0092-8674(02)00971-6
49. Monick MM, Powers L, Butler N, et al. Interaction of matrix with integrin receptors is required for optimal LPS-induced MAP kinase activation. Am J Physiol Lung Cell Mol Physiol. 2002;283:L390–402. doi:10.1152/ajplung.00437.2001
50. Redondo-Muñoz J, Ugarte-Berzal E, García-Marco JA, et al. Alpha4beta1 integrin and 190- kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood. 2008;112:169–178. doi:10.1182/blood-2007-08-109249
51. Su G, Hodnett M, Wu N, et al. Integrin alphavbeta5 regulates lung vascular permeability and pulmonary endothelial barrier function. Am J Respir Cell Mol Biol. 2007;36:377–386. doi:10.1165/rcmb.2006-0238OC
52. Su G, Atakilit A, Li JT, et al. Absence of integrin αvβ3 enhances vascular leak in mice by inhibiting endothelial cortical actin formation. Am J Respir Crit Care Med. 2012;185:58–66. doi:10.1164/rccm.201108-1381OC
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.