")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Thapsigargin induces apoptosis in adrenocortical carcinoma by activating endoplasmic reticulum stress and the JNK signaling pathway: an in vitro and in vivo study
Authors Wu L, Huang X, Kuang Y , Xing Z , Deng X, Luo Z
Received 25 March 2019
Accepted for publication 25 July 2019
Published 12 August 2019 Volume 2019:13 Pages 2787—2798
DOI https://doi.org/10.2147/DDDT.S209947
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Lili Wu,1 Xuemei Huang,2 Yaqi Kuang,2 Zengmiao Xing,2 Xiujun Deng,2 Zuojie Luo2
1Department of Integrated Medicine, The Affiliated Tumor Hospital of Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China; 2Department of Endocrinology, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China
Objective: Thapsigargin (TG) is a natural product that exists in most parts of the plant Thapsia garganica L. and possesses potential anticancer activities against variety tumor cell lines. TG induces endoplasmic reticulum (ER) stress and apoptosis by inhibiting cancer growth. However, the antineoplastic effect of TG in human adrenocortical carcinoma (ACC) cells is still unknown.
Methods: In this study, two human ACC cell lines including SW-13 and NCI-H295R were employed to explore the potential role of TG in ACC. A mouse xenograft model of SW-13 cells was established to verify the role of TG in vivo. The cell viability was tested using Cell Counting Kit-8 and Transwell assays. Flow cytometry and Hoechst 33,258 staining were employed to analyze cell apoptosis. RT-qPCR and Western blot (WB) were performed to explore the underlying mechanism of TG-induced apoptosis in ACC cells.
Results: The results indicated that TG dose-dependently inhibited proliferation, migration and invasion in human ACC cells. TG significantly increased the mitochondrial rate of apoptosis and ER stress activity in ACC cells and suppressed ACC xenograft growth in vivo. In addition, the expression of Jun N-terminal kinase (JNK) signaling-related genes and proteins was upregulated by the treatment with TG.
Conclusion: Our findings suggest that TG inhibits the viability of ACC cells by inducing apoptosis through the activation of JNK signaling. Thus, TG is expected to be a potential candidate for the treatment of ACC.
Keywords: thapsigargin, adrenocortical carcinoma, apoptosis, Jun N-terminal kinase signaling, endoplasmic reticulum ER stress
Introduction
Adrenocortical carcinoma (ACC) is a rare endocrine tumor with a poor prognosis. The occurrence rate of ACC ranges from approximately 1 to 2 cases per million persons per year.1 Among early stage cases, after complete surgical resection, the 5-year survival rate of ACC patients is 32% to 50%, but the risk of recurrence after surgery is as high as 50% to 85%.2–4 Late stage ACC patients who present with metastatic disease have a 5-year survival rate of 15%5 and a median survival of less than 1 year.6 The most commonly associated endocrine disorder is Cushing’s syndrome;7 however, other syndromes, including Lynch syndrome, multiple endocrine neoplasia type 1, Beckwith-Wiedemann syndrome, and other tumor-related syndromes, are also related to ACC.1 Mitotane (o, p’-dichlorodiphenyldichloroethane, o, p’-DDD) is the most commonly used drug; nevertheless, its efficacy is limited by its narrow therapeutic index and toxicity.8 Other systemic therapies, such as radiotherapy and chemotherapy, are also restricted by their limited efficacy.9 Therefore, new therapeutic approaches for ACC are urgently needed. The SW-13 cell line was extracted from human ACC10 and is currently used in anticancer drug and signaling research experiments. The H295R cell line, which also originated from human ACC, exhibits a secretion function and has been established as an appropriate model for determining the molecular mechanisms regulating ACC.11
Thapsigargin (TG) has recently emerged as an outstanding anticancer lead compound, and prodrug derivatives of TG, such as mipsagargin (G-202), are currently in clinical trials for the treatment of multiple cancers, including hepatocellular carcinoma and prostate cancer, neovascular tissues and a wide range of other cancer cells.12 TG is an irreversible paralyzer of the important Sarco-/Endoplasmic Reticulum Calcium ATPase (SERCA), which blocks the reabsorption of calcium from the cytosol to the Endoplasmic Reticulum (ER).13 This important step leads to an increase in the cytosolic calcium concentration and ultimately results in cell apoptosis.14
The ER, which is an important organelle in cells, participates in the synthesis, folding, trafficking and secretion of a variety of chaperone proteins. However, in the presence of internal environmental disturbance in the body, the calcium ion steady state is destroyed, proteins unfold and misfold, and external stimuli in the ER could result in ER stress (ERS), therefore leading to cell apoptosis.15,16 Serious ERS can ultimately induce cell apoptosis.
To alleviate the progression of cell apoptosis, the unfold-protein reaction (UPR)17 is subsequently activated by stimulating three transcription factors, including inositol requiring 1 (IRE1/ERN1), activating transcription factor 4 (ATF4),18 and PEK, such as ER kinase (PERK/PEK). PERK is a specific protein kinase that controls protein translation and mediates the terminal UPR through the regulation of ATF4, which is a vital element in the promotion of apoptosis in response to ERS and is accompanied by the upregulation of C/EBP homologous protein (CHOP).15,19,20 Jun N-terminal kinase (JNK) is a major stress response factor and an important regulator of cell growth and death in many Drosophila tissues.21–23 JNK is a key stress reaction factor and an important modulator of cell growth and death similar to mahj,24 myc,25 APC 6 and Minute competition26 and has been proposed to play a role in the late stage induction of cell death. Studies have demonstrated that mitogen-activated protein kinases (MAPKs) are crucial regulators of cell migration and transformation in cancer. Within the MAPK family, extracellular signal-regulated kinase (ERK), JNK and p38 kinases are the most widely studied. However, the relationship between JNK signaling and ACC development remains unclear.
In the present study, we explored the effect of TG on ACC in vitro and in vivo. Our findings reveal that TG dose-dependently suppresses proliferation and motility in ACC cell lines in vitro and inhibits tumor growth in vivo. TG activates ERS and apoptosis. Moreover, the JNK signaling pathway is the potential mechanism underlying TG-induced apoptosis, which was confirmed by the fact that ER-related factors significantly decreased apoptosis after TG treatment in vitro and in vivo.
Materials and methods
Cell culture
SW-13 and NCI-H295R human ACC cells were purchased from Shanghai Cell Bank, the Chinese Academy of Sciences (Shanghai, China). The SW-13 Cells were maintained in DMEM 10% FBS (Gibco, USA), 1 mM glutamine, and 100 U/ml penicillin-streptomycin (Solarbio, Beijing, China). The NCI-H295R cells were cultured in DMEM/F12 (Sigma-Aldrich, Japan) supplemented with 5% FBS, 100 U/ml penicillin-streptomycin and 0.1% ITS (BD Biosciences, USA).
Preparation and treatment with TG
TG was purchased from Shanghai Amquar Ltd. (Shanghai, China). A TG solution at a concentration of 5 mM was prepared by dissolving TG in DMSO (Solarbio, Beijing, China) and then stored at −20 °C. The TG stock solution was diluted into different concentrations for further experiments.
Cell morphological analysis
The cell morphology was analyzed by using Crystal Violet Staining Solution (Leagene Biotechnology, China). The images were obtained under an upright microscope (Olympus BX53; Japan).
Cell viability assay
TG was diluted with DMEM to concentrations of 0, 0.5, 1, 2, 4, 8 and 16 µM. After incubation with TG for 48 h, the SW-13 and NCI-H295R cells were treated with 100 µL DMEM and 10 µL Cell Counting Kit-8 (Gibco, USA). The plates were incubated in the dark for 1 h at 37 °C. The optical density (OD) value was evaluated using a SynergyH1 reader (BioTek, USA) at 450 nm.
Migration and invasion assays
The SW13 and NCI-H295R cells were treated with 2, 4 or 8 mM TG. The invasive captivity and motility of the cells were detected using a Transwell assay. The SW13 and NCI-H295R cells were serum starved overnight, and the cells were loaded onto the upper chambers (Corning Incorporated, USA) at a concentration of 2×105 cells per well. Then, 500 µL DMEM were added to the lower chambers. The upper chambers were loaded with Matrigel matrix (Corning Incorporated, USA) before the addition of the cells for the invasion analysis. After culturing for 24 h, the numbers of cells were counted and captured under an inverted microscope (Nikon Corporation, Japan).
Apoptosis assay
The SW-13 and NCI-H295R cells were cultured with different concentrations of TG or JNK inhibitor SP600125 (AbMole, USA) for 48 h, followed by harvesting and suspending in 1× Binding Buffer; the cells were incubated with 5 µL Annexin V-FITC and 5 µL PI (BD Biosciences, USA). The apoptotic SW-13 and NCI-H295R cells were analyzed by BD FACS Verse (BD Biosciences, USA).
The apoptotic cells were also analyzed by Hoechst 33,258 staining. Briefly, 48 h after incubation, the SW-13 and NCI-H295R cells were immobilized with 70% ethanol for 20 min; then, the 70% ethanol was removed, and Hoechst 33,258 (Beyotime, China, 500 µL/well) was added for 10 min. The pictures were captured under a fluorescence microscope (Nikon Corporation, Japan).
Real-time quantitative polymerase chain reaction analysis
The RNA purity was detected after extracting the total RNA from ACC cells treated with TG by RNAiso Plus Reagent (TaKaRa, Shiga, Japan). Oligo dT primer and AMV reverse transcriptase were used to synthesize cDNA. The RT-PCR reactions were performed using a SYBR Green (TaKaRa, Japan) assay on an ABI 7500 (Applied Biosystems, USA). The primers used are shown in Table 1. The fold changes of each gene were calculated using the 2−ΔΔCt method. GAPDH was used as an internal reference.
 |
Table 1 Primers for RT-PCR performance |
Western blot analysis
The cells were cultured with TG at concentrations of 0, 2, 4, or 8 µM for 48 h, and nude mouse xenograft tissues were obtained after treatment for TG for 15 days. The protein was separated by SDS-PAGE (Bio‑Rad, USA) and then transferred onto a PVDF membrane (Millipore, USA), which was incubated with 5% nonfat dry milk-TBST. The membranes were probed with primary antibodies (anti-JNK, anti-p-JNK, anti-ERK, anti-p-ERK, anti-AMPK, anti-p-AMPK, anti-PERK, anti-p-PERK, IRE1 and anti-GRP78, Cell Signaling Technology, USA). Then, the membrane was incubated with a horseradish peroxidase-conjugated secondary antibody (Signalway Antibody, China) for 1 h, followed by visualization using LI-COR (Odyssey, USA) according to the manufacturer’s instructions. An anti-GAPDH antibody (Signalway Antibody, China) was used as an internal control.
Nude mouse xenograft assay
Male BALB/c nude mice were obtained from the Chang Zhou Cavens Laboratory Animal Center. The animal protocol was approved by the Animal Ethics and Welfare Committee of Guangxi Medical University (201901007), and followed the Guiding Opinions on the Treatment of Laboratory Animals issued by the Ministry of Science and Technology of the People’s Republic of China and the Laboratory Animal Guidelines for Ethical Review of Animal Welfare issued by the National Standard GB/T35892-2018 of the People’s Republic of China. Five male nude mice aged 4–5 weeks and weighing 20–22 g were used per group. The mice were injected with SW-13 cells (2×106 in 200 µL of medium) under their left shoulder. Once the subcutaneous tumors were approximately 0.2×0.2 cm2 in size, the mice were intraperitoneally injected with vehicle alone (0.5% methylcellulose) or TG (1 mg/kg) daily. We recorded the body weights and the tumors of the mice. The tumor volume (V) was calculated as follows: V =π/6 (1/2(A + B)).3 The inhibition rate (IR) was calculated as follows: IR =1-mean tumor weight of the experimental group/mean tumor weight of the control group×100%.27
Statistical analysis
Graphs were generated using GraphPad Prism 6.0. The results are expressed as the mean ± SD and were analyzed with SPSS 22.0 software. Student’s t-test and one-way ANOVA were selected to analyze the differences among the groups. p<0.05 was considered statistically significant.
Results
Cell lines identification
The identification for cell lines was analyzed using a crystal violet staining solution. As shown in Figure 1A and B, the typical morphological characteristics of the ACC cell lines SW-13 and NCI-H295R could be clearly observed.
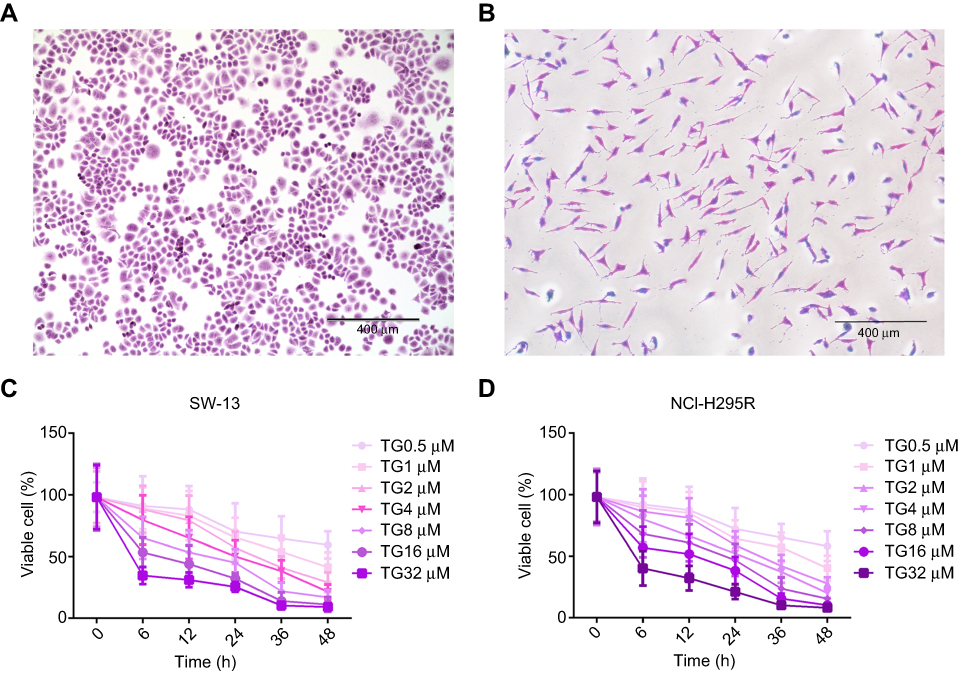 |
Figure 1 TG inhibited viability of ACC cell lines. (A and B) Crystal violet staining of SW-13 and NCI-H295R (×10, scale bar =400 μm). (C and D) SW-13 and NCI-H295R cells were incubated with TG at different concentrations (0, 0.5, 1, 2, 4, 8, 16 and 32 µM) for 0, 6, 12, 24, 36 and 48 h. Each experiment was performed independently in triplicate, and the data is represented as means ± SD. |
TG inhibits the viability of human ACC cells
The CCK-8 results showed that the cell viability was decreased in both SW-13 and NCI-H295R following culture with TG at concentrations higher than 1 µM (Figure 1C and D). The half maximal inhibition concentration of TG in the SW-13 and NCI-H295R cell lines was ~4 µM. These results suggest that TG suppresses the cell viability of ACC cells in vitro.
TG suppresses the migration and invasion ability of ACC cells
After incubation with different concentrations of TG (0, 2, 4, or 8 µM), the SW-13 and NCI-H295R cells were observed. As shown in Figure 2A and B, the migration and invasion abilities were dose-dependently inhibited by TG. The cells in both chambers were counted. TG, particularly at 8 µM, suppressed the motility of the ACC cells (Figure 2C and D).
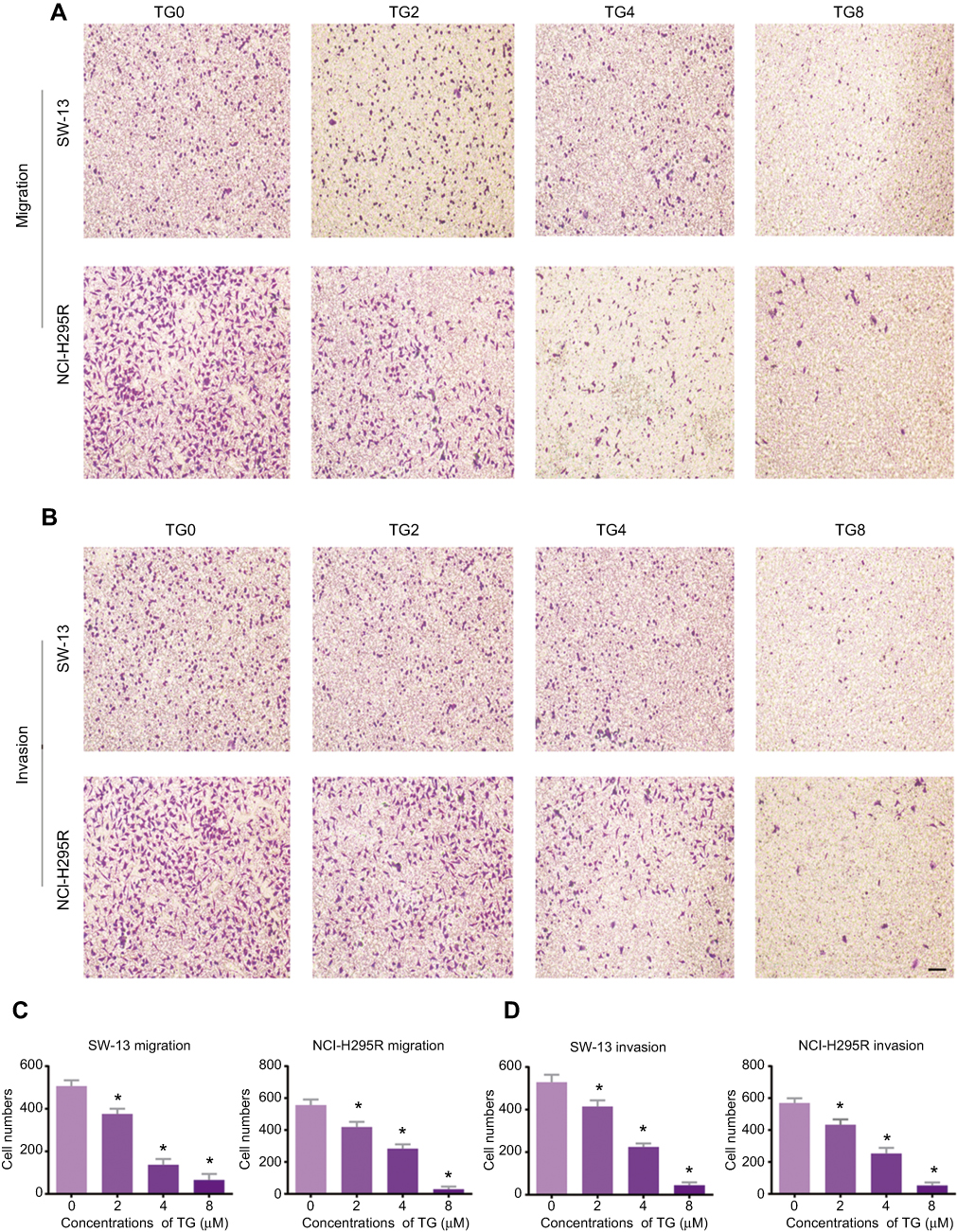 |
Figure 2 TG suppresses the migration and invasion ability of ACC cells. The migration (A) and invasion (B) of ACC cells after treatment with or without TG (×10, scale bar =400 μm). (C and D) The number of migrated and invaded cells were quantified, and the data is represented as means ± SD (n=6). *p<0.05 vs the control group. |
TG induces mitochondrial apoptosis in ACC cells
To determine whether TG inhibits cell viability by leading to cell apoptosis in ACC cell lines, the rate of apoptosis in the TG-treated cells was evaluated by flow cytometry. As shown in Figure 3A–C, after incubation with various concentrations of TG (0, 0.5, 1, 2, 4, 8, 10, or 16 µM) for 48 h, the apoptosis rate of the cells increased as the concentration of TG increased. These results suggest that the treatment with TG resulted in apoptosis in the ACC cells in vitro, which is consistent with the results of the CCK-8 assay showing that TG suppressed the viability of ACC cells (Figure 1C and D).
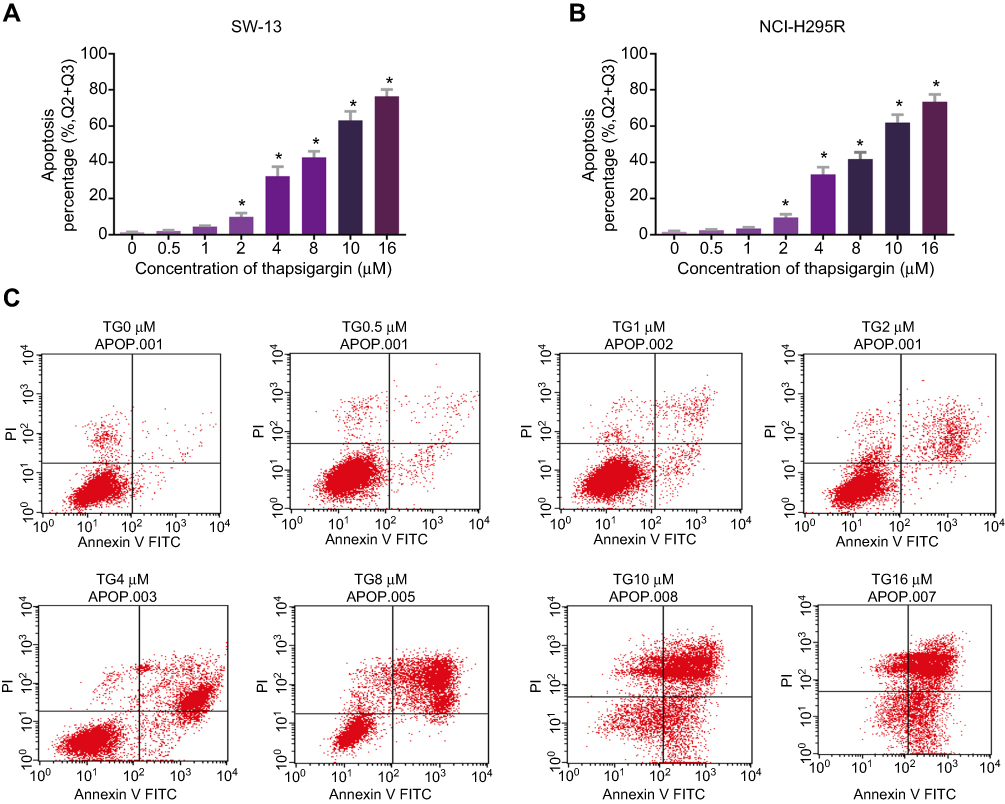 |
Figure 3 TG induced apoptosis of ACC cells. SW-13 (A) and NCI-H295R (B) cells were treated with TG at different concentrations (0, 0.5, 1, 2, 4, 8, 10 and 16 µM) for 48 h. Flow cytometry was used to analyze TG-induced apoptosis of ACC cells. (C) Early and late apoptotic events in SW-13 cells and NCI-H295R cells in the present or absent of different doses of TG were detected by flow cytometry. The top left quadrant indicates non-apoptotic cells; the top right quadrant represents late apoptosis events; the bottom right quadrant represents early apoptosis cells; and the bottom left quadrant represents living cells. Each experiment was performed in triplicate, and the data represent the means ± SD. *p<0.05 vs the control group. |
The Hoechst 33258 staining further confirmed the effect of TG on apoptosis in ACC cells. A large number of apoptotic cells, which were detected based on nuclear morphology changes, such as chromatin condensation and fragmentation, were present following the treatment with TG (Figure 4A), indicating that TG significantly induced apoptosis in the ACC cells. In contrast, the control group cells showed a grated nucleus with almost no apoptotic morphological changes.
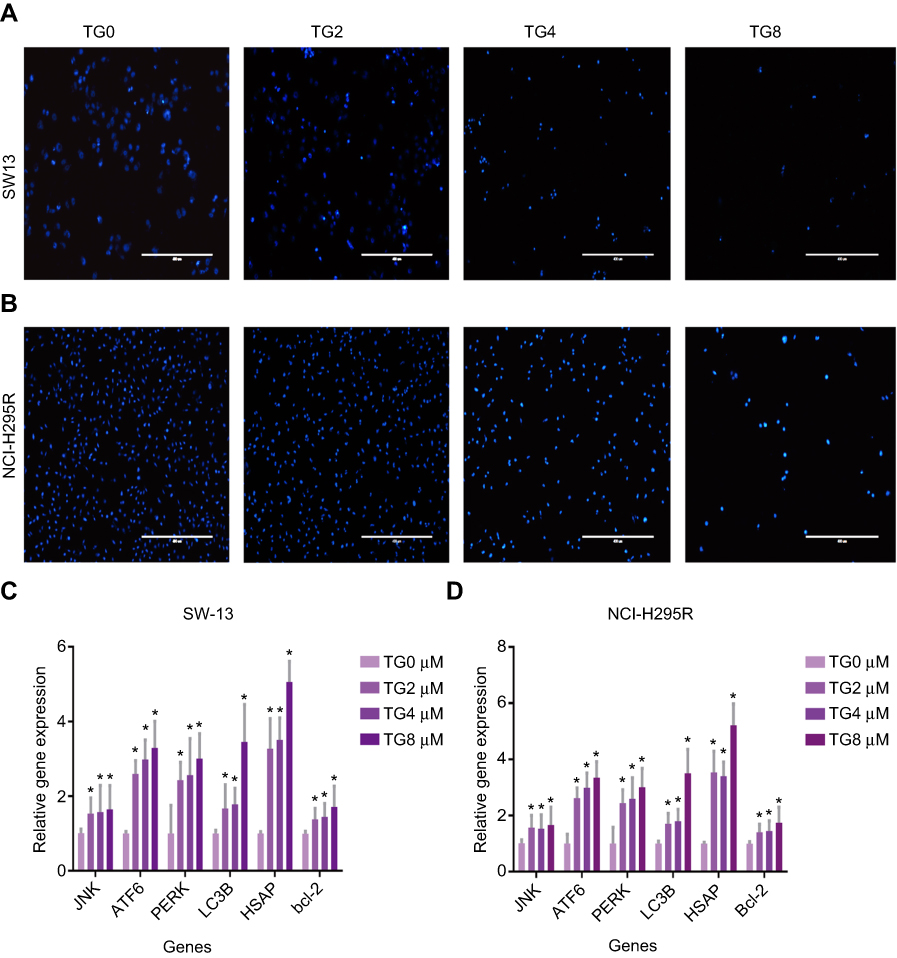 |
Figure 4 TG induced apoptosis of ACC cells by activating ERS and JNK signaling pathways. Apoptosis of cells that induced by TG was stained with Hoechst 33,258 (A and B). The gene expression levels of JNK, ATF6, PERK, LC3B, HSAP and Bcl-2 were detected by qRT-PCR (C and D). The data is represented asmeans ± SD (n=3). *p<0.05 vs the control group. |
TG inhibits cell viability and induces cell apoptosis by activating JNK signaling
To further clarify the molecular mechanism of the anti-viability and apoptosis-inducing effects of TG in ACC cells, the apoptosis-related JNK signaling pathway and endoplasmic reticulum (ER) stress markers were investigated after the TG treatment. The expression levels of JNK signaling-related genes, including JNK, ATF6, PERK, LC3B, and HSAP, were obviously promoted by TG (Figure 4C and D). TG at a concentration of 8 µM exhibited the best performance with respect to the upregulation of the expression levels of the JNK, ATF6, PERK, LC3B and HSAP genes. The apoptosis-associated gene Bcl-2 was also detected by qPCR. Bcl-2 was increased in the TG8 groups compared with that in the TG0 group (P<0.05) (Figure 4C and D). The expression of the JNK and p-JNK proteins was also significantly upregulated by TG (Figure 5A–D).
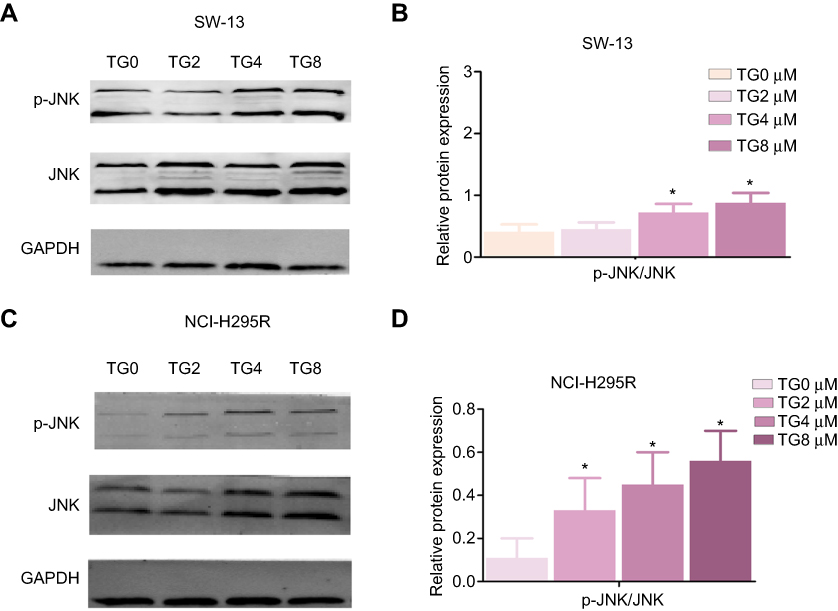 |
Figure 5 Expression levels of the JNK signaling-related proteins, JNK and p-JNK, after TG treatment in SW-13 (A and B) and NCI H295R (C and D) cells. The data is represented as means ± SD (n=3). *p<0.05 vs the control group (TG 0 µM group). |
TG induces cell apoptosis by activating JNK signaling
To further verify the molecular mechanism of JNK signaling in the TG-mediated ER stress-induced apoptosis in ACC cells, the JNK inhibitor SP600125 were used. The rate of apoptosis in the TG-treated cells with JNK inhibitor SP600125 was evaluated by flow cytometry. As shown in Figure 6A–E, the apoptosis rate of the cells and the expression of JNK had not significant difference in the treatment of SP600125. Suggesting the JNK signaling is the main molecular mechanism in the TG-mediated ER stress-induced apoptosis.
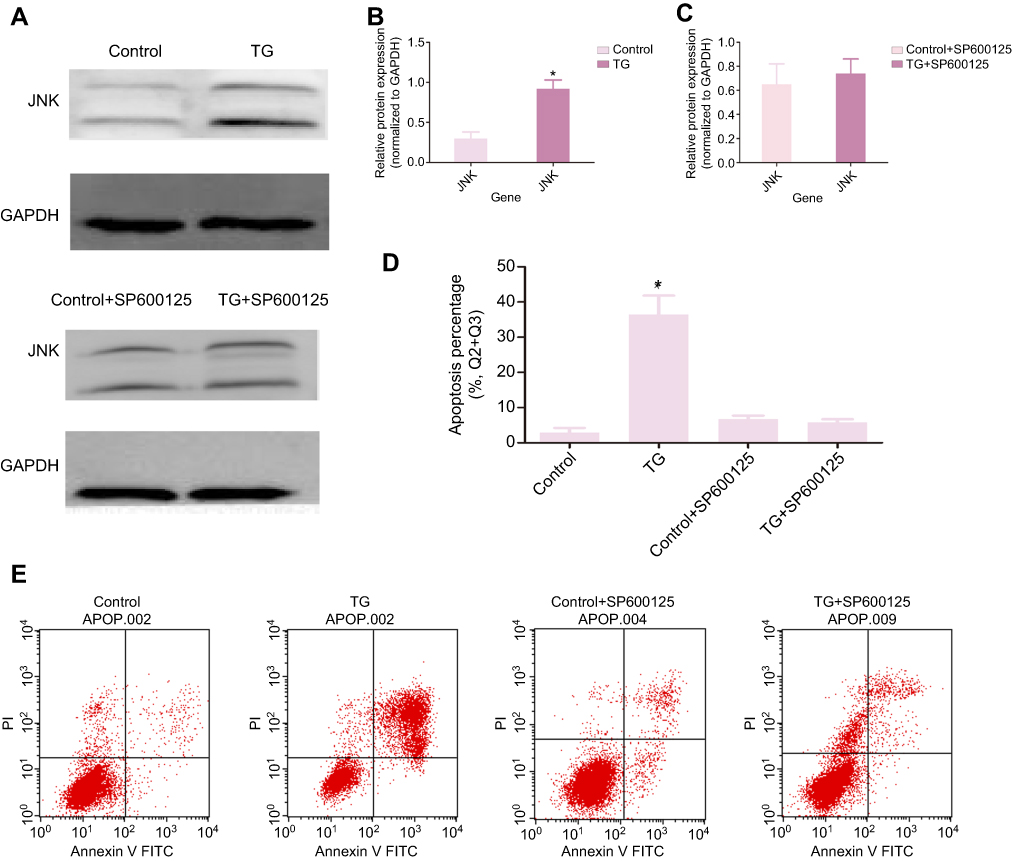 |
Figure 6 TG induces cell apoptosis by activating JNK signaling. SW-13 was treated in the present or absent of TG or JNK inhibitor SP600125 (µM) for 48 h. The expression of JNK and p-JNK was detected by WB (A - C). Flow cytometry was used to analyze the apoptosis rate in ACC (D and E). Each experiment was performed in triplicate and the data is represented as means ± SD. *p<0.05 vs the control group. |
TG inhibited ACC tumor growth in nude mice
To investigate the effects of TG on ACC in vivo, SW-13 xenograft tumors were generated in nude mice. As shown in Figure 7A–F, the treatment with TG suppressed the growth of the SW-13 xenograft tumors in both the tumor volumes and weights. Furthermore, the mouse body weights in the TG group and control group were similar, suggesting that TG did not cause serious toxicity in the mice (Figure 7D).
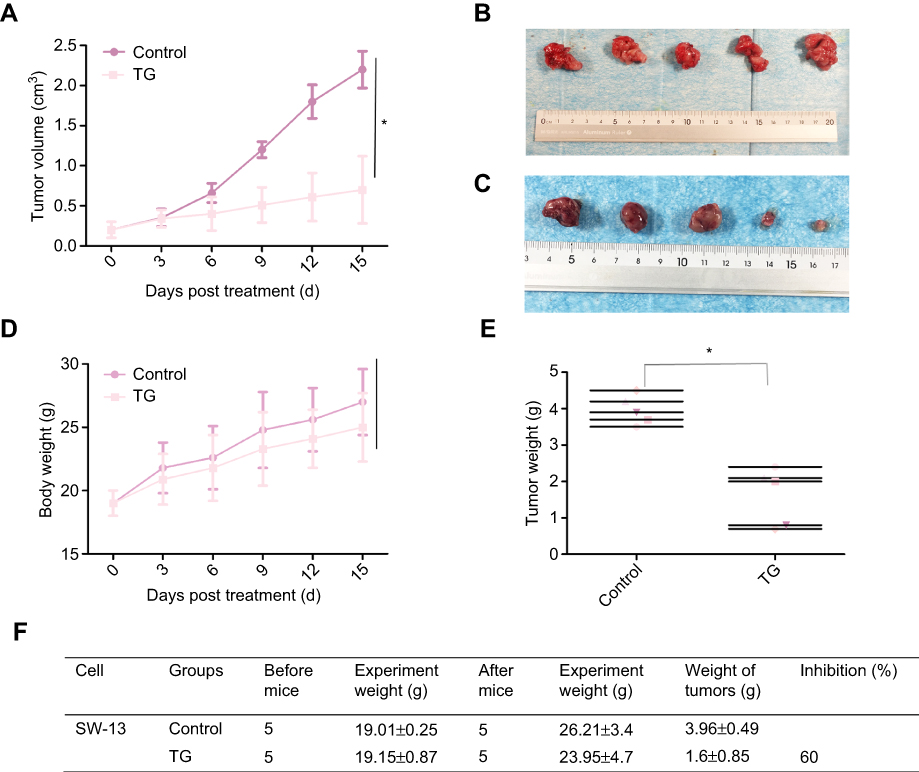 |
Figure 7 TG inhibited ACC tumor growth in nude mice. Each mouse was injected subcutaneously with SW-13 cells (2×106 in 200 µL of medium) under their left shoulder. When the subcutaneous tumors were approximately 0.2×0.2 cm in size, the mice were randomized divided into two groups and received intraperitoneal injections of vehicle alone (0.5% methylcellulose) or TG (1.0 mg/kg) three times a week. Body weights and tumor volumes were recorded every three days. After the experiment, the mice were anesthetized, and the tumor tissues were recieved from the mice and weighed. Tumor volumes (A), original tumors (B and C), body weights (D), tumor weights (E), and summarized data (F) are shown. The data is represented the as means ± SD (n=5). *P<0.05 vs corresponding control. |
JNK signaling in ACC xenografts in nude mice treated with TG
The activation of JNK pathways plays a central role in the proliferation and apoptosis of cancer cells. We investigated whether JNK, ERK1/2, MAPK, PERK, IRE1 and GRP78 signaling pathway modulation accounted for the antitumor activity of TG. As shown in Figure 8, after 14 days of treatment with TG, increases in GRP78, IRE1 and the phosphorylation levels of JNK, ERK, PERK and MAPK were detected in the ACC xenograft tumors, whereas the total JNK, ERK, PERK and MAPK protein levels remained unchanged. These results suggest that the inhibition of tumors by TG is mediated through JNK, ERK and MAPK signaling pathway inactivation in ACC in vivo.
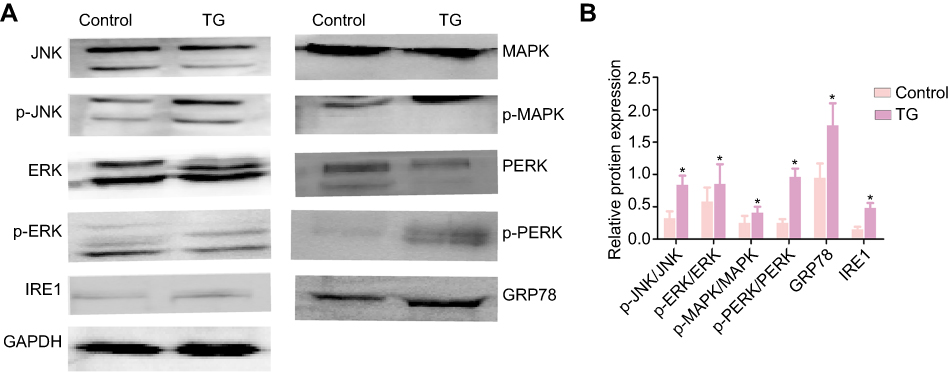 |
Figure 8 Expression of the JNK signaling-related proteins JNK, p-JNK, ERK, p-ERK, MAPK, p-MAPK, PERK, p-PERK, IRE1 and GRP78 in tumers in nude mice that treated with or without TG were analyzed by WB (A and B). The data is represented as means ± SD (n=5). *p<0.05 vs the control group. |
Discussion
ACC is rare, and typically, the tumor is characterized by invasive malignancy with a poor prognosis. To date, surgical treatment of the primary site has remained the mainstay of therapy, but the prognosis is poor. Recent studies investigating the treatment of ACC have mainly focused on exploring effective therapeutic agents. Thapsigargin (TG) is a natural product that has been reported to inhibit the function of the transmembrane portion of the sarcoplasmic/ER calcium adenosine triphosphatase pump, subsequently inducing apoptosis.28,29 TG initiates ER stress via the UPR, which is initiated by 3 ER transmembrane proteins, namely, protein ER kinase (PERK), inositol-requiring enzyme 1 (IRE1) and ATF6.30,31 TG has been considered a promising approach for the treatment of various cancers, such as breast cancer, pancreatic adenocarcinoma and melanoma.30,32
Our results show that TG decreases the proliferation (Figure 1), migration and invasion (Figure 2) of SW-13 and NCI-H295R. TG also inhibited ACC xenograft tumor growth in vivo (Figure 7). In addition, TG promoted apoptosis in ACC cells (Figures 3, 4A and B). Wang et al33 revealed that TG can induce apoptosis in human lung adenocarcinoma cells through cofilin-1 and paxillin, which is consistent with our present study.
The detailed mechanism studies revealed that TG promoted JNK signaling and ERS, thus promoting the downstream genes of pro-apoptosis regulators. JNK signaling-related markers, such as JNK, ATF6, PERK, LC3B, HSAP and Bcl-2, were also enhanced by TG (Figure 4B). TG significantly increased the JNK and p-JNK protein levels in vitro (Figure 5) and the JNK, p-JNK, ERK, p-ERK, MAPK, p-MAPK, PERK, p-PERK, IRE and GRP78 protein levels in vivo (Figure 8). When treated with JNK inhibitor SP600125, the apoptosis rate of the ACC cells had not significant difference (Figure 6). It has been reported that apoptosis mediated by the ERS pathway plays a key role in many diseases.34–36 To date, three important apoptotic pathways related to ER stress have been reported. First, the gene CHOP (C/EBP homologous protein)/GADD153, which regulates cellular bioenergetics and cell-death, is obviously induced by ER stress and plays an important role in ER stress-induced apoptosis.37 The second pathway is the JNK pathway.38 The third pathway involves caspase-12.39,40 Mutations occurring in the JNK signaling pathway (eg, MAP3K1, MAP2K4, and MAP2K7) are implicated in the pathology of breast cancer.41 Hiroaki Konishi et al42 revealed that apoptosis is mediated by JNK activation. JNK, MAPK and P38 are famously known as stress-activated protein kinases because they can be activated by multiple stress stimuli. In addition, the activation of JNK, MAPK and p38 plays an important role in natural compound-induced apoptosis. It has been reported that many anticancer compounds activate MAPK signaling, ultimately leading to cancer cell apoptosis.43 Consistent with these reports, TG can increase the phosphorylation levels of JNK, MAPK and ERK. Furthermore, the levels of the ERS-related protein GRP78 were increased after the treatment with TG. These results suggest that TG regulates apoptosis in ACC by activating the JNK/MAPK/ERK signaling pathway.
In conclusion, this study indicates that TG could play a significant role in the treatment of ACC. Our research demonstrated that TG repressed proliferation and invasion in human ACC cells in vitro and inhibited ACC xenograft growth in vivo. Moreover, TG induced mitochondrial apoptosis and triggered the synergistic cytotoxicity of ERS and apoptosis, potentially through JNK signaling, to treat ACC.
Acknowledgments
The National Natural Science Foundation of China (grant numbers: 81660138 and 81860146); Innovation Project of Guangxi Graduate Education (grant number: YCBZ2018037); and Guangxi Medical and Health Self-Financing Project (grant number: Z20180636).
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Else T, Kim AC, Sabolch A, et al. Adrenocortical carcinoma. Endocr Rev. 2014;35(2):282–326. doi:10.1210/er.2013-1029
2. Bilimoria KY, Shen WT, Elaraj D, et al. Adrenocortical carcinoma in the United States: treatment utilization and prognostic factors. Cancer. 2008;113(11):3130–3136. doi:10.1002/cncr.23886
3. Terzolo M, Baudin AE, Ardito A, et al. Mitotane levels predict the outcome of patients with adrenocortical carcinoma treated adjuvantly following radical resection. Eur J Endocrinol. 2013;169(3):263–270. doi:10.1530/EJE-13-0242
4. Gratian L, Pura J, Dinan M, et al. Treatment patterns and outcomes for patients with adrenocortical carcinoma associated with hospital case volume in the United States. Ann Surg Oncol. 2014;21(11):3509–3514. doi:10.1245/s10434-014-3931-z
5. Sidhu S, Sywak M, Robinson B, Delbridge L. Adrenocortical cancer: recent clinical and molecular advances. Curr Opin Oncol. 2004;16(1):13–18.
6. Libe R, Borget I, Ronchi CL, et al. Prognostic factors in stage III-IV adrenocortical carcinomas (ACC): an European Network for the Study of Adrenal Tumor (ENSAT) study. Ann Oncol. 2015;26(10):2119–2125. doi:10.1093/annonc/mdv329
7. Berruti A, Baudin E, Gelderblom H, et al. Adrenal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(Suppl 7):vii131–vii138. doi:10.1093/annonc/mds231
8. Puglisi S, Perotti P, Cosentini D, et al. Decision-making for adrenocortical carcinoma: surgical, systemic, and endocrine management options. Expert Rev Anticancer Ther. 2018;18(11):1125–1133. doi:10.1080/14737140.2018.1510325
9. Berruti A, Fassnacht M, Haak H, et al. Prognostic role of overt hypercortisolism in completely operated patients with adrenocortical cancer. Eur Urol. 2014;65(4):832–838. doi:10.1016/j.eururo.2013.11.006
10. Leibovitz A, McCombs WM
11. Rainey WE, Bird IM, Mason JI. The NCI-H295 cell line: a pluripotent model for human adrenocortical studies. Mol Cell Endocrinol. 1994;100(1–2):45–50.
12. Andersen TB, Lopez CQ, Manczak T, Martinez K, Simonsen HT. Thapsigargin–from Thapsia L. to mipsagargin. Molecules. 2015;20(4):6113–6127.
13. Xu C, Ma H, Inesi G, Al-Shawi MK, Toyoshima C. Specific structural requirements for the inhibitory effect of thapsigargin on the Ca2+ ATPase SERCA. J Biol Chem. 2004;279(17):17973–17979. doi:10.1074/jbc.M313263200
14. Jakobsen CM, Denmeade SR, Isaacs JT, Gady A, Olsen CE, Christensen SB. Design, synthesis, and pharmacological evaluation of thapsigargin analogues for targeting apoptosis to prostatic cancer cells. J Med Chem. 2001;44(26):4696–4703. doi:10.1021/jm010985a
15. Tao YK, Yu PL, Bai YP, Yan ST, Zhao SP, Zhang GQ. Role of PERK/eIF2alpha/CHOP endoplasmic reticulum stress pathway in oxidized low-density lipoprotein mediated induction of endothelial apoptosis. Biomed Environ Sci. 2016;29(12):868–876. doi:10.3967/bes2016.116
16. Hauck AK, Bernlohr DA. Oxidative stress and lipotoxicity. J Lipid Res. 2016;57(11):1976–1986. doi:10.1194/jlr.R066597
17. Wang X, Eno CO, Altman BJ, et al. ER stress modulates cellular metabolism. Biochem J. 2011;435(1):285–296. doi:10.1042/BJ20101864
18. Hetz C, Martinon F, Rodriguez D, Glimcher LH. The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol Rev. 2011;91(4):1219–1243. doi:10.1152/physrev.00001.2011
19. Werner N, Nickenig G, Laufs U. Pleiotropic effects of HMG-CoA reductase inhibitors. Basic Res Cardiol. 2002;97(2):105–116.
20. Porter KE, Turner NA. Statins and myocardial remodelling: cell and molecular pathways. Expert Rev Mol Med. 2011;13:e22. doi:10.1017/S1462399411001931
21. Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the wingless signaling pathways. Dev Cell. 2004;7(4):491–501. doi:10.1016/j.devcel.2004.08.019
22. Bosch M, Serras F, Martin-Blanco E, Baguna J. JNK signaling pathway required for wound healing in regenerating Drosophila wing imaginal discs. Dev Biol. 2005;280(1):73–86. doi:10.1016/j.ydbio.2005.01.002
23. McEwen DG, Peifer M. Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development. 2005;132(17):3935–3946. doi:10.1242/dev.01949
24. Tamori Y, Bialucha CU, Tian AG, et al. Involvement of Lgl and Mahjong/VprBP in cell competition. PLoS Biol. 2010;8(7):e1000422. doi:10.1371/journal.pbio.1000418
25. Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117(1):117–129. doi:10.1016/s0092-8674(04)00262-4
26. Kolahgar G, Suijkerbuijk SJ, Kucinski I, et al. Cell competition modifies adult stem cell and tissue population dynamics in a JAK-STAT-dependent manner. Dev Cell. 2015;34(3):297–309. doi:10.1016/j.devcel.2015.06.010
27. Yuan ML, Li P, Xing ZH, et al. Inhibition of WEE1 suppresses the tumor growth in laryngeal squamous cell carcinoma. Front Pharmacol. 2018;9:1041. doi:10.3389/fphar.2018.01041
28. Toyoshima C, Nomura H, Sugita Y. Crystal structures of Ca2+-ATPase in various physiological states. Ann N Y Acad Sci. 2003;986:1–8. doi:10.1111/j.1749-6632.2003.tb07131.x
29. Winther AM, Liu H, Sonntag Y, et al. Critical roles of hydrophobicity and orientation of side chains for inactivation of sarcoplasmic reticulum Ca2+-ATPase with thapsigargin and thapsigargin analogs. J Biol Chem. 2010;285(37):28883–28892. doi:10.1074/jbc.M110.136242
30. Rajapaksa G, Nikolos F, Bado I, Clarke R, Gustafsson JA, Thomas C. ERbeta decreases breast cancer cell survival by regulating the IRE1/XBP-1 pathway. Oncogene. 2015;34(31):4130–4141. doi:10.1038/onc.2014.343
31. Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40(3):141–148.
32. Romero-Ramirez L, Cao H, Regalado MP, et al. X box-binding protein 1 regulates angiogenesis in human pancreatic adenocarcinomas. Transl Oncol. 2009;2(1):31–38. doi:10.1593/tlo.08211
33. Wang F, Liu DZ, Xu H, et al. Thapsigargin induces apoptosis by impairing cytoskeleton dynamics in human lung adenocarcinoma cells. The Scientific World Journal. 2014;2014:619050.
34. Omura T, Kaneko M, Okuma Y, et al. A ubiquitin ligase HRD1 promotes the degradation of Pael receptor, a substrate of Parkin. J Neurochem. 2006;99(6):1456–1469. doi:10.1111/j.1471-4159.2006.04155.x
35. Gao B, Lee SM, Chen A, et al. Synoviolin promotes IRE1 ubiquitination and degradation in synovial fibroblasts from mice with collagen-induced arthritis. EMBO Rep. 2008;9(5):480–485. doi:10.1038/embor.2008.37
36. Lei X, Zhang S, Barbour SE, et al. Spontaneous development of endoplasmic reticulum stress that can lead to diabetes mellitus is associated with higher calcium-independent phospholipase A2 expression: a role for regulation by SREBP-1. J Biol Chem. 2010;285(9):6693–6705. doi:10.1074/jbc.M109.084293
37. Wang XZ, Lawson B, Brewer JW, et al. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol Cell Biol. 1996;16(8):4273–4280. doi:10.1128/mcb.16.8.4273
38. Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi:10.1126/science.287.5453.664
39. Nakagawa T, Zhu H, Morishima N, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403(6765):98–103. doi:10.1038/47513
40. Araki E, Oyadomari S, Mori M. Endoplasmic reticulum stress and diabetes mellitus. Internal Med. 2003;42(1):7–14. doi:10.2169/internalmedicine.42.7
41. Nik-Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534(7605):47–54. doi:10.1038/nature17676
42. Konishi H, Fujiya M, Tanaka H, et al. Probiotic-derived ferrichrome inhibits colon cancer progression via JNK-mediated apoptosis. Nat Commun. 2016;7:12365. doi:10.1038/ncomms12365
43. Zikaki K, Aggeli IK, Gaitanaki C, Beis I. Curcumin induces the apoptotic intrinsic pathway via upregulation of reactive oxygen species and JNKs in H9c2 cardiac myoblasts. Apoptosis. 2014;19(6):958–974. doi:10.1007/s10495-014-0979-y
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.